

SUMMARY
The Endoplasmic Reticulum (ER) stress and unfolded protein response are energetically challenging under nutrient stress conditions. However, the regulatory mechanisms that control the energetic demand under nutrient and ER stress are largely unknown. Here we show that ER stress and glucose deprivation stimulate mitochondrial bioenergetics and formation of respiratory supercomplexes (SCs) through the eukaryotic translation initiation factor 2-alpha kinase 3 (PERK). Genetic ablation or pharmacological inhibition of PERK suppresses nutrient and ER stress mediated increases in SC levels and reduces oxidative phosphorylation-dependent ATP production. Conversely, PERK activation augmented respiratory SCs. PERK-eIF2α-ATF4 axis increases the SC assembly factor 1 (SCAF1 or COX7A2L) promoting SCs and enhanced mitochondrial respiration. Remarkably, PERK activation is sufficient to rescue bioenergetic defects caused by complex I missense mutations derived from mitochondrial disease patients. These studies have identified an energetic communication between the ER and mitochondria with implications in cell survival and diseases associated with mitochondrial failures.
Keywords: Mitochondria, Respiratory Chain Supercomplexes, ER stress, Nutrient stress, PERK, Mitochondrial diseases
Graphical Abstract
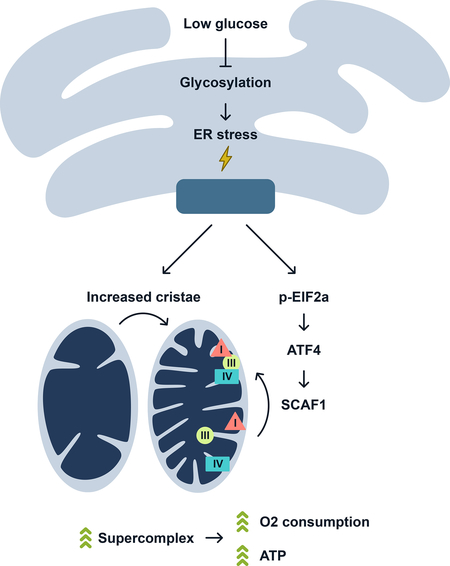
eTOC Blurb
ER and nutrient stress cause mitochondrial energetic demands to maintain cell survival that can be compromised in diseases associated with mitochondrial failures. Balsa, Soustek, et al. use a series of metabolic and proteomic analyses to identify the ER and nutrient stress activated PERK that promotes formation of respiratory supercomplexes through the assembly factor SCAF1.
INTRODUCTION
Cellular ATP can be produced through glycolysis in the cytosol or via oxidative phosphorylation in the mitochondria. Eukaryotic cells exhibit metabolic flexibility and are capable of switching from glycolysis to mitochondrial respiration (Birsoy et al., 2014). While cytosolic ATP production relies mainly on glucose, the mitochondrial oxidative phosphorylation (OXPHOS) system can be fueled by alternative substrates such as fatty acids and amino acids that feed into the Krebs cycle to produce reducing equivalents that drive electron transport (Boroughs and DeBerardinis, 2015). Consequently, cells have evolved to quickly transition between these two conserved energetic pathways to adapt to fluctuations in nutrient availability. Warburg’s effect or aerobic glycolysis describes how many cancer and proliferating cells display a decreased respiration rate and an enhancement lactate production rate in the presence of glucose, in spite of oxygen availability (Vander Heiden et al., 2009). Multiple regulatory events, including HIF-dependent transcription and tumor suppressor/oncogene-driven metabolic rewiring, have been implicated in glucose uptake and glycolysis (Denko, 2008; Jones and Thompson, 2009). Conversely, regulation of mitochondrial oxidative metabolism has been mainly studied at the transcriptional level and encompasses key transcription factors and coactivators such as ERRα, NRF1–2, and PGC1α/β (Scarpulla et al., 2012). Strikingly, non-transcriptional regulation of mitochondrial function has been less investigated and is often ascribed to changes in mitochondrial dynamics or post-translational modifications and activity of key OXPHOS components (Liesa and Shirihai, 2013; Van Vranken et al., 2018).
During starvation or nutrient deprivation the mitochondrial network becomes hyperfused since fission rates decrease, contributing to increased mitochondrial bioenergetic efficiency (Gomes et al., 2011). However, whether this remodeling expands to different components of mitochondrial function such as the electron transport chain (ETC) architecture or cristae morphology has not been fully addressed. To explain ETC architecture, the “fluid model” depicted the organization of the mitochondrial respiratory chain as free complexes (CI, CII, CIII and CIV) moving within the inner mitochondrial membrane with electrons flowing between these complexes through the mobile electron carriers CoQ and cytochrome c. This model has been challenged by the “plasticity model”, where individual complexes co-exist with super-assembled, quaternary structures called supercomplexes (SCs) (Acin-Perez et al., 2008). Studies in multiple mammalian cell lines and tissues using blue native polyacrylamide gel electrophoresis (BN-PAGE) enabled the resolution and visualization of SCs that exist in a wide variety of stoichiometries including I+III2+IV1, I+III2, and III2+IV1–2 (Cogliati et al., 2016a). The assembly of SCs is now widely accepted, however, their functional relevance is under debate and how SCs are regulated is unknown.
Multiple physiological and pathological conditions, including nutrient deprivation, can disrupt protein folding and glycosylation in the ER causing ER stress. To overcome an imbalance in protein folding capacity, eukaryotic cells have evolved a conserved pathway called the unfolded protein response (UPR) aimed to re-establish ER homeostasis (Frakes and Dillin, 2017a). In mammals, three ER membrane-associated proteins act as ER stress sensors: protein kinase R (PKR)-like ER kinase (PERK), inositol-requiring enzyme (IRE1), and activating transcription factor (ATF6) (Senft and Ronai, 2015). These factors initiate an adaptive response to maintain protein folding and ER activity (Frakes and Dillin, 2017b; Walter and Ron, 2011). PERK signaling is initiated in response to ER stress through a mechanism involving PERK dimerization and autophosphorylation of the cytosolic PERK kinase domain. Once activated, PERK phosphorylates eukaryotic translation initiation factor 2 alpha (eIF2α) to decrease overall translation while increasing the specific translation of genes including the activating transcription factor ATF4 (Walter and Ron, 2011). ATF4 induces transcriptional expression of genes involved in amino acid metabolism, redox control and proteostasis. Interestingly, the ATF4 pathway is a hallmark of mitochondrial dysfunction and its activation has been documented both in vitro and in vivo upon induction of mitochondrial stress (Bao et al., 2016).
Communication between the mitochondria and the ER is important for calcium homeostasis, regulation of mitochondrial fission, autophagy, inflammasome formation, and lipid metabolism (Rainbolt et al., 2014). The ER and mitochondria also form physical contact sites termed mitochondria-ER associated membranes (MAMs) and recent studies have revealed the significance of ER-mitochondrial crosstalk in pathophysiological situations (Annunziata et al., 2018). Nevertheless, the metabolic and bioenergetic events taking place after UPR activation remain largely undefined, specifically, how the ER communicates with the OXPHOS system to increase ATP supply and promote protein homeostasis upon episodes of energetic demands. Nutrient stress imposed by glucose deprivation requires a cellular energetic shift from cytosolic glycolysis to mitochondrial OXPHOS in order to maintain survival and growth (Gohil et al., 2010; Rossignol et al., 2004). Experimentally, this shift is modeled by culturing cells in media containing galactose instead of glucose (Barrow et al., 2016). In fact, cells exhibiting mitochondrial bioenergetic defects such as those with mutations derived from mitochondrial disease patients, are vulnerable to cell death under these conditions since they are reliant on glycolysis for energetic and metabolic requirements (Ghelli et al., 2003).
We have identified a novel mechanism whereby the ER communicates with the mitochondria in conditions of nutrient stress. We found that the PERK arm of the UPR coordinate changes in cristae density and respiratory chain SCs assembly to boost oxidative metabolism to meet energetic and metabolic demands when glycolysis is compromised. Importantly, we show that the activation of this pathway poses a promising therapeutic target to combat mitochondrial disorders associated with CI dysfunction.
RESULTS
Glucose deprivation enhances mitochondrial respiration, respiratory chain SCs and cristae density.
Despite the established mitochondrial energetic dependency during nutrient stress and glucose deprivation, the regulatory mechanisms and components that drive mitochondrial respiration under metabolic and energetic stress conditions are largely unknown. Thus, we decided to investigate how cells under glucose deprivation activate mitochondrial respiration to cope with the energetic demands and maintain survival and growth. Consistent with previous studies (MacVicar and Lane, 2014), we observed an increase in respiration in cells cultured for 48 hours under either low glucose (1 mM glucose) or glucose-free (10 mM galactose) media when compared to high glucose (25 mM glucose) conditions (Figure 1A). To determine if this energetic shift in respiration was due to intrinsic changes in mitochondrial function rather than enhanced flux of metabolites, mitochondria were isolated from high glucose or galactose-grown cells and both basal and state 3 respiration were measured. Mitochondria from galactose-cultured cells exhibited increased oxygen consumption driven by pyruvate and malate (complex I substrates), as well as an increase in complex I (CI), combined complex I+III and complex IV (CIV) enzymatic activity. Conversely, oxygen consumption driven by succinate (complex II substrate), complex II (CII) activity and combined complex II+III activity were unchanged (Figures 1B and C). We observed a striking rearrangement of the ETC architecture after galactose challenge, with increased super SCs levels and activity (most notably SC I+III2+IVn). Interestingly, only minor changes on free complexes III2, IV or II (Figures 1D and E) were observed, which is coherent with the specific increase in CI driven respiration. Similar increases in SC levels were also seen in other human and mouse cell lines (Figure S1A), suggesting that increases in SC levels are likely a conserved energetic and metabolic adaptation to glucose deprivation. These respiratory changes occurred independently of transcriptional differences in nuclear or mitochondrial encoded genes or the rate of mitochondrial protein translation (Figures S1B and C). Interestingly, chloramphenicol, a specific inhibitor of mitochondrial protein translation, completely abolished SC levels when cells were grown in glucose; however, SC levels were still maintained in galactose media (Figure S1D). Proteomic analysis in these conditions showed that mitochondrial proteins levels that form part of CI, CIII, CIV and CV were enriched in isolated mitochondria (Figure S1E). These increases in respiration and SCs were accompanied by remodeled mitochondrial ultrastructure with densely packed cristae (Figure 1F) without changes in mitochondrial mass (Figure 1G). Together, these results show that energetic and metabolic demands imposed by nutrient stress are met through elevated mitochondrial respiratory activity including increased cristae formation, OXPHOS proteins and SC levels. Importantly, galactose-induced SCs favor the oxidation of CI substrates over CII, likely due to the strong increase in SC I+III2+IVn levels, indicating that the configuration of the electron transport chain can preferentially dictate nutrient utilization(Guaras et al., 2016). AMP-activated protein kinase (AMPK) is known to promote energy generating processes under conditions of nutrient stress(Hardie et al., 2012). To determine whether AMPK regulates SC formation, we treated cells with the AMPK activator, AICAR, in glucose conditions or the AMPK inhibitor, Compound C, in galactose conditions. Figure S1F shows that neither AMPK activation or inhibition altered SC assembly levels indicating that this kinase does not mediate increases in SC levels in galactose conditions.
Figure 1: Limited glucose availability promotes mitochondrial oxygen consumption, Respiratory Chain Supercomplexes and Cristae density.

U2OS cells grown in glucose or galactose for 48h. Oxygen consumption analysis of (A) intact cells, or (B) isolated mitochondria, using Pyruvate/Malate or Succinate as specific substrates respectively. (C) Mitochondrial enzymatic activities of CI, CIV, CII, CI+III and CII+III normalized to citrate synthase (CS) levels. (D) Upper panel; Immunodetection of the indicated proteins representing CI, CIII, CIV and CII after BNGE of digitonin-solubilized mitochondria from glucose or galactose (48h). Right; Bands were quantified using ImageJ software and fold induction under galactose conditions is represented. Lower panel; Superposition profiles of the different supercomplexes and free complexes using GelEval software. (E) Complex I in-gel activity of digitonin-solubilized mitochondria from 48h glucose or galactose cultured cells. (F) Mitochondrial cristae morphology, abundance and density assayed by electron microscopy (EM) in glucose and galactose-grown U2OS cells at 24h or 48h. (G), Mean fluorescence corresponding to mitochondrial content in cells cultured in glucose or galactose measured by flow cytometry. SDHA was used as a loading control. Immunoblots shown are representative of >3 independent experiments and all other experiments are represented as mean ± s.e.m., n>3. Asterisks denote *p<0.05 or **p<0.01. For two comparisons a two-tailed t-test was used, for multiple comparisons, one-way ANOVA with Bonferroni post-test was applied. gluc/g, glucose. Galac/G, galactose.
ER stress increases mitochondrial respiration
In order to gain further insight into how glucose restricted conditions increase SC amounts and mitochondrial respiration we performed comprehensive metabolomics analysis of intracellular metabolites comparing glucose-rich versus glucose-free cultured U2OS cells. As expected, levels of glycolytic intermediates such as dihydroxyacetone-phosphate and lactate were decreased in cells grown in galactose media (Figures 2A and S1G). Interestingly, we identified UDP-N-acetyl-glucosamine (UDP-GlcNAc) as the most downregulated metabolite after glucose deprivation (Figure S1G). Since UDP-GlcNAc is part of the hexosamine biosynthesis pathway used for protein glycosylation in the ER (Figure 2B), we analyzed changes in both O-linked and N-linked protein glycosylation. Global O-GlcNAcylation was reduced in cells cultured in galactose media as well as N-glycosylation of the ER-resident protein Cathepsin C (Figures 2C, D and S2A). These changes correlated with an increase in ER stress markers in multiple cell lines (Figure 2E and S2B), suggesting that under glucose deprivation, cells face strong energetic demands imposed by the inability to synthesize glycolytic ATP and an increase in chaperone activity to maintain protein folding. Thus, it is conceivable that ER stress may directly drive the increases in respiration and SC formation under glucose deprivation. To test this and uncouple it from nutrient stress, we used low doses of tunicamycin to block protein glycosylation and trigger ER stress in glucose conditions without causing cell death (Figure S2C). Similar to glucose deprivation, tunicamycin activated ER stress (Figure S2D), increased mitochondrial respiration, CI and CIV enzymatic activities, SC levels and activity (notably I+III2+IVn), and cristae density (Figures 2F to I and S2E). We used an additional inducer of ER stress, Brefeldin A, and observed a similar increase in SCs levels compared to tunicamycin treatment (Figure S2F). Collectively, these results indicate that ER stress increases mitochondrial OXPHOS, cristae formation and SCs to maintain energy demands imposed by ER function and protein folding.
Figure 2: ER stress induction leads to an increase in OXPHOS performance.

(A) Volcano plot of global metabolomics analysis comparing galactose vs glucose-grown cells. The most down-regulated metabolite, UDP-N-acetylgucosamine, is highlighted in red. (B) Simplification of the hexosamine pathway. (C) Immunoblot showing total O-linked glycosylation in cells cultured either in glucose or galactose. (D) Immunoblots showing a shift in migration of Cathepsin C due to impaired ER protein glycosylation in U2OS cells. (E) Western blot analysis of ER stress markers: GRP78/BiP, CHOP and ATF4 in U2OS cells cultured in galactose at the indicated time points. (F) Oxygen consumption rates, (G) mitochondrial enzymatic activities of CI, CIV and CII normalized to CS and (H) SC levels in isolated mitochondria from tunicamycin treated cells after 48 hours. (I) Complex I in-gel activity of digitonin (DIG) or DDM-solubilized mitochondria from glucose, galactose and tunicamycin treated cells. SDHA was used as a loading control. Immunoblots shown are representative of >3 independent experiments and all other experiments are represented as mean ± s.e.m., n>3. Asterisks denote *p<0.05 or **p<0.01. For two comparisons a two-tailed t-test was used, for multiple comparisons, one-way ANOVA with Bonferroni post-test was applied. gluc/g, glucose. Galac/G, galactose. Tunica/T, tunicamycin. UDP-GlcNac, UDP-N-acetetylglucosamine. DHAP, dihydroxyacetone-phosphate.
Activation of ER stress sensor PERK enhances OXPHOS capacity and promotes electron transfer chain SC assembly both in vitro and in vivo.
To determine the role of the ER stress sensor proteins in glucose deprivation-induced SC formation we used CRISPR/Cas9 editing technology to deplete X-box-binding protein 1 (Xbp1, which is downstream of IRE1), PERK and ATF6 (Figure S3A). BN-PAGE analysis using cells depleted of these factors showed that only cells with PERK ablation failed to increase SC levels in galactose media (Figure 3A). In addition, genetic disruption of PERK abolished the increase in mitochondrial respiration induced by galactose medium or tunicamycin treatment (Figures 3B and C). Similar results were obtained when PERK kinase activity was inhibited using GSK2606414 (Figures S3B, C and D). PERK inhibition also decreased ATP levels in cells grown in galactose media, which translated into a marked decrease in proliferation (Figures 3D and S3E and F). Conversely, two different small molecule PERK activators, DHBDC and CCT020312, increased SC levels with no apparent increases in free CIV or CII complexes in human U2OS cells (Figures 3E and S3G) and mouse fibroblasts (Figure S3H). PERK activation mediated increases in SCs were accompanied by elevated mitochondrial respiration in cells expressing PERK, but not in PERK depleted cells (Figure 3F). Furthermore, PERK activation also resulted in changes in cristae formation similar to that of tunicamycin and galactose (Figure S3I). Since the Eukaryotic Initiation Factor 2 alpha (eIF2α) is the canonical downstream target of PERK, we next tested whether it mediates the increases in SCs caused by glucose deprivation. A selective inhibitor, ISRIB (Sidrauski et al., 2015), was used to block eIF2α phosphorylation in cells cultured in galactose (Figure S3J). Figure 3G shows that similar to PERK inhibition, ISRIB strongly attenuated increases in SC levels. Additionally, two eIF2α mutants in serine 52 (PERK phosphorylation residue) were further employed to support the role of eIF2α in this process. Cells were transfected with either wildtype (WT), phospho-mimetic (SD) or phospho-deficient (SA) eIF2α and SCs were analyzed. Consistent with PERK activation, only the SD mutant showed an increase in SC levels (Figure S4A). We next examined whether PERK activation in vivo is also sufficient to increase mitochondrial respiration and SC levels. Similar to the effects seen in cultured cells, mice treated with the PERK activator (CCT020312) significantly increased SC levels without changing free CIV and CII levels in liver tissue (Figure 3H). These changes in SCs translated into functional increases in mitochondrial respiration (Figure 3I). Moreover, tunicamycin-injected mice also showed an increase in SC levels (Figure S4B). These results indicate that activation of the PERK/eIF2α axis increases SCs and mitochondrial respiration to maintain the energetic demands imposed by ER and nutrient stress responses.
Figure 3: PERK increases mitochondrial respiration and electron transport chain supercomplexes in vitro and in vivo.

(A) BNGE analysis of CRISPR mediated ablation of ER stress effectors XBP1, PERK and ATF6. (B-C) Oxygen consumption rates of isolated mitochondria from PERK depleted cells cultured in glucose or galactose (B) or treated with tunicamycin (C) for 24h. (D) ATP/ADP ratio of PERK KO (sgPERK) and negative control (sgNeg) cells after 48 hours in galactose. (E) BNGE immunoblot of SC levels in cells treated with PERK activators DHBDC or CCT020312 for 72h. (F) Mitochondrial respiration of mitochondria isolated from control cells or PERK deficient cells after treatment with PERK activators for 72h. (G) BNGE immunoblot after inhibition of eIF2α phosphorylation with ISRIB. (H) SC levels assessed by BNGE and (I) oxygen consumption measurements of isolated mitochondria from mouse livers 4 days post CCT020312 injections. Immunoblots shown are representative of >3 independent experiments using CII (anti-SDHA) as a loading control. All other experiments are represented as mean ± s.e.m., n>3. Asterisks denote *p<0.05 or **p<0.01. For two comparisons a two-tailed t-test was used, for multiple comparisons, one-way ANOVA with Bonferroni post-test was applied. gluc, glucose. Galac, galactose. Tunica, tunicamycin. CCT, CCT020312.
PERK/eIF2α/ATF4 axis transcriptionally controls SCAF1 levels to mediate mitochondrial respiration and electron transfer chain SC formation.
We subsequently investigated how PERK activation increases mitochondrial respiration and SCs. The understanding of the regulatory mechanisms of SC assembly and its components is largely unknown (Chen et al., 2012; Cogliati et al., 2016a; Cogliati et al., 2013). In addition, the contribution of SCs to cellular energetics is currently controversial (Milenkovic et al., 2017). COX7A2L or Supercomplex assembly factor (SCAF1) was identified as a specific protein that mediates the interaction between CIII and CIV (Lapuente-Brun et al., 2013) and thus is crucial for anchoring complexes III and IV in SCs III2+IV and I+III2+IVn (Cogliati et al., 2016a). To determine whether SCAF1 is involved in PERK-mediated SC assembly, we analyzed SCAF1 transcript and protein levels in conditions of PERK activation. Time-course experiments showed that SCAF1, but not its paralog Cox7a2, is transcriptionally upregulated along with ER-stress markers, in cells cultured in galactose (Figure 4A) and with tunicamycin treatment (Figures S4C and D). Concomitant with elevated mRNA levels, SCAF1 protein was induced in galactose media or with tunicamycin treatment (Figures S4E and S4F). In addition, mice injected with tunicamycin also increased SCAF1 mRNA and protein levels in liver samples (Figure S4G). PERK and p-eIF2α inhibition blocked SCAF1 induction in galactose media (Figures 4B and S4H), while PERK activation increased SCAF1 gene expression and protein levels (Figures 4C, S4I and J). To assess the impact of SCAF1 in nutrient stress-induced mitochondrial respiration, we depleted SCAF1 using CRISPR/Cas9 gene editing. Under galactose conditions, cells without SCAF1 exhibited reduced SC levels, mitochondrial oxygen consumption, ATP levels, and impaired proliferation without disrupting mitochondrial cristae morphology (Figures 4D to G and Figure S4K and 5SA). Interestingly, mouse adult fibroblast obtained from C57B/L6 mice that harbor a spontaneous non-functional mutated version of SCAF1 (Lapuente-Brun et al., 2013) strongly induced SCAF1 expression in galactose media over the induction of a mouse fibroblast harboring the wild type version of SCAF1 (Figure S5B). In agreement with the CRISPR/Cas9 ablation of SCAF1, C57B/L6 carrying mutant SCAF1 cells had compromised proliferation in galactose media (Figure S5C). Since ATF4 is downstream of the PERK-eIF2α axis, we tested whether SCAF1 mRNA was controlled by this transcription factor. ATF4 depleted cells failed to induce SCAF1 transcripts under galactose conditions. Furthermore, we detected direct binding of ATF4 to the SCAF1 promoter (Figures S5D and E). Functionally, ATF4 depleted cells phenocopied the bioenergetic defects of SCAF1 null cells, displaying reduced SCs, mitochondrial respiration, ATP levels and compromised proliferation in conditions of glucose deprivation (Figures 4H to K and Figure S5F). To extend our quantitative metabolite analysis we employed LC-MS to profile metabolite levels in ATF4 and SCAF1 depleted cells. Consistent with Figures 4F and 4J, ATP was significantly reduced in both ATF4 and SCAF1 deficient cell lines, indicating that these cells face an energetic decline under galactose conditions (Figure S6A). This decline was also extended to some TCA cycle intermediates including oxaloacetate, was decreased in both depleted cell lines. There are, however, metabolite changes that are specific to ATF4 deficiency (asparagine, glutamine and alpha-ketoglutarate) or SCAF1 deficiency (tryptophan, succinate, succinyl-CoA, isocitrate, citrate, nucleotides) suggesting that other metabolites, in addition to ATP, are likely to contribute to decreases in cell growth. Interestingly, ectopic SCAF1 expression was able to normalize SCs levels and increase proliferation under galactose conditions in ATF4 null cells, but not in cells where PERK activity was inhibited (Figure 4L and S6B).
Figure 4: SCAF1 is transcriptionally controlled by the PERK/eIF2α/ATF4 axis to mediate mitochondrial respiration and electron transfer chain supercomplex formation.

(A) mRNA levels of CHOP, Cox7a2l/SCAF1 or Cox7a2 measured by qPCR in U2OS cells cultured under galactose at the indicated time points. (B) Galactose-induced mRNA expression levels of SCAF1 in cells cultured in the presence of PERK or eIF2α phosphorylation (ISRIB) inhibitors. (C) SCAF1 mRNA levels upon PERK activation with DHBDC or CCT020312. (D) SC levels, (E) mitochondrial respiration, (F) ATP/ADP ratios and (G) cell proliferation in galactose in CRISPR-Cas9 SCAF1 ablated cells. (H) SC levels, (I) mitochondrial respiration, (J) ATP/ADP ratios, and (K) cell proliferation of ATF4 ablated cells cultured in galactose media. (L) SC levels (left) and cell proliferation (right) under galactose, in ATF4 or PERK depleted cells (for 4 days) with or without ectopic overexpression of SCAF1. Immunoblots shown are representative of >3 independent experiments using CII (anti-SDHA) as a loading control. All other experiments are the mean ± s.e.m., n>3. Asterisks denote *p<0.05, **p<0.01 or ***p<0.001. For two comparisons a two-tailed t-test was used, for multiple comparisons, one-way ANOVA with Bonferroni post-test was applied. gluc, glucose. Galac, galactose. CCT, CCT020312.
Mitochondrial cristae are necessary for nutrient and ER stress induced electron transfer chain SC formation.
A tight correlation between increased cristae density and enhanced SC formation is observed. Since mitochondrial cristae are the platforms that house the SCs and have been previously implicated in the assembly and stability of SCs, we tested whether galactose-driven alterations in cristae density are essential for the increase observed in SC levels. Two cell lines with alter mitochondrial cristae were generated by depleting OPA1 or MIC60 using CRISPR technology. These cells displayed a mild decrease in the abundance of I+III2+IVn and III2+IV in glucose conditions but most importantly, OPA1 or MIC60 loss of function led to impaired increase in SCs levels under galactose (Figure 5A and S6C). Moreover, and consistent with ETC disrupted architecture, OPA1 or MIC60 depleted cells showed lower proliferation rates when cultured in glucose-free media (Figure 5A). This indicates that intact cristae structure is required to boost SCs formation in conditions of ER and nutrient stress. Next, we aimed to investigate whether SCAF1 overexpression would be sufficient to restore SC levels in the context of impaired cristae morphology. We observed that cells expressing increased levels of SCAF1 were unable to upregulate SCs and restore proliferation under galactose in OPA1 or MIC60 ablated cells (Figure 5B and S6D), suggesting that SCAF1 upregulation and cristae morphology must be coupled in order to increase SC formation. All together these data indicate that PERK activation increases cristae density and induces SCAF1 levels to promote SCs formation and OXPHOS capacity during ER and nutrient stress conditions.
Figure 5: Nutrient and ER stress induced Supercomplex formation rely on mitochondrial cristae integrity.

(A) OPA1 or MIC60 depleted cells were assessed for SC levels by BNGE (left) and cell proliferation under galactose conditions (right). (B) SCAF1 protein was overexpressed in OPA1 and MIC60 depleted cells. SC levels (left) and cell proliferation (right) was analyzed under galactose conditions. Immunoblots shown are representative of >3 independent experiments using CII (anti-SDHA) as a loading control. All other experiments are the mean ± s.e.m., n>3. Asterisks denote *p<0.05, **p<0.01 or ***p<0.001. For two comparisons a two-tailed t-test was used.
PERK activation partially rescues bioenergetic defects in cells with human Complex I mutations.
The most common human respiratory deficiencies involve CI activity. Some CI mutations decrease the stability of this complex due to missense mutations in structural components or the depletion of assembly factors. Other CI mutations cause the inability to assemble a functional complex, i.e., the lack of a structural subunit due to a non-sense mutation. Since PERK activation led to an increase in stability of CI due to an increase in SC formation (Figure 3E) we hypothesize that PERK activation may be beneficial for mutations that result in a loss of CI stability, but that still allow sufficient CI activity when the complex is formed. Based on the fact that cells with defective mitochondrial OXPHOS function are unable to survive in glucose deprivation conditions (Hao et al., 1999; Robinson et al., 1992) we decided to test whether PERK activation could rescue the mitochondrial bioenergetic defects in cells with CI missense mutations. Human cybrid cells carrying a mutation (3796 A>G, found in adult onset dystonia) in the mitochondrial-encoded protein ND1 or in ND6 (14459 G>A, found in Leber Hereditary Optic Neuropathy) were treated with tunicamycin for 72h. BN-PAGE analysis revealed partial rescue in defective CI (primarily found in SCs) assembly, which correlated with an increase in oxygen consumption (Figures 6A, B and S6E). Moreover, consistent with this data, tunicamycin pretreatment in glucose prior to culturing the cells in galactose also conferred resistance to cell death in both cell lines (Figure 6C). Similar to control cells, we observed restoration of CI into SCs in ND1 and ND6 mutant cells after treatment with the PERK activator (Figures 6D and S6F), and found that treated cells had elevated CI activity, oxygen consumption levels and increased cell survival in galactose media (Figures 4E, F, G and S6G). Similar results were obtained in fibroblasts derived from patients harboring a mutation in ACAD9, a chaperone protein necessary for CI assembly (Figures 6H and I). Since PERK activation was sufficient to rescue galactose-induced cell death in CI mitochondrial missense mutant cells, we next analyzed the role of SCAF1 in this adaptation. We found that knocking out SCAF1 in ND1 mutant cybrid cells partially prevented the full effects of PERK activation in cell survival and CI restoration (Figure 6J and S6H). Interestingly, we observed elevated SCAF1 incorporated in the SCs of cells treated with a PERK activator (Figure S6I). We next tested whether PERK activation could increase SC assembly in mouse ND4 (delA10227) or ND6 (delC13887) non-missense mutant cells where no CI is detected. As predicted, no elevation in defective CI levels or rescue in galactose-induced cell death were observed, indicating that PERK activation requires a residual amount of assembled complex to rescue CI mutations (Figures 6K and S6J). Consistent with these results, PERK activation failed to protect against cell death in Rieske KO and COX10 KO fibroblasts as well as MELAS cybrid cells (mitochondrial encephalomyopathy and stroke-like episodes A3243G-Leucine tRNA) with complete ETC deficiency (Figure S6K). These results indicate that PERK activation can restore SC formation in human CI cells harboring mutations that lead to instability of the complex, thereby increasing mitochondrial function and cell survival.
Figure 6: PERK activation ameliorates the bioenergetic deficiencies cause by human Complex I mutations.

(A) SC levels assessed by BNGE of ND1 and ND6 mutant cybrid cells treated for 72h with tunicamycin. (B) Oxygen consumption measurements in control cybrids and ND1 tunicamycin treated cells. (C) cell survival of ND1 and ND6 cells pretreated for 72h with DMSO or tunicamycinin in glucose then cultured in galactose media an additional 72 hours. (D) SC levels of ND1 and ND6 mutant cybrid cells treated for 72h with CCT020312. (E) Complex I activity and (F) Oxygen consumption levels in control and ND1 cybrids treated with DMSO or CCT020312. (G) Cell survival of ND1 and ND6 cells treated with DHBDC or CCT020312 in galactose for 72h. (H) SC levels and (I) cell survival of patient-derived fibroblast harboring a mutation in ACAD9 treated with DMSO or PERK activators. (J) ND1 SCAF1-negative and SCAF1-positive cell survival in galactose with DMSO or CCT020312 treatment after 72h. (K) Cell survival in galactose of WT, ND4d, or ND6d mouse fibroblast treated with DMSO or CCT020312 for 72h (L) Model depicting how glucose deprivation leads to an increase in mitochondrial ATP generation. During glucose-deprivation, PERK is activated stimulating increases in mitochondrial cristae density. In parallel, the PERK/eIF2α/ATF4 axis transcriptionally increases SCAF1 levels to assist in the formation of CIII and CIV containing supercomplexes. Overall, these molecular changes are aimed to stimulate the OXPHOS system and increase ATP production. Immunoblots shown are representative of >3 independent experiments using CII (anti-SDHA) as a loading control. All other experiments are represented as mean ± s.e.m., n>3. Asterisks denote *p<0.05, **p<0.01 or ***p<0.001. For two comparisons a two-tailed t-test was used, for multiple comparisons, one-way ANOVA with Bonferroni post-test was applied. Tunica, tunicamycin. CCT, CCT020312.
DISCUSSION
Nutrient stress conditions of limited glucose availability force cells to use mitochondrial oxidative metabolism to maintain cellular bioenergetic and metabolic-dependent viability. Under these conditions cells utilize glutaminolysis to drive the TCA cycle and increase ETC activity (Rossignol et al., 2004). Yet, molecular drivers that contribute to this metabolic and energetic rewiring have not been identified. The studies presented here provide a regulatory molecular mechanism of how cells activate bioenergetics during ER and nutrient stress. The ER stress response components, PERK and eIF2α drive mitochondrial energetics through increased cristae density, SC levels, and respiration. Downstream of PERK and eIF2α, the transcription factor ATF4 increases SCAF1, promoting SC assembly and mitochondrial respiration. This protein remodeling of the ETC complexes enhances respiration and ATP generation and metabolic-dependent cell growth and viability (Figure 6L).
We show here that the ER is a nutrient sensing organelle due to its requirement of the glucose-derived hexosamine pathway for ER protein glycosylation. When glucose becomes limiting for glucosamine synthesis and protein glycosylation, the ER launches a metabolic/energetic response. This process, mediated by PERK, does not completely remodel the mitochondrial proteome, but rather efficiently reorganizes the ETC complexes in combination with expansion of the mitochondrial cristae. This mechanism exemplifies a cellular adaptation to efficiently harness energy when nutrients are scarce or ER stress is present to maintain cellular integrity and homeostasis. PERK integrates transcriptional and translational signaling to promote mitochondrial molecular quality control pathways. Our results expand the scope of PERK’s function in fine-tuning mitochondrial function during ER stress showing that it coordinates changes in cristae morphology and ETC architecture to maximize substrate utilization in conditions of limited nutrients. Recent studies showed that ER stress induces dynamic remodeling of mitochondrial morphology by promoting protective stress-induced mitochondrial hyperfusion (Lebeau et al., 2018). Together with our studies, it is tempting to speculate that changes in fusion rates and cristae shape might be connected to SC formation and oxidative metabolism performance. Moreover, maintenance and increased cristae morphology has been shown to mediate antiapoptotic effects by preventing cytochrome c release (Frezza et al., 2006; Scorrano et al., 2002). There are several complexes that control cristae biogenesis, shape and function including OPA1, MICOS and dimers of ATP synthase (Cogliati et al., 2016b). Previous studies failed to observe processing of the inner membrane GTPase OPA1 in conditions of ER stress (Lebeau et al., 2018). We show that PERK promotes cristae formation, however, whether it impacts these cristae remodeling complexes is unknown. Moreover, our data suggest that eIF2α activity plays an important role in controlling oxidative metabolism. In addition to PERK, other kinases have been demonstrated to phosphorylate eIF2 at serine 51 as a result of specific stresses such as amino acid deprivation (GCN2), the presence of dsRNA (PKR) or heme deficiency (HRI) (Taniuchi et al., 2016). Thus, it would be interesting to determine whether activation of these eIF2α kinases can stimulate OXPHOS function to the same extent.
The contribution of SCAF1 and the role of the SCs in respiration efficiency has been proposed (Lapuente-Brun et al., 2013) and reinforced recently in humans (Greggio et al., 2017). Our results support the notion that assembling isolated respiratory complexes into SCs is sufficient to increase mitochondrial respiration efficiency, at least, under ER and nutrient stress conditions. PERK-induced SCs formation produces an energy supply during these conditions, without changing the transcriptional or translational rates of hundreds of nuclear and mitochondrial encoded genes. Noteworthy, SCAF1 KO cells displayed minimal bioenergetic phenotype when culture under glucose-rich conditions. However, detrimental phenotypes manifest in SCAF1 KO cells during glucose deprivation including reduced SCs, mitochondrial respiration, ATP and metabolite levels, and impaired proliferation. This suggest that SCAF1 might be dispensable in conditions where glycolysis is the predominant source of ATP but becomes highly relevant when cells are forced to heavily rely on the OXPHOS system for energy production. Our current knowledge of how SCs biogenesis is regulated and what the biological function of these entities remains somewhat limited; this is partially due to lack of precise genetic and pharmacological tools available to modulate SC formation. The work presented here sheds light on how nutrient and ER stress impact SC formation and function.
Importantly, the PERK-dependent ER-mitochondria interorganelle signaling mechanism rescues bioenergetic defects in patient-derived missense CI mutant cells through increasing the assembly of active respiratory SCs. Noteworthy, ATF4 induction has been observed in animal models of mitochondrial dysfunction (Khan et al., 2017; Kuhl et al., 2017), which might indicate a compensatory mechanism aimed to increase SCs and restore OXPHOS function. Since mammalian CI is primarily found assembled in SCs, we speculate that PERK-induced SC formation helps to stabilize the low, but detectable CI harboring mutated subunits that eventually result in increased steady-state levels and activity. Consistent with our results, OPA1 overexpression was able to ameliorate detrimental phenotypes in mouse models of mitochondrial dysfunction (COX15 and NDUFS4 KO) (Civiletto et al., 2015), highlighting the therapeutic potential of boosting cristae density and SCs levels. These studies suggest that modulation of the components of the ER and nutrient stress pathways, including PERK activation, may be potential therapeutic targets in the future treatment of mitochondrial diseases.
STAR Methods
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pere Puigserver (pere_puigserver@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Treatments.
All cell lines were maintained in DMEM high glucose, 10% FBS and 1% P/S at 37°C with 5% CO2. For galactose experiments, cells were seeded in 6-well plates and grown in DMEM high glucose, 10% FBS and 1% P/S at 37°C and 5% CO2 for 24h to allow cells to adhere. Cells were then washed twice with PBS and media was changed to DMEM with no glucose, supplemented with 4mM glutamine and 10 mM galactose, 10% FBS, and 1% P/S. Cells were incubated in galactose-containing media with media changes daily. Cells were then trypsinized and quantified using a hemocytometer. Transfections for gain- and loss-of-function studies were performed according to the manufacturer’s instructions using the polyfect reagent. PERK activators CCT020312 and DHBDC were used at 1 and 10μ M respectively, while PERK inhibitor GSK and p-eIF2α inhibitor ISRIB were used at 500nM and 200nM respectively. Tunicamycin was used at 25–50nM. Brefeldin A was used at 0.5 ug/ml. AICAR was used at 2.5 mM and Compound C at 10uM. Guide sequences for CRISPR/Cas9 were cloned in the lenti-CRISPR v2 vector and are available in table 1.
Table 1.
List of Oligonucleotides
| Human primers | |
| Ndufs4 F | TGCTCGCAATAACATGCAGTC |
| Ndufs4 R | GATCAGCCGTTGATGCCCAA |
| Uqcrc1 F | GGGAGTGTGGATTGATGTTGG |
| Uqcrc1 R | TGTTCCCTTGAAAGCCAGATG |
| Cox5a F | ATCCAGTCAGTTCGCTGCTAT |
| Cox5a R | CCAGGCATCTATATCTGGCTTG |
| Cox4 F | GAGAAAGTCGAGTTGTATCGCA |
| Cox4 R | GCTTCTGCCACATGATAACGA |
| Cycs F | CTTTGGGCGGAAGACAGGTC |
| Cycs R | TTATTGGCGGCTGTGTAAGAG |
| MT-COI F | TCCCCGCATAAACAACATAAG |
| MT-COI R | CAGTAGCGAGCAGGAGTAGGA |
| MT-COII F | CGACTGGTTCTAGGAATAATGG |
| MT-COII R | CACCGACTACGGCGGACTAA |
| MT-NDI F | TTAGTAGAAGAGCGATGGTGAGAGCTA |
| MT-NDI R | ACGCCATAAAACTCTTCACCAAAG |
| MT-ND2 F | TGGTGGGGATGATGAGGCTA |
| MT-ND2 R | AGCACCACGACCCTACTACT |
| MT-CytB F | GCTCCGCTTTCTCTCCGAG |
| MT-CytB R | TGTCAATCCCATAAGTGCTGTTG |
| CHOP F | GGAAACAGAGTGGTCATTCCC |
| CHOP R | CTGCTTGAGCCGTTCATTCTC |
| Cox7a2 F | CTCGGAGGTAGTTCCGGTTC |
| Cox7a2 R | TCTGCCCAATCTGACGAAGAG |
| SCAF1 F | CACCAACTAAACTGACCTCCG |
| SCAF1 R | GGGCACACCATCAGCTTTCT |
| ChIP primers | |
| PSAT1 F | GTGGAGAAGCACACCCATTT |
| PSAT1 R | GATAGGGTTGATGGCAGGAA |
| SCAF1 F | CGTGTTAGCCAGGATGGTTT |
| SCAF1 R | GGAACAGTGAGGCAGTTGGT |
| Mouse primers | |
| SCAF1 F | GTTTAGCAGTTTCACGCAGAAG |
| SCAF1 R | GGCAAATATGATAGGTGGTGCT |
| ATF4 F | CTCTTGACCACGTTGGATGAC |
| ATF4 R | CAACTTCACTGCCTAGCTCTAAA |
| GRP78 (BIP) F | TTCAGCCAATTATCAGCAAACTCT |
| GRP78 (BIP) R | TTTTCTGATGTATCCTCTTCACCAGT |
| Human guides sequences for CRISPR/Cas9 | |
| sgXBP1 | CCCGTCGGCCGGGTTCGGCG |
| sgPERK | GCAGCCCCTCACCTGCCGCG |
| sgATF6 | CACACAGCTCCCTAATCACG |
| sgATF4 | TCTCTTAGATGATTACCTGG |
| sgSCAF1 | GGTGTGGCAAATATGATAGG |
| sgOPA1 | 5’-ATATGGTTCTCCTTCCATGA |
| sgMIC60 | 5’-CTTGGCTTTGAGAAGGGCAT |
Bacterial strains
Cloning was done using One Shot™ Stbl3™ Chemically Competent E. coli.
METHOD DETAILS
Proteomics protein extraction and protease digestion.
Mitochondrial extracts were combined 1:1 with SDS lysis buffer (4.0 % SDS w/v, 250 mM NaCl, PhosStop phosphatase inhibitors, EDTA free protease inhibitor cocktail and 50 mM HEPES, pH 8.5). Extracts were reduced with 5 mM DTT (57 °C for 30 minutes) and cysteine residues alkylated with iodoacetamide (14 mM) in the dark (45 min). Extracts were purified by methanol-chloroform precipitation and subsequent ice-cold acetone washes. Pellets were resuspended in 8 M urea containing 50 mM HEPES, pH 8.5, and protein concentrations were measured by BCA assay prior to protease digestion. 200 μg of protein extracts were diluted to 4 M urea and digested with LysC in a 1/200 enzyme/protein ratio overnight. Digests were diluted further to a 1.5 M urea concentration and trypsin was added to a final 1/250 enzyme/protein ratio for 6 hours at 37 °C. Digests were acidified with 20 μL of 20% formic acid (FA) to a pH ~2 and subjected to C18 solid-phase extraction (SPE) (50 mg, Sep-Pak, Waters).
Isobaric tag labeling.
Isobaric labeling of peptides was performed using a 6-plex tandem mass tag (TMT) reagents. TMT reagents (5 mg) were dissolved in 250 μl dry acetonitrile (ACN) and 10 μl was added to 100 μg of peptides dissolved in 100 μl of 200 mM HEPES, pH 8.5. After 1 hour (RT), the reaction was quenched by adding 8 μl of 5% hydroxylamine. Labeled peptides were combined, acidified with 20 μL of 20 % FA (pH ~2) and concentrated via C18 SPE on Sep-Pak cartridges (50 mg).
Basic pH reversed-phase HPLC (bpHrp).
TMT labeled peptides were solubilized in buffer A (5% ACN, 10 mM ammonium bicarbonate, pH 8.0) and separated by an Agilent 300 Extend C18 column (5 μm particles, 4.6 mm ID and 220 mm in length). Using an Agilent 1100 binary pump coupled with a degasser and a photodiode array (PDA) detector, a 45 minute linear gradient from 18% to 40% acetonitrile in 10 mM ammonium bicarbonate pH 8 (flow rate of 0.8 mL/min) separated the peptide mixtures into a total of 96 fractions (37 seconds). 96 Fractions were consolidated into 12 samples in a checkerboard manner, acidified with 10 μL of 20% formic acid and vacuum dried. Each sample was re-dissolved in 5% FA, desalted via StageTips, dried via vacuum centrifugation, and reconstituted for LC-MS/MS analysis.
Oribtrap Fusion parameters.
All MS analysis was performed on an Orbitrap Fusion Lumos (Thermo Fischer Scientific) coupled to a Proxeon nLC-1200 ultra high-pressure liquid chromatography (UPLC) pump (Thermo Fisher Scientific). Peptides were separated onto a packed 100 μM inner diameter column containing 0.5 cm of Magic C4 resin (5 μm, 100 Å, Michrom Bioresources) followed by 40 cm of Sepax Technologies GP-C18 resin (1.8 μm, 120 Å) and a gradient consisting of 6–30% (ACN, 0.125% FA) over 125 min at ~450 nl/min. The instrument was operated in data-dependent mode with a 60 s (+/− 7 ppm window) expiration time, with FTMS1 spectra collected at 120,000 resolution with an AGC target of 500,000 and a max injection time of 100 ms. The ten most intense ions were selected for MS/MS and precursors were filtered according to charge state (required > 1 z). Monoisotopic precursor selection was enabled. Isolation width was set at 0.7 m/z. ITMS2 spectra were collected at an AGC of 18,000, max injection time of 120 ms and CID collision energy of 35%. For the FTMS3 acquisition, the Orbitrap was operated at 30,000 resolution with an AGC target of 50,000 and a max injection time of 250 ms and an HCD collision energy of 55%. Synchronous-precursor-selection (SPS) was enabled to include 10 MS2 fragment ions in the FTMS3 spectrum.
Data processing and MS2 spectra assignment.
A compendium of in-house software was used to convert .raw files to mzXML format, as well as to correct monoisotopic m/z measurements and erroneous charge state assignments. Assignment of MS/MS spectra was performed using the Sequest algorithm. A protein sequence database containing Human Uniprot database (downloaded 1½015) as well as known contaminants such as human keratins and reverse protein sequences were appended. Sequest searches were performed using a 20 ppm precursor ion tolerance, requiring trypsin protease specificity, while allowing up to two missed cleavages. TMT tags on peptide N termini/lysine residues (+229.162932 Da) and carbamidomethylation of cysteine residues (+57.02146 Da) were set as static modifications while methionine oxidation (+15.99492 Da) was set as variable modifications. An MS2 spectra assignment false discovery rate (FDR) of less than 1% was achieved by applying the target-decoy database search strategy and filtered using an in-house linear discrimination analysis algorithm with the following peptide ion and MS2 spectra metrics: XCorr, peptide ion mass accuracy, charge state, peptide length and missed-cleavages. Peptides were further filtered a 1% protein-level false discovery rate for the final dataset.
Calculation of TMT reporter ion intensities.
For quantification, a 0.03 m/z (6-plex TMT) window centered on the theoretical m/z value of each reporter ion, with the maximum signal intensity from the theoretical m/z value was recorded. Reporter ion intensities were adjusted based on the overlap of isotopic envelopes of all reporter ions as per manufacturer specifications. Total signal to noise values for all peptides were summed for each TMT channel (300 minimum) and all values were normalized to account for variance in sample handling.
Immunoblot.
Cells were harvested in RIPA buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 0.1% SDS, 140 mM NaCl, 1X protease inhibitor cocktail, 1mM PMSF) and protein concentrations were quantified using the BCA assay. Images were acquired with the Amersham Typhoon 5 Biomolecular Imager from GE Healthcare Life Sciences. Densitometry was performed using ImageJ v1.49 and GelEval 1.37 software.
Gene Expression.
Total RNA was isolated with Trizol. Two micrograms of RNA were used to generate complementary DNA (cDNA) with a High Capacity cDNA Reverse Transcription Kit following the manufacturer’s protocol. For gene expression analysis, cDNA samples were mixed with Sybr Green qPCR mastermix and were analyzed by a CFX 384 Real-Time system (Bio-Rad). All primer sequences are available in table 1.
Chromatin Immunoprecipitation.
A minimum of 1.0×107 glucose or galactose-grown cells were crosslinked with 1% formaldehyde (Sigma) for 10 min followed by quenching with 0.125 M glycine for 5 min. Cells were lysed with ChIP lysis buffer (50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, and Complete protease inhibitors and sonicated in a Diagenode Bioruptor for 5 cycles for 5 min with a duty cycle of 30 s “on” and 30 s “off.” Samples were immunoprecipitated with specific antibodies to anti-ATF4 D4B8 and normal rabbit IgG. ATF4 enriched chromatin was analyze by qPCR.
Oxygen Consumption.
In intact cells: 1.0×105 of the indicated cell type were seeded in an XFE-24 Seahorse plate (Seahorse Biosciences, 102340) and allowed to adhere for 24 hours at 37°C with 5% CO2. Media was then removed, and cells were washed with pre-warmed unbuffered DMEM without bicarbonate (Sigma, D5030) supplemented with 15 mM glucose, 2 mM sodium pyruvate and 1 mM glutamine. After the wash, 600 μL of the same buffer was added and cells were transferred to a 37°C non-CO2 incubator for 1h. The Seahorse 24 optical fluorescent analyzer cartridge was prepared in the interim by adding 5 μM oligomycin, 0.5 μM FCCP, and 2 μM rotenone to each cartridge port. Oxygen consumption rates (pmol/min) were then measured for each treatment condition at 37°C using the Seahorse Bioanalyzer instrument. After measurement, media was removed and 20 μL of RIPA buffer was added and protein concentrations were measured using BCA assay. OCR measurements were then normalized to total protein content.
In isolated mitochondria: To minimize variability between wells, mitochondria were first diluted 10x in cold 1x MAS (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1.0 mM EGTA and 0.2 % (w/v) fatty acid-free BSA, pH 7.2 at 37 °C). Stock substrates 0.5 M malic acid, 0.5 M pyruvic acid or 0.5 M succinate and 0.2mM ADP, were subsequently diluted to the concentration required for plating. Next, while the plate was on ice, 50 μL of mitochondrial suspension (containing 25 μg of mitochondria) was delivered to each well (except for background correction wells). The XF24 cell culture microplate was then transferred to a centrifuge equipped with a swinging bucket microplate adaptor, and spun at 2,000 g for 20 minutes at 4 °C. The Seahorse 24 optical fluorescent analyzer cartridge was prepared in the interim by adding 4mM ADP and 10 μM rotenone to each cartridge port.
After centrifugation, 450 μL of prewarmed (37 °C) 1x MAS + substrates Pyruvate/malate (10mM/2mM) or succinate (10mM) were added to each well. In the case of succinate driven respiration, 100 μM Rotenone was also added to the MAS buffer. The mitochondria were viewed briefly under a microscope at 20x to ensure consistent adherence to the well. The plate was then transferred to the Seahorse XFe/XF24 Analyzer, and the experiment initiated.
Metabolomics.
5.0×106 U2OS cells were seeded in a 10 cm plate (Corning) and cultured either in glucose or galactose medium for 24h. Following incubation, cells were harvested on dry ice with 1 mL pre-chilled 80% HPLC-grade methanol. The cell mixture was incubated for 15 min on dry ice prior to centrifugation at 18,000 × g for 10 min at 4°C. The supernatant was retained and the remaining cell pellet was resuspended in 800 μL chilled 80% methanol and centrifuged. Supernatant was combined with the previous retention and was lyophilized using a SpeedVac. Lyophilized samples were resuspended in 20 μL ultrapure water and subjected to metabolomics profiling using the AB/SCIEX 5500 QTRAP triple quadrupole instrument.
Blue native gel electrophoresis (BNGE) and In Gel activity.
Digitonin (DIG) at 4 g/g mitochondrial protein or n-Dodecyl β-D-maltoside (DDM) at a final concentration 1% was used to solubilize the electron transfer chain complexes and 50 μg were applied and run on pre-cast 3%–12% gradient BN gel according to manufacturer’s instructions. After electrophoresis, the complexes were electroblotted onto PVDF membranes and sequentially probed with complex specific antibodies. To assess complex I in gel activity, the gel was incubated with 0.14 mM NADH and 1 mg/ml nitro blue tetrazolium in 100 mM Tris–HCl (pH 7.4) at 378C until the color developed.
Complex Activities.
CIV activity was measured spectrophotometrically with 10 μg of isolated mitochondria. 1 mg/ml of reduced cytochrome C (Sigma, C2506) was added and the decrease in absorbance at 550 nm was measured for 3 minutes at 37°C in a 96-well plate. Rotenone sensitive NADH-dehydrogenase activity (CI activity) was measured at 340 nm (ε= 6.22 mM-1cm-1) in a mix containing buffer CI/CII (25 mM K2HPO4 pH 7.2, 5 mM MgCl2, 3 mM KCN, 2.5 mg/ml BSA), 0.13 mM NADH, 0.13 mM UQ1, and 0.2 μg/ml antimycin A. Rotenone sensitivity was measured under the same conditions with the addition of 5 μM rotenone. For complex II measurements, succinate dehydrogenase activity (CII activity) was measured using 100 μg of isolated mitochondrial resuspended in 950 μL CI/CII buffer (25 mM K2HPO4 pH 7.2, 5 mM MgCl2, 3 mM KCN, 2.5 mg/ml BSA, 10 mM succinate, 0.03 mM DCPIP, 2 μg/ml antimycin A and 5 μM rotenone). Isolated mitochondria were incubated for 10 min at 37°C. The reaction was initiated by adding 15 μL of 10 mM ubiquinone, and the decrease in absorbance at 600 nm was measured for 4 minutes at 37°C in a 96 well plate spectrophotometrically. CI+III activity was measured at 550nM incubating mitochondria in buffer containing 25 mM K2HPO4 pH 7.2, 5 mM MgCl2, 0.5 mM, 2.5 mg/ml BSA, 1mg/ml oxydized cytochrome C and 1mg/ml NADH. For CII+III a buffer containing 25 mM K2HPO4 pH 7.2, 5 mM MgCl2, 0.5 mM KCN, 5 μM Rotenone 2.5 mg/ml BSA, 1mg/ml oxidized cytochrome C and 1mg/ml Succinate, was used.
Lastly, Citrate synthase (CS) activity was measured resuspending 10 μg of isolated mitochondrial in buffer containing 10 Tris (100 mM, pH 8.0), 10 μM of DTNB and Ac CoA (30 μM). Reaction was started by adding 50 μl of 10 mM oxaloacetic acid and the increase in absorbance at 412 nm was monitored for 3 min.
Mitochondrial cristae morphology.
Cells were cultured on coverslips for the indicated amount of time (24h or 48h) and were then fixed for 1hr (.5% Glutaraldehyde 2% Paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4), postfixed in 1% Osmium tetroxide (OsO4)/1.5% Potassiumferrocyanide (KFeCN6) at room temperature for 30 min. Samples were then washed in water 3x and incubated in 1% aqueous uranyl acetate for 30min followed by 2 washes in water and subsequent dehydration in grades of alcohol (5min each; 50%, 70%, 95%, 2× 100%). Cells were embedded in plastic by inverting a gelatin capsule filled with Epon/Araldite on top of the coverslip and polymerized at 60 degrees C for 24hrs. Ultrathin sections (about 60nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids stained with lead citrate and examined in a JEOL 1200EX Transmission electron microscope or a TecnaiG² Spirit BioTWIN. Images were recorded with an AMT 2k CCD camera. For quantification mitochondria were categorized based on cristae abundance ranging from low to high. For morphometric analysis of mitochondrial cristae: cristae width and density were measured using the Image J Multimeasure plug-in.
ATP/ADP levels.
ATP and ADP levels were measured in a 96 well plate (Corning, 3603) using ADP/ATP Ratio Bioluminescence Assay Kit, ApoSENSOR kit according to manufacturer’s instructions (BioVision K255–200)
Mitochondrial mass.
To determine mitochondrial mass MitoTracker™ Green was used according to the manufacturer’s instructions.
Cell death quantification.
Propidium iodide cell exclusion assays were carried out as a measure of cell death. Cells were incubated with propidium iodide (PI) and analyzed by the Flow Cytometry. Sub-G1 peak was selected as indicative of apoptosis.
PERK activator and Tunicamycin preparation for in vivo experiments.
Five milligrams of compound CCT020312 was first dissolved in 100 μl of sterile DMSO and then diluted with sterile saline (0.9% m/v NaCl) to achieve a stock solution of 0.5 mg/ml. Mice (129SVE strain) received i.p. injections of vehicle or 4mg/kg/day CCT020312 for 4 days for PERK activation experiments. For ER stress induction, mice received i.p injections of either 0.5ug/kg/day tunicamycin or dextrose (vehicle) for 2 days. Mice were euthanized, and liver samples were collected. All animal studies were approved in accordance with the guidelines set forth by Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee (IACUC).
Mitochondrial Translation.
U2OS cells cultured in glucose or galactose for 24h were washed in PBS and incubated for 30 min in 1mL of labeling medium (10% dFBS, 1mM sodium pyruvate, and 50 mg/mL uridine in DMEM without methionine/cysteine). Emetine was added to a final concentration of 200 mg/mL, and cells were incubated for 5 min before addition of 50 mCi 35S-labeled methionine/ cysteine mixture and incubation for 1 hr. Cells were recovered and washed twice in PBS before lysis and protein quantification. 50 mg of total protein was loaded on a 10%– 20% SDS-PAGE. In all experiments, cells treated with chloramphenicol (50 mg/mL) was used as a negative control to ensure the mitochondrial origin of the 35S signal. The protein identification associated with each band is proposed based on their relative abundance and molecular weight as determined by prior studies.(Fernandez-Silva et al., 2007)
Statistical Analysis
All statistics are described in Figure legends. In general, for two experimental comparisons, a two-tailed unpaired Student’s t-test was used. For multiple comparisons, one-way ANOVA with Bonferroni post-test was applied. Three replicates per treatment were chosen as an initial sample size unless otherwise stated in the Figure legends. All western blot analysis were repeated at least three times. Statistical significance is represented by asterisks corresponding to *P < 0.05, **P < 0.01 or ***P < 0.001.
Supplementary Material
Table S1. Proteomics data related to Figure 1.
Table S2. Metabolomics data related to Figure 2.
Table S3. Metabolomics data related to Figure 6.
Highlights.
ER and nutrient stress increase mitochondrial oxidative phosphorylation
PERK increases formation of mitochondrial cristae and respiratory supercomplexes
PERK/ATF4 promotes assembly of respiratory supercomplexes through SCAF1
PERK activation rescues bioenergetic failures in human complex 1 mutant cells
ACKNOWLEDGMENTS
We thank all the members of the Puigserver lab for discussions regarding this project. ND6-mutated LHON cybrids were a generous gift from Carlos Moraes at the University of Miami whereas ND1 mutant cybrid were kindly obtained from Rutger Vogel at Radboud Center for Mitochondrial Medicine, Netherlands. ACAD9-mutated patient derived fibroblasts were obtained from Michio Hirano at Columbia University. The SCAF1 expressing vector was a kind gift from Jose Antonio Enriquez. We would like to thank Maria Ericsson and Kyle Smith at Harvard Medical School EM Facility and BIDMC Electron Microscopy (EM) Core respectively for technical assistance analyzing electron microscopy images. This work was supported by the National Institute of Health, Grants RO1 CA181217 NCI and RO1 GM121452 NIGMS to P.P. J.A.E. was funded through MINECO: SAF2015–65633-R; SAF2015–71521-REDC; CNIC is supported by MINECO and Pro-CNIC Foundation and is a SO-MINECO (award SEV-2015–0505). E.B. was supported in part by an EMBO postdoctoral fellowship and MDA Development Grant. M.S.S. was supported by 5 F32 DK105679–03.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
Additional unprocessed datasets have been deposited on Mendeley: http://dx.doi.org/10.17632/7259h7tv8p.1
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, and Enriquez JA (2008). Respiratory active mitochondrial supercomplexes. Mol Cell 32, 529–539. [DOI] [PubMed] [Google Scholar]
- Annunziata I, Sano R, and d’Azzo A (2018). Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis 9, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, et al. (2016). Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow JJ, Balsa E, Verdeguer F, Tavares CD, Soustek MS, Hollingsworth L.R.t., Jedrychowski M, Vogel R, Paulo JA, Smeitink J, et al. (2016). Bromodomain Inhibitors Correct Bioenergetic Deficiency Caused by Mitochondrial Disease Complex I Mutations. Mol Cell 64, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, and Sabatini DM (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs LK, and DeBerardinis RJ (2015). Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Taylor EB, Dephoure N, Heo JM, Tonhato A, Papandreou I, Nath N, Denko NC, Gygi SP, and Rutter J (2012). Identification of a protein mediating respiratory supercomplex stability. Cell Metab 15, 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, and Zeviani M (2015). Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab 21, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Calvo E, Loureiro M, Guaras AM, Nieto-Arellano R, Garcia-Poyatos C, Ezkurdia I, Mercader N, Vazquez J, and Enriquez JA (2016a). Mechanism of super-assembly of respiratory complexes III and IV. Nature 539, 579–582. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Enriquez JA, and Scorrano L (2016b). Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem Sci 41, 261–273. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko NC (2008). Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8, 705–713. [DOI] [PubMed] [Google Scholar]
- Fernandez-Silva P, Acin-Perez R, Fernandez-Vizarra E, Perez-Martos A, and Enriquez JA (2007). In vivo and in organello analyses of mitochondrial translation. Methods Cell Biol 80, 571–588. [DOI] [PubMed] [Google Scholar]
- Frakes AE, and Dillin A (2017a). The UPR(ER): Sensor and Coordinator of Organismal Homeostasis. Mol Cell 66, 761–771. [DOI] [PubMed] [Google Scholar]
- Frakes AE, and Dillin A (2017b). The UPRER: Sensor and Coordinator of Organismal Homeostasis. Mol Cell 66, 761–771. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. [DOI] [PubMed] [Google Scholar]
- Ghelli A, Zanna C, Porcelli AM, Schapira AH, Martinuzzi A, Carelli V, and Rugolo M (2003). Leber’s hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J Biol Chem 278, 4145–4150. [DOI] [PubMed] [Google Scholar]
- Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, et al. (2010). Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol 28, 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, and Scorrano L (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio C, Jha P, Kulkarni SS, Lagarrigue S, Broskey NT, Boutant M, Wang X, Conde Alonso S, Ofori E, Auwerx J, et al. (2017). Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab 25, 301–311. [DOI] [PubMed] [Google Scholar]
- Guaras A, Perales-Clemente E, Calvo E, Acin-Perez R, Loureiro-Lopez M, Pujol C, Martinez-Carrascoso I, Nunez E, Garcia-Marques F, Rodriguez-Hernandez MA, et al. (2016). The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep 15, 197–209. [DOI] [PubMed] [Google Scholar]
- Hao H, Morrison LE, and Moraes CT (1999). Suppression of a mitochondrial tRNA gene mutation phenotype associated with changes in the nuclear background. Hum Mol Genet 8, 1117–1124. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, and Hawley SA (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, and Thompson CB (2009). Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 23, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivela R, Pessia A, Velagapudi V, and Suomalainen A (2017). mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab 26, 419–428 e415. [DOI] [PubMed] [Google Scholar]
- Kuhl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A, and Larsson NG (2017). Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, Latorre-Pellicer A, Colas C, Balsa E, Perales-Clemente E, Quiros PM, Calvo E, Rodriguez-Hernandez MA, et al. (2013). Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570. [DOI] [PubMed] [Google Scholar]
- Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC, and Wiseman RL (2018). The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep 22, 2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, and Shirihai OS (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar TD, and Lane JD (2014). Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci 127, 2313–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic D, Blaza JN, Larsson NG, and Hirst J (2017). The Enigma of the Respiratory Chain Supercomplex. Cell Metab 25, 765–776. [DOI] [PubMed] [Google Scholar]
- Rainbolt TK, Saunders JM, and Wiseman RL (2014). Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab 25, 528–537. [DOI] [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, and Wallace DC (1992). Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem Med Metab Biol 48, 122–126. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, and Capaldi RA (2004). Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64, 985–993. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, and Kelly DP (2012). Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, and Korsmeyer SJ (2002). A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2, 55–67. [DOI] [PubMed] [Google Scholar]
- Senft D, and Ronai ZA (2015). UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, McGeachy AM, Ingolia NT, and Walter P (2015). The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi S, Miyake M, Tsugawa K, Oyadomari M, and Oyadomari S (2016). Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci Rep 6, 32886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vranken JG, Nowinski SM, Clowers KJ, Jeong MY, Ouyang Y, Berg JA, Gygi JP, Gygi SP, Winge DR, and Rutter J (2018). ACP Acylation Is an Acetyl-CoA-Dependent Modification Required for Electron Transport Chain Assembly. Mol Cell 71, 567–580 e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Proteomics data related to Figure 1.
Table S2. Metabolomics data related to Figure 2.
Table S3. Metabolomics data related to Figure 6.