

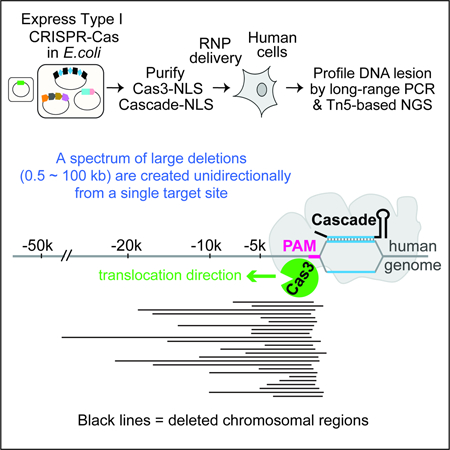
SUMMARY
CRISPR-Cas systems enable microbial adaptive immunity and provide eukaryotic genome-editing tools. These tools employ a single effector enzyme of Type II or V CRISPR to generate RNA-guided, precise genome breaks. Here we demonstrate the feasibility of using Type I CRISPR-Cas to effectively introduce a spectrum of long-range chromosomal deletions with a single RNA guide in human embryonic stem cells and HAP1 cells. Type I CRISPR systems rely on the multi-subunit ribonucleoprotein (RNP) complex Cascade to identify DNA targets, and the helicase-nuclease enzyme Cas3 to degrade DNA processively. With RNP delivery of T. fusca Cascade and Cas3, we obtained 13%−60% editing efficiency. Long-range PCR- and high-throughput sequencing- based lesion analyses reveal that a variety of deletions, ranging from a few hundred base-pairs to 100 kilobases, are created upstream of the target site. These results highlight the potential utility of Type I CRISPR-Cas for long-range genome manipulations and deletion screens in eukaryotes.
eTOC
Dolan et al. demonstrate that T. fusca Type I CRISPR-Cas can generate a spectrum of large genome deletions in human cells. Cascade and Cas3 together induce heterogeneous DNA lesions upstream of a single CRISPR-targeted site, highlighting their potential utilities for long-range genome manipulation and deletion screen.
Graphical Abstract
INTRODUCTION
The majority of prokaryotes rely on CRISPR-Cas systems to establish adaptive immunity against foreign genetic elements (Barrangou et al., 2007; Bolotin et al., 2005; Makarova et al., 2006; Marraffini and Sontheimer, 2008; Mojica et al., 2005; Pourcel et al., 2005). The diverse set of CRISPR-Cas systems rely on the basic principle of CRISPR RNA-guided nucleic acid destruction facilitated by one or more CRISPR-associated (Cas) proteins. CRISPR-Cas systems can be divided into two major classes: Class 1 systems utilize a multi-subunit effector complex to search and destroy nucleic acid targets, whereas Class 2 systems use a singleprotein effector; each Class is further classified into at least three Types, based on the cas operon composition (Makarova and Koonin, 2015; Shmakov et al., 2015). The utilization of Cas9 and Cas12 (Class 2, Type II and V effectors, respectively) for RNA-guided eukaryotic genome editing has revolutionized biomedical research and precision medicine (Cong et al., 2013; Jinek et al., 2012; Knott and Doudna, 2018; Mali et al., 2013; Zetsche et al., 2015). Directed by the guide RNA, these enzymes introduce a DNA double-strand break (DSB) at the targeted site, which is typically repaired via the error-prone non-homologous end joining (NHEJ) pathway, leading to nucleotide insertion/deletions (indels) and gene disruption. Homology-directed repair (HDR) can also occur when a DNA repair template is present, leading to gene conversion and knock-in. A wide variety of Class 2 CRISPR-based tools have been developed for gene regulation, high-throughput genetic screening, epigenome modification, and programmable base editing, etc (Knott and Doudna, 2018; Komor et al., 2017).
Class 2 systems are far less abundant than Class 1 in nature, accounting for only ~10% of all CRISPR systems identified in sequenced microbial genomes (Makarova et al., 2015). The most widespread and diverse form of CRISPR-Cas is the Class 1 Type I system, which employs a very different interference mechanism from that of Cas9. Rather than introducing a single DSB at the targeted site, Type I systems shred the DNA target processively through a multi-step process. First, a multi-subunit ribonucleoprotein (RNP) called Cascade (CRISPR associated complex for antiviral defense) uses a CRISPR RNA (crRNA) to recognize a complimentary target flanked by a 5’ protospacer adjacent motif (PAM) (Brouns et al., 2008; Westra et al., 2012; Wiedenheft et al., 2011). This results in stable R-loop formation and triggers a large conformational change in Cascade (Hochstrasser et al., 2014; Huo et al., 2014; Wiedenheft et al., 2011; Xiao et al., 2017). The helicase-nuclease fusion enzyme Cas3 is then specifically recruited to the R-loop-forming Cascade, nicks the non-target strand (NTS) DNA, and processively degrades its upstream region (PAM-proximal side) (Hochstrasser et al., 2014; Mulepati and Bailey, 2013; Sinkunas et al., 2013). Cas3 further degrades the target strand (TS) DNA, although the detailed mechanism remains unclear (Kunne et al., 2016; Mulepati and Bailey, 2013; Sinkunas et al., 2013). Among different subtypes I-A through I-G, the best-understood are the Type I-E systems from E. coli (Brouns et al., 2008; Hochstrasser et al.,2014; Jackson et al., 2014; Mulepati and Bailey, 2013; Mulepati et al., 2014; Rutkauskas et al., 2015; Sashital et al., 2012; Sinkunas et al., 2013; Westra et al., 2012; Wiedenheft et al., 2011; Zhao et al., 2014) and Thermobifida fusca (Tfu) (Dillard et al., 2018; Hayes et al., 2016; Huo et al., 2014; Xiao et al., 2018; Xiao et al., 2017).
So far, all CRISPR-based eukaryotic gene editing tools were harnessed from single effectors of Class 2 systems. The more prevalent and sophisticated Class 1 CRISPR systems have only been exploited for applications in bacteria and archaea. These include the use of Cascade from Type I systems for programmable transcription repression of bacterial genes (Luo et al., 2015; Rath et al., 2015), and the adaptation of Cascade and Cas3 for the selective targeting of plasmids or E. coli genomic material for degradation (Caliando and Voigt, 2015). Moreover, the endogenous Type I and III systems of Sulfolobus islandicus have been repurposed for counter selection of HDR-mediated editing events in the native host (Li et al., 2016). In this study, we explored the feasibility of achieving RNA-guided genome editing in human embryonic stem cells (hESCs) using Type I CRISPR-Cas. Guided by structural and mechanistic insights from previous studies, we were able to achieve 13% gene editing in hESCs and 30–60% editing in HAP1 cells, through transient delivery of purified T. fusca Type I-E RNP complexes. Both the Cas3 enzyme and a cognate Cascade are required to achieve gene disruption. Strikingly, genome targeting by Cascade and Cas3 led to a spectrum of long-range chromosomal deletions, ranging from a few hundred nucleotides to over 50 kb, at regions upstream of a single-CRISPR-targeted site. The ability of Type I CRISPR to edit long regions of the human genome and the unique repair outcome are not attainable by the current gene-editing platforms (e.g. Cas9 and Cas12), therefore carry the promise for practical usage in long-range genome manipulations and deletion screens in eukaryotes.
RESULTS
Design of T. fusca Type I-E CRISPR-Cas for Genome Editing in hESCs
We chose the T. fusca Type I-E system to develop eukaryotic genome editing tools (Fig. 1A) for its clearly defined mechanisms and the highly active Cas3 nuclease. Several modifications were introduced to adapt this system for potential use in hESCs. First, the optimal growth temperature for T. fusca is 55 °C and R-loop formation by TfuCascade exhibits a strong temperature dependency (Xiao et al., 2017), which presents a potential technical hurdle for its adoption for mammalian use. Although robust in vivo interference activity was observed at 37 °C from T. fusca Type I-E CRISPR system functioning inside the E. coli cells (Huo et al., 2014), as a precaution, we screened a number of structure-guided mutations aimed at weakening the thermostability features of TfuCascade using in vitro approaches. TfuCascade bearing an N23A mutation in the Cse2 subunit (Xiao et al., 2017) was found to be more specific in DNA-binding and equally efficient in R-loop formation at mesothermic temperature (Fig. S1A). More importantly, this same mutant was more efficient in recruiting TfuCas3 for DNA nicking and degradation at 37 °C (Fig. S1B).
Figure 1. Type I CRISPR-Cas can enable RNA-guided genome editing in human ES cells.

(A) Schematic diagram of the T. fusca Type I-E CRISPR-cas locus. Black rectangles and blue diamonds represent CRISPR repeats and spacers; colored boxes, cas genes. (B) Procedure of the genome editing experiments in hESCs. (C) Schematic of the hESC dual-reporter line bearing EGFP and tdTomato at the DNMT3B locus. Protospacers for the three reporter-targeting crRNAs are indicated in blue, and corresponding PAMs in magenta. (D-F) Flow cytometry analysis of the dual-reporter hESC line 4–5 days after RNP delivery. Percentages of EGFP-negative/tdTomato-positive cells (boxed) are indicated in (D) (E), and percentages of EGPF-positive/tdTomato-negative cells (boxed) are indicated in (F).
We decided to deliver this mutant version of TfuCascade and wild type TfuCas3 into hESCs via electroporation. RNP delivery was chosen over a plasmid-based expression method, partly to bypass the optimization steps needed for expressing and assembling a multi-subunit RNP complex in hESCs, and partly to avoid the possible off-target editing or cellular toxicity typically associated with long-term expression of CRISPR-Cas (Kim et al., 2014). Moreover, RNP delivery has been reported to be less stressful to hESCs (Kim et al., 2014). We attached nuclear localization signals (NLSs) to the C-terminus of TfuCas3 and the C-terminus of each of the six Cas7 subunits in TfuCascade to promote nuclear import. This NLS tagging scheme did not affect the stability of Cascade, nor its ability to target DNA for degradation in conjunction with Cas3 (Fig. S1C–D). To assay for genome editing activity (Fig. 1B), we created a hESC dual reporter line (H9-DNMT3B-tdTomato/EGFP) bearing knock-ins of a tandem dimer tomato fluorescent protein (tdTomato) gene and an enhanced green fluorescent protein (EGFP) gene at the two alleles of the highly expressed DNMT3B locus (Fig. 1C), leading to high levels of dual fluorescence. RNA-guided gene disruption of the EGFP reporter would lead to the accumulation of EGFP-negative/tdTomato-positive cells, and vice versa for the tdTomato gene disruption. Human ES cells were chosen for this study over cancer-derived cell lines for their normal karyotype and DNA repair mechanisms.
Cascade and Cas3 Enable Programmable RNA-guided Gene Disruption in hESCs
We first programmed TfuCascade with a 61-nt crRNA containing guide sequence G1 against a 32 base pair (bp) region in EGFP that was flanked by an interference-enabling PAM 5’-AAG (Fig. 1C), purified this TfuCascade-G1 RNP, and electroporated it together with TfuCas3 into the hESC dual-reporter line. A sub-population (7.3%) of EGFP-negative and tdTomato-positive cells became detectable by flow cytometry after 4–5 days (Fig. 1D). Negligible levels of EGFP-negative/tdTomato-positive cells were detected in control transfections that included TfuCascade-G1 alone, or TfuCas3 alone, or a non-targeting (NT) TfuCascade together with TfuCas3 (Fig. 1D). A very small fraction of cells lacking both EGFP and tdTomato fluorescence were observed for each reaction, even when no CRISPR components were delivered. This was most likely caused by spontaneous hESC differentiation that leads to rapid repression of the DNMT3B locus (Sperger et al., 2003), which results in the simultaneous loss of both EGFP and tdTomato expression in our reporter line. No apparent cell toxicity was observed for any combination of Cas3 and/or Cascade delivery. Collectively, these results suggest that the T. fusca Type I-E CRISPR-Cas system can induce RNA-guided gene disruption in hESCs, and that both the nuclease-helicase effector Cas3 and a cognate Cascade are required.
To demonstrate that the editing is programmable, we designed two additional TfuCascade RNPs. Co-delivery of a TfuCascade-G2 targeting the opposite strand of EGFP (Fig. 1C) together with TfuCas3 lead to the accumulation of 2.7% EGFP-negative and tdTomato-positive cells (Fig. 1E). Moreover, electroporation of a tdTomato-targeting (Td) TfuCascade (Fig. 1C) in conjunction with TfuCas3 resulted in a 2.7% tdTomato-negative and EGFP-positive cell population (Fig. 1F). These results further demonstrated that this Type I CRISPR-based novel gene editing platform is re-programmable.
Since Cascade alone has been shown to silence the targeted gene in bacteria by sterically blocking transcription (Luo et al., 2015; Rath et al., 2015), we felt compelled to distinguish whether the silencing mechanism was due to DNA editing or transcriptional repression. Several lines of evidence argue that the loss of EGFP fluorescence in our experiment was not due to Cascade-mediated transcriptional silencing. First, hESCs that received Cascade-G1 RNP alone exhibited zero EGFP-negative/tdTomato-positive events (Fig. 1D), suggesting that the DNA degradation factor Cas3 is indispensable for GFP silencing. Second, editing at the DNA level would persist through generations, whereas transcriptional effects enabled by RNP delivery would be titrated away as cells divide. When we cultured the Cascade-G1/Cas3 treated cells continuously and retrieved samples on days 2, 4 and 14 post RNP delivery for flow cytometry analysis, a 7.9% EGFP-negative/tdTomato-positive sub-population appeared within two days and remained at ~ 9% on days 4 and 14 (Fig. S2A). The background EGFP intensity in the EGFP- /tdTomato+ cells further diminished with extended culturing, probably because the existing EGFP proteins degraded over time (Fig. S2A). These observations again are consistent with permanent DNA changes rather than transient transcriptional suppression. Lastly, we monitored Cas protein stability in hESCs after RNP delivery using western blot. The vast majority of HA-tagged TfuCas3 was degraded rapidly within the first 24 hrs (Fig. S2B), which is on par with the reported persistence time of Cas9-sgRNA RNP in human cells (Kim et al., 2014). Although an antibody was not in hand to track Cascade stability, we had no reason to suspect that Cascade-caused transcriptional blockage, if any, would still persist after 14 days of cell growth and divisions.
Editing Efficiency is Limited by the Activity of Cascade, but not Cas3
We attempted to optimize this editing platform by varying the amount of Cascade and Cas3 delivered. The efficiency of EGFP disruption positively correlated with TfuCascade abundance, increasing from 3.3% to 13.1% when the amount of TfuCascade-G1 RNP delivered via a 10 μL electroporation reaction was increased from 20 to 80 pmole, with TfuCas3 kept constant at 20 pmole (Fig. 2A). A similar correlation was observed between the amount of electroporated TfuCascade-G2 RNP and the editing efficiency (Fig. 2B). In contrast, doubling, tripling, or quadrupling the amount of TfuCas3 while keeping Cascade constant did not improve the editing efficiency (Fig. 2C). These findings suggest that the editing efficiency in hESCs might be currently limited by the target-searching activity or the chemical stability of TfuCascade, rather than DNA degradation by TfuCas3.
Figure 2. Optimization of genome editing efficiency.

(A-C) Flow cytometry analysis of dual-reporter hESC line 4 days after RNP delivery. Increasing amount of TfuCascade-G1 or TfuCascade-G2 was used in conjunction with constant TfuCas3 in (A) and (B). Increasing amount of TfuCas3 was used in conjunction with constant amount of TfuCascade-G1 in (C). Percentages of EGFP-negative/tdTomato-positive cells (boxed) are indicated. Amounts of Cascade or Cas3 delivered via 10 μL electroporation reactions were indicated.
Type I CRISPR-Cas Editing Induces a Spectrum of Large Chromosomal Deletions
Based on prior knowledge of DNA interference by Type I CRISPR-Cas (Mulepati and Bailey, 2013; Sinkunas et al., 2013), we speculated that chromosomal deletions may be induced upstream (i.e. PAM-proximal direction) of the target site. To understand the genomic lesions that underlie EGFP disruption, we extracted genomic DNA from the TfuCascade-G1/Cas3 edited hESCs before and after fluorescence activated cell sorting (FACS), and PCR-amplified a ~5.1 kb region using two primers spanning a region 4.7 kb upstream and 400 bp downstream of the target site (Fig. 3A, −4.4kF and R1). The untransfected cells and the TfuCascade-NT/Cas3 treated cells served as two controls, and both produced a single PCR band of 5.1 kb, suggesting that the DNMT3B-EGFP locus was intact (Fig. 3B, lanes 1–2). The amplicons from the unsorted total cells after the TfuCascade-G1/Cas3 treatment contained a faint ladder of smaller bands in addition to the full-length product, indicating that a fraction of these cells harbor deletions of varying lengths at the DNMT3B-EGFP locus (Fig. 3B, lane 3). Notably, PCR amplifications from the sorted EGFP-negative/tdTomato-positive population were highly enriched with a distribution of smaller products, ranging from 5 kb to ~1 kb in size. The lack of a discernible full-length product (~5.1 kb) also implies that small indel-mediated EGFP disruption was rare during Type I CRISPR editing (Fig. 3B, lane 4). Speculating that some deletions might extend beyond the 4.7 kb detection limit, we repeated the experiment using a different forward primer annealing further upstream of the target site (Fig. 3A, −8.2kF). The resulting PCR band pattern indeed suggests that the chromosomal deletions were well-represented all the way up to ~7.5 kb (Fig. 3B, lanes 5–8). Control PCRs amplifying a 5.5 kb region downstream of EGFP detected no genomic deletions (Fig. 3B, lanes 9–12), in agreement with the idea that Cas3 is a highly processive helicase-nuclease that translocates uni-directionally towards the PAM-proximal direction. The observed lesion profile for Cascade/Cas3 is in stark contrast to that of eukaryotic gene editing by the Cas9 or Cas12 nucleases, which typically lead to small indels at the target site.
Figure 3. Long-range PCR based characterization of genomic lesions induced by Type I CRISPR-Cas.

(A) (C) Schematic of the DNMT3B-EGFP locus and annealing sites for five PCR primers used in (B) or ten PCR primers used in (D). Positions relative to the EGFP translation start site (+1) are indicated. Recognition sites (2nd nt of the PAM) for Cascade- G1 are marked by the dashed blue line. Blue arrowhead, direction of Cas3 translocation. (B) Characterization of genomic lesions by long-range PCRs. A collection of DNA lesions was introduced upstream of GFP by a GFP-targeting Cascade-G1 and Cas3, in the sorted EGFP-negative/tdTomato-positive population, as well as in unsorted total cells. NT, total cells treated with a non-targeting Cascade and Cas3. PCR primers used are indicated and their annealing sites depicted in (A). (D) Heterogeneous large genomic deletions were introduced by Cascade-G1/Cas3 in the ~20 kb region upstream of GFP, revealed by serial long-range PCRs with tiling forward primers. NT, non-targeting Cascade. PCR primers used are indicated and their annealing sites depicted in (C). M, DNA size markers.
Because the DNA lesion pattern could not be comprehensively captured in any single PCR reaction, we performed a series of long-range PCRs using a common reverse primer annealing 2 kb downstream, paired up with one of the nine forward primers tiling along a 22 kb region upstream of the EGFP targeted site (Fig. 3C, +2.3kR and nine tiling primers A through I). FACS-sorted, Cascade-G1/Cas3-edited cells from six independent experiments were pooled together, and nine individual PCR amplifications from this “pooled” genomic DNA all gave rise to a collection of smaller products of varying sizes (Fig. 3D, lanes 1–9), indicating that heterologous large deletions were induced across the 22 kb upstream region. Control PCR amplifications using the same nine primer pairs on untransfected cells (Fig. 3D, right) generated either the expected full-length product (lanes 10–13), discrete non-specific bands (lanes 11–12 and 14–17), or no product (lane 18).
A recent study showed that in addition to the desired small indels, CRISPR-Cas9 may also cause rare complex distal deletions (kilobases in size) in mouse embryonic stem cells (mESCs) (Kosicki et al., 2018), and the observed deletions could be bi-directional from the Cas9 cut site, which is distinct from the uni-directional deletion pattern observed for Type I editing events (Figs. 3A–B). We further investigated whether deletion events spanning both PAM-proximal and PAM-distal regions may exist among the edited human cells, due to Type I-editing induced genome instability. First, we noticed that the smallest PCR products in lanes 1 through 9 of Fig. 3D were all around or slightly above 2 kb, which matches the genomic distance between the targeting site and the annealing site of the common downstream primer. This suggests that the deletion events did not extend to the PAM-distal (downstream) region. Secondly, we performed additional PCRs using a common forward primer (−2.7kF) but varying the reverse primer annealing site to be ~0.9 kb, 2.0 kb, or 3.0 kb 3’ of the target site (Fig. S3A). The minimum amplicon size in each reaction varied in accordance with the distance between the target site and the annealing position for the reverse primer used (Fig. S3B, lanes 1 −3). These observations together suggest that our Type I editing experiments rarely led to complex bidirectional deletion events spanning the target site, as seen in the Cas9 study (Kosicki et al., 2018).
An Unusual Pattern of Type I CRISPR-mediated Genomic Lesions
To map out the precise boundaries of the Cascade-G1/Cas3-induced deletions, we first employed a Sanger sequencing based low-throughput method that can reveal DNA lesions at single-nucleotide resolution. The amplicons from lanes 1 through 9 in Fig. 3D were pooled and TOPO-cloned. Two hundred and eleven positive clones were randomly chosen for Sanger sequencing using the GFP reverse primer R1 to identify the chromosomal junctions; an additional fifteen random clones from the TOPO-cloned PCR products from lanes 4 or 8 of Fig. 3B were also sequenced. 215 out of the 226 sequenced clones yielded good quality sequencing traces. A total of 180 unique chromosomal lesions were identified, and they can be categorized into four major groups based on the features of their junctions (Figs. 4B, 4D–E and S4, a complete list in Table S2). Group I is the most prevalent, consisting of 140 cases (78% of 180) where the 5’ and 3’ regions flanking the deletions were re-ligated seamlessly, presumably via the NHEJ pathway in human cells. This finding suggests that the T. fusca Type I CRISPR-Cas likely induced at least two DSBs in the upstream region; more DSBs possibly occurred in between but were masked by the terminal DSBs. We were not able to distinguish if any small deletions were further generated at the junction during NHEJ repair, because the precise locations of the DSBs and the nature of the resulting DNA ends (blunt or recessive) are unclear.
Figure 4. Type I CRISPR induced deletion/repair junctions are revealed at single nucleotide resolution by sanger-sequencing.

(A) Representative deletion locations at the DNMT3B-EGFP locus, revealed by cloning of pooled tiling PCRs in Fig. 3D and Sanger sequencing. The entire PCR in lanes 4 or 8 of Fig. 3B was also analyzed, the detected lesions are shown at the bottom. R1, reverse primer used for Sanger sequencing. Black lines, deleted genomic regions. Orange, magenta, green, and the lack of dots on the right indicate Groups II, III, IV, and I deletion junctions as described in (B). (B) A pie chart of the four groups of deletion junctions identified. Group I (light blue), one seamless junction; Group II (orange), one deletion with an small insertion or partial inversion; Group III (pink), one deletion with point mutations); Group IV (green), two deletions. (C) Scatter plot for the 180 unique junctions, showing the upstream (5’, X-axis) and downstream (3’, Y-axis) end points of the chromosomal deletions, relative to the EGFP translation start site (+1, marked by the dashed green line) at the DNMT3B locus. Dashed blue line, recognition site for Cascade-G1. (D) Schematics of the gene structure of representative groups I, II, III, and IV lesion junctions. Black horizontal line, reference and sequenced alleles; gray shadow; direct match between the reference and sequenced alleles. Lack of grey shadow at the reference locus represents a deletion in the sequenced allele. Orange bar, an insertion or partial inversion; pink line, point mutation(s). Cascade-G1 recognition site (+273, relative to the beginning of EGFP ORF +1) is marked by the dashed blue line in (A) and (C). (E) Example Sanger sequencing chromatograms showing representative lesion events. Dashed black line, deletion/repair junctions. Genomic positions of the flanking nucleotides, relative to the EGFP translation start site (+1), are indicated on the top. Sizes of the deletions are shown in parentheses. Orange, a 2 bp insertion; pink, a T to G point mutation.
Group II contains 25 cases (14% of 180) of a single large deletion combined with a short insertion at the repair junction (Figs. 4B, 4D–E, S4, and Table S2). Among them, fifteen deletions were associated with a small insertion less than 18 bp; while ten deletions were associated with an insertion of a few hundred bp (seven of which could be mapped back to part of the deleted genomic sequence in reverse orientation. i.e. inverted). Group III only has 4 examples containing a large deletion combined with a point mutation near the junction (Figs. 4B, 4D–E S4, and Table S2). Groups II and III products are presumably formed by the mutagenic NHEJ repair pathway(s).
Interestingly, eleven group IV cases exist, each of which contains two large deletions separated by an intervening chromosomal sequence of a few hundred base pairs (Figs. 4B, 4D–E, S4, and Table S2). One potential cause of the group IV events may be the re-insertion of a segment of the originally deleted genome during the repair of one large deletion, and it is possible that the re-inserted segments are Cas3-generated dsDNA fragments. Notably, for 95% of all unique lesions (171/180), the Cascade recognition site and its flanking PAM remained intact after editing and repair (Fig. 4A, and Table S2), and in theory might be able to support additional rounds of editing if free Cascade and Cas3 are available. Therefore, we cannot rule out the possibility that group IV events resulted from two successive rounds of editing events.
The 5’ deletion boundaries, which likely reflect the last DSB generated by Cas3 before its dissociation from DNA, are distributed across the ~20 kb upstream region, highlighting the heterogeneous nature of the long-range lesions induced by Cas3 and a single-CRISPR-programmed Cascade. An unexpected finding was that the 3’ boundaries of these deletions, which possibly represent the first DSB by Cas3, did not line up precisely with Cas3’s first nicking site, which is 9–11 nt after the PAM, into the R-loop (or protospacer) region (Xiao et al., 2017). Instead, they spread out along a ~400 bp window upstream of the target site, which included the first ~300 bp of EGFP coding sequence and the preceding 100 bp sequence in the upstream intron (Figs. 4A and 4C). More editing events may have started further upstream, but would not be enriched by cell sorting if the deletions were limited to the intronic region and did not affect EGFP expression. This observation suggests that Cas3 does not necessarily elicit DSBs during the very initial phase of its DNA translocation. Previous single molecule studies revealed that after recruitment by Cascade, Cas3 nicks the non-target strand DNA, then initially remains associated with Cascade and reels dsDNA towards itself repeatedly, and eventually dissociates from Cascade and translocates alone for kilobases along the DNA (Dillard et al., 2018); in both phases NTS DNA was sporadically erased, leading to the exposure of short TS single-stranded DNA (ssDNA) tracts (Dillard et al., 2018; Redding et al., 2015). DSB formation was not frequently observed at the single molecule level (Dillard et al., 2018; Redding et al., 2015). However, dsDNA targets were found to be shredded by Cas3 into pieces in bulk biochemical experiments (Dillard et al., 2018; Kunne et al., 2016; Redding et al., 2015).
To define the genome lesion at single cell level, we isolated fifteen single cell clones from the sorted GFP-negative cells from the Cascade G1/Cas3 editing experiment in Figure 1D. All clones appear healthy. Their genomic DNAs were subjected to the tiling PCR and Sanger sequencing analysis described in Figures 3D and 4A, to identify potential lesions within the 20 kb PAM-proximal DNMT3B region. Nine clones each contains an identifiable, unique long-range deletion (listed in Table S2); it is unclear what kind of DNA lesions exist for the other six clones.
Heterogeneous Large Deletions Introduced on a Second Chromosomal Target Site.
To understand if the formation of a spectrum of large deletions is a generalized feature for Type I CRISPR editing, we did lesion analysis for another target site on the opposite strand of EGFP specified by Cascade-G2 (Fig. 1C). Because Cas3 was oriented by Cascade-G2 to translocate in the opposite direction as Cascade-G1, we anticipated that chromosomal deletions would occur downstream of GFP accordingly. We extracted genomic DNA from the sorted EGFP-negative/tdTomato-positive cells from the experiment in Fig. 1E and PCR-amplified a 6.5 kb region using primers bracketing the EGFP coding sequence and 4.9 kb downstream of GFP (Fig. S5A, F and +6.5KR). As expected, a spectrum of PCR products smaller than 6.5 kb was amplified from the sorted cells; whereas two negative control PCRs from untransfected cells or TfuCascade-NT/Cas3 treated cells both produced a single 6.5 kb band (Fig. S5B, lanes 1–3). A similar pattern was observed when the PCR was repeated using a reverse primer annealing 3.5 kb further downstream; while control PCRs amplifying a 4.4 kb region upstream of the target revealed no genomic lesions (Figs. S5A, S5B, lanes 4–9).
We TOPO-cloned the amplicons in lanes 3 or 6 of Fig. S5B, and randomly picked 53 clones for Sanger sequencing. 26 unique lesion events were identified, among which 22 were seamless junctions (Group I), one was a deletion plus a 2 bp small insertion (Group II), one was a deletion with a point mutation 3 bp nearby (Group III), and two had double deletion junctions (Group IV) (Figs. S5C–D, a complete list in Table S2). The 5’ deletion boundaries here, likely reflecting the first DSB by Cas3, spread out in a ~390 bp window right after the target site; and the recognition site for Cascade-G2 remained intact for all 22 lesion cases. The 3’ endpoints of lesions, which possibly represent the last DSB by Cas3, were distributed across a 9.7 kb region downstream of the target site (Fig. S5C). Collectively, these results further demonstrated that Type I CRISPR-Cas could be reprogrammed to induce a spectrum of large deletions on the PAM-proximal side of a single CRISPR-targeted site.
Comprehensive Lesion Analysis by Tn5-based Next-Generation Sequencing (NGS)
To define Type I CRISPR-induced lesions more comprehensively, we developed a Tn5 tagmentation and NGS based method (Fig. 5A). The genomic DNAs of FACS-sorted, Cascade-G1/Cas3 edited EGFP-/tdTomato+ hESCs from six independent experiments were pooled together and treated with adapter-loaded Tn5 transposase, which randomly fragments DNA and attaches a single type of adapter onto the fragmented ends. We then did a multi-step PCR using nested EGFP primers and a primer specific for the Tn5 adapter to enrich for sequences spanning the lesion junctions (Fig. 5A). The resulting NGS library was sequenced on an Illumina MiSeq using 50×450 bp paired end sequencing, and the long R2 reads were analyzed to determine the extent of the corresponding deletions, as described in Methods. We reasoned that since 97% (174/180) of all unique lesions detected by Sanger sequencing in Fig. 4 started within ~330 bp from the Cascade binding site, we should be able to cover most of the junctions using the 450 bp MiSeq reads from the EGFP-specific primer used for library construction. Bioinformatic analysis of the NGS dataset detected DNA lesions in 33.2% of the 278,074 aligned MiSeq reads obtained, and identified roughly 3,376 unique junctions (it is unclear whether the repeated instances of identical junctions reflect PCR duplicates or genuine repeated biological events). Out of the total set of reads, 95.3% of the reads have at least 95% of their length aligned to the ~130 kb region consisting of the DNMT3B-GFP locus and its upstream sequence. The remainder (4.7%) could represent either sequencing errors, cases where insertions were drawn from other portions of the genome, or deletions too large to be identified in our reference sequence – we excluded all such reads from further consideration. Among the considered set of lesion reads, consistent with Sanger sequencing results, the vast majority (86%) contain Group I events, with one junction between the 5’ and 3’ flanking regions of a large chromosomal deletion (Fig. 5B). Their 3’ deletion endpoints occur within a ~400 bp window upstream of the targeted EGFP sequence, whereas the locations of the 5’ endpoints are far more spread out and can be tens of kilobases upstream (Fig. 5C). The sizes of these Type IE CRISPR-induced Group I deletions are concentrated within a 30 kb range, however, a portion of deletions exists even above 50 kb (Fig. 5D). In roughly 12% of the considered set of lesion reads, we observed an inverted segment of the human DNMT3B locus forming a junction with what is presumably the 3’ endpoint of a Cas3-induced deletion. Due to the sequencing length limitation, we cannot see the deletion’s 5’ breakpoint, but nonetheless still classified these events into Group II lesions (a deletion with an insertion, in this case inverted) (Fig. 5B). Finally, 2% of the considered lesion reads contain Group IV cases with two large deletions (Fig. 5B). We have also observed that a portion of the analyzed reads appear to have small un-mappable insertions between the two alignable ends of the deletion junction. Due to NGS sequencing error rates and the possibility of minor alignment error, we decided to not call point mutations or small insertions less than 10 bp at the repair junctions, and all such reads were classified into Group I lesions in Fig. 5B. Deletion junctions with an un-mappable insertion larger than 9 bp only account for <2% of all the lesion reads, and are classified into Group I or II events.
Figure 5. Tn5- and NGS- based characterization of Type I CRISPR-induced genome deletions.

(A) Schematic of Tn5 tagmentation procedure and NGS library construction. (B) Pie chart of three main types of lesion-containing reads identified by Miseq analysis. Light blue, Group I (one deletion junction); orange, Group II (a deletion with a large [>=9 bp] insertion, including partial inversion); green, Group IV (two deletions). The coloring scheme and categorization for deletion junctions are consistent with Fig. 4B. (C) Scatter plot for all Group I lesion-containing reads, showing the upstream (5’, X-axis) and downstream (3’, Y-axis) end points of the chromosomal deletions, relative to the EGFP translation start site (+1) at the DNMT3B locus. Dashed green line, and light blue lines, the (+1) start of the EGFP ORF; dashed blue line, recognition site for Cascade-G1. Kernel density estimates for the marginal distributions are shown along the axes, revealing the narrow range of downstream deletion endpoints and very long-tailed distribution of upstream deletion endpoints. (D) Histogram showing the distribution of deletion lengths observed for all the Group I reads in (B). The inset view on the top right corner is a zoom-in on deletions smaller than 10 kb.
To test if Type I CRISPR can be exploited to engineer an endogenous locus in a different cell line, we programmed Cascade with two crRNAs targeting the promoter region ~280 or ~460 nt upstream of the HPRT gene (HPRT-G1 and HPRT-G2, Fig. 6A), and tested them in the near-haploid chronic myeloid leukemia derived cell line HAP1. Genomic deletions caused by CRISPR targeting and Cas3 translocation towards the coding sequence would disrupt HPRT production, leading to resistance to 6-thioguanine (6-TG). After RNP delivery, we estimated the editing efficiency by comparing the single cell colony forming capability in the presence or absence of 6-TG in the culture media (Fig. 6B). When co-delivered with Cas3, Cascade HPRT-G1 or HPRT-G2 enabled 67% and 32% targeting, respectively (Figs. 6C–D). Importantly, a single nucleotide mutation introduced at the beginning of the crRNA spacer (HPRT-G2*, Fig. 6A) prevented editing (Figs. 6C–D), suggesting that genome targeting by the Tfu Type I-E CRISPR is stringent. For comparison, a SpyCas9 RNP recognizing a site within HPRT exon 1 exhibited 44% editing in the single clone 6-TG cytotoxicity assay (Figs. 6A, 6C–D), consistent with the indel formation rates assessed using the tracking of indels by decomposition (TIDE) analysis (Brinkman et al., 2014) and T7E1 assay (Guschin et al., 2010) (Figs. S6A–B).
Figure 6. Efficient Editing at the Endogenous HPRT1 Locus in HAP1 Cells.

(A) Schematic of the HPRT locus and annealing sites for six PCR primers used in (E). Protospacers for the two crRNAs are indicated in blue, and corresponding PAMs in magenta. Positions relative to HPRT translation start site (+1) are indicated. Recognition sites (2nd nt of the PAM) for HPRT G1 and G2 are marked by the dashed blue line. Blue arrowhead, direction of Cas3 translocation. Recognition site (1st G in PAM) for the SpyCas9 is marked by the dashed green line. The G2 guide sequence is shown at the bottom, with the single nt mutation in HPRTG2* in orange. (B) Procedure of the genome editing experiments in HAP1 cells. (C) Estimate HPRT targeting efficiency by single clone 6-TG cytotoxicity assay. (D) Bar graph plotting the HAP1 colony counts obtained in (C). The average 6-TG survival rates from three independent experiments are indicated. Error bar, standard deviation. (E) A collection of DNA lesions was induced in the HAP1 cells treated with Cas3 and Cascades HPRT-G1 or HPRT-G2, but not the HPRT-G2* mutant. PCR primers used are indicated and their annealing sites depicted in (A). M, DNA size markers. Lanes from two different agarose gels are separated by the dashed black line. (F) Locations of the HPRT genomic deletions. Black lines, deleted genomic regions. Orange, green, and the lack of dots on the right indicate Groups II, IV, and I deletion junctions as described in Fig. 4B.
Genomic lesions caused by HPRT-G1/Cas3 and HPRT-G2/Cas3 were characterized by long-range PCRs using a forward primer and one of the reverse primers tiling along the HPRT locus (Fig. 6A). Wild type genomic DNA failed to produce amplicons, possibly because a GC-rich region in exon 1 prevented PCR amplification; whereas the genomic DNA from the edited cells produced heterogeneous deletions, evidenced by the ladder of smaller PCR products (Fig. 6E). Consistent with the 6-TG cytotoxicity results, HPRT-G2* mutant failed to induce DNA lesions because no amplicons were produced (Fig. 6E). Next, we pooled amplicons from lanes 6–10 of Figure 6E for TOPO-cloning and picked 20 clones for Sanger sequencing. Each clone revealed a unique uni-directional DNA lesion, and Group I, II and IV deletions/junctions events were all observed (Fig. 6F), suggesting that the editing pattern by Type I CRISPR is likely not cell type-or locus-specific.
DISCUSSION
A unique lesion/repair profile for Type I CRISPR enabled eukaryotic gene editing
In its native environment, Type I CRISPR interference typically eradicates the targeted foreign DNA completely, and may cause cell death if accidentally programmed against the prokaryotic host genome. In this study, the large chromosome size and strong intrinsic NHEJ activity of human cells allowed us to observe the unique deletion/repair outcome for Type I CRISPR in a heterologous eukaryotic context. The phenomenon of Cas3-mediated human genome editing over a long distance is quite different from the localized editing by Cas9 and Cas12 at the CRISPR-targeted site. Perhaps most unexpectedly, Cas3 and a single guide-programmed Cascade together lead to a spectrum of large chromosomal deletions in a hESC population. The heterogeneity manifests in the number of unique lesion junctions observed (180 out of 217 by Sanger sequencing in Fig. 4), as well as the wide distribution of deletion sizes and distal endpoints (Figs. 3–5, S5, and Table S2). The onsets of deletions were not uniform either, spreading out within a predictable ~400 bp region upstream of target site. Moreover, Cas3-induced lesions are predominantly large deletions, with very rare small indels (Figs. 3–4, S5, and Table S2).
In comparison, current gene-editors Cas9 and Cas12 cause precise genomic breaks inside the CRISPR-complementary target site. NHEJ repair of these breaks will eventually lead to small indels that change the original target sequence and therefore may prevent any further targeting by the same guide. Intriguingly, we found that the vast majority of the DNA deletions by Type I CRISPR do not affect the sequence integrity of the target site (Figs. 4 and S5, Table S2). This implies that prolonged exposure to Type I CRISPR machinery will likely enable iterative rounds of DNA deletion and repair, which could serve as a tool to create extremely large chromosomal deletions. This may also allow tuning of the final deletion outcome, through the control of Type I CRISPR gene expression in cells, or through the choice of different delivery methods.
It was recently reported that in addition to small indels, CRISPR-Cas9 may also cause rare distal deletions and complex chromosomal rearrangements around the target site in mESCs (Kosicki et al., 2018). It needs to be pointed out that the large genomic deletions from our Type I CRISPR edited hESCs are clearly of a different nature. They are unidirectional, occur at high frequency, are not accompanied by small indel formation, and have a predictable range of onset points, all in agreement with the known native mechanism of Type I CRISPR function.
Insights into the DNA interference mechanism by Type I system
Because of its long-range impact, the final products of Type I CRISPR mediated DNA degradation were difficult to define in prokaryotes. Precious insights have been obtained from single molecule studies (Blosser et al., 2015; Dillard et al., 2018; Krivoy et al., 2018; Loeff et al., 2018; Redding et al., 2015; Rutkauskas et al., 2015; Szczelkun et al., 2014). A single molecule DNA curtain study of the E. coli Type I-E system revealed two modes of action after Cas3 has been recruited to the R-loop region by Cascade and nicked the non-target strand DNA (Redding et al., 2015). First, Cas3 reels DNA towards itself while bound to Cascade. It then dissociates from Cascade and translocates along dsDNA for kilobases (Redding et al., 2015). ssDNA is exposed to various extents during these processes, however, DSB formation was not observed (Redding et al., 2015). A recent study of the T. fusca Type I-E system also revealed a two-action-mode behaviour for Cas3 (Dillard et al., 2018). ssDNA exposure was observed during the DNA reeling mode, but not in the translocation mode. An important new insight is that DSB formation was observed in real time for the first time, which occurs at low frequency along the translocation path of Cas3, and was observed more frequently when Cas3 is stalled by a protein roadblock (Dillard et al., 2018). It was speculated that stalling may cause the HD nuclease in Cas3 to iteratively nick the DNA until both strands are cleaved at close proximity to give rise to a DSB (Dillard et al., 2018). An alternative mechanism may be that the translocating Cas3 processively degrades regions of non-target strand DNA, exposing stretches of ssDNA on the target-strand, which are subsequently cleaved by cellular nucleases, leading to DSB formation.
Our work suggest that in human cells, Cas3 presumably generates multiple DSBs during DNA translocation, and the distal deletion boundary likely reflects the last DSB by Cas3. How DSB formation and long-range deletion may be influenced by the local chromatin environment awaits future investigation. The most unexpected finding from a mechanistic perspective, was that the onsets of the deletions were never exactly at the first Cas3 nicking site, which is on the nontarget strand DNA inside the R-loop region (Mulepati and Bailey, 2013; Sinkunas et al., 2013; Xiao et al., 2017). Rather, they were distributed within a ~400 bp window in the PAM-proximal region, indicating that Cas3 may not elicit the first DSBs during the very initial phase of its translocation on human genome.
Expanding the CRISPR-based gene editing toolbox
We report ~13% genome editing efficiency ex vivo in hESCs and 30–60% efficiencies in HAP1 cells through transient Type I RNP delivery. Even higher editing efficiencies may be achieved by further improving TfuCascade’s activity at 37 °C through structure-guided engineering or directed evolution, or by employing plasmid/mRNA-based delivery to increase effector concentration and persistence. Additional Type I systems can also be screened for special features such as higher target-searching activity in Cascade, and more robust translocation capability in Cas3. Furthermore, significantly more streamlined Type I CRISPRs (e.g. I-C systems) exist that require only four cas genes to achieve DNA interference, and therefore may facilitate easier delivery into eukaryotic cells. One potential limitation for our current RNP-based Type I editing platform is that an intact Cascade has to be purified for each genomic targeting experiment. However, we envision that mRNA- or plasmid-based delivery approaches would enable easy Cascade reprogramming and high-throughput Type I applications.
Off-target effect is a concern for all genome editing applications. A previous study suggests that tolerance of mismatches by TfuCascade only gradually increases beyond the first 8-nt “seed” region (Jung et al., 2017). This behaviour is similar to that of Cas12 (Strohkendl et al., 2018), but stands in contrast to Cas9 (Boyle et al., 2017). Off-targeting for Type I CRISPR is further suppressed at the Cas3 recruitment step, by a large conformational change in Cascade upon full R-loop formation (Hochstrasser et al., 2014; Wiedenheft et al., 2011; Xiao et al., 2018). Our data support the notion that Type I CRISPR-mediated genome editing is quite stringent. The robust HPRT targeting by Cascade/Cas3 in HAP1 cells is completely abrogated by a point mutation at the 5’ end of the crRNA spacer (Fig. 6E). Furthermore, informatic prediction suggests that off-targeting in the human genome is highly unlikely in our editing experiments. It is well-established that Cascade specifies 27 out of the 32 nucleotides in the crRNA spacer region while ignoring the five nucleotides at special positions (6th, 12th, 18th, 24th, 30th) (Fineran et al., 2014; Jackson et al., 2014; Mulepati et al., 2014; Zhao et al., 2014). High-throughput studies reported no tolerance of more than five mismatches in non-special positions, and typically two of such mismatches are more than enough to abolish Type I CRISPR interference (Fineran et al., 2014; Semenova et al., 2011). Based on these prior knowledge and using a more relaxed NAG PAM, we performed in silico prediction of off-target sites in the human genome for all five RNA guides used in this study (two against HPRT, two against EGFP, and one for TdTomato). The top ten potential off-target sites for each guide (Table S3) contain 6–9 nt mismatches at non-seed and non-special positions in the protospacer. Such degree of mismatches render off-targeting a remote possibility.
More than 98% of the human genome is non-coding, containing cis-elements important for gene regulation and diseases. Yet genetic tools to characterize these large regions are limited. Large genome deletion is typically achieved by programming CRISPR-Cas9 with a pair of sgRNA guides dictating the deletion boundaries (Canver et al., 2014; Chen et al., 2014; Cong et al., 2013). Cas9-based screening methods also allow high-throughput functional interrogation of the non-coding genome, which typically involves the laborious design of a tiling library of sgRNA or sgRNA pairs (Diao et al., 2017; Fulco et al., 2016; Komor et al., 2017). The ability of Type I CRISPR to generate such a diverse range of large deletions from a single CRISPR-targeted site could potentially enable long-range CRISPR screens that are more simple and cost-effective to execute, because far fewer guides are needed and each guide leads to a library of deletion mutants. It could conceivably be adapted to erase parasitic or diseased genetic elements, or to introduce long-range epigenetic modifications. Such Type-I CRISPR-based applications would greatly expand the genome engineering toolkit.
STAR Methods
Dolan and Hou et al.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yan Zhang (yzhangbc@med.umich.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Escherichia coli BL21 (DE3)
E. coli BL21 (DE3) cells were used for protein production. Cells were grown in Lysogeny Broth (LB) or M9 medium supplemented with appropriate antibiotics.
Escherichia coli DH5alpha
This strain was used for cloning. Cells were grown at 37°C in LB supplemented with appropriate antibiotics.
Human embryonic stem cell (hESC) culture
Human ESC line H9 (sex: female) were cultured in E8 medium on matrigel (Corning) coated tissue culture plates at 37°C and 5% CO2 in a humidified incubator, with daily media change. Cells were split every 4–5 days with 0.5 mM EDTA in 1x PBS.
HAP1 cell culture
Human HAP1 cells (Horizon Discovery) were cultured in IMDM (Gibco) supplemented with 10% FBS (Corning) at 37°C and 5% CO2 in a humidified incubator, with daily media change. Cells were split every 2 to 3 days using TrypLE Express (Gibco).
METHOD DETAILS
Expression and purification of TfuCas3 and TfuCascade
T. fusca Cascade and Cas3 was purified as described previously (Xiao et al., 2018), with minor modifications. TfuCascade was recombinantly expressed in E. coli BL21 cells in LB media using a three-plasmid co-expression system. Cse1 is encoded on one vector (pET19b) with an N-terminal 6xHis-TwinStrep-SUMO tag. The rest of the Cascade components (Cse2, Cse4, Cas5e, and Cse3) were encoded polycistronically in another vector (pCDF-Duet1) with a C-terminal NLS tag on Cse4. The crRNA was expressed from a synthetic CRISPR array containing three repeats and two spacers in ORF1 position of pRSF-Duet1. Cells were grown at 37°C until the OD600 is between 0.6 and 1.0. Protein and RNA expression were induced by adding IPTG to a final concentration of 0.5mM, and allowing the cell to grow overnight at 22°C. 12 liters of cells were harvested and lysed by sonication in lysis buffer containing 30 mM HEPES pH 7.5 and 500 mM NaCl. The supernatant after centrifugation was loaded onto ~5 mL of StrepTactin resin and 2 mg Avidin per L of cells was supplemented to prevent cellular biotin from binding to the column. The column was washed with 3×15 ml of lysis buffer, and the protein eluted with 10 ml of lysis buffer supplemented with 5 mM Desthiobiotin. After cleaving the TwinStrep-SUMO tag with SUMO protease overnight at 4 °C, TfuCascade was concentrated and buffer-exchanged to a buffer containing 30 mM HEPES pH 7.5 and 200 mM NaCl, and further purified on MonoQ. The pooled fractions were further purified by size-exclusion chromatography (Superdex 200 Increase 10/300 GL, GE Healthcare). The final RNP was buffer-exchanged to 30 mM HEPES pH8.0 and 150 mM NaCl, sterilized with a syringe filter, concentrated to >20 μM, and flash-frozen for −80°C storage. To account for the nucleic acid component of TfuCascade, nanodrop UV 260/280 measurements were taken alongside a Bradford Assay standard curve. A conversion ratio was determined to more accurately estimate the concentration of the protein components.
TfuCas3 was expressed from M9 minimal media with an N-terminal TwinStrep-PreScission tag and a C-terminal 2xHA-NLS tag from a pET52b plasmid. A 5 ml starting culture was grown from LB media overnight at 37°C, propagated to a 100 mL M9 culture overnight at 37 C, then used to inoculate 3×2 L of M9 media. The trace metal supplement was left out of the standard M9 media to prevent Fe2+ incorporation into the Cas3 active site. 100 μM final concentration of cobalt chloride was added to the cell culture 30 minutes prior to IPTG induction, when the OD600 reached 0.6. Protein expression was induced by 1 mM IPTG overnight at 20°C. The cells were harvested, resuspended in lysis buffer (30 mM HEPES pH 7.5 and 500 mM NaCl), lysed by sonication, and purified with a Strep-Tactin column similar to TfuCascade purification. The eluted protein was treated with PreScission protease overnight at 4°C to remove the TwinStrep tag. Cas3 was further purified over a HiLoad Superdex 200 size-exclusion column (SEC) equilibrated with 30 mM HEPES 7.5 and 150 mM NaCl. The main peak fractions were pooled and concentrated, flash-frozen in liquid nitrogen, and stored at −80°C until needed.
Construction of hESC dual-reporter line and DNMT3b targeting plasmids
Cells for transfection were harvested 2 days post passaging using TrypLE (Life Technologies) and resuspended in OptiMem (Life Technologies) at a final concentration of 5 × 106 cells/mL. 500 μL of cell suspension was added to a 0.4cm cuvette containing 30 μg of the linearized DNMT3B-EGFP vector. Cells were electroporated using condition 320 V, 200 μF, then plated on a 10 cm matrigel-coated dish in E8 media supplemented with 10 μM Y-27632 (Cayman Chemical). 0.5 μg/mL puromycin was added to the medium 3 days post-transfection, and drug-resistant colonies exhibiting uniform EGFP expression were identified by fluorescent microscopy. A single EGFP+ clone was expanded and the puromycin selection cassette removed following electroporation of CRE recombinase mRNA. A subsequent round of targeting was performed as described above using the DNMT3B-tdTomato vector. Individual colonies expressing both tdTomato and EGFP reporters were identified, isolated and expanded. Successful biallelic targeting of the endogenous DNMT3B was confirmed by genotyping PCR using primers flanking the DNMT3B start codon.
To create DNMT3B targeting constructs (hES-2A-DNMT3B-EGFP and hES-2A-DNMT3B-tdTomato), a BAC clone (CTD-2608L15) containing the complete DNMT3B coding region was obtained from CalTech Human BAC Library (Life Technologies). Red-ET recombination was used to insert a DNA cassette encoding a tdTomato or EGFP reporter gene adjacent to a loxP-flanked PGK promoter driven puromycin resistance gene at the DNMT3B start codon in exon 2. The ~40 kb SbfI fragment containing the modified DNMT3B locus was then subcloned into the copy number inducible BAC vector, hES-2A. Prior to transfection, these DNMT3B targeting constructs were linearized by SwaI.
RNP Electroporation of hESCs
The H9-DNMT3B-tdTomato/EGFP dual-reporter cells were electroporated using the Neon Transfection system (ThermoFisher) according to the manufacturer’s instructions. Briefly, reporter cells were individualized with Accutase (ThermoFisher), washed once with DMEM/F12 (ThermoFisher) and resuspended in Neon buffer R to a concentration of 2×106 cells/mL. 20–120 pmoles of NLS-TfuCascade and 20–60 pmoles of NLS TfuCas3 were mixed with approximately 105 cells in buffer R in a total volume of 10 μL. This mixture was then electroporated with a 10 μL Neon tip (1100V, 20ms, 2 pulses) and plated in 24-well matrigel-coated plates containing 500 μL E8 medium supplemented with 10 μM Y-27632. The media was changed to regular E8 medium 24 hrs post electroporation. Cells were cultured in E8 with daily media change until analysis.
RNP Electroporation of HAP1 cells and single cell 6-TG cytotoxicity assay
The HAP1 cells were electroporated using the Neon Transfection system (ThermoFisher) according to the manufacturer’s instructions. Briefly, HAP1 cells were individualized with TrypLE Express (Gibco), washed once with IMDM, 10% FBS and resuspended in Neon buffer R to a concentration of 2×106 cells/mL. 20–60 pmoles of NLS-TfuCascade and 20 pmoles of NLS-TfuCas3 were mixed with approximately 105 cells in buffer R in a total volume of 10 μL. Each mixture was then electroporated with a 10 μL Neon tip (1575V, 10ms, 3 pulses) and plated in 24-well tissue culture plates containing 500 μL IMDM, 10% FBS. Cells were individualized 2 days after electroporation and seeded into 6-well plates at a density of ~200 cells per well. 6-TG (6-Thioguanine, Sigma) was added to the media 2 days after cell seeding at a final concentration of 15 μM. 6-TG selection was carried out for 6 days. The cells were then fixed with ice cold 90% methanol for 30min, washed once with 1x PBS and stained with 0.5% crystal violet at RT for 5min. After destain with water, the plates were allowed to air-dry at RT overnight. The number of surviving colonies on the plate was then counted by OpenCFU (Geissmann, 2013).
Flow Cytometry analysis, FACS sorting and single cell isolation
Cells were individualized with Accutase 4–5 days after electroporation and resuspended in DMEM/F12 media immediately before experiments. For analysis, individualized cells were analyzed on an LSR Fortessa (BD) using 488nm laser for EGFP and 561nm laser for tdTomato. Data analysis was performed using FlowJo® v10.4.1. For FACS sorting, individualized cells were put on a SH800 cell sorter (Sony) fitted with a 130 μm chip and GFP negative cells were sorted directly into a well of a 24-well plate coated with matrigel and filled with 1.5 ml E8 media supplemented with 10 μM Y-27632 and 25 μg/mL recombinant human albumin (Sigma). Sorted cells were then cultured in tissue culture incubator with 5% CO2 at 37°C. Media was changed to regular E8 one day after sorting and daily media change with E8 was carried out thereafter. For isolating single cell clones, GFP-negative and tdTomato-positive cells were sorted directly into 96-well plate (one cell per well) coated with matrigel and filled with 150 μL E8 media supplemented with 10 μM Y-27632 and 25 μg/mL recombinant human albumin (Sigma). Media was changed to regular E8 two days after sorting and media change with E8 was carried out every two days thereafter.
DNA lesion analysis by long-range PCR genotyping
Genomic DNAs of hESCs or HAP1 cells were isolated using Gentra Puregene Cell Kit (Qiagen) per manufacturer protocol. Long-range PCRs in Figs. 3B, 3D, 6E, S3B, and S5B were all done using Q5 DNA Polymerase (NEB). Products were resolved on 1% agarose gel stained by SYBR Safe (Invitrogen) and visualized with Chemidoc MP imager (Biorad). See Supplemental Table 1 for all primers used for long-range PCRs.
To define lesion junctions shown in Figs. 4, 6F, S5 and Supplemental Table 3, lesion PCR reactions were purified using QIAquick PCR Purification Kit (Qiagen), and cloned into PCR-BluntII-TOPO vector (Invitrogen). Colony PCR with M13 forward and reverse primers were carried out from the resulting colonies, and randomly selected positive clone amplicons were Sanger sequenced (Eurofin) using an EGFP reverse primer HZG511 or HPRT primer oYZ960. Sanger sequencing results were analyzed using Snapgene and BLASTN search.
Tn5 tagmentation-based NGS library construction
Tn5 transposase was purified and loaded with one pre-annealed oligo pair ME-A/ME-rev as previously described (Picelli et al., 2014). Tagmentation was performed in 10 mM Tris pH8.5, 5mM MgCl2 and 50% DMF using 300 ng of genomic DNA and 1.4 μg of loaded Tn5, in a total volume of 40 μL. After 7 min incubation at 55°C, tagmentation reactions were stopped by addition of 1 μL Protease K (20mg/ml) and incubation at 55°C for 7 min and 95°C for 10 min. Tagmented DNA was purified with 32 μl AMPure beads and eluted with 15 μL 10 mM T ris pH 8.0. For NGS library construction, 1st step PCR amplification was carried out using Q5 DNA Polymerase for 15 cycles with oligos OYZ510+478, and then treated with Exonuclease I (NEB) to digest excess primers. 2nd step of nested-PCR was done for another 15 cycles using Q5 with OYZ510+511. After Exonuclease I treatment, the 3rd step PCR was carried out for 10 cycles with OYZ510 and index primers. The final NGS libraries were purified using Select-a-Size DNA Clean & Concentrator MagBead Kit (Zymo Research) using a 400 bp cutoff, eluted in 10 mM Tris pH 8.5 and sequenced on Illumina MiSeq with a 500 cycles Nano kit for 50×450 bp paired-end reads. 450 cycles were performed for R2 and 50 cycles for R1.
NGS data analysis
MiSeq R2 sequencing reads were first subjected to adapter trimming using cutadapt 1.8.1 (Martin, 2011) to ensure that all reads began with the expected sequence immediately following the GFP sequencing primer, and trimming the Tn5 adapter sequence from the ends of reads in case of read-through. Reads were then quality trimmed using Trimmomatic v0.33 (Bolger et al., 2014) filter settings “TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:10”, and then aligned to a defined window of the human genome spanning ~130 kb, which covered the entire DNMT3B locus with EGFP sequence inserted along with 91 kb upstream of DNMT3B transcription start site.
Alignment was performed using nucmer 4.0.0beta2 (Kurtz et al., 2004) with a minimum match length of 10 and minimum cluster length of 20. nucmer alignments were then filtered using an in-house python program (see Supplemental Data S1) first to prune any alignments which overlapped by more than 10% of their length with another, longer alignment of the same read, thus removing redundant alignments which would otherwise occur, and then to remove any alignments that were not properly anchored to the expected start site based on the sequencing primer used. Python and bash programs were subsequently used to extract and plot read counts and locations.
Purification and assembly of SpyCas9 RNP
NLS-tagged SpyCas9 was purified using a modified protocol as described previously (Zuris et al., 2015). Briefly, BL21 (DE3) cells were grown at 37°C until the 0D600 reaches 0.6. Protein expression was induced by adding IPTG to a final concentration of 0.5 mM, and allowing the cell to grow overnight at 18°C. Cells were collected by centrifugation, resuspended in 1XPBS with 350 mM NaCl and lysed by sonication. Cleared lysate was mixed with Ni resin at 4°C for 1 hour. After washing with lysis buffer, bound proteins were eluted with 1XPBS, 350 mM NaCl and 0.5 M Imidazole. Proteins eluted from Ni resin was then loaded onto a 5 ml Heparin column (GE Healthcare) and eluted with a step gradient of NaCl (1xPBS with 600 mM NaCl, 850 mM NaCl and 2 M NaCl). Cas9 containing fractions were pooled, concentrated and dialyzed into 1XPBS, 20% glycerol overnight. Dialyzed proteins were filter sterilized and stored at −80°C until use.
The HPRT1-targeting sgRNA for SpyCas9 was generated using GeneArt Precision gRNA Synthesis Kit (ThermoFisher) following manufacture’s instruction. For SpyCas9 RNP assembly, 3 μg (19 pmoles) of NLS-SpyCas9 and 1.2 μg (37 pmoles) of sgRNA were mixed in buffer R to 5 μL. After 10 min incubation at RT, this reaction was mixed with 105 cells in buffer R to a final volume of 10 μL for electroporation.
T7E1 Assay
50 ng genomic DNA was used for PCR amplification using Q5 DNA polymerase supplemented with 1x GC enhancer (NEB) with oligos oYZ954+oYZ955 that flank the targeted site. 20 μL of each PCR product was heated to 95°C for 1min and cooled down to 23°C at a rate of 0.1°C/sec. 10 μL annealed PCR product was digested with 1 μL T7 endonuclease I (NEB) reaction in 1X NEBuffer 2.1 at 37°C for 1hr, and resolved on a 2.5% agarose/1XTAE gel. The gels were imaged with ChemiDoc MP and quantified using Image Lab (BioRad). Editing efficiency (% lesion) was calculated using the formula: 100 x (1 - (1- fraction cleaved)1/2)(Guschin et al., 2010).
In silico off-target prediction
To predict potential off-target sites, we searched the entire human genome for sequences that match the intended target site as closely as possible. We demanded a ‘NAG’ PAM (−3, −2, −1 positions) and a perfectly matched seed-proximal region (positions 1–5, 7–11), while allowing all possible mismatches to the kinked positions (6th, 12th, 18th, 24th, and 30th positions). Top predicted off-targets sites with minimum number of mismatches at the non-seed, non-kinked positions are listed in Table S3. The search and scoring described above were implemented using elementary string operations in python 2.7, with sequence input provided by Biopython, and applied to the hg38 reference sequence (see Supplemental Data S2).
DATA AND SOFTWARE AVAILABILITY
The Tn5-NGS data set has been deposited at the SRA as submission # SRR8146241. Custom python programs are included in the supplemental files.
Supplementary Material
Data S1: Python codes used for lesion analysis. Related to Star Methods.
Data S2: Python codes used for off target prediction. Related to Star Methods.
Table S2: Sanger sequencing results of Type I CRISPR editing outcome. Related to Figures 4 and S5
Highlights.
T. fusca Type I CRISPR-Cas enables long-range genome deletions in human cells
The target binding complex Cascade and helicase-nuclease Cas3 are both required
Genome engineering by Type I CRISPR is crRNA-guided and programmable
Heterogeneous deletions up to 100 kb is induced upstream of a genomic target site
Acknowledgements
This work is supported by National Institutes of Health (NIH) grants GM128637 to P.L.F., GM118174 and GM102543 to A.K, GM117268 to Y.Z., and University of Michigan institutional fund and Biological Scholar Award to Y.Z. The authors thank J. Budhathoki and R.A. Battaglia for helpful discussions, and Xufei Zhou for graphical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
A patent application has been filed describing the invention reported herein.
References
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Blosser TR, Loeff L, Westra ER, Vlot M, Kunne T, Sobota M, Dekker C, Brouns SJJ, and Joo C (2015). Two distinct DNA binding modes guide dual roles of a CRISPR-Cas protein complex. Mol Cell 58, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, and Ehrlich SD (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- Boyle EA, Andreasson JOL, Chircus LM, Sternberg SH, Wu MJ, Guegler CK, Doudna JA, and Greenleaf WJ (2017). High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proceedings of the National Academy of Sciences of the United States of America 114, 5461–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic acids research 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliando BJ, and Voigt CA (2015). Targeted DNA degradation using a CRISPR device stably carried in the host genome. Nature communications 6, 6989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, Maeda T, Paw BH, and Orkin SH (2014). Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. The Journal of biological chemistry 289, 21312–21324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu F, Zhu C, Ji J, Zhou X, Feng X, and Guang S (2014). Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci Rep 4, 7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, Lin KC, Huang H, Liu T, Marina RJ, et al. (2017). A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nature methods 14, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillard KE, Brown MW, Johnson NV, Xiao Y, Dolan A, Hernandez E, Dahlhauser SD, Kim Y, Myler LR, Anslyn EV, et al. (2018). Assembly and Translocation of a CRISPR-Cas Primed Acquisition Complex. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Gerritzen MJ, Suarez-Diez M, Kunne T, Boekhorst J, van Hijum SA, Staals RH, and Brouns SJ (2014). Degenerate target sites mediate rapid primed CRISPR adaptation. Proceedings of the National Academy of Sciences of the United States of America 111, E1629–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann Q (2013). OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS One 8, e54072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, and Rebar EJ (2010). A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649, 247–256. [DOI] [PubMed] [Google Scholar]
- Hayes RP, Xiao Y, Ding F, van Erp PB, Rajashankar K, Bailey S, Wiedenheft B, and Ke A (2016). Structural basis for promiscuous PAM recognition in type I-E Cascade from E. coli. Nature 530, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Bhat P, Guegler CK, Sternberg SH, Nogales E, and Doudna JA (2014). CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proceedings of the National Academy of Sciences of the United States of America 111, 6618–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Nam KH, Ding F, Lee H, Wu L, Xiao Y, Farchione MD Jr., Zhou S, Rajashankar K, Kurinov I, et al. (2014). Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nature structural & molecular biology 21, 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Golden SM, van Erp PB, Carter J, Westra ER, Brouns SJ, van der Oost J, Terwilliger TC, Read RJ, and Wiedenheft B (2014). Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science 345, 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Hawkins JA, Jones SK Jr., Xiao Y, Rybarski JR, Dillard KE, Hussmann J, Saifuddin FA, Savran CA, Ellington AD, et al. (2017). Massively Parallel Biophysical Analysis of CRISPR-Cas Complexes on Next Generation Sequencing Chips. Cell 170, 35–47 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, and Kim JS (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, and Doudna JA (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, and Liu DR (2017). CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 169, 559. [DOI] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, and Bradley A (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nature biotechnology 36, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivoy A, Rutkauskas M, Kuznedelov K, Musharova O, Rouillon C, Severinov K, and Seidel R (2018). Primed CRISPR adaptation in Escherichia coli cells does not depend on conformational changes in the Cascade effector complex detected in Vitro. Nucleic acids research 46, 4087–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunne T, Kieper SN, Bannenberg JW, Vogel AI, Miellet WR, Klein M, Depken M, Suarez-Diez M, and Brouns SJ (2016). Cas3-Derived Target DNA Degradation Fragments Fuel Primed CRISPR Adaptation. Molecular cell 63, 852–864. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, and Salzberg SL (2004). Versatile and open software for comparing large genomes. Genome biology 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Pan S, Zhang Y, Ren M, Feng M, Peng N, Chen L, Liang YX, and She Q (2016). Harnessing Type I and Type III CRISPR-Cas systems for genome editing. Nucleic acids research 44, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeff L, Brouns SJJ, and Joo C (2018). Repetitive DNA Reeling by the Cascade-Cas3 Complex in Nucleotide Unwinding Steps. Molecular cell 70, 385–394 e383. [DOI] [PubMed] [Google Scholar]
- Luo ML, Mullis AS, Leenay RT, and Beisel CL (2015). Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic acids research 43, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, and Koonin EV (2006). A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, and Koonin EV (2015). Annotation and Classification of CRISPR-Cas Systems. Methods in molecular biology 1311, 47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nature reviews Microbiology 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal; Vol 17, No 1: Next Generation Sequencing Data Analysis. [Google Scholar]
- Mojica FJ, García-Martínez J, and Soria E (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. Journal of molecular evolution 60, 174–182. [DOI] [PubMed] [Google Scholar]
- Mulepati S, and Bailey S (2013). In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. The Journal of biological chemistry 288, 22184–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, Heroux A, and Bailey S (2014). Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science 345, 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Reinius B, Sagasser S, Winberg G, and Sandberg R (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, and Vergnaud G (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. [DOI] [PubMed] [Google Scholar]
- Rath D, Amlinger L, Hoekzema M, Devulapally PR, and Lundgren M (2015). Efficient programmable gene silencing by Cascade. Nucleic acids research 43, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding S, Sternberg SH, Marshall M, Gibb B, Bhat P, Guegler CK, Wiedenheft B, Doudna JA, and Greene EC (2015). Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell 163, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkauskas M, Sinkunas T, Songailiene I, Tikhomirova MS, Siksnys V, and Seidel R (2015). Directional R-Loop Formation by the CRISPR-Cas Surveillance Complex Cascade Provides Efficient Off-Target Site Rejection. Cell reports. [DOI] [PubMed] [Google Scholar]
- Sashital DG, Wiedenheft B, and Doudna JA (2012). Mechanism of foreign DNA selection in a bacterial adaptive immune system. Molecular cell 46, 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, and Severinov K (2011). Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proceedings of the National Academy of Sciences of the United States of America 108, 10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, et al. (2015). Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Molecular cell 60, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkunas T, Gasiunas G, Waghmare SP, Dickman MJ, Barrangou R, Horvath P, and Siksnys V (2013). In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J 32, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperger JM, Chen X, Draper JS, Antosiewicz JE, Chon CH, Jones SB, Brooks JD, Andrews PW, Brown PO, and Thomson JA (2003). Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proceedings of the National Academy of Sciences of the United States of America 100, 13350–13355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ, and Russell R (2018). Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol Cell 71, 816–824 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, and Seidel R (2014). Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proceedings of the National Academy of Sciences of the United States of America 111, 9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PB, Kunne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, et al. (2012). CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJ, van der Oost J, Doudna JA, and Nogales E (2011). Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477, 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo M, Dolan AE, Liao M, and Ke A (2018). Structure basis for RNA-guided DNA degradation by Cascade and Cas3. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, and Ke A (2017). Structure Basis for Directional R-loop Formation and Substrate Handover Mechanisms in Type I CRISPR Cas System. Cell 170, 48–60 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sheng G, Wang J, Wang M, Bunkoczi G, Gong W, Wei Z, and Wang Y (2014). Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature 515, 147–150. [DOI] [PubMed] [Google Scholar]
- Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, and Liu DR (2015). Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 33, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Python codes used for lesion analysis. Related to Star Methods.
Data S2: Python codes used for off target prediction. Related to Star Methods.
Table S2: Sanger sequencing results of Type I CRISPR editing outcome. Related to Figures 4 and S5
Data Availability Statement
The Tn5-NGS data set has been deposited at the SRA as submission # SRR8146241. Custom python programs are included in the supplemental files.