

Summary
MITF and MYC are well-known oncoproteins and members of the basic helix-loop-helix leucine zipper (bHLH-Zip) family of transcription factors (TFs) recognizing hexamer E-box motifs. MITF and MYC not only share the core binding motif, but are also the two most highly expressed bHLH-Zip transcription factors in melanocytes, raising the possibility that they may compete for the same binding sites in select oncogenic targets. Mechanisms determining the distinct and potentially overlapping binding modes of these critical oncoproteins remain uncharacterized. We introduce computational predictive models using local sequence features, including a boosted convolutional decision tree framework, to distinguish MITF vs. MYC-MAX binding sites with up to 80% accuracy genome wide. Select E-box locations that can be bound by both MITF and MYC-MAX form a separate class of MITF binding sites characterized by differential sequence content in the flanking region, diminished interaction with SOX10, higher evolutionary conservation, and less tissue-specific chromatin organization.
Keywords: MITF, MYC-MAX, Co-binding Factors, Machine Learning, Boosted Convolutional Decision Tree
Introduction
Microphthalmia-associated transcription factor (MITF) is a critical regulator of melanocyte biology and serves multifarious roles in the development, survival, and malignant transformation of melanocytes (Levy & Fisher, 2011). On the one hand, it promotes melanocyte differentiation and controls the expression of melanocyte-specific genes needed for skin pigmentation. On the other hand, it also regulates cell cycle progression and cell survival by modulating the transcription of key gatekeeper genes expressed across multiple cell lineages. MITF’s melanocyte-specific control over these non-lineage-specific genes may lead to its oncogenic functions in melanomas, contributing to cancer proliferation and increased potential for tumorigenic transformation (Garraway et al., 2005). Furthermore, MITF has been recently implicated in conferring drug resistance to BRAF inhibitors (Haq, Shoag, et al., 2013; Haq, Yokoyama, et al., 2013; Webster et al., 2014). MITF thus represents a central hub in the complex regulatory network governing several salient aspects of melanocyte biology.
Another well-known transcription factor (TF) in the same phylogenetic clade as MITF is MYC (Skinner, Rawls, Wilson-Rawls, & Roalson, 2010), one of the earliest discovered and most potent oncogenes. A large body of evidence links MYC to the generation, progression, and maintenance of a wide range of cancers. Similar to MITF, MYC robustly potentiates tumorigenic transformation, promotes cancer proliferation, and confers cancer drug resistance.
MITF and MYC both belong to the same family of basic helix-loop-helix leucine zipper (bHLH-Zip) TFs that bind a consensus hexamer E-box motif CANNTG. The bHLH domain contains the DNA binding function, while the leucine zipper domain mediates dimerization with another bHLH TF. MITF has been shown to form heterodimers with the related MiT family proteins TFE3, TFEB, and TFEC, but none of these transcription factors show a high expression level (15.5, 3.7, 0.0 FPKM respectively, compared to 272.6 FPKM for MITF) in the melanocyte lineage, where MITF thus likely forms a homodimer. Likewise, MYC forms a MYC-MAX heterodimer complex (Jones, 2004). MAX itself can form homodimers, but such homodimers poorly bind DNA and are transcriptionally inert (Amati et al., 1992; Kretzner, Blackwood, & Eisenman, 1992). In addition, the expression level of MAX in melanocytes is 5-fold less than MYC, likely leading to a significantly lower stoichiometric concentration of MAX-MAX homodimers compared to MYC-MAX heterodimers.
MITF and MYC are the two most highly expressed bHLH-Zip transcription factors in melanocytes (273 FPKM and 254 FPKM, respectively; Supplementary Methods). Since they share the core DNA binding motif, unraveling the mechanisms determining the genome-wide distinct and shared binding modes of these oncoproteins remains a major challenge. In particular, it is important to understand which local genomic features allow MITF and MYC-MAX to find their respective regulatory targets and to what extent these genomic features can moderate their competition for a single binding site. Similar questions regarding the discriminatory sequence features associated with specific bHLH-Zip family members were previously investigated in regulatory regions of select genes (Aksan & Goding, 1998; F. Fisher et al., 1993; F. Fisher & Goding, 1992). To systematically address these questions in a genome-wide analysis, we constructed tree-based models of increasing complexity to predict experimentally detected MITF vs. MYC-MAX binding activities genome wide in human melanoma cells. First, we constructed a Random Forest model based on scanning sequences with known TF position-specific scoring matrices, two nucleotides flanking the core E-box motif, and local GC content to assess the performance of a simple model using information about (1) cooperating factors that may help recruit MITF and MYC-MAX to specific genomic loci, and (2) subtle differences in the binding motif of MITF and MYC-MAX. We then improved upon this approach by constructing a Boosted Decision Tree (BDT) model to sequentially minimize prediction errors. Finally, we developed a Boosted Convolutional Decision Tree (BCDT) algorithm that learns classifying genomic features from raw DNA sequences. Just as in a convolutional neural network, the main idea behind BCDT is to detect translationally invariant sequence features; but, BCDT has the advantage that existing tree-based analysis methods can be readily applied to yield interpretable results.
Our models revealed several discriminating features associated with either MITF or MYC-MAX binding. Most prominent among these is a T nucleotide flanking the 5’ end of an E-box. This finding represents a genome-wide generalization of the previously reported differential inhibition between CPF-1 and PHO4 bHLH-Zip proteins in S. cerevisiae by a 5’ T (F. Fisher et al., 1993; F. Fisher & Goding, 1992) and the inhibition of MYC binding by a 5’ T or a 3’ A flanking CACGTG at select loci (F. Fisher et al., 1993; Solomon, Amati, & Land, 1993). Another discriminating feature is the proximal presence of SOX10 co-binding factor. By contrast, sequences that lack specific features preferred by either MITF or MYC-MAX form a distinct subclass that may be equally bound by either.
Methods
Transcription factor and histone modification ChIP-seq experiments
Performing ChIP-seq for MYC using commercially available antibodies is challenging, and the difficulty is also apparent in the ENCODE data (Encode Project Consortium, 2012). Therefore, we instead performed MAX ChIP-seq in the COLO829 cell line as a proxy for assessing the genome-wide locations of MYC-MAX complex. Corresponding MITF ChIP-seq in COLO829 was obtained from a publicly available dataset (Webster et al., 2014) and normalized using our own Input data in COLO829. We obtained SOX10 ChIP-seq in 501-mel from a publicly available dataset (Laurette et al., 2015).
siRNA transfection
Primary human melanocytes were transfected with control siRNA pool (siGENOME Non-Targeting siRNA #2 (Dharmacon)) or siRNA pool against MITF (siGENOME MITF siRNA (Dharmacon)) using lipidoid as described before (Li et al., 2012). qPCR and RNA-seq quantification of the level of MITF in siMITF vs. control cells showed a 4-fold suppression of MITF (Supplementary Fig. S1).
Primary human melanocyte culture
Primary human melanocytes were cultured in TIVA medium (Ham’s F-10 (Corning), 7.5% fetal bovine serum (Atlanta Biologicals), 50 ng/ml 12-O-tetradecanoylphorbol-13-acetate, 0.1 mM 3-isobutyl-1-methylxanthine, 1 μM sodium orthovanadate, 1 mM N6,2’-O-Dibutyryladenosine 3’,5’-cyclic monophosphate sodium salt (Sigma-Aldrich)). All methods using the primary melanocytes were carried out in accordance with the ethical guidelines and regulations of the institutional review board (IRB #2013P000093) at Harvard Medical School, and all experimental protocols were approved by the board. Tissues were obtained with written informed consent according to Partner’s Healthcare IRB guidelines.
Gene expression profiling
We performed RNA-seq experiments in primary melanocytes under siMITF and siControl conditions. We used Tophat2 and CuffDiff2 (Trapnell et al., 2013) to quantify the FPKM level of genes.
Decision Tree Methods
A comprehensive description of our computational approaches is available in Supplementary Methods.
Results
Distribution of MITF and MYC-MAX ChIP-seq Peaks
Supplementary Table S1 describes all experimental data used in this manuscript. We have assessed the quality of MAX ChIP-seq data in COLO829 using published methods that are designed to separate background from ChIP-enriched regions (Diaz, Nellore, & Song, 2012; Diaz, Park, Lim, & Song, 2012; Li et al., 2012). We found that 6% of the genome had statistically significant enrichment of MAX ChIP-seq signal. Supplementary Fig. S2 shows representative MAX and MITF binding locations and a clustering heatmap of the read densities of MITF, MAX, H3K27ac and H3K4me3 ChIP-seq data. Furthermore, the ChIP-seq signal showed localization around gene transcription start sites (Supplementary Fig. S3a). Analysis using the software GREAT (McLean et al., 2010) showed ‘translational initiation’ as the most enriched GO term (q-value 4.7×10−26). At 5% FDR, MAX ChIP-seq signal has 6415 peaks (Supplementary Methods). We divided MAX ChIP-seq peaks into quintiles based on the binding strength. Motif analysis using MEME and DREME (Bailey et al., 2009) showed E-box motifs to be enriched in each quintile (Supplementary Fig. S3b).
The ENCODE project (Encode Project Consortium, 2012) provides MAX ChIP-seq data in 9 different cell types and conditions. Out of the 6415 MAX peaks in our dataset, 3803 peaks were present in at least one of the ENCODE cell, while 2612 peaks were unique to our dataset. The number of MAX peaks in our dataset was 58% of the median of ENCODE Project MAX datasets. Furthermore, 5% of MITF peaks in COLO829 and 39% of MYC-MAX peaks overlapped with CpG islands. H3K27ac histone modifications were found in 62% of MAX peaks, 87% of MITF only sites, and 91% of MITF and MYC-MAX overlapping sites.
Random Forest predictor detects SOX10 as a discriminatory cooperating factor of MITF
To assess the degree to which MITF and MYC-MAX binding of E-boxes in the melanoma cell line COLO829 can be distinguished by basic machine learning methods, we first conducted a pilot study using a Random Forest (RF) classifier trained to classify DNA sequences centered around MITF-bound and MYC-MAX-bound canonical E-boxes (CACGTG, CATGTG, CACATG) (Hemesath et al., 1994). We here restricted our attention to canonical E-boxes so as to focus our first model on well-characterized binding motifs of MITF and MYC-MAX. Our subsequent models contained increasing levels of complexity and accommodated other E-box motifs as well as de novo motif discovery. Whether a canonical E-box was bound or not was determined by ChIP-seq experiments in the melanocyte lineage (Supplementary Table S1 and Methods). The training features of this RF classifier were as follows: (1) Presence or absence of known TF motifs in a 200 bp window centered at a canonical E-box (Supplementary Methods). Some of these motifs differentially enriched between MITF and MYC-MAX binding regions could indicate the presence of a cooperating TF that may either directly interact with MITF or MYC-MAX, or cause protein-induced DNA bending that stabilizes the binding of either MITF or MYC-MAX (Travers, 1997). (2) The two nucleotides flanking the 5’ and 3’ ends of a canonical E-box. These flanking nucleotides could reflect a preference stemming from the slight differences in the DNA binding domains of MITF and MYC-MAX (Aksan & Goding, 1998). (3) GC and CpG content in a 100bp window centered at an E-box. GC and CpG content are known to affect DNA flexibility and DNA configuration (Olson, Gorin, Lu, Hock, & Zhurkin, 1998), which may play a role in creating differentially affinity for MITF vs. MYC-MAX.
The 5-fold cross-validation accuracy of the RF classifier on a balanced set of experimentally detected 2975 MITF and 2975 MYC-MAX mutually exclusive binding sites was 73%. The predictive power of our RF classifier showed that the DNA sequence flanking a canonical E-box motif was highly predictive of MITF vs. MYC binding. The most important features used by the classifier, as measured by the mean decrease in Gini index of node impurity, were (Supplementary Table S2): (1) the presence of MYC-MAX motif from TRANSFAC, but not the experimentally inferred motif, and closely related bHLH-Zip TF motifs, (2) the presence of a T nucleotide at the 5’ end of an E-box, (3) GC and CpG content, and (4) the presence of a SOX10 motif (Fig. 1c). We will subsequently describe a method using a new computational framework of boosted convolutional decision trees that learns the first three features from raw binding sequences of MITF and MYC-MAX.
Figure 1:
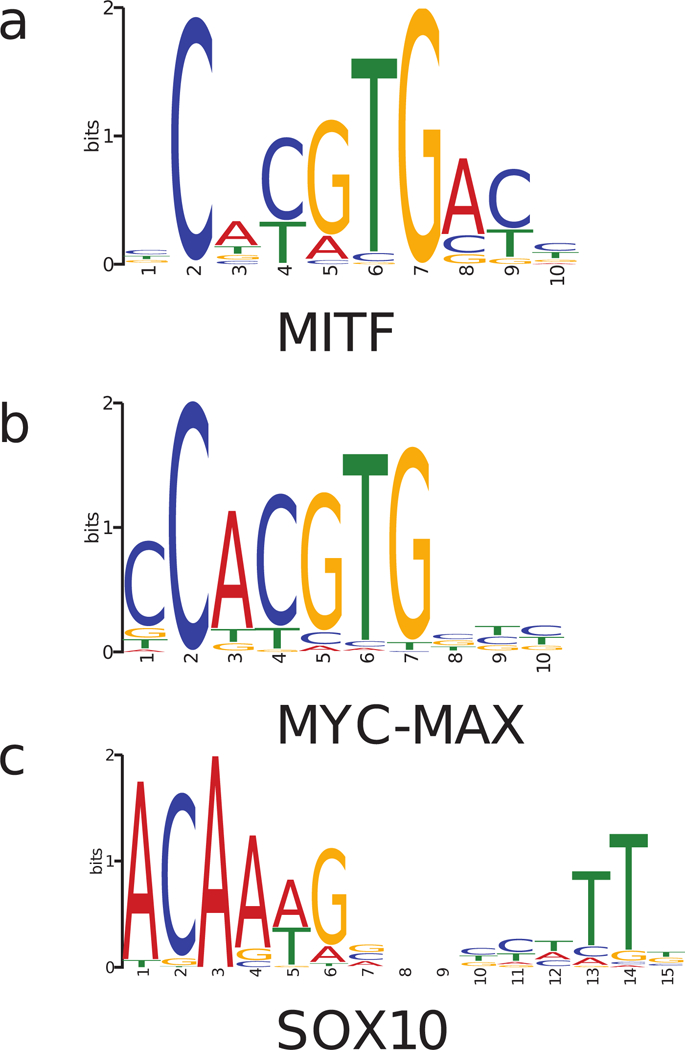
Motifs of (a) MITF, (b) MYC-MAX, and (c) SOX10 inferred from their respective ChIP-seq data (Supplementary Methods).
SOX10, a TF previously reported to co-localize with MITF and regulate the cellular functions of melanocytes and melanoma (Laurette et al., 2015; Seberg, Van Otterloo, & Cornell, 2017), was the TF with the highest importance score among all non bHLH-Zip TF motifs. SOX10 is a high-mobility-group TF expressed in the neural crest and neural crest-derived cells. Given that SOX10 was among the most highly expressed TFs in melanocytes and melanoma and that the presence of its motif showed high importance in classification between MITF and MYC-MAX binding sites, we further examined the co-localization pattern of MITF and SOX10 using both motif analysis and ChIP-seq data.
SOX10 motifs showed a strong enrichment around 30–150 bps from MITF-bound E-boxes, but this enrichment was absent in MYC-MAX binding sites (Fig. 2a). Such bimodal co-localization was not exhibited by any other motif analyzed (205 motifs of JASPAR core vertebrates and 834 motifs of TRANSFAC database). Although SOX10 has been previously shown to bind DNA either as a monomer or a dimer (Peirano & Wegner, 2000), the SOX10 motif inferred from ChIP-seq data suggested a dimer function in melanoma (Fig. 1c). In addition to the motif enrichment, the ChIP-seq read density of SOX10 in the melanoma cell line 501-mel also exhibited a ~5-fold enrichment around MITF ChIP-seq peaks, but not MYC-MAX peaks (Fig. 2b). In terms of peak numbers, 7.8% of MITF ChIP-seq peaks with a canonical E-box in COLO829 overlapped a SOX10 ChIP-seq peak in 501-mel, while only 1.0% of MYC-MAX peaks in COLO829 overlapped a SOX10 peak. These findings together provided strong evidence for preferential co-localization of SOX10 with MITF over MYC.
Figure 2:
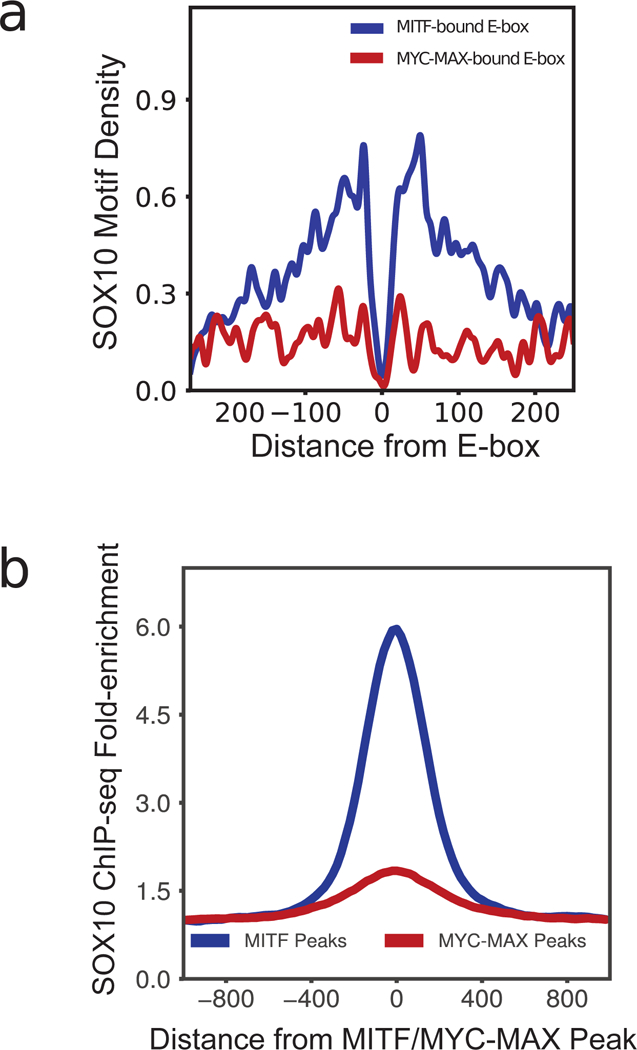
(a) Density of SOX10 motifs around MITF-bound and MYC-MAX-bound E-boxes. SOX10 motif shows a strong co-localization 30–150 bps from MITF-bound E-boxes, but does not co-localize with MYC-MAX E-boxes. (b) ChIP-seq read density of SOX10 enrichment around MITF ChIP-seq peaks and MAX ChIP-seq peaks.
MITF binding sites have two subclasses distinguished by sequence features and epigenetic signatures
Analysis of ChIP-seq data revealed that while the majority of E-boxes were bound exclusively by MITF or MYC-MAX, approximately 23% of E-boxes bound by MITF were also bound by MYC-MAX. The E-boxes that were bound by both MITF and MYC-MAX had sequence characteristics that clearly distinguished them from those bound by MITF exclusively, thus forming a distinct subclass of MITF binding sites. The distinguishing characteristics of the overlapping class included a lack of co-localization with SOX10 (binomial test p-value = 1.6×10−23), higher GC (Wilcoxon rank-sum test p-value = 1.4 ×10−117) and CpG content (Wilcoxon rank-sum test p-value = 1.1 ×10−84), and higher evolutionary conservation (binomial test p-value = 4.1 ×10−9) (Fig. 3; Supplementary Methods). Furthermore, knocking down MITF in melanocytes showed that H3K27ac and H3K4me3 modifications, markers of an active promoter, showed a higher response to MITF depletion in sites bound exclusively by MITF (binomial test p-value = 2.2 ×10−19 and 5.9 ×10−10, respectively). Median fold-enrichment of H3K27ac and H3K4me3 at responsive sites was 5.58 and 2.91, respectively (Supplementary Table S3). In concordance with these epigenetic signatures, the transcription level of genes with TSS within 10 kb of an MITF-bound E-box also showed a higher response to MITF depletion in sites bound exclusively by MITF (binomial test p-value = 0.04) (Fig. 3c). Distribution of MITF and MYC-MAX peaks shows a trend of increasing MITF occupancy with decreasing MYC-MAX occupancy and vice versa and increasing responsiveness of H3K27ac to siMITF knockdown with increasing MITF binding strength (Supplementary Fig. S3c). The subset of MITF-bound E-boxes also bound by MYC-MAX thus forms a distinct subclass characterized by chromatin organization substantially less dependent on the expression level of MITF, possibly due to MYC-MAX being able to substitute for MITF at these sites.
Figure 3:
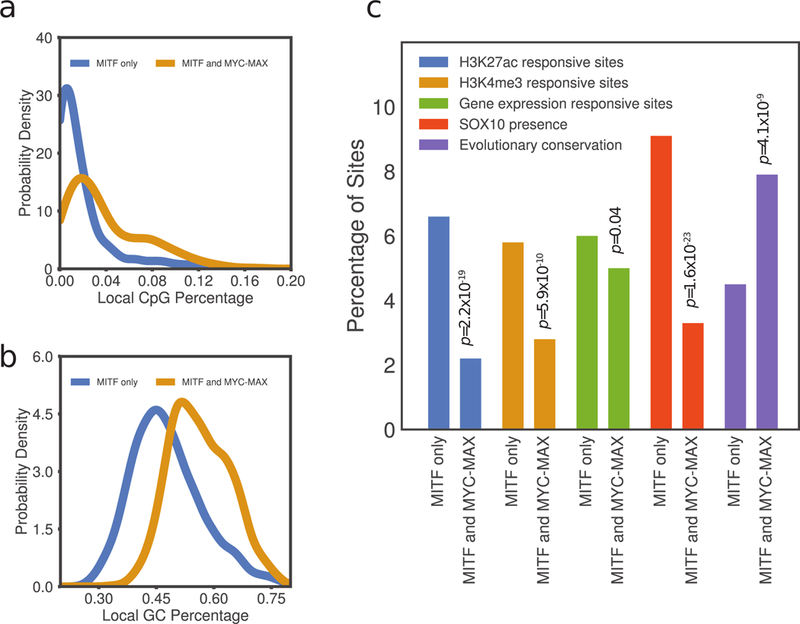
(a) Distribution of average GC nucleotide frequency in 100 bp windows centered at E-boxes bound by both MITF and MYC and those bound by MITF exclusively. (b) Distribution of average CpG di-nucleotide frequency in 100 bp windows around E-boxes bound by both MITF and MYC and those bound by MITF exclusively. (c) Distinguishing characteristics of the overlapping E-box class, including H3K27ac, H3K4me3 histone modifications, gene expression response to siMITF knockdown, SOX10 co-localization, and E-box evolutionary conservation. Binomial test p-values for testing the difference between the two subclasses of E-boxes are indicated (Supplementary Methods).
Using 1kb as a cutoff distance for proximal promoters, we found that E-boxes bound by both MITF and MYC-MAX and associated with H3K27ac markers are 31% in proximal and 69% in distal sites. E-boxes that were bound by MITF alone and associated with H3K27ac markers were found 12% in proximal and 88% in distal sites. Local CpG content based on 100pb window was 2.7% and 7.8% in distal and proximal sites, respectively, for the MITF and MYC-MAX co-bound E-boxes, and 1.3% and 6.8% in distal and proximal sites, respectively, for MITF only E-boxes. Conservation score was 1.4x higher in the proximal sites vs. the distal sites in the MITF and MYC-MAX co-bound E-boxes and 3.2x higher in proximal vs. distal sites in the MITF only class.
Predictive sequence features distinguish between MITF- and MYC-MAX-bound sequences
To relax the previously imposed condition that the input DNA sequences be centered around a canonical E-box (CACGTG, CACATG, or CATGTG), a non-convolutional boosted decision tree (BDT) model was trained to classify between MITF-bound and MYC-MAX-bound sequences, each centered around a ChIP-seq peak summit and represented as a binary indicator vector of TF motif presence (Supplementary Methods). All bHLH TF motif counts were not included in the feature set, thereby enforcing the model to focus on discovering potential cooperating factors of MITF and MYC-MAX. Using only these non-E-box motif features, an area under the ROC curve (AUC) of 0.82 (Fig. 4a) was achieved by the BDT classifier.
Figure 4:
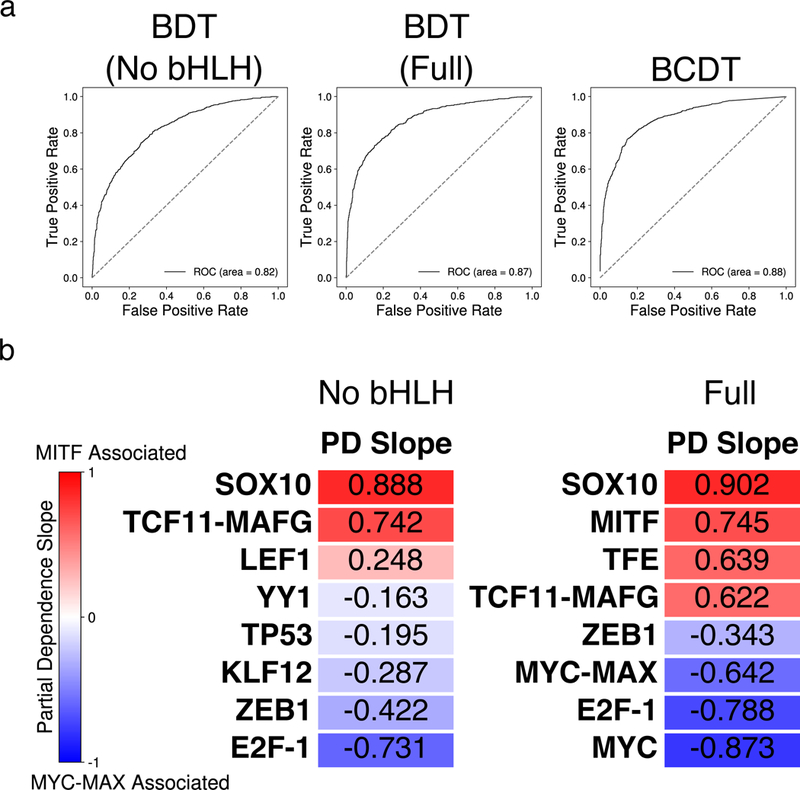
(a) Area under the receiver-operating characteristic (ROC) curve for three different models trained to classify between MITF- and MYC-MAX-bound sequences. From left to right: BDT model with bHLH motifs removed; BDT model with full set of motifs; BCDT model. (b) Heatmap of the partial dependence slopes, ordered by slope, of the eight features with greatest importance for the BDT models with bHLH motifs removed (left) and using the full set of motif features (right). Positive (negative) slope values indicate a positive (negative) association between the presence of a particular motif and the model’s prediction that a sequence is bound by MITF (MYC-MAX). The relevant TRANSFAC IDs are as follows: (LEF1:M00805), (YY1:M00793), (TP53:M00761), (KLF12:M00468), (E2F-1:M00428), (TFE:M01029), (SREBP-1:M00220), (MYC: M00799).
To interpret the trained BDT model and assess feature importance for making predictions, we examined partial dependence plots. A partial dependence plot shows the marginal average output of a predictive model as a function of a single feature and thus evaluates the overall effect that each feature has on the model’s output. In our study, a positive slope for a particular motif’s partial dependence plot implies that the presence of that motif is preferentially associated with MITF binding. We first measured the importance of a feature based on how often that feature is used in the BDT’s decision-making process and ranked the features based on their estimated importance. Fig. 4b and Supplementary Fig. S4 show the partial dependence slopes and plots, respectively, for the eight most important features. Notably, SOX10 was the third most important feature; and, the partial dependence plot of the SOX10 motif indicated that the presence of a SOX10 motif was positively associated with MITF. Furthermore, even though other motifs from the SOX family were also present in the feature set, the SOX10 motif had a much greater feature importance score than any other SOX family motif, supporting the role of SOX10 specifically as a cooperating factor important for determining MITF binding. Besides SOX10, our model also detected LEF1 as an important feature promoting MITF localization, in agreement with its previously reported physical interaction with MITF (Yasumoto et al., 2002). Furthermore, the importance ranking of LEF1 only decreased from 8th to 9th when our model was trained on a dataset with SOX10-associated (either overlapping a SOX10 ChIP-seq peak or containing a SOX10 motif) MITF and MYC-MAX sequences removed, suggesting that there remained a significant co-localization effect between LEF1 and MITF even after taking into account potential co-localization of LEF1 and SOX10. LEF1 motif was still significantly more common in non-SOX10-associated MITF sequences than in non-SOX10-associated MYC-MAX sequences (binomial test p-value =1.14×10−20, Supplementary Table S4).
Likewise, our model found known co-localizing factors of MYC, YY1 (Shrivastava et al., 1993) and E2F1 (Leung, Ehmann, Giangrande, & Nevins, 2008), to be important features favoring the MYC-MAX binding class over MITF. Although YY1 has also been shown to physically interact with MITF (Li et al., 2012; Seberg et al., 2017), our finding demonstrates that YY1 is more enriched in MYC-MAX binding sites than MITF binding sites genome wide. Furthermore, we analyzed the distributions of the output value of the BDT model without bHLHL motifs for sites bound by only MITF, only MYC-MAX, and both MITF and MYC-MAX (Supplementary Fig. S5). We observed that the BDT model tended to output an intermediate prediction value when attempting to classify shared sites bound by both MITF and MYC-MAX, demonstrating that these sites contained mixed sequence characteristics of both TFs. Finally, to check for other important motif features that might have been correlated with SOX10, we removed SOX10 from our feature set and repeated our BDT analysis. We found that the remaining seven of the original eight most important features still comprised the seven new most important features, while ZIC2 moved from 9th to 8th in importance ranking. The relatively small changes suggest that our method is robust to additions and subtractions of features.
This BDT method based on scanning with known TF motifs relied on previous annotations that contained an incomplete set of TF binding motifs. In order to carry out a more unbiased analysis, we developed a boosted convolutional decision tree (BCDT) model for de novo motif discovery and classification. This new model was trained on raw one-hot-encoded DNA sequences, thus requiring no prior information to distinguish between MITF-bound and MYC-MAX-bound sequences (Supplementary Methods). The trained BCDT model achieved high accuracy with an AUC of 0.88 (Fig. 4a), demonstrating that it could successfully distinguish between MITF-bound and MYC-MAX-bound sequences by learning informative motifs and sequence features from the set of training sequences.
To begin interpreting the sequence patterns learned by the BCDT, we first measured the effect of GC content on the output of our model by regressing the predicted probability that a sequence was MYC-MAX-bound against its percent GC content. The linear regression analysis yielded an R2 and p-value of 0.49 and 3.4×10−296, respectively (Fig. 5a), demonstrating that our model learned a strong preference of MYC-MAX for sequences with higher GC content than the sequences bounded by MITF. Supporting the model’s learned GC bias in MYC-MAX-bound sequences, the distribution of percent GC content of MYC-MAX-bound sequences was highly shifted compared to MITF-bound sequences and randomly drawn sequences from DNase I hypersensitive regions (Fig. 5b).
Figure 5:
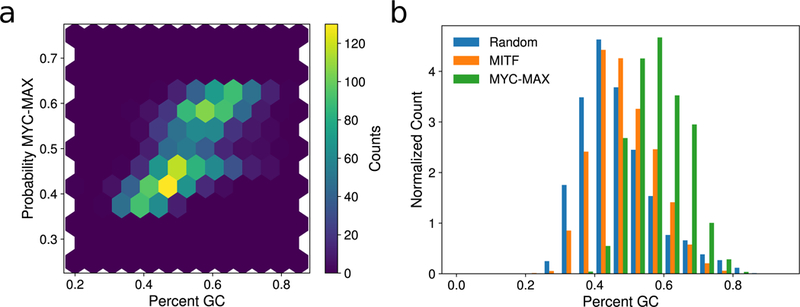
(a) Density plot showing a positive correlation (Pearson r = 0.70) between percent GC content and the probability of a sequence being MYC-MAX-bound according to our BCDT model trained to classify between MITF- and MYC-MAX-bound sequences. (b) Normalized histogram of GC content percentage for MITF-bound, MYC-MAX-bound, and random DNase I hypersensitive sequences, demonstrating relative GC enrichment in MYC-MAX-bound sequences.
MITF and MYC-MAX have different preferences for their binding motif
Within each sequence, a subsequence was labeled as either “pro-MITF” or “pro-MYC-MAX” based on whether its presence increased or decreased the BCDT’s predicted probability of MITF binding, respectively. MEME was then used to align the resulting sets of “pro-MITF” and “pro-MYC-MAX” subsequences separately and discover significantly enriched motifs in each category (Bailey et al., 2009). In both cases, variants of the core E-box element were found, but distinct differences could be observed in the learned motifs (Supplementary Fig. S6a-b).
First, the two central nucleotides in the canonical E-box had more flexibility for MITF. That is, while MYC-MAX generally preferred the canonical E-box pattern CACGTG, a large fraction of MITF-bound sequences contained E-box variants with the two central nucleotides deviating from the expected ‘CG’. Specifically, the variants CATGTG and CACATG were abundant in MITF-bound sequences (36.7% for MITF vs. 19.9% for MYC, Supplementary Fig. S6c). Furthermore, both MITF and MAX rarely bound the motif CATATG that had both middle nucleotides deviating from the canonical hexamer (Supplementary Fig. S6c). Second, the flanking nucleotides on each side of the canonical hexamer E-box were important for distinguishing between MITF vs. MYC-MAX binding. Specifically, the E-boxes bound by MITF tended to be flanked by T and A nucleotides, while the MYC-MAX-bound E-boxes were preferentially flanked by a C or G nucleotide on either side (Supplementary Fig. S6a-b). This pattern was verified by ranking all possible octamers by the percentage of MITF-bound and MYC-MAX-bound sequences containing that octamer: in MITF-bound sequences, the three most common octamers were TCACATGA, ATCACATG, and GTCACATG (including reverse complements). In sharp contrast, the three most common octamers for MYC-MAX were CACGTGGC, CACGTGGG, and CCACGTGG.
To confirm the preferences in E-box variations of MITF vs. MYC-MAX, a non-convolutional BDT model was trained to classify between MITF vs. MYC-MAX using the full list of motif-count features (Supplementary Methods), this time including bHLH TF motifs but not the MITF and MYC-MAX motifs learned by BCDT. This BDT model achieved an AUC of 0.87 (Fig. 4a), an improvement from the BDT model with bHLH TF motifs removed from its feature set. This increase in performance indicated that information about the presence or absence of various bHLH TF motifs added distinguishing power to the BDT trained without the use of bHLH TF motifs. We then ranked all the features by their relative importance. Fig. 4b and Supplementary Fig. S4 show the partial dependence slopes and plots for the eight most important features for this BDT model, the first, second, and eighth being the MITF-specific E-box motif that we constructed (Fig. 1a), the TRANSFAC MYC E-box motif, and the MYC-MAX specific E-box motif that we constructed (Fig. 1b), respectively. The partial dependence plot of the MITF-specific E-box motif showed a strong positive slope while those of the MYC-MAX-specific E-box and TRANSFAC MYC E-box motif showed the opposite trend. Furthermore, the differences between the position-specific scoring matrices defining the MITF-specific E-box motif and the MYC-MAX-specific E-box motif shared key characteristics to the ones learned by the BCDT model, verifying the E-box variations that the BCDT model learned during training. First, the middle two nucleotides of MITF’s central E-box hexamer deviated away from the canonical CACGTG, towards the variants CACATG and CATGTG. Second, the 3’ nucleotide flanking the E-box core hexamer in the MITF-specific position-specific scoring matrix showed a strong bias towards an A while the nucleotide in the same position in the MYC-specific position-specific scoring matrix showed a bias towards a G or C. Additionally, we analyzed the distributions of the output value of the full BDT model for sites bound by only MITF, only MYC-MAX, and both MITF and MYC-MAX (Supplementary Fig. S5). Similar to the BDT model without bHLH motifs, the full BDT model made intermediate predictions when classifying shared sites bound by both MITF and MYC-MAX, suggesting that these shared sites contained mixed sequence characteristics of both TFs. We further corroborated this finding by ranking all possible octamers by the percentage of shared binding sites containing them and found the most common one to be CACGTGAC, containing the canonical E-box hexamer CACGTG preferred by MYC-MAX, as well as the flanking 3’ AC preferred by MITF.
Discussion
The results of our machine learning models demonstrate that local sequence information reliably predicts the binding specificity of two important members of the bHLH-Zip family. In particular, the bases flanking the core E-box motif have the largest discriminative capacity for determining MITF vs. MYC-MAX binding, with the presence of a T nucleotide at the 5’ end favoring MITF over MYC-MAX. By contrast, a 5’ C and a 3’ G tend to flank the core E-box preferentially bound by MYC-MAX. Similarly, we have shown that different variants of the E-box motif are enriched in MITF vs. MYC-MAX binding sites: while MYC-MAX prefers the canonical CACGTG E-box hexamer, MITF frequently binds the variants CATGTG and CACATG throughout the genome (Fig. 4b and Supplementary Fig. S6a-c). These results suggest that MYC-MAX may competitively displace MITF from CACGTG motifs, despite the TFE protein family having ~10-fold higher affinity to CACGTG compared to CATGTG in vitro (D. E. Fisher, Carr, Parent, & Sharp, 1991). From a structural perspective, a nonpolar side chain interaction in the MITF protein between Ile212, that is not present in MYC-MAX, and the flanking T of the closely related M-box (TCATGTG) provides an explanation for why E-box elements with a flanking 5’ flanking T would favor MITF over MYC-MAX (Pogenberg et al., 2012). Furthermore, (Nair & Burley, 2003) has identified key interactions between the canonical CACGTG E-box hexamer and MYC-MAX that may shed light on the relative affinity of MITF and MYC-MAX to the canonical E-box. Although this study did not focus on binding sites shared by MITF and MYC-MAX, studying the structure and physical interactions involved in MITF and MYC-MAX binding may also help explain the slightly higher MITF ChIP enrichment in binding sites shared by MITF and MYC-MAX compared to those bound solely by MITF (Supplementary Fig. S5b).
Partial dependence plots of our BDT models have also demonstrated that the presence of a SOX10 motif is one of the most important features for distinguishing between sequences bound by MITF vs. MYC-MAX (Fig. 4b and Supplementary Fig. S4). In-silico addition of a SOX10 motif to MYC-MAX-bound sequences containing the MYC-MAX-preferring E-box CACGTGG increased the BDT model’s prediction probability that the sequences may be MITF-bound by 10.7%. Studying the effects of competing factors such as these would be an interesting area for future work. Furthermore, recent studies demonstrating that MITF and SOX10 can co-localize and recruit the chromatin remodeler BRG1 supports our findings (Laurette et al., 2015; Marathe et al., 2017). Finally, our BCDT model has learned that MYC-MAX generally prefers more GC-rich genomic regions compared to MITF (Fig. 5). These results strongly support the idea that subtle but predictive variations in binding sequences may have an important effect on the physical binding affinity of TFs sharing a common DNA binding domain and allow for their binding specificity. In this study, we only considered the MITF-M isoform of MITF, the isoform expressed in melanocytes. Different MITF isoforms could potentially exhibit different binding patterns and are an interesting direction for future study.
Analysis of ChIP-seq experiments for MITF and MAX revealed that a subset of E-boxes is bound by both MITF and MYC-MAX complex. These doubly-regulated E-boxes appear in more GC- and CpG-rich regions, have diminished interaction with SOX10, and higher evolutionary conservation. H3K27ac and H3K27me3 chromatin marks in the vicinity of these E-boxes show a lower response to MITF depletion indicating less tissue-specific chromatin organization and function.
Gene ontology analysis with GREAT (McLean et al., 2010) showed ‘melanosome’ as the most significant GO term for the MITF only class (p-value = 1.3×10−9), followed by ‘Waardenburg syndrome’ (p-value = 1.1×10−6) and ‘TOR signaling cascade’ (p-value = 7.5×10−6). Similarly, ‘colon carcinoma’ is the most significant GO term for the MITF and MYC-MAX overlapping class (p-value = 3.0×10−7), followed by ‘large Intestine adenocarcinoma’ (p-value = 4.1×10−7) and ‘regulation of cytokine production’ (p-value = 4.5×10−7). Furthermore, from a list of 2028 genes implicated in cancer (Sadelain, Papapetrou, & Bushman, 2011), 105 genes are expressed at a higher level than 1 FPKM in melanocytes and have a transcription start site within 10kb of a MITF-bound E-box. Despite the overlapping MITF and MYC-MAX class constituting only 23% of MITF-bound E-boxes, 37 of the 105 genes (35%) are associated with this overlapping class (binomial test p-value = 2.8×10−3). Given the oncogenic role of MYC across a large spectrum of cancers, future studies will help reveal whether the E-boxes bound by both MYC-MAX and MITF can account for some of the oncogenic activities of MITF.
Better understanding the mechanisms that MITF and MYC-MAX utilize to bind distinct and overlapping genomic locations may help improve therapeutic approaches to combating melanoma and other cancers. Our computational approach introduces a biologically interpretable framework for addressing this problem, and it may also facilitate studying the binding pattern of other set of highly similar TFs, such as the ETS family TFs that regulate diverse aspects of cancer (Bell et al., 2015).
Supplementary Material
Significance.
Our work improves the understanding of genetic information used by MITF and MYC-MAX to find their genomic targets and reveals the factors that determine their distinct and overlapping binding patterns. This knowledge may help improve therapeutic approaches to combating melanoma and other cancers in the future. Our computational approach introduces a biologically interpretable framework for studying the binding pattern of other highly similar TFs, such as the oncogenic ETS family TFs. High accuracy of predictive models introduced in this work demonstrates that specific combinatorial local sequence features that interact with MITF and MYC-MAX binding sites play an important role in differentially recruiting MITF vs. MYC to target genes.
Acknowledgements
This work was supported by funds from the NIH (2R01CA163336) and the Grainger Engineering Breakthroughs Initiative to J.S.S. and the NIH (5P01CA163222, 5R01AR043369), the Melanoma Research Alliance, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to D.E.F.
Footnotes
Accession codes
All sequencing data have been deposited in the Gene Expression Omnibus under the accession code GSE115845.
Conflict of Interest
The authors declare no conflict of interest.
References
- Aksan I, & Goding CR (1998). Targeting the microphthalmia basic helix-loop-helix-leucine zipper transcription factor to a subset of E-box elements in vitro and in vivo. Mol Cell Biol, 18(12), 6930–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, & Land H (1992). Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature, 359(6394), 423–426. doi: 10.1038/359423a0 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, . . . Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res, 37(Web Server issue), W202–208. doi: 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RJ, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, . . . Costello JF (2015). Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science, 348(6238), 1036–1039. doi: 10.1126/science.aab0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A, Nellore A, & Song JS (2012). CHANCE: comprehensive software for quality control and validation of ChIP-seq data. Genome Biol, 13(10), R98. doi: 10.1186/gb-2012-13-10-r98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A, Park K, Lim DA, & Song JS (2012). Normalization, bias correction, and peak calling for ChIP-seq. Stat Appl Genet Mol Biol, 11(3), Article 9. doi: 10.1515/1544-6115.1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encode Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. doi: 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher DE, Carr CS, Parent LA, & Sharp PA (1991). Tfeb Has DNA-Binding and Oligomerization Properties of a Unique Helix Loop Helix Leucine-Zipper Family. Genes & Development, 5(12a), 2342–2352. doi:DOI 10.1101/gad.5.12a.2342 [DOI] [PubMed] [Google Scholar]
- Fisher F, Crouch DH, Jayaraman PS, Clark W, Gillespie DA, & Goding CR (1993). Transcription activation by Myc and Max: flanking sequences target activation to a subset of CACGTG motifs in vivo. EMBO J, 12(13), 5075–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher F, & Goding CR (1992). Single amino acid substitutions alter helix-loop-helix protein specificity for bases flanking the core CANNTG motif. EMBO J, 11(11), 4103–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, . . . Sellers WR (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature, 436(7047), 117–122. doi: 10.1038/nature03664 [DOI] [PubMed] [Google Scholar]
- Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, . . . Widlund HR (2013). Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell, 23(3), 302–315. doi: 10.1016/j.ccr.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Yokoyama S, Hawryluk EB, Jonssond GB, Frederick DT, McHenry K, . . . Fisher DE (2013). BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proceedings of the National Academy of Sciences of the United States of America, 110(11), 4321–4326. doi: 10.1073/pnas.1205575110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemesath TJ, Steingrimsson E, Mcgill G, Hansen MJ, Vaught J, Hodgkinson CA, . . . Fisher DE (1994). Microphthalmia, a Critical Factor in Melanocyte Development, Defines a Discrete Transcription Factor Family. Genes & Development, 8(22), 2770–2780. doi:DOI 10.1101/gad.8.22.2770 [DOI] [PubMed] [Google Scholar]
- Jones S (2004). An overview of the basic helix-loop-helix proteins. Genome Biol, 5(6), 226. doi: 10.1186/gb-2004-5-6-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzner L, Blackwood EM, & Eisenman RN (1992). Myc and Max proteins possess distinct transcriptional activities. Nature, 359(6394), 426–429. doi: 10.1038/359426a0 [DOI] [PubMed] [Google Scholar]
- Laurette P, Strub T, Koludrovic D, Keime C, Le Gras S, Seberg H, . . . Davidson I (2015). Transcription factor MITF and remodeller BRG1 define chromatin organisation at regulatory elements in melanoma cells. Elife, 4. doi: 10.7554/eLife.06857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JY, Ehmann GL, Giangrande PH, & Nevins JR (2008). A role for Myc in facilitating transcription activation by E2F1. Oncogene, 27(30), 4172–4179. doi: 10.1038/onc.2008.55 [DOI] [PubMed] [Google Scholar]
- Levy C, & Fisher DE (2011). Dual roles of lineage restricted transcription factors: the case of MITF in melanocytes. Transcription, 2(1), 19–22. doi: 10.4161/trns.2.1.13650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Song JS, Bell RJ, Tran TN, Haq R, Liu H, . . . Fisher DE (2012). YY1 regulates melanocyte development and function by cooperating with MITF. PLoS Genet, 8(5), e1002688. doi: 10.1371/journal.pgen.1002688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marathe HG, Watkins-Chow DE, Weider M, Hoffmann A, Mehta G, Trivedi A, . . . de la Serna IL (2017). BRG1 interacts with SOX10 to establish the melanocyte lineage and to promote differentiation. Nucleic Acids Res, 45(11), 6442–6458. doi: 10.1093/nar/gkx259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, . . . Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol, 28(5), 495–501. doi: 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SK, & Burley SK (2003). X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell, 112(2), 193–205. [DOI] [PubMed] [Google Scholar]
- Olson WK, Gorin AA, Lu XJ, Hock LM, & Zhurkin VB (1998). DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A, 95(19), 11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirano RI, & Wegner M (2000). The glial transcription factor Sox10 binds to DNA both as monomer and dimer with different functional consequences. Nucleic Acids Research, 28(16), 3047–3055. doi:DOI 10.1093/nar/28.16.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogenberg V, Ogmundsdottir MH, Bergsteinsdottir K, Schepsky A, Phung B, Deineko V, . . . Wilmanns M (2012). Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev, 26(23), 2647–2658. doi: 10.1101/gad.198192.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Papapetrou EP, & Bushman FD (2011). Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer, 12(1), 51–58. doi: 10.1038/nrc3179 [DOI] [PubMed] [Google Scholar]
- Seberg HE, Van Otterloo E, & Cornell RA (2017). Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res, 30(5), 454–466. doi: 10.1111/pcmr.12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A, Saleque S, Kalpana GV, Artandi S, Goff SP, & Calame K (1993). Inhibition of transcriptional regulator Yin-Yang-1 by association with c-Myc. Science, 262(5141), 1889–1892. [DOI] [PubMed] [Google Scholar]
- Skinner MK, Rawls A, Wilson-Rawls J, & Roalson EH (2010). Basic helix-loop-helix transcription factor gene family phylogenetics and nomenclature. Differentiation, 80(1), 1–8. doi: 10.1016/j.diff.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DL, Amati B, & Land H (1993). Distinct DNA binding preferences for the c-Myc/Max and Max/Max dimers. Nucleic Acids Res, 21(23), 5372–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, & Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol, 31(1), 46–53. doi: 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers A (1997). DNA-protein interactions: IHF--the master bender. Curr Biol, 7(4), R252–254. [DOI] [PubMed] [Google Scholar]
- Webster DE, Barajas B, Bussat RT, Yan KJ, Neela PH, Flockhart RJ, . . . Khavari PA (2014). Enhancer-targeted genome editing selectively blocks innate resistance to oncokinase inhibition. Genome Research, 24(5), 751–760. doi: 10.1101/gr.166231.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumoto K, Takeda K, Saito H, Watanabe K, Takahashi K, & Shibahara S (2002). Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. Embo Journal, 21(11), 2703–2714. doi:DOI 10.1093/emboj/21.11.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.