

Abstract
Background and Purpose
Sunitinib is a small‐molecule TK inhibitor associated with hepatotoxicity. The mechanisms of its toxicity are still unclear.
Experimental Approach
In the present study, mice were treated with 60, 150, and 450 mg·kg−1 sunitinib to evaluate sunitinib hepatotoxicity. Sunitinib metabolites and endogenous metabolites in liver, serum, faeces, and urine were analysed using ultra‐performance LC electrospray ionization quadrupole time‐of‐flight MS‐based metabolomics.
Key Results
Four reactive metabolites and impaired clearance of sunitinib in liver played a dominant role in sunitinib‐induced hepatotoxicity. Using a non‐targeted metabolomics approach, various metabolic pathways, including mitochondrial fatty acid β‐oxidation (β‐FAO), bile acids, lipids, amino acids, nucleotides, and tricarboxylic acid cycle intermediates, were disrupted after sunitinib treatment.
Conclusions and Implications
These studies identified significant alterations in mitochondrial β‐FAO and bile acid homeostasis. Activation of PPARα and inhibition of xenobiotic metabolism may be of value in attenuating sunitinib hepatotoxicity.
Abbreviations
- 12‐carnitine
lauroylcarnitine
- 14‐carnitine
myristoylcarnitine
- 16‐carnitine
palmitoylcarnitine
- 18‐carnitine
stearoylcarnitine
- ABT
1‐aminobenzotriazole
- ALP
alkaline phosphatase
- ALT
alanine transaminase
- AST
aspartate transaminase
- CA
cholic acid
- CPT
carnitine palmitoyltransferase
- DCA
deoxycholic acid
- GPX
GSH peroxidase
- HLM
human liver microsomes
- LPC
lyso‐phosphocholine
- LPE
lyso‐phosphatidylethanolamine
- MLM
mouse liver microsomes
- PCA
principal component analysis
- SHP
small heterodimer partner
- TDCA
taurodeoxycholic acid
- UPLC‐ESI‐QTOFMS
ultra‐performance LC electrospray ionization quadrupole time‐of‐flight MS
- β‐FAO
fatty acid β‐oxidation
What is already known
Sunitinib is associated with hepatotoxicity in the clinic.
What this study adds
Sunitinib hepatotoxicity is a result of metabolism and impaired clearance.
Sunitinib disrupted mitochondrial fatty acid β‐oxidation and bile acid homeostasis.
What is the clinical significance
Activation of PPARα and inhibition of xenobiotic metabolism might help to attenuate sunitinib hepatotoxicity.
1. INTRODUCTION
Sunitinib is a low MW tyrosine kinase (TK) inhibitor approved for the treatment of advanced renal cell carcinoma, imatinib‐refractory gastrointestinal stromal tumours, and pancreatic neuroendocrine tumours (Goodman et al., 2007). In addition, sunitinib is in development for the treatment of patients with other solid tumours, including breast, colorectal, and neuroendocrine (Speed et al., 2012). Sunitinib inhibits several receptor TKs, such as those associated with the VEGF receptor, PDGF receptor , Fms‐like TK‐3 receptor (FLT3), and stem cell factor receptor (Mendel et al., 2003; Sun et al., 2003).
However, sunitinib treatment shows some side effects, including hand–foot skin reaction, hypertension, and cardiac dysfunction (Faivre et al., 2006; Goodman et al., 2007). Furthermore, sunitinib carries a black box warning for potentially life‐threatening hepatotoxicity (Amaya et al., 2018). A recent meta‐analysis of sunitinib adverse events in metastatic renal cell carcinoma revealed that elevated liver enzymes were found in 40% of 5,658 patients (Ibrahim, Kazkaz, Abouelkhair, Bayer, & Elmasri, 2013). Another meta‐analysis of sunitinib adverse events in primary hepatocellular carcinoma and gastrointestinal stromal tumours showed that liver function impairment was found in 33% and 23% of patients respectively (Fu, Wei, Lin, Xu, & Liang, 2018). Furthermore, liver injury was reported in 3–4% of patients taking sunitinib, and liver failure was reported in 0.3% of patients (Amaya et al., 2018). A population‐based cohort study also found that severe liver injury occurred infrequently during exposure to sunitinib: 7.4–9.3% patients had doubled alanine transaminase (ALT) elevation; 8.6–18.1% patients had elevated bilirubin (Shantakumar et al., 2016). Furthermore, many clinical cases were reported on liver failure following sunitinib administration (Guillen, Meijer, & de Jongh, 2016; Mermershtain, Lazarev, Shani‐Shrem, & Ariad, 2013; Mueller, Rockey, & Rashkin, 2008; Taran, Ignatov, Smith, Costa, & Bischoff, 2009; Weise, Liu, & Shields, 2009). Recent studies reported that mitochondrial damage, inhibition of glycolysis, and metabolic activation contributed to sunitinib hepatotoxicity (Amaya et al., 2018; Paech, Bouitbir, & Krahenbuhl, 2017).
Ultra‐performance LC electrospray ionization quadrupole time‐of‐flight MS (UPLC‐ESI‐QTOFMS)‐based metabolomics was successfully applied to investigate the toxicity mechanisms of TK inhibitors, including pazopanib (Wang et al., 2018), sorafenib (Jensen, Parry, Huang, Beak, et al., 2017), and gefitinib (Liu et al., 2015). Metabolomics has proven to be a powerful tool to investigate drug metabolism and toxicity. Using metabolomics approach, the goals of the present study were as follows: (a) to determine the metabolic map of sunitinib, (b) to identify the endogenous metabolites that were altered in liver, serum, faeces, and urine, and (c) to understand its mechanism of hepatotoxicity and therapeutic strategy.
2. METHODS
2.1. In vitro metabolism of sunitinib
In vitro microsomal incubations were carried out in PBS (pH 7.4), containing 50 μM sunitinib, 0.5 mg·ml−1 mouse liver microsomes (MLM), 0.5 mg·ml−1 human liver microsomes (HLM), and 2 pmol·ml−1 of each recombinant human CYP in a final volume of 200 μl. The reaction was initiated by adding 20 μl of NADPH (10 mM). After a 40‐min incubation at 37°C, the reaction was quenched by adding 200 μl of ice‐cold acetonitrile. The absence of NADPH and absence of sunitinib in the microsomal system were separately incubated as control groups.
2.2. Animals
All animal care and experimental procedures were carried out in accordance with the Institutional Animal Care and Use Committee of the Kunming Institute of Botany, Chinese Academy of Sciences. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Six‐ to 8‐week‐old male C57BL/6J mice (20–22 g, RRID:IMSR_JAX:000664) were purchased from the SLACCAS Laboratory Animal Co., Ltd. (Hunan, China). The mice were maintained under a standard 12‐hr dark/light cycle with standard chow and water ad libitum. Mice were randomized for treatment. Mice were anaesthetized with CO2 and killed by cervical dislocation at indicated time points.
2.2.1. Experiment 1
A previous study reported that serum aspartate transaminase (AST) and ALT were slightly increased, and minimal periportal inflammation changes were detected after treatment with 140 mg·kg−1 sunitinib in male ICR mice (Tan, Chakravarthi, Judson, Haleagrahara, & Segarra, 2013). To evaluate the dose effects of sunitinib on hepatotoxicity, two doses (60 and 150 mg·kg−1 in normal saline) were orally administered to C57BL/6J mice (n = 5). Control mice were gavaged with normal saline. Samples of urine and faeces were collected using metabolic cages (Techniplast, Buguggiate, Italy). Samples of serum were collected at 0, 3, and 24 hr and liver samples at 3 and 24 hr, rapidly frozen on dry ice, and stored at −80°C until analysis.
2.2.2. Experiment 2
To evaluate the severe hepatotoxicity of sunitinib. Ten C57BL/6J mice were randomly divided into two groups (n = 5). Sunitinib, 450 mg·kg−1 in normal saline, was orally administered to C57BL/6J mice for two consecutive days. Control mice were treated with normal saline. Samples of serum, at 0, 3, and 48 hr, and of liver at 48 hr were collected, rapidly frozen on dry ice, and stored at −80°C until analysis.
2.2.3. Experiment 3
To evaluate the hepatoprotective effects of a broad‐spectrum CYP inhibitor 1‐aminobenzotriazole (ABT). The mice were randomly divided into three groups (n = 5): control group, 450 mg·kg−1 sunitinib group, and 450 mg·kg−1 sunitinib + ABT group. The 450 mg·kg−1 sunitinib + ABT group mice were intraperitoneally injected with ABT (100 mg·kg−1 dissolved in normal saline) 1 hr prior to oral administration of sunitinib for two consecutive days (Wang et al., 2018). The 450 mg·kg−1 group mice were injected with normal saline prior to the administration of sunitinib. The control group mice were injected with normal saline prior to the administration of normal saline, and 48 hr serum and liver samples were harvested, rapidly frozen on dry ice, and stored at −80°C until analysis.
2.2.4. Experiment 4
To investigate the protective effect of a PPARα agonist, fenofibrate (Feno), in sunitinib‐induced toxicity, the mice were randomly assigned into three groups (n = 5): control group, sunitinib group, and sunitinib + Feno group. Sunitinib + Feno group mice were treated with fenofibrate (200 mg·kg−1 dissolved in 0.5% sodium carboxymethylcellulose) for three consecutive days (Zhao, Yang, Wang, et al., 2017). After treatment with fenofibrate for 3 days, sunitinib and the sunitinib + Feno groups were given a single oral dose of sunitinib (200 mg·kg−1), and 3 hr serum and liver samples were harvested, rapidly frozen on dry ice, and stored at −80°C until analysis.
2.3. UPLC‐ESI‐QTOFMS analysis
The preparations of liver, plasma, faeces, and urine samples were carried out using the methods described previously (Zhao, Li, Liu, Gonzalez, & Li, 2018; Zhao, Yang, Wang, et al., 2017). The UPLC system consisted of a 1290 Autosampler and a Quat Pump (Agilent, Santa Clara, CA, USA) equipped with a reverse‐phase XDB‐C18 column (2.1 × 100 mm, 1.8 μM). Column temperature was kept at 45°C. The flow rate was set at 0.3 ml·min−1 with a gradient ranging from 2% to 98% acetonitrile containing 0.01% formic acid over the next 16 min. The Agilent 6530 QTOFMS (Agilent, Santa Clara, CA, USA) was operated at m/z ranging from 50 to 800. Nebulizer pressure was set at 35 psi. Capillary voltage was kept at 3.5 kV. Drying gas temperature was set at 350°C.
Samples were subjected to chromatography on a Poroshell 120 HILIC column (2.1 × 100 mm, 2.7 μM; Agilent, Santa Clara, CA, USA) to analyse the hydrophilic endogenous metabolites. Their retention times can be found in Table S1. The flow rate and mobile phase were consistent with the reverse‐phase column. The gradient ranged from 98% to 65% acetonitrile at 0.5–3 min, next decreased to 40% acetonitrile at 8 min, then returned to 2% acetonitrile for 3 min and increased to 98% acetonitrile to 16 min. Data were collected at m/z ranging from 50 to 950. Quantitative determination of GSH and GSSG levels in tissue and serum was performed using external standards by means of a six‐point calibration curve.
2.4. Data processing and multivariate data analysis
The MassHunter Workstation software (Agilent, Santa Clara, CA, USA) was used for chromatographic and spectral data collection. Multivariate data analysis was performed using Mass Profinder software (Agilent, Santa Clara, CA, USA, RRID:SCR_017026) to generate a multivariate data matrix. Principal component analysis (PCA) and orthogonal projection to latent structure‐discriminant analysis were performed using SIMCA‐P + 13.0 software (Umetrics, Kinnelon, NJ, USA). Clustering of a serious of endogenous metabolites in liver, serum, urine, and faeces was performed using Cluster and TreeView software (Zhao, Yang, Wang, et al., 2017).
2.5. Identification of sunitinib and endogenous metabolites
The identification of xenobiotic and endogenous metabolites was performed as previously described (Zhao et al., 2018; Zhao, Yang, Wang, et al., 2017). The chemical structures of sunitinib metabolites were elucidated on the basis of their MS/MS fragmentation patterns (see Figure S1). The structures of endogenous metabolites were searched in HMDB and METLIN databases and further identified by comparing retention times and MS/MS fragmentation patterns with those of authentic standards (Table S1 and Figure S2).
2.6. Histological and biochemical assessment
Haematoxylin and eosin staining was carried as detailed in a previous report (Zhao, Yang, Wang, et al., 2017). AST, ALT, and alkaline phosphatase (ALP) activities were measured following the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The assignment of tissue to histological assessment was randomized. Ten sections per preparation were analysed blindly by a pathologist.
2.7. Gene expression analysis
QPCR was carried as detailed in a previous report (Zhao, Yang, Wang, et al., 2017). Total RNA was extracted from 100‐mg liver using TRIzol reagent (Life Technologies, Carlsbad, CA, USA). qPCR was carried out using SYBR green PCR master mix (Takara, Dalian, China) in a CFX Connect Real‐Time System (Bio‐Rad Laboratories, Hercules, CA, USA). Actb mRNA was measured as an internal control for each sample. qPCR primer sequences were listed in Table S2.
2.8. Western blot and elisa analyses
Protein was prepared using RIPA (PC1001, Beyotime), and the level of the protein was evaluated by BCA protein assay kit (P0010S, Beyotime Biotech, Shanghai, China). Samples of protein (50 μg) were subjected to SDS‐PAGE gel (Bio‐Rad), removed to PVDF membranes (Bio‐Rad), and probed with primary antibodies followed by secondary antibodies. The following antibodies were used: GAPDH (Cell Signaling Technology, Cat# 3683), IL‐1β (Cell Signaling Technology, Cat# 12507), IL‐6 (Cell Signaling Technology, Cat# 12912), and anti‐rabbit peroxidase‐conjugated second antibody (Proteintech, Cat# SA00001‐2, RRID:AB_2722564). Serum IL‐6 levels were measured following the manufacturer's instructions (Cayman, Cat# 583371). The assignment of tissue to Western blot was randomized.
2.9. Synthesis and chemical reduction of sunitinib N‐oxide metabolites
For purpose of validation of sunitinib N‐oxide in mice, authentic sunitinib N‐oxide was prepared by a modification of a published method (Li et al., 2012; Nery, 1971). A solution of sunitinib malate (200 mg) in 50‐ml diluted ammonium hydroxide (pH 9) was extracted with chloroform (2 × 20 ml). The extracts were combined and dried down in nitrogen at room temperature. Part of the residue (20 mg) was dissolved in chloroform (2 ml) and treated with 39% (vol/vol) peroxyacetic acid (40 mg) dropwise during 30 min. To further determine the synthetic N‐oxide metabolites of sunitinib, TiCl3 was used for chemical reduction of sunitinib N‐oxide. A 100‐μl aliquot was treated with ice‐cold TiCl3 in HCl (10 μl) and shaken at room temperature for 1 hr. The mixture was analysed by UPLC‐ESI‐QTOFMS following 1:1,000 dilution.
2.10. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. All data and images were analysed blindly. The raw data were assessed by two co‐authors to ensure the correctness of the conclusions. The investigators treating the mice were not aware of the pharmacological treatments of each group; a number was assigned for each group during the assessment. The data are shown as mean ± SEM. Statistical analysis was carried out using the software GraphPad Prism (v6, GraphPad Software, USA, RRID:SCR_002798) and SPSS Statistics, version 23 (IBM, Beijing, China, RRID:SCR_002865). Differences between two groups were tested using the Student's t test. Differences among multiple groups were tested using one‐way ANOVA followed by Dunnett's post hoc comparisons. Post hoc tests were conducted only if F was significant and there was no variance inhomogeneity. P values less than 0.05 were considered significant. Correlation factor (r) was estimated with Pearson's correlation analysis.
2.11. Materials
Sunitinib was obtained from LC Laboratories (Woburn, MA, USA). NADPH, chlorpropamide, lauroylcarnitine (12‐carnitine), myristoylcarnitine (14‐carnitine), palmitoylcarnitine (16‐carnitine), stearoylcarnitine (18‐carnitine), cholic acid (CA), deoxycholic acid (DCA), taurocholic acid, taurodeoxycholic acid (TDCA), lyso‐phosphocholine14:0 (LPC14:0), LPC16:0, LPC18:0, LPC18:1, AMP, inosine, l‐lactic acid, malic acid, and hippuric acid were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Tauro‐β‐muricholic acid and ω‐muricholic acid were bought from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Tauroursodeoxycholic acid and lithocholic acid were obtained from MedChemExpress (Monmouth Junction, NJ, USA). Tauro‐α‐muricholic acid was purchased from Steraloids (Newport, RI, USA). Lyso‐phosphatidylethanolamine16:0 (LPE16:0) and LPE18:0 were purchased from Avanti Polar Lipids (Birmingham, AL, USA). MLM and HLM were bought from Bioreclamationivt Inc. (Hicksville, NY, USA), and recombinant human CYPs were provided from Xenotech, LLC (Kansas City, KS, USA). GSH was purchased from Sigma (St. Louis, MO, USA), and GSSG was provided by Macklin (Shanghai, China). All other reagents were of the highest grade.
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro, et al., 2017a,b; Alexander, Kelly, et al., 2017).
3. RESULTS
3.1. Sunitinib‐induced liver injury in mice
In the livers from mice treated with 150 mg·kg−1 sunitinib, scattered haemocytes and inflammatory infiltration were observed, accompanied by neutrophils as the major increased immune cells (Figure 1a). AST was significantly increased at 3 hr after 60 mg·kg−1 sunitinib treatment, and AST, ALT, and ALP levels were significantly increased at 3 hr after 150 mg·kg−1 sunitinib treatment (Figure 1b). In 24 hr serum, two doses of sunitinib resulted in a significant increase of ALP (Figure 1b). Furthermore, the mRNA levels of proinflammatory cytokines Il1b, Il6, and Tnfa were dramatically increased after 150 mg·kg−1 sunitinib treatment (Figure 1c). The protein levels of IL‐6 were significantly increased after two doses of sunitinib treatment (Figure 1d), whereas no significant increase of serum IL‐6 levels was found (Figure S3a). A significant decrease in body weight was observed in 150 mg·kg−1 sunitinib‐treated mice compared with control mice (Figure 1e), which was consistent with results in rats and humans (Blanca et al., 2016; Motzer et al., 2009). Furthermore, the faeces weight was significantly decreased after 150 mg·kg−1 sunitinib treatment (Figure S3b). The urine volume was unchanged by two doses of sunitinib (Figure S3b). These results showed that 150 mg·kg−1 sunitinib caused a low level of liver injury in the mouse.
Figure 1.
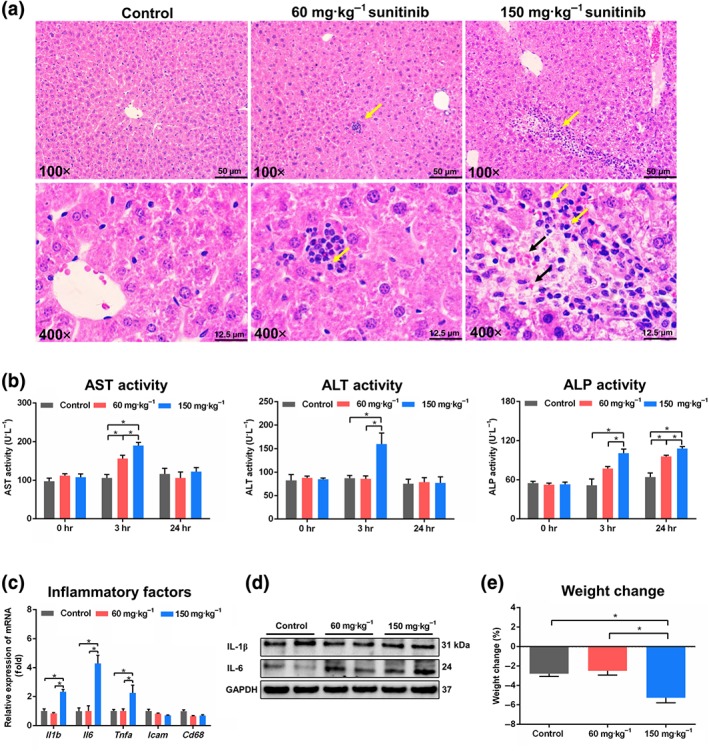
Sunitinib induced liver injury in mice. (a) Haematoxylin and eosin staining of liver. Neutrophils were shown by yellow arrowheads in 60 and 150 mg·kg−1 sunitinib groups. Scattered haemocytes were shown by black arrowheads in 150 mg·kg−1 sunitinib groups. (b) Aspartate transaminase (AST), alanine transaminase (ALT), and alkaline phosphatase (ALP) enzymes activity in 0, 3, and 24 hr serum. (c) Real‐time PCR analysis of inflammatory factors in liver. Il1b, Il6, and Tnfa levels were significantly increased after 150 mg·kg−1 sunitinib treatment. Values represented fold change after normalization to control. (d) Western blot was used to measure IL‐1β and IL‐6. IL‐6 level was significantly increased after 60 and 150 mg·kg−1 sunitinib treatment. (e) Body weight was significantly decreased after 150 mg·kg−1 sunitinib treatment. All data plotted are means ± SEM (n = 5). *P < 0.05, significantly different as indicated
3.2. Metabolomics profiling of sunitinib metabolites in vivo and in vitro
The in vivo metabolism of sunitinib was examined in C57BL/6J mice by global metabolite analysis using PCA. The liver, serum, urine, and faeces samples of sunitinib‐treated group were well separated from the vehicle group by PCA (Figures 2a, 3a,b, and S4). Thirty‐nine metabolites (M1–M20, M22, M26–M29, M31, M34–M37, M40–M45, and M47–M49) were altered in liver samples from sunitinib‐treated mice; the parent sunitinib was also detected (Figure S5a and Table 1). Twenty‐seven metabolites were altered from 24 hr serum samples from sunitinib‐treated mice including M1–M7, M11–M13, M15, M17–M20, M26, M28–M31, M36, M37, M39, M44–M46, and M48, and parent sunitinib was also detected (Figure S5b and Table 1). Thirty‐eight metabolites (M1–M13, M15, M17–M22, M26, M27, M32–M35, M37–M39, and M41–M49) with the parent sunitinib were found in faeces samples from sunitinib‐treated mice (Figure S5c and Table 1). Parent sunitinib and 37 metabolites (including M1–M9, M12, M13, M15–M18, M21, M23, M26, M28–M39, M41–M43, and M46–M49) were found in urine (Figure S5d and Table 1).
Figure 2.
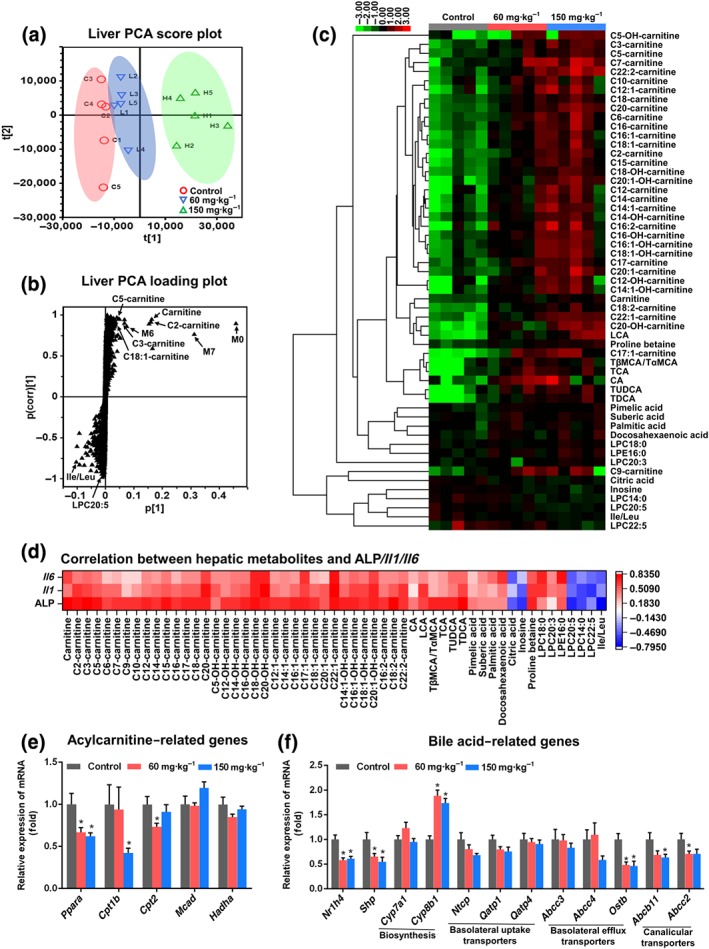
Sunitinib altered the liver metabolome. Principle component analysis (PCA) score plot (a) and scatter plot (b) derived from ultra‐performance LC electrospray ionization quadrupole time‐of‐flight MS data of liver ions. Each point represented an individual mouse liver sample (top) and an ion in the sample (bottom). Metabolites were labelled in the loading plot, which included three xenobiotic metabolites and seven endogenous metabolites. (c) Heatmap of 55 significantly changed endogenous metabolites (P < 0.05) in liver. (d) Correlation analysis between 55 changed endogenous metabolites and serum alkaline phosphatase (ALP) and hepatic Il1b and Il6 mRNAs. All these metabolites showed a good correlation with serum ALP and hepatic Il1b, and Il6 mRNAs. (e) Real‐time PCR analysis was performed to measure the levels of mRNA‐encoding enzymes associated with fatty acid synthesis and metabolism. Acylcarnitine‐related genes were significantly inhibited after sunitinib treatment. (f) Real‐time PCR analysis of the hepatic mRNAs associated with bile acid synthesis and transport. Bile acid‐related genes were significantly inhibited after sunitinib treatment. Values represented fold change after normalization to control. All data plotted are means ± SEM (n = 5). *P < 0.05 significantly different from control. CA, cholic acid; Ile, isoleucine; Leu, leucine; LCA, lithocholic acid; LPC, lyso‐phosphocholine; LPE, lyso‐phosphatidylethanolamine; TCA, taurocholic acid; TDCA, taurodeoxycholic acid; TUDCA, tauroursodeoxycholic acid; TαMCA, tauro‐α‐muricholic acid; TβMCA, tauro‐β‐muricholic acid
Figure 3.
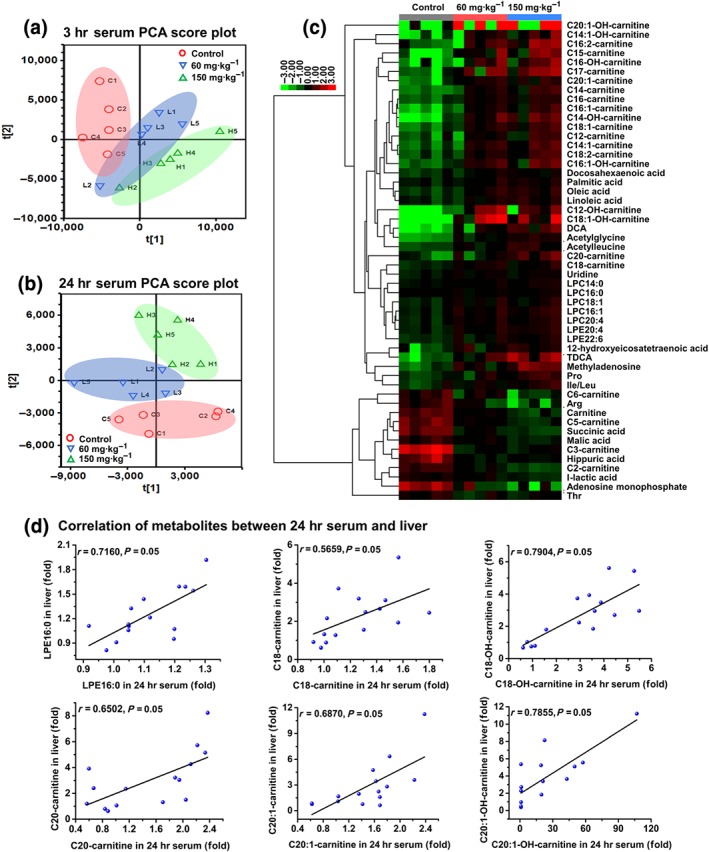
Sunitinib altered the serum metabolome. Principal component analysis (PCA) score plot derived from ultra‐performance LC electrospray ionization quadrupole time‐of‐flight MS data of 3 hr serum (a) and 24 hr serum (b) ions. Each point represented an individual mouse sample. (c) Heatmap of 52 significantly changed endogenous metabolites in 3 hr serum. (d) Correlation of metabolites between 24 hr serum and 24 hr liver. Six endogenous metabolites showed a good correlation between 24 hr serum and 24 hr liver (n = 5). Arg, arginine; DCA, deoxycholic acid; Ile, isoleucine; Leu, leucine; LPC, lyso‐phosphocholine; LPE, lyso‐phosphatidylethanolamine; Pro, proline; TDCA, taurodeoxycholic acid; Thr, threonine
Table 1.
Summary of sunitinib metabolites produced in vivo and in vitro metabolism
| ID | RT (min) | Observed m/z | Mass error (ppm) | Formula | Identity | Source |
|---|---|---|---|---|---|---|
| M0 | 6.47 | 399.2193 | 0.5 | C22H27FN4O2[H+] | Sunitinib | S·L·U·F·M·H |
| M1 | 5.18 | 415.2142 | 0.5 | C22H27FN4O3[H+] | Sunitinib + O | S·L·U·F·M·H |
| M2 | 5.68 | 415.2148 | 1.9 | C22H27FN4O3[H+] | Sunitinib + O | S·L·U·F·M·H |
| M3 | 5.83 | 415.2160 | 4.8 | C22H27FN4O3[H+] | Sunitinib + O | S·L·U·F·M·H |
| M4 | 6.01 | 415.2157 | 4.1 | C22H27FN4O3[H+] | Sunitinib + O | S·L·U·F·M·H |
| M5 | 6.49 | 415.2144 | 1.0 | C22H27FN4O3[H+] | Sunitinib + O | S·L·U·F·M·H |
| M6 | 5.08 | 371.1885 | 1.9 | C20H23FN4O2[H+] | Sunitinib—C2H4 | S·L·U·F·M·H |
| M7 | 6.15 | 371.1885 | 1.9 | C20H23FN4O2[H+] | Sunitinib—C2H4 | S·L·U·F·M·H |
| M8a | 4.40 | 431.2085 | −0.9 | C22H27FN4O4[H+] | Sunitinib + O2 | L·U·F·M·H |
| M9a | 5.56 | 431.2096 | 1.6 | C22H27FN4O4[H+] | Sunitinib + O2 | L·U·F·M·H |
| M10a | 6.46 | 431.2091 | 0.5 | C22H27FN4O4[H+] | Sunitinib + O2 | L·F·M |
| M11a | 5.96 | 417.2294 | −0.7 | C22H29FN4O3[H+] | Sunitinib + O + H2 | S·L·F·M·H |
| M12 | 5.28 | 397.2238 | 1.0 | C22H28N4O3[H+] | Sunitinib—F + OH | S·L·U·F·M·H |
| M13 | 5.91 | 343.1564 | −0.3 | C18H19FN4O2[H+] | Sunitinib—C2H4—C2H4 | S·L·U·F·M·H |
| M14 | 9.19 | 401.2311 | −9.0 | C22H29FN4O2[H+] | Sunitinib + H2 | L·M·H |
| M15 | 7.40 | 358.1190 | −2.2 | C18H16FN3O4[H+] | Sunitinib—C6H14N + C2H3O2 | S·L·U·F·M·H |
| M16a | 6.61 | 447.2040 | 0.4 | C22H27FN4O5[H+] | Sunitinib + O3 | L·U·M·H |
| M17 | 4.90 | 387.1808 | −4.9 | C20H23FN4O3[H+] | Sunitinib—C2H4 + O | S·L·U·F·M·H |
| M18 | 5.28 | 387.1842 | 3.9 | C20H23FN4O3[H+] | Sunitinib—C2H4 + O | S·L·U·F·M·H |
| M19 | 5.61 | 387.1823 | −1.0 | C20H23FN4O3[H+] | Sunitinib—C2H4 + O | S·L·F·M·H |
| M20 | 7.00 | 387.1837 | 2.6 | C20H23FN4O3[H+] | Sunitinib—C2H4 + O | S·L·F·M·H |
| M21a | 5.79 | 413.2204 | 5.1 | C22H28N4O4[H+] | Sunitinib + O + (—F + OH) | U·F·M·H |
| M22a | 5.00 | 369.1925 | 1.1 | C20H24N4O3[H+] | Sunitinib—C2H4 + (—F + OH) | L·F·M·H |
| M23a | 5.59 | 429.2142 | 2.1 | C22H28N4O5[H+] | Sunitinib + O2 + (—F + OH) | U·M·H |
| M24a | 7.30 | 371.2096 | 4.8 | C20H26N4O3[H+] | Sunitinib—C2H4 + H2 + (—F + OH) | M·H |
| M25a | 7.74 | 413.1991 | 1.7 | C22H25FN4O3[H+] | Sunitinib + O—H2 | M·H |
| M26a | 6.06 | 385.1645 | −6.8 | C20H21FN4O3[H+] | Sunitinib—C2H4—H2 + O | S·L·U·F·M·H |
| M27a | 7.21 | 344.1390 | −4.4 | C18H18FN3O3[H+] | Sunitinib—C6H14N + C2H5O | L·F·M·H |
| M28a | 4.11 | 563.2144 | −0.7 | C26H31FN4O9[H+] | Sunitinib—C2H4 + O + Glu | S·L·U |
| M29a | 4.53 | 563.2173 | 4.4 | C26H31FN4O9[H+] | Sunitinib—C2H4 + O + Glu | S·L·U |
| M30a | 4.68 | 563.2155 | 1.2 | C26H31FN4O9[H+] | Sunitinib—C2H4 + O + Glu | S·U |
| M31a | 4.88 | 563.2127 | −3.7 | C26H31FN4O9[H+] | Sunitinib—C2H4 + O + Glu | S·L·U |
| M32a | 4.39 | 547.2193 | −1.1 | C26H31FN4O8[H+] | Sunitinib—C2H4 + Glu | U·F |
| M33a | 5.05 | 547.2185 | −2.6 | C26H31FN4O8[H+] | Sunitinib—C2H4 + Glu | U·F |
| M34a | 5.09 | 495.1733 | 5.0 | C22H27FN4O6S[H+] | Sunitinib + O + SO3 | L·U·F |
| M35a | 5.47 | 495.1737 | 5.9 | C22H27FN4O6S[H+] | Sunitinib + O + SO3 | L·U·F |
| M36a | 4.83 | 591.2477 | 2.7 | C28H35FN4O9[H+] | Sunitinib + O + Glu | S·L·U |
| M37a | 5.16 | 591.2489 | 4.7 | C28H35FN4O9[H+] | Sunitinib + O + Glu | S·L·U·F |
| M38 | 4.67 | 575.2512 | 0.0 | C28H35FN4O8[H+] | Sunitinib + Glu | U·F |
| M39 | 5.23 | 575.2529 | 3.0 | C28H35FN4O8[H+] | Sunitinib + Glu | S·U·F |
| M40a | 5.54 | 704.2831 | −5.8 | C32H42FN7O8S[H+] | Sunitinib + GSH | L |
| M41a | 5.32 | 676.2514 | −6.7 | C30H38FN7O8S[H+] | Sunitinib—C2H4 + GSH | L·U·F |
| M42a | 5.22 | 477.1800 | −0.4 | C22H28N4O6S[H+] | Sunitinib—F + OH + SO3 | L·U·F |
| M43a | 5.15 | 467.1396 | 0.2 | C20H23FN4O6S[H+] | Sunitinib—C2H4 + O + SO3 | L·U·F |
| M44a | 5.93 | 429.2267 | −2.3 | C23H29FN4O3[H+] | Sunitinib + CH3O | S·L·F |
| M45a | 6.31 | 429.2286 | −2.3 | C23H29FN4O3[H+] | Sunitinib + CH3O | S·L·F |
| M46a | 5.41 | 560.2334 | −0.5 | C27H34FN5O5S[H+] | Sunitinib + N—acetylcysteine | S·U·F |
| M47a | 5.30 | 479.1774 | 3.1 | C22H27FN4O5S[H+] | Sunitinib + SO3 | L·U·F |
| M48a | 5.23 | 577.2645 | −4.0 | C28H37FN4O8[H+] | Sunitinib + O + Glu | S·L·U·F |
| M49a | 5.35 | 558.2373 | −1.4 | C27H35N5O6S[H+] | Sunitinib—F + OH + N‐acetylcysteine | L·U·F |
Notes: F, faeces; Glu, glucose; H, human liver microsomes; L, liver; M, mouse liver microsomes; RT, retention time; S, serum; U, urine.
Novel metabolites identified in this study.
To predict the metabolic differences between humans and mice, phase I metabolism of sunitinib was assessed using MLM and HLM incubation systems. Overall, 27 sunitinib metabolites (M1–M27) and 26 sunitinib metabolites (M1–M9 and M11–M27) were identified from MLM and HLM incubation systems respectively (Figure S6a and Table 1). The sunitinib metabolites produced by HLM were similar to those from MLM (Figure S6a). The metabolic rate of parent sunitinib was 12.3% in MLM and 33.7% in HLM, indicating that HLM showed higher metabolic rates than MLM (Figure S6b).
In the present study, 49 metabolites of sunitinib were identified, and 32 metabolites were new metabolites, including M8–M11, M16, M21–M37, and M40–M49 (Table 1). The MS/MS of these metabolites is shown in Figure S1. M1 and M5 were proposed to be N‐oxide metabolites of sunitinib and were confirmed with a synthetic standard (Figure S7). M1 and M5 were lost following treatment with TiCl3 (Figure S7d). Many sunitinib metabolites were isomerized because of the exocyclic double bond, such as the two sunitinib N‐oxide metabolites M1 and M5. The metabolic map of sunitinib is shown in Figure S8.
3.3. Screening the CYPs involved in the metabolism of sunitinib
Sunitinib was incubated with a panel of CYPs, including CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP2E1, CYP3A4, CYP3A5, and CYP4A11. Among these recombinant CYPs, CYP1A1 and CYP1A2 were largely responsible for the formation of sunitinib N‐oxide (M1 and M5) and sunitinib diethyl‐amine‐oxide (M2 and M4; Table S3). CYP1A1, CYP1B1, and CYP2D6 were involved in the formation of sunitinib indolylidene/dimethylpyrrole‐oxide (M3; Table S3). CYP2C8, CYP3A4 and CYP3A5 were responsible for the generation of the N‐dealkylation (M6, M7, and M13; Table S3). CYP1A1, CYP1A2, CYP2D6, and CYP3A4 were responsible for the metabolite derived from oxidative defluorination (M12; Table S3). CYP3A4 and CYP3A5 were responsible for the formation of M15 and M27, which underwent deamination of N,N′‐diethylamine (Table S3).
3.4. Metabolic activation of sunitinib and its impaired clearance
Among 49 sunitinib metabolites, two new GSH‐conjugated metabolites (M40 and M41) and two new N‐acetylcysteine‐conjugated metabolites (M46 and M49) were of interest because they were associated with pathways of sunitinib bioactivation. The structure of these adducts was elucidated by MS/MS analysis (Figure 4a,b). Furthermore, GSH and GSSG levels were depleted in both 3 hr serum and 3 hr liver from sunitinib‐treated mice (Figures 4c and S9a). No significant decrease of GSH and GSSG levels was found in the 24 hr serum and 24 hr liver (Figures 4d and S9a). The exhausted GSH in 3 hr serum was negatively correlated with a hepatic GSH‐conjugated metabolite, M40 (r = −0.8068; Figure S9b). The mRNA‐encoding enzymes that catalyse the detoxification of electrophilic compounds by conjugation with GSH were also evaluated. GSH peroxidase 1 (Gpx1), Gpx3, and Gpx4 mRNAs were significantly increased after sunitinib treatment (Figure S10). These data indicated that sunitinib exposure disrupted the homeostasis of GSH in vivo.
Figure 4.
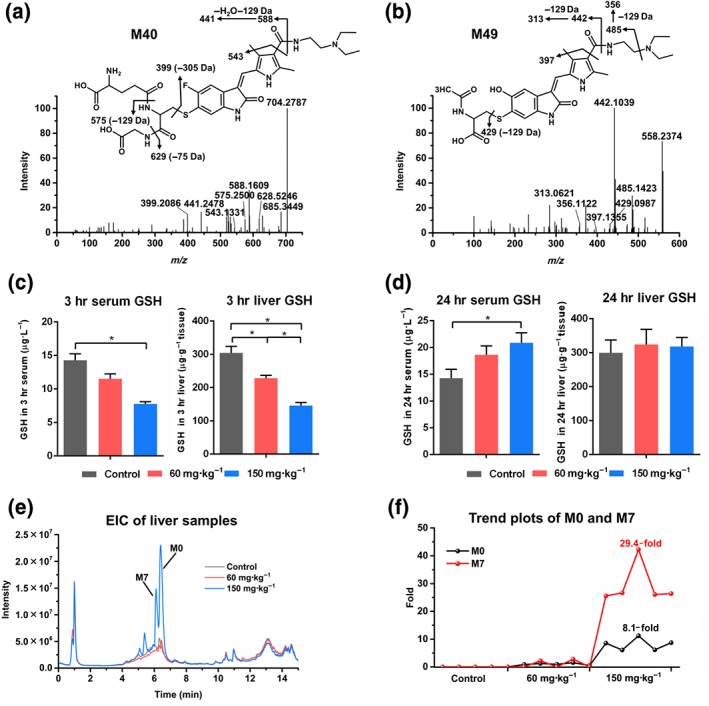
Reactive metabolites and impaired clearance of sunitinib. (a) The MS/MS of GSH‐conjugated metabolite M40. (b) The MS/MS of N‐acetylcysteine‐conjugated metabolite M49. (c) GSH levels were significantly decreased in 3 hr serum and 3 hr liver. (d) GSH levels had no significant change in 24 hr serum and 24 hr liver (e) Extracted ion chromatogram (EIC) of liver samples. Sunitinib (M0) and its metabolite M7 in liver were significantly increased after 150 mg·kg−1 sunitinib treatment. (f) Trend plots of M0 and M7 in liver. Compared with the 60 mg·kg−1 group, the content of M0 and M7 increased 8.1‐fold and 29.4‐fold respectively. All data were plotted as mean ± SEM (n = 5). *P < 0.05, significantly different as indicated
Another interesting finding was that sunitinib and its metabolites, especially M7, were significantly increased in liver after 150 mg·kg−1 sunitinib treatment (Figure 4e). Compared with the 60 mg·kg−1 group, the content of sunitinib (M0) and metabolite M7 increased 8.1‐fold and 29.4‐fold respectively (Figure 4f). The accumulated sunitinib (M0) was positively correlated with a GSH‐conjugated metabolite, M40 (r = 0.9151), which indicated that the accumulation of sunitinib was related to its metabolic activation (Figure S9c). Furthermore, partial metabolites, including M3, M7, M11, M13, M20, M26, M31, and M46, also significantly accumulated in 24 hr serum after 150 mg·kg−1 sunitinib treatment compared with 3 hr serum (Figure S11). Taken together, the formation of reactive metabolites and the impaired clearance of sunitinib may contribute to induce liver injury.
3.5. Sunitinib alters the liver metabolome
PCA analysis demonstrated clear differences in the liver metabolome between the control and sunitinib groups (Figure 2a). Orthogonal projection to latent structure‐discriminant analysis revealed ions contributing to this group separation. Ten metabolites, including three xenobiotic metabolites (M0, M6, and M7) and seven endogenous metabolites (carnitine, C2‐carnitine, C3‐carnitine, C5‐carnitine, C18:1‐carnitine, isoleucine/leucine, and LPC20:5), were found to be derived from the ion cloud in the loading scatter plot (Figure 2b).
Fifty‐five changed endogenous metabolites were identified in the liver by untargeted analysis (Figure 2c). The 35 increased acylcarnitines implied that hepatic mitochondrial fatty acid β‐oxidation (β‐FAO) was impaired (Figure 2c). The increase of pimelic acid and suberic acid suggested up‐regulation of hepatic endoplasmic reticulum ω‐FAO (Zhang et al., 2012; Figure 2c). The increased lithocholic acid, Tβ/αMCA, taurocholic acid, CA, tauroursodeoxycholic acid, and TDCA levels showed that sunitinib disrupted bile acid homeostasis in liver (Figure 2c). The increased LPC18:0, LPC20:3, LPE16:0, palmitic acid, and docosahexaenoic acid and the decreased LPC14:0, LPC20:5, and LPC22:5 implied that lipid homeostasis was disrupted by sunitinib (Figure 2c). These results showed that sunitinib may disrupt hepatic mitochondrial β‐FAO, endoplasmic reticulum ω‐FAO, bile acid homeostasis, and lipid homeostasis. More importantly, these changed metabolites were correlated with hepatic ALP, Il1b, and Il6 mRNAs levels after 24 hr sunitinib treatment (Figure 2d). Therefore, they may be the potential hepatic biomarker of sunitinib‐induced liver injury.
As 35 of the 55 metabolites were acylcarnitines, the expression of mRNAs encoded by acylcarnitine‐related genes was measured (Figure 2e). Ppara and its target genes carnitine palmitoyltransferases 1b and 2 (Cpt1b and Cpt2) were significantly down‐regulated by sunitinib treatment (Figure 2e).
The expression of mRNAs encoded by bile acid‐related genes was also evaluated. The level of Cyp8b1 mRNA‐encoding sterol 12α‐hydroxylase involved in bile acid synthesis was increased by two doses of sunitinib (Figure 2f). Ostb mRNA encoding the basolateral efflux transporter, organic solute transporter subunit β, was decreased by two doses of sunitinib (Figure 2f). Two canalicular transporters mRNAs Abcb11 and Abcc2 encoding bile salt export pump and multidrug resistance protein 2 were decreased (Figure 2f). It is well known that farnesoid X receptors (FXR, NR1H4) controls bile acid homeostasis. The expression of Fxr mRNA and its target gene small heterodimer partner (Shp) mRNA was significantly decreased by two doses of sunitinib (Figure 2f). These results showed that FXR signalling was inhibited by sunitinib, resulting in the disruption of bile acid homeostasis.
3.6. Sunitinib alters the serum metabolome
PCA was used to analyse 3 and 24 hr serum samples from control and 60 and 150 mg·kg−1 sunitinib‐treated groups. The sunitinib group deviated from the control group, indicating that sunitinib treatment significantly altered the content of metabolites in 3 and 24 hr serum (Figure 3a,b). Fifty‐two metabolites were identified in 3 hr serum (Figure 3c). The 20 increased acylcarnitines implied that mitochondrial β‐FAO was impaired (Figure 3c). The increased DCA and TDCA showed that bile acids were increased in 3 hr serum (Figure 3c). The increased LPC14:0, LPC16:0, LPC16:1, LPC18:1, LPC20:4, LPE20:4, LPE22:6, palmitic acid, oleic acid, stearic acid, docosahexaenoic acid, and 12‐hydroxyeicosatetraenoic acid revealed that lipids were increased in 3 hr serum (Figure 3c). The increased threonine, proline, isoleucine/leucine, acetylglycine, and acetylleucine and the decreased arginine implied that amino acid homeostasis was disrupted (Figure 3c). The increased methyladenosine and uridine and the decreased AMP implied that nucleotide homeostasis was disrupted (Figure 3c). Decreases in malic acid and succinic acid indicated that the tricarboxylic acid cycle was impaired (Figure 3c). Similarly, 31 metabolites, including acylcarnitines, lipids, and amino acids, were identified in 24 hr serum (Figure S12).
Fifteen metabolites were the same biomarkers in both liver and 24 hr serum (Table S1). Correlation analysis found that only LPE16:0, C18‐carnitine, C18‐OH‐carnitine, C20‐carnitine, C20:1‐carnitine, and C20:1‐OH‐caritine showed a good correlation between liver and serum (Figure 3d). These six metabolites could be used as biomarkers for sunitinib‐induced liver injury.
3.7. Sunitinib disturbed faeces and urine metabolome in mice
As shown in Figure S4a,c, the 150 mg·kg−1 sunitinib group deviated from the control group by PCA, indicating that sunitinib treatment significantly altered the content of metabolites in faeces and urine. Fifteen metabolites were identified in faeces (Figure S4b). The increase of ω‐muricholic acid, CA, DCA, and sulfocholic acid implied that the bile acid metabolism was disrupted by sunitinib (Figure S4b). The increase of LPC18:1, LPE14:0, oleic acid, docosahexaenoic acid, tetracosahexaenoic acid, arachidonic acid, and hydroxyoctadecanoic acid showed that lipid metabolism was affected by sunitinib (Figure S4b). Additionally, 10 metabolites were identified in the urine, and five were significant increased, including N2,N2‐dimethylguanosine, acetylleucine, phenylacetylglycine, sebacic acid, and decenedioic acid. Five of the 10 metabolites were significantly decreased, including carnitine, indoleacrylic acid, indolelactic acid, hippuric acid, and N‐acetylarylamine (Figure S4d). The change of faeces and urinary metabolome also provided evidence that the disorder of bile acid, fatty acid, and amino acid metabolism was the result of sunitinib exposure.
3.8. Severe hepatotoxicity was observed after 450 mg·kg−1 sunitinib exposure
An obvious congestion was observed in the liver when mice were treated with 450 mg·kg−1 sunitinib (Figure 5a), accompanied by a 2.6 g decrease in body weight after 2‐day treatment (Figure 5b). Biochemical analysis showed that 450 mg·kg−1 sunitinib exposure dramatically increased serum AST, ALT, and ALP levels at both 3 and 48 hr (P < 0.05; Figure 5c). Additionally, sunitinib treatment significantly increased cytokines Il1b, Il6, and Tnfa mRNAs, and intercellular adhesion molecule (Icam), and CD68 antigen (Cd68) mRNA levels (Figure 5d). Hepatic IL‐1β and IL‐6 protein expression and serum IL‐6 protein expression were significantly increased (Figure 5e,f). Furthermore, with the increase of sunitinib and its metabolites after the 450 mg·kg−1 sunitinib treatment, 48 hr liver GSH and GSSG levels were further depleted (Figure S13a,b). Compared with the changes in the 150 mg·kg−1 group, 450 mg·kg−1 sunitinib increased the content of acylcarnitines and decreased the expression of PPAR‐α target genes (Figure S13c,d). Correlation analysis found that acylcarnitine still had a good correlation with serum AST, ALT, and ALP and liver Il1b and Il6 mRNAs (Figure S13e). Therefore, 450 mg·kg−1 sunitinib induced obvious and severe liver injury.
Figure 5.
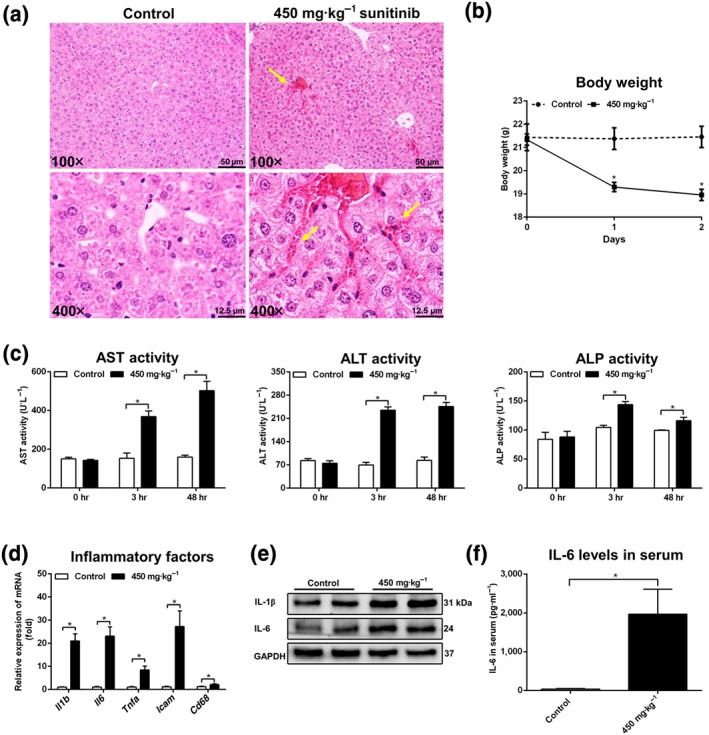
High‐dose sunitinib induced severe liver injury. (a) Haematoxylin and eosin staining of liver treated with 450 mg·kg−1 sunitinib. Haemocytes were shown by yellow arrowheads in 450 mg·kg−1 sunitinib groups. (b) Body weight was significantly decreased after 450 mg·kg−1 sunitinib treatment. (c) Aspartate transaminase (AST), alanine transaminase (ALT), and alkaline phosphatase (ALP) enzyme activity was significantly increased in 3 and 48 hr serum. (d) Real‐time PCR analysis of inflammatory factors in liver. All inflammatory factors were significantly increased after 450 mg·kg−1 sunitinib treatment. Values represented fold change after normalization to control. (e) Western blotting was used to measure IL‐1β and IL‐6. IL‐1β and IL‐6 levels were significantly increased after 450 mg·kg−1 sunitinib treatment. (f) IL‐6 levels in 48 hr serum were significantly increased. All data were plotted as mean ± SEM (n = 5). *P < 0.05, significantly different as indicated
3.9. ABT and fenofibrate protected against sunitinib‐induced liver damage
ABT could attenuate liver injury induced by sunitinib, as revealed by lowered serum AST, ALT, and acylcarnitines levels (Figure 6). Although the sunitinib content was not increased after using ABT, sunitinib metabolites, such as M7 and M40, were significantly decreased (Figure 6c). Correlation analysis revealed that M40 showed a good correlation with serum AST and ALT levels (Figure 6d), indicating that the hepatotoxicity of sunitinib was correlated with its metabolism. The question arose whether an improvement of β‐FAO by treatment of mice with a PPARα agonist might protect from sunitinib‐induced hepatotoxicity. The data suggested that fenofibrate could significantly attenuate liver injury induced by sunitinib, as shown by lowered AST and acylcarnitine levels (Figure 6e,f).
Figure 6.
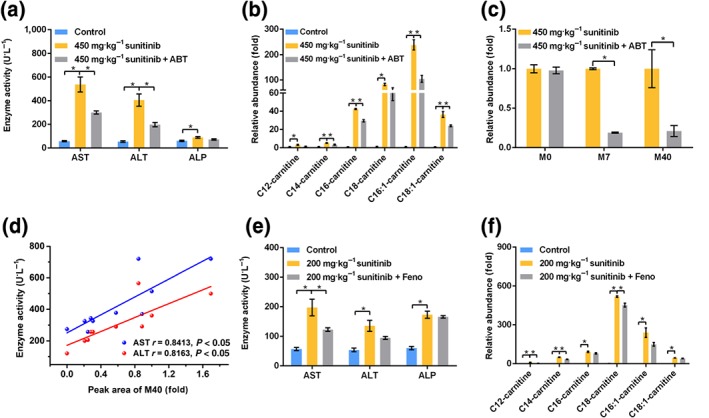
1‐Aminobenzotriazole (ABT) and fenofibrate (Feno) attenuated liver injury induced by sunitinib. (a) Aspartate transaminase (AST) and alanine transaminase (ALT) levels were significantly decreased after ABT treatment. (b) Acylcarnitine levels were significantly decreased after ABT treatment. (c) Xenobiotic metabolites M0, M7, and M40 levels in 450 mg·kg−1 sunitinib and 450 mg·kg−1 sunitinib + ABT groups. M7 and M40 levels were significantly decreased after ABT treatment. (d) Correlation analysis showed that M40 level had a good correlation with AST and ALT enzyme activities. (e) AST levels were significantly decreased after fenofibrate treatment. (f) Acylcarnitine levels were significantly decreased after fenofibrate treatment. All data were plotted as mean ± SEM (n = 5). *P < 0.05, significantly different as indicated
4. DISCUSSION
Recent studies of 31 Food and Drug Administration‐approved low MW kinase inhibitors indicated that sunitinib did not damage mitochondrial function, as assessed by use of isolated rat liver mitochondria (Zhang et al., 2017). However, it was reported that sunitinib caused mitochondrial toxicity after acute and long‐term exposure in vivo (Paech et al., 2017). Low concentrations of sunitinib were found to reduce mitochondrial transmembrane potential (ΔΨm) resulting in mitochondrial dysfunction (Porceddu et al., 2012). Severe mitochondrial structural abnormalities were also found in the heart of a patient treated with sunitinib (Kerkela et al., 2009). In the present study, the levels of acylcarnitines were significantly elevated in the serum and liver of sunitinib‐treated mice, suggesting that sunitinib exposure caused liver mitochondrial toxicity in mice (Chen, Krausz, Shah, Idle, & Gonzalez, 2009; McGill et al., 2014).
Two new GSH adducts (M40 and M41) and two new N‐acetylcysteine conjugates (M46 and M49) were found in the liver of mice treated with sunitinib. These metabolites result from conjugation of electrophilic intermediates that are commonly considered as mediators of drug‐induced toxicity, leading to mutagenesis, apoptosis, necrosis, and carcinogenicity in both animals and cultured cells (Williams et al., 2002). Further analysis indicated that sunitinib exposure disrupted the GSH homeostasis. Several GSH conjugates of sunitinib were found in previous studies (Amaya et al., 2018; Kenny et al., 2012). Urinary N‐acetylcysteine conjugates of xenobiotics were identified as markers reflecting the electrophilic burden of environmental chemicals on humans (Jian, Yao, Zhang, & Zhu, 2009). Therefore, metabolic activation may play a dominant role in sunitinib‐induced hepatotoxicity.
One intriguing finding was that the clearance of sunitinib was impaired at the high dose, and sunitinib and its metabolites were increased in the liver. After a single oral administration of sunitinib, the plasma terminal elimination half‐lives of sunitinib were 8 hr in rats, 17 hr in monkeys, and 51 hr in humans (Speed et al., 2012). In mice, sunitinib and its metabolites were found in liver at 24 hr after 150 mg·kg−1 sunitinib treatment. The abundance of metabolites in 24 hr serum was higher than in 3 hr serum, including M3, M7, M11, M13, M20, M26, M31, and M46. Interestingly, impaired clearance of sunitinib was observed in the 150 and 450 mg·kg−1 groups and not in the 60 mg·kg−1 group. Although the clinical dose of sunitinib (50–150 mg·day−1) is not too high, sunitinib is given to patients continuously for several months, which might cause accumulation of sunitinib and its reactive metabolites. This might explain why sunitinib does not induce liver injury after the first round of use, with liver injury typically occuring during later cycles of sunitinib (Guillen et al., 2016; Mermershtain et al., 2013; Mueller et al., 2008; Taran et al., 2009; Weise et al., 2009).
The current study suggests that sunitinib hepatotoxicity results from its metabolism. When sunitinib metabolism was lowered by the CYP inhibitor ABT, its hepatotoxicity was significantly decreased companied by a decrease of its metabolites. These data suggest an important role of sunitinib metabolism in liver injury. In previous studies, ABT could reduce pazopanib‐induced acute hepatotoxicity through the inhibition of pazopanib metabolism (Wang et al., 2018). ABT could reduce paracetamol‐induced cytotoxicity through inhibition of the production of paracetamol–protein adducts (Miyakawa et al., 2015).
Using a metabolomics approach, several metabolic pathways were found to be affected by sunitinib exposure, including amino acids, lipids, and bile acids. A recent metabolomics study indicated that sunitinib could increase the levels of several endogenous metabolites in FVB/N mice, such as disaccharides and ethanolamine (Jensen, Parry, Huang, Ilaiwy, et al., 2017). Among these changed metabolites, sunitinib had a dominant influence on lipid metabolism. Free fatty acids, including long‐chain fatty acids and medium‐chain dicarboxylic acids, were increased in the sunitinib‐treated group, and conjugated fatty acids, including long‐chain acylcarnitines and LPCs, were increased in serum. In agreement with the current results, clinical observations also found that sunitinib treatment caused hyperlipidaemia in patients with metastatic renal cell carcinoma (Tassi, Baldazzi, Lapini, Carini, & Mazzanti, 2015). Earlier studies revealed that the accumulated long‐chain acylcarnitines such as C16‐carnitine produced oxidative stress in the liver (Zhao, Yang, Wang, et al., 2017) and elevated LPCs such as LPC18:0 and LPC18:1 can activate NF‐κB (Fang et al., 2017). Several studies reported that increased acylcarnitines were associated with liver toxicity of drugs such as paracetamol and cocaine (Chen et al., 2009; Shi, Yao, Gosnell, & Chen, 2012). However, this is the first report that low MW kinase inhibitors cause the accumulation of acylcarnitines. The activation of PPAR‐α and its target genes Cpt1b and Cpt2 was inhibited after sunitinib treatment, suggesting that the abnormal lipid metabolism was due to the suppression of PPAR‐α signalling. Bile acids, which were synthesized in the liver, were also increased by sunitinib. However, increased bile acids may cause liver injury as a result of cholestasis (Higuchi & Gores, 2003; O'Brien et al., 2013). CP‐724714, a HER2 TK inhibitor, was discontinued from clinical development due to hepatocellular injury and hepatobiliary cholestatic mechanisms (Feng et al., 2009). Further studies found that this compound inhibited the activity of ABCB11 and multidrug resistance protein 1 (ABCC1). Here, the expression levels of Abcb11, Abcc2, and Ostb mRNAs were suppressed by sunitinib exposure. Furthermore, the elevated bile acids inhibited the expression of Fxr and Shp mRNAs. Therefore, activation of PPAR‐α or FXR may be of value in attenuating sunitinib‐induced liver injury.
PPAR‐α may be a potential therapeutic target for the treatment of various liver injury, including alcoholic liver disease, liver fibrosis, and d‐glactosamine/LPS‐induced acute liver failure (Chen et al., 2015; Jiao et al., 2014; Nan, Wang, & Fu, 2014). Some pilot studies demonstrated the efficacy of fenofibrate and ursodeoxycholic acid combination in patients with primary biliary cirrhosis (Ghonem & Boyer, 2013; Zhang et al., 2015). Furthermore, fenofibrate could protect ethinylestradiol and chlorpromazine‐, bile duct‐ligated‐, α‐naphthylisothiocyanate‐, and lithocholic acid‐induced cholestasis (Cindoruk et al., 2007; Dai et al., 2017; El‐Sisi, Hegazy, & El‐Khateeb, 2013; Zhao, Yang, Liu, et al., 2017). In the present study, fenofibrate protected against sunitinib‐induced liver injury.
Numerous studies have demonstrated that the side effects of drugs may be closely related to drug metabolites. Through the combination of high‐resolution LC–MS technology and OPLS analysis, 32 novel metabolites of sunitinib (M8–M11, M16, M21–M37, and M40–M49) were identified in this study. As seen from the metabolic map (Figure S8), the phase I metabolic pathway of sunitinib includes N‐dealkylation, hydroxylation, dehydrogenation, oxidative defluorination, hydrogenation, and deamination of N,N′‐diethylamine. Consistent with a previous study, N‐dealkylation was found to be the main metabolic pathway in vitro and in vivo (Speed et al., 2012). The phase II metabolic pathway was glucuronide conjugation, sulfate conjugation, methylation, glucose conjugation, GSH conjugation, and N‐acetylcysteine conjugation. Multiple CYPs involved in the formation of these metabolites, including CYP1B1, 2C8, and 2D6. Two highly abundant quaternary ammonium N‐oxide metabolites M1 and M5 were generated by the oxidation of this tertiary amine in sunitinib. Long‐term therapy with procainamide may cause lupus erythematosus in 25–30% of patients in which the N‐oxidized metabolites of procainamide may be responsible for systemic lupus erythematosus development (Li et al., 2012). An earlier study reported that sunitinib can cause lupus erythematosus and hand–food skin reaction in 29% of 5,658 patients (Motzer et al., 2009). Clearly, the clinical side effects of sunitinib may be associated with its metabolism.
Previous studies found the defluorinated metabolites, M12, could be further oxidized to an electrophilic quinone imine (Amaya et al., 2018). Therefore, M12 was more electrophilic than sunitinib, and its GSH conjugate was found in a previous study (Amaya et al., 2018). In the present study, an N‐acetylcysteine conjugate of M12, M49, was found in liver, urine, and faeces. A number of reports suggested that variation in genes related to sunitinib metabolism and transport significantly affected drug toxicity (Diekstra et al., 2014; van Erp et al., 2009). CYP1A2 was the main enzyme involved in the formation of M12, and thus, CYP1A2 may play an important role in the bioactivation of sunitinib in vitro.
In conclusion, metabolomics was used to profile metabolic changes that occur during sunitinib exposure in mice. Alterations in endogenous metabolites revealed sunitinib hepatotoxicity, which may be attributed to metabolic activation of sunitinib and its impaired clearance. Further, the regulation of PPAR‐α and inhibition of xenobiotic metabolism may be used to attenuate sunitinib‐induced liver injury.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Q.Z., T.Z., X.‐R.X., and J.‐F.H. performed the experiments and analysed the data. Q.Z. and F.L. wrote the paper. F.L. and F.J.G. designed the experiments. All authors approved the final version of the paper.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Data S1.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by the National Key Research and Development Program of China (2017YFC1700906 and 2017YFC0906903), CAS “Light of West China” Program (Y72E8211W1), Kunming Institute of Botany, Chinese Academy of Sciences (Y76E1211K1 and Y4662211K1), State Key Laboratory of Phytochemistry and Plant Resources in West China (52Y67A9211Z1), and the Open Fund of State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University (KF‐GN‐201705).
Zhao Q, Zhang T, Xiao X‐R, et al. Impaired clearance of sunitinib leads to metabolic disorders and hepatotoxicity. Br J Pharmacol. 2019;176:2162–2178. 10.1111/bph.14664
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. , Peters, J. A. , Benson, H. E. , et al. (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174, S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174, S360–S446. 10.1111/bph.13883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya, G. M. , Durandis, R. , Bourgeois, D. S. , Perkins, J. A. , Abouda, A. A. , Wines, K. J. , … Jackson, K. D. (2018). Cytochromes P450 1A2 and 3A4 catalyze the metabolic activation of sunitinib. Chemical Research in Toxicology, 31, 570–584. 10.1021/acs.chemrestox.8b00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanca, A. J. , Ruiz‐Armenta, M. V. , Zambrano, S. , Miguel‐Carrasco, J. L. , Arias, J. L. , Arevalo, M. , … Vázquez, C. M. (2016). Inflammatory and fibrotic processes are involved in the cardiotoxic effect of sunitinib: Protective role of l‐carnitine. Toxicology Letters, 241, 9–18. 10.1016/j.toxlet.2015.11.007 [DOI] [PubMed] [Google Scholar]
- Chen, C. , Krausz, K. W. , Shah, Y. M. , Idle, J. R. , & Gonzalez, F. J. (2009). Serum metabolomics reveals irreversible inhibition of fatty acid β‐oxidation through the suppression of PPARα activation as a contributing mechanism of acetaminophen‐induced hepatotoxicity. Chemical Research in Toxicology, 22, 699–707. 10.1021/tx800464q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Li, L. , Chen, J. , Li, L. , Zheng, Z. , Ren, J. , & Qiu, Y. (2015). Oleoylethanolamide, an endogenous PPAR‐α ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget, 6, 42530–42540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cindoruk, M. , Kerem, M. , Karakan, T. , Salman, B. , Akin, O. , Alper, M. , … Ünal, S. (2007). Peroxisome proliferators‐activated alpha agonist treatment ameliorates hepatic damage in rats with obstructive jaundice: An experimental study. BMC Gastroenterology, 7, 44 10.1186/1471-230X-7-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, M. , Yang, J. , Xie, M. , Lin, J. , Luo, M. , Hua, H. , … Liu, A. (2017). Inhibition of JNK signalling mediates PPARα‐dependent protection against intrahepatic cholestasis by fenofibrate. British Journal of Pharmacology, 174, 3000–3017. 10.1111/bph.13928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekstra, M. H. , Klumpen, H. J. , Lolkema, M. P. , Yu, H. , Kloth, J. S. , Gelderblom, H. , … Mathijssen, R. H. (2014). Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clinical Pharmacology and Therapeutics, 96, 81–89. 10.1038/clpt.2014.47 [DOI] [PubMed] [Google Scholar]
- El‐Sisi, A. , Hegazy, S. , & El‐Khateeb, E. (2013). Effects of three different fibrates on intrahepatic cholestasis experimentally induced in rats. PPAR Research, 2013. 781348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erp, N. P. , Eechoute, K. , van der Veldt, A. A. , Haanen, J. B. , Reyners, A. K. , Mathijssen, R. H. , … Gelderblom, H. (2009). Pharmacogenetic pathway analysis for determination of sunitinib‐induced toxicity. Journal of Clinical Oncology, 27, 4406–4412. 10.1200/JCO.2008.21.7679 [DOI] [PubMed] [Google Scholar]
- Faivre, S. , Delbaldo, C. , Vera, K. , Robert, C. , Lozahic, S. , Lassau, N. , … Raymond, E. (2006). Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. Journal of Clinical Oncology, 24, 25–35. 10.1200/JCO.2005.02.2194 [DOI] [PubMed] [Google Scholar]
- Fang, Z. Z. , Tanaka, N. , Lu, D. , Jiang, C. T. , Zhang, W. H. , Zhang, C. Z. , … Gonzalez, F. J. (2017). Role of the lipid‐regulated NF‐κB/IL‐6/STAT3 axis in alpha‐naphthyl isothiocyanate‐induced liver injury. Archives of Toxicology, 91, 2235–2244. 10.1007/s00204-016-1877-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, B. , Xu, J. J. , Bi, Y. A. , Mireles, R. , Davidson, R. , Duignan, D. B. , … Wang, H. F. (2009). Role of hepatic transporters in the disposition and hepatotoxicity of a HER2 tyrosine kinase inhibitor CP‐724,714. Toxicological Sciences, 108, 492–500. 10.1093/toxsci/kfp033 [DOI] [PubMed] [Google Scholar]
- Fu, Y. , Wei, X. , Lin, L. , Xu, W. , & Liang, J. (2018). Adverse reactions of sorafenib, sunitinib, and imatinib in treating digestive system tumors. Thorac Cancer, 9, 542–547. 10.1111/1759-7714.12608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonem, N. S. , & Boyer, J. L. (2013). Fibrates as adjuvant therapy for chronic cholestatic liver disease: Its time has come. Hepatology, 57, 1691–1693. 10.1002/hep.26155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, V. L. , Rock, E. P. , Dagher, R. , Ramchandani, R. P. , Abraham, S. , Gobburu, J. V. , … Pazdur, R. (2007). Approval summary: Sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clinical Cancer Research, 13, 1367–1373. 10.1158/1078-0432.CCR-06-2328 [DOI] [PubMed] [Google Scholar]
- Guillen, S. S. , Meijer, M. , & de Jongh, F. E. (2016). Lethal acute liver failure in a patient treated with sunitinib. BML Case Reports. 10.1136/bcr-2015-213624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi, H. , & Gores, G. J. (2003). Bile acid regulation of hepatic physiology: IV. Bile acids and death receptors. Am J Physiol Gastr L, 284, G734–G738. [DOI] [PubMed] [Google Scholar]
- Ibrahim, E. M. , Kazkaz, G. A. , Abouelkhair, K. M. , Bayer, A. M. , & Elmasri, O. A. (2013). Sunitinib adverse events in metastatic renal cell carcinoma: A meta‐analysis. International Journal of Clinical Oncology, 18, 1060–1069. 10.1007/s10147-012-0497-2 [DOI] [PubMed] [Google Scholar]
- Jensen, B. C. , Parry, T. L. , Huang, W. , Beak, J. Y. , Ilaiwy, A. , Bain, J. R. , … Willis, M. S. (2017). Effects of the kinase inhibitor sorafenib on heart, muscle, liver and plasma metabolism in vivo using non‐targeted metabolomics analysis. Brit J Pharmacol, 174, 4797–4811. 10.1111/bph.14062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, B. C. , Parry, T. L. , Huang, W. , Ilaiwy, A. , Bain, J. R. , Muehlbauer, M. J. , … Willis, M. (2017). Non‐targeted metabolomics analysis of the effects of tyrosine kinase inhibitors sunitinib and erlotinib on heart, muscle, liver and serum metabolism in vivo. Metabolites, 7, 31 10.3390/metabo7030031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian, W. , Yao, M. , Zhang, D. , & Zhu, M. (2009). Rapid detection and characterization of in vitro and urinary N‐acetyl‐l‐cysteine conjugates using quadrupole–linear ion trap mass spectrometry and polarity switching. Chemical Research in Toxicology, 22, 1246–1255. 10.1021/tx900035j [DOI] [PubMed] [Google Scholar]
- Jiao, M. , Ren, F. , Zhou, L. , Zhang, X. , Zhang, L. , Wen, T. , … Duan, Z. (2014). Peroxisome proliferator‐activated receptor α activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death & Disease, 5, e1397 10.1038/cddis.2014.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny, J. R. , Mukadam, S. , Zhang, C. , Tay, S. , Collins, C. , Galetin, A. , & Khojasteh, S. C. (2012). Drug–drug interaction potential of marketed oncology drugs: In vitro assessment of time‐dependent cytochrome P450 inhibition, reactive metabolite formation and drug–drug interaction prediction. Pharmaceutical Research, 29, 1960–1976. 10.1007/s11095-012-0724-6 [DOI] [PubMed] [Google Scholar]
- Kerkela, R. , Woulfe, K. C. , Durand, J. B. , Vagnozzi, R. , Kramer, D. , Chu, T. F. , … Force, T. (2009). Sunitinib‐induced cardiotoxicity is mediated by off‐target inhibition of AMP‐activated protein kinase. Clinical and Translational Science, 2, 15–25. 10.1111/j.1752-8062.2008.00090.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Patterson, A. D. , Krausz, K. W. , Dick, B. , Frey, F. J. , Gonzalez, F. J. , & Idle, J. R. (2012). Metabolomics reveals the metabolic map of procainamide in humans and mice. Biochemical Pharmacology, 83, 1435–1444. 10.1016/j.bcp.2012.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Lu, Y. , Guan, X. , Dong, B. , Chavan, H. , Wang, J. , … Li, F. (2015). Metabolomics reveals the formation of aldehydes and iminium in gefitinib metabolism. Biochemical Pharmacology, 97, 111–121. 10.1016/j.bcp.2015.07.010 [DOI] [PubMed] [Google Scholar]
- McGill, M. R. , Li, F. , Sharpe, M. R. , Williams, C. D. , Curry, S. C. , Ma, X. C. , & Jaeschke, H. (2014). Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Archives of Toxicology, 88, 391–401. 10.1007/s00204-013-1118-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel, D. B. , Laird, A. D. , Xin, X. , Louie, S. G. , Christensen, J. G. , Li, G. , … Cherrington, J. M. (2003). In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clinical Cancer Research, 9, 327–337. [PubMed] [Google Scholar]
- Mermershtain, W. , Lazarev, I. , Shani‐Shrem, N. , & Ariad, S. (2013). Fatal liver failure in a patient treated with sunitinib for renal cell carcinoma. Clin Genitourin Canc, 11, 70–72. 10.1016/j.clgc.2012.09.005 [DOI] [PubMed] [Google Scholar]
- Miyakawa, K. , Albee, R. , Letzig, L. G. , Lehner, A. F. , Scott, M. A. , Buchweitz, J. P. , … Roth, R. A. (2015). A cytochrome P450‐independent mechanism of acetaminophen‐induced injury in cultured mouse hepatocytes. The Journal of Pharmacology and Experimental Therapeutics, 354, 230–237. 10.1124/jpet.115.223537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer, R. J. , Hutson, T. E. , Tomczak, P. , Michaelson, M. D. , Bukowski, R. M. , Oudard, S. , … Figlin, R. A. (2009). Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, 27, 3584–3590. 10.1200/JCO.2008.20.1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, E. W. , Rockey, M. L. , & Rashkin, M. C. (2008). Sunitinib‐related fulminant hepatic failure: Case report and review of the literature. Pharmacotherapy, 28, 1066–1070. 10.1592/phco.28.8.1066 [DOI] [PubMed] [Google Scholar]
- Nan, Y. M. , Wang, R. Q. , & Fu, N. (2014). Peroxisome proliferator‐activated receptor alpha, a potential therapeutic target for alcoholic liver disease. World J Gastroentero, 20, 8055–8060. 10.3748/wjg.v20.i25.8055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery, R. (1971). The metabolic interconversion of arecoline and arecoline 1‐oxide in the rat. The Biochemical Journal, 122, 503–508. 10.1042/bj1220503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, K. M. , Allen, K. M. , Rockwell, C. E. , Towery, K. , Luyendyk, J. P. , & Copple, B. L. (2013). IL‐17A synergistically enhances bile acid‐induced inflammation during obstructive cholestasis. The American Journal of Pathology, 183, 1498–1507. 10.1016/j.ajpath.2013.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paech, F. , Bouitbir, J. , & Krahenbuhl, S. (2017). Hepatocellular toxicity associated with tyrosine kinase inhibitors: Mitochondrial damage and inhibition of glycolysis. Frontiers in Pharmacology, 8, 367 10.3389/fphar.2017.00367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porceddu, M. , Buron, N. , Roussel, C. , Labbe, G. , Fromenty, B. , & Borgne‐Sanchez, A. (2012). Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicological Sciences, 129, 332–345. 10.1093/toxsci/KFS197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shantakumar, S. , Nordstrom, B. L. , Djousse, L. , Hall, S. A. , Gagnon, D. R. , Fraeman, K. H. , … Nelson, J. (2016). Occurrence of hepatotoxicity with pazopanib and other anti‐VEGF treatments for renal cell carcinoma: An observational study utilizing a distributed database network. Cancer Chemoth Pharm, 78, 559–566. 10.1007/s00280-016-3112-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, X. , Yao, D. , Gosnell, B. A. , & Chen, C. (2012). Lipidomic profiling reveals protective function of fatty acid oxidation in cocaine‐induced hepatotoxicity. Journal of Lipid Research, 53, 2318–2330. 10.1194/jlr.M027656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed, B. , Bu, H. Z. , Pool, W. F. , Peng, G. W. , Wu, E. Y. , Patyna, S. , … Kang, P. (2012). Pharmacokinetics, distribution, and metabolism of [14C]sunitinib in rats, monkeys, and humans. Drug Metabolism and Disposition, 40, 539–555. 10.1124/dmd.111.042853 [DOI] [PubMed] [Google Scholar]
- Sun, L. , Liang, C. , Shirazian, S. , Zhou, Y. , Miller, T. , Cui, J. , … Tang, C. (2003). Discovery of 5‐[5‐fluoro‐2‐oxo‐1,2‐dihydroindol‐(3Z)‐ylidenemethyl]‐2,4‐dimethyl‐1H‐pyrrole‐3‐carboxylic acid (2‐diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet‐derived growth factor receptor tyrosine kinase. Journal of Medicinal Chemistry, 46, 1116–1119. [DOI] [PubMed] [Google Scholar]
- Tan, J. R. , Chakravarthi, S. , Judson, J. P. , Haleagrahara, N. , & Segarra, I. (2013). Potential protective effect of sunitinib after administration of diclofenac: Biochemical and histopathological drug–drug interaction assessment in a mouse model. N‐S Arch Pharmacol, 386, 619–633. 10.1007/s00210-013-0861-4 [DOI] [PubMed] [Google Scholar]
- Taran, A. , Ignatov, A. , Smith, B. , Costa, S. D. , & Bischoff, J. (2009). Acute hepatic failure following monotherapy with sunitinib for ovarian cancer. Cancer Chemoth Pharm, 63, 971–972. 10.1007/s00280-008-0814-7 [DOI] [PubMed] [Google Scholar]
- Tassi, R. , Baldazzi, V. , Lapini, A. , Carini, M. , & Mazzanti, R. (2015). Hyperlipidemia and hypothyroidism among metastatic renal cell carcinoma patients taking sunitinib malate. Related or unrelated adverse events? Clin Genitourin Canc, 13, e101–e105. 10.1016/j.clgc.2014.08.009 [DOI] [PubMed] [Google Scholar]
- Wang, Y. K. , Yang, X. N. , Liang, W. Q. , Xiao, Y. , Zhao, Q. , Xiao, X. R. , … Li, F. (2018). A metabolomic perspective of pazopanib‐induced acute hepatotoxicity in mice. Xenobiotica, 1–16. 10.1080/00498254.2018.1489167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise, A. M. , Liu, C. Y. , & Shields, A. F. (2009). Fatal liver failure in a patient on acetaminophen treated with sunitinib malate and levothyroxine. The Annals of Pharmacotherapy, 43, 761–766. 10.1345/aph.1L528 [DOI] [PubMed] [Google Scholar]
- Williams, D. P. , Kitteringham, N. R. , Naisbitt, D. J. , Pirmohamed, M. , Smith, D. A. , & Park, B. K. (2002). Are chemically reactive metabolites responsible for adverse reactions to drugs? Current Drug Metabolism, 3, 351–366. 10.2174/1389200023337423 [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Salminen, A. , Yang, X. , Luo, Y. , Wu, Q. , White, M. , … Shi, Q. (2017). Effects of 31 FDA approved small‐molecule kinase inhibitors on isolated rat liver mitochondria. Archives of Toxicology, 91, 2921–2938. 10.1007/s00204-016-1918-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Li, F. , Patterson, A. D. , Wang, Y. , Krausz, K. W. , Neale, G. , … Schuetz, J. D. (2012). Abcb11 deficiency induces cholestasis coupled to impaired β‐fatty acid oxidation in mice. The Journal of Biological Chemistry, 287, 24784–24794. 10.1074/jbc.M111.329318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Li, S. , He, L. , Wang, F. , Chen, K. , Li, J. , … Guo, C. (2015). Combination therapy of fenofibrate and ursodeoxycholic acid in patients with primary biliary cirrhosis who respond incompletely to UDCA monotherapy: A meta‐analysis. Drug Des Dev Ther, 9, 2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. , Li, X. M. , Liu, H. N. , Gonzalez, F. J. , & Li, F. (2018). Metabolic map of osthole and its effect on lipids. Xenobiotica, 48, 285–299. 10.1080/00498254.2017.1306660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. , Yang, R. , Liu, F. , Wang, J. , Hu, D.‐D. , Yang, X.‐W. , & Li, F. (2017). Metabolomics reveals that PPARα activation protects against lithocholic acid‐induced liver injury. RSC Advances, 7, 49849–49857. 10.1039/C7RA08823J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. , Yang, R. , Wang, J. , Hu, D. D. , & Li, F. (2017). PPARα activation protects against cholestatic liver injury. Scientific Reports, 7, 9967 10.1038/s41598-017-10524-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Supporting information