

Abstract
Background and Purpose
Inactivation of the gene for adenosine A2A receptors (ADORA2A for humans and Adora2a for rodents) protects against brain injury in experimental stroke. However, the cell‐specific pathogenic effects of A2A receptors in thromboembolic stroke and the underlying mechanisms remain undefined. Here, we tested the hypothesis that inhibition of endothelial A2A receptors after thromboembolic stroke improves post‐stroke outcomes via down‐regulation of inflammation.
Experimental Approach
Thromboembolic stroke was induced by embolic middle cerebral artery occlusion in mice. Post‐stroke outcomes were determined with neurological deficit scoring, infarct volume, inflammatory marker expression, brain leukocyte infiltration, blood–brain barrier (BBB) leakage, and oedema assessment. Anti‐inflammatory effects of silencing the gene for A2A receptors or pharmacological antagonism of these receptors were assessed in vitro.
Key Results
Thromboembolic stroke induced Adora2a expression in the brain. Mice globally deficient in Adora2a (Adora2a −/−) were resistant to stroke injury. Mice specifically deficient in endothelial Adora2a (Adora2a ΔVEC) showed reduced leukocyte infiltration, BBB leakage, and oedema after stroke, along with attenuated downstream proinflammatory markers, both in vivo and in vitro. The A2A receptor antagonist, KW 6002, also reduced brain injury and inflammation after stroke. Inactivation of ADORA2A inhibited endothelial inflammation via suppression of the NLRP3 inflammasome, down‐regulating cleaved caspase 1 and IL‐1β expression.
Conclusions and Implications
Specific inactivation of endothelial A2A receptors mitigated ischaemic brain injury and improved post‐stroke outcomes, at least partly, through anti‐inflammatory effects via blockade of NLRP3 inflammasome activity. Our findings may open new approaches to vascular protection after ischaemic stroke.
Abbreviations
- ADORA2A (Adora2a for rodents)
gene for adenosine A2A receptor
- ASC
apoptosis‐associated speck‐like protein containing a C‐terminal caspase activation and recruitment domain
- BBB
blood–brain barrier
- BMECs
brain microvascular endothelial cells
- eMCAo
embolic middle cerebral artery occlusion
- huBMEC
human brain microvascular endothelial cell
- ICAM1
intercellular adhesion molecule 1
- NLRP3
nucleotide‐binding domain and leucine‐rich repeat containing family, pyrin domain containing 3
- PFA
paraformaldehyde
- TTC
2,3,5‐triphenyltetrazolium chloride
- VCAM1
vascular adhesion molecule 1
- WT
wild‐type
What is already known
The level of extracellular adenosine is increased dramatically in ischaemic brain.
A2A receptors on diverse types of cells play different roles in brain ischaemic injury.
What this study adds
A2A receptors on brain endothelial cells mediate inflammatory responses in thromboembolic stroke.
Genetic deficiency and pharmacological blockade of endothelial A2A receptors improve post‐stroke outcomes through anti‐inflammatory effects.
What is the clinical significance
Inhibition of A2A receptors provides a new approach to vascular protection in ischaemic stroke.
1. INTRODUCTION
Ischaemic stroke, which accounts for approximately 87% of all stroke cases, is one of the most common cerebrovascular diseases (Owens, 2014). Neuroinflammation, mediated by proinflammatory cytokines such as IL‐1β and TNF‐α, plays a pivotal role in the progression of post‐stroke pathogenesis resulting in long‐term adverse prognosis (Anrather & Iadecola, 2016). Specifically, brain microvascular endothelial cells (BMECs) express adhesion molecules, including selectins, vascular adhesion molecule 1 (VCAM1), and intercellular adhesion molecule 1 (ICAM1), that mediate the rolling, adhesion, and trans‐endothelial migration of leukocytes into the ischaemic brain parenchyma (Jin, Yang, & Li, 2010), resulting in irreversible neuronal injury. These inflammatory reactions are preceded by disruption of the blood–brain barrier (BBB) and oedema, collectively recognized as the major cause of post‐stroke mortality (Wang, Tang, & Yenari, 2007). Therefore, vascular protection is urgently needed after stroke to minimize neuron damage.
The nucleotide‐binding domain and leucine‐rich repeat containing family, pyrin domain containing 3 (NLRP3) inflammasome, which consists of the NLRP3 scaffold, the adaptor molecule apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (ASC), and caspase‐1, is the best characterized member of the NOD‐like receptor family. Activation of the NLRP3 inflammasome induces caspase‐1 cleavage, leading to the secretion of IL‐1β and IL‐18, thus triggering initiation and amplification of various inflammatory processes (Lamkanfi & Dixit, 2014). Hyperactivation of the NLRP3 inflammasome has been implicated in neuroinflammatory diseases including stroke and dementia (Yang et al., 2014; Yin et al., 2018); thus, suppression of NLRP3 inflammasome activity reduces inflammation in ischaemic stroke.
Adenosine is an essential neuromodulator that plays an important role in various pathophysiological conditions (Melani, Pugliese, & Pedata, 2014). Cellular signalling by adenosine occurs through four GPCRs: adenosine A1, A2A, A2B and A3 receptors, coded for by the corresponding genes, ADORA1, ADORA2A, ADORA2B, and ADORA3. Among them, A1 and A2A receptors have stronger affinities for adenosine, and the abundance of these two receptors in the brain is much greater than that of A2B and A3 receptors (Gomes, Kaster, Tome, Agostinho, & Cunha, 2011). Previous studies have shown that activation of A1 receptors is protective in cerebral ischaemia, but the serious side effects, including sedation, bradycardia, and hypotension, greatly limit the therapeutic value of this action (Melani et al., 2014). The role of A2A receptors in ischaemic stroke has become a focus for much research. Some studies indicate that A2A receptors contribute to brain injury in different animal models of cerebral ischaemia (Chen et al., 2007). However, the effects are complex and vary in the different brain ischaemic injury models or in different stages of the same model (Chen et al., 1999; Gao & Phillis, 1994; Melani, Corti, Cellai, Vannucchi, & Pedata, 2014; Melani et al., 2003; Sheardown & Knutsen, 1996; Von Lubitz, Lin, & Jacobson, 1995). Also, A2A receptors play different roles in diverse types of cells in ischaemic injury (Fredholm, Cunha, & Svenningsson, 2003). As endothelial cells are an essential component of blood vessels in inflammation and because the therapeutic benefits of the inactivation of A2A receptors following thromboembolic stroke, the most clinically relevant stroke model, remains understudied, we tested the hypothesis that the inhibition of A2A receptors, especially those on the endothelial cells, in ischaemic stroke is critical for neurovascular protection and improved outcomes.
2. METHODS
2.1. Animals
All animal care and experimental procedures complied with the according to the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) of Peking University and Augusta University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Global homozygous Adora2a (Adora2a −/−) knockout mice were generated as previously described (Chen et al., 1999). Breeding pairs of Adora2a Flox/Flox (Adora2a F/F) mice were kindly provided by Dr Joel Linden (La Jolla Institute for Allergy and Immunology, CA). Endothelium‐specific Adora2a‐deficient (Adora2a ΔVEC) mice were obtained by cross‐breeding Cdh5–Cre transgenic mice (IMSR Cat# JAX:006137, RRID:IMSR_JAX:006137) with Adora2a F/F mice, all on a C57BL/6J background. Wild‐type (WT) C57BL/6J mice (IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664) were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were housed in individually ventilated caging systems in groups of five mice per cage and kept under controlled environmental conditions (12‐hr light/12‐hr dark cycle, room temperature 21–23°C, and humidity 50–60%) with free access to standard laboratory food pellets and water.
2.2. Thromboembolic stroke model
Mouse thromboembolic stroke induced by embolic middle cerebral artery occlusion (eMCAo) is accepted as a stroke model in studying the effect of certain molecules in stroke (Hoda et al., 2011). The emboli were prepared prior to surgery. Briefly, arterial blood from WT C57BL/6J mice was supplemented with fibrinogen (2 mg·ml−1; Cat # 341578. Millipore, Billerica, MA) and immediately filled in a PE‐50 tube for 6 hr at room temperature followed by storage at 4°C. Before use, the clot (~50 mm) was flushed out from the tube into a sterile Petri dish containing normal saline. The clot was then transferred into a modified PE‐10 tube filled with sterile saline and was subsequently transferred to a Petri dish containing PBS and left for further retraction for 4 hr at room temperature. A single 9.0 ± 0.5‐mm‐long clot was transferred to a modified PE‐10 tube for the following procedure.
For the induction of cerebral ischaemia in mice, adult male mice (14–16 weeks old, 28–32 g) were anaesthetized with 3.5% isoflurane and maintained with 2.0% isoflurane during the surgery. Body temperature was maintained with a heating pad at 37.0 ± 0.5°C during surgery. Under sterile conditions, the neck vessels were exposed with a midline incision, and the right common carotid artery, external carotid artery, and internal carotid artery were carefully isolated. A modified PE‐10 catheter containing a clot was inserted into the internal carotid artery through a small hole on the external carotid artery, and the clot was gently injected with 100‐μl PBS. After that, the catheter was removed immediately. Mice in the sham group were treated using the same surgical procedures but injected with an equal volume of PBS without a clot. Buprenorphine (0.1 μg·g−1) was given as a post‐operative analgesic agent. Ringer's lactate solution (1 mL) was injected subcutaneously to prevent dehydration. After recovering from anaesthesia, mice were returned to the regular sterilized cages with some wetted food. All reasonable attempts were made to minimize the animals' suffering and to decrease the numbers of mice used.
To test the effect of A2A receptor antagonism in cerebral ischaemia, istradefylline (KW 6002), a potent A2A receptor antagonist, was used in the C57BL/6J mouse model. KW6002 (10 mg·kg−1) was injected intraperitoneally at 10 min after eMCAo. This dose of KW6002 has been used in our studies and is effective in specifically blocking the A2A receptor‐mediated effects in rodent disease models (Chen et al., 2001; Zhou et al., 2018). Mice were randomly divided into the KW 6002 treatment group and vehicle control group. Researchers were blinded to the assignment of treatments, during surgeries and evaluation of the experimental outcomes.
2.3. Neurological examination
Before the end of the experiment, a neurological examination was performed on the mice using a modified Bederson's method (Bederson et al., 1986; Yang et al., 1994). The 4‐point scoring scale was as follows: 0, no observable neurological deficit (normal); 1, mildly injured grade (forelimb flexion); 2, moderately injured grade (decreased resistance to lateral push and forelimb flexion); 3, severely injured grade (unidirectional circling); and 4, most severely injured grade (unidirectional circling and decreased level of consciousness). All tests were evaluated in a blinded fashion with regard to the animal groups. Only animals with Grades 1–4 of neurological deficits were included.
2.4. Assessment of infarct volume
At 24 hr after eMCAo, mice were killed by CO2 asphyxiation, and the brains were removed quickly and frozen at −20°C for 20 min. Sections (2.0‐mm thick) were cut and stained with 2% 2,3,5‐triphenyltetrazolium chloride (TTC; 93140, Sigma‐Aldrich, St. Louis, MO) at 37°C for 30 min in the dark. To ensure even staining, the sections were turned over several times. The well‐dyed brain slices were fixed in 4% paraformaldehyde (PFA) at 4°C until imaging. After staining, ischaemic and non‐ischaemic brain regions were stained white and red respectively.
2.5. Determination of cerebral water content
Mice were killed by CO2 asphyxiation 24 hr after eMCAo. Brains were removed immediately and dissected into ipsilateral ischaemic hemispheres and contralateral non‐ischaemic hemispheres through the interhemispheric fissure. The bilateral hemispheres (without the brain stem and cerebellum) were weighed rapidly (wet weight) and then dried in an oven at 110°C for 48 hr and weighed again (dry weight). The brain water content (%) of each hemisphere was calculated as follows: brain water content (%) = (wet weight − dry weight)∕wet weight × 100%.
2.6. Evaluation of BBB permeability
The integrity of the BBB was evaluated by the leakage of Evans Blue (EB) dye into the brain after systemic injection. EB dye (4% in 0.9% saline, 4 ml·kg−1; Sigma‐Aldrich, E2129) was administered via the tail vein, 2 hr before mice were killed. At the end of the experiment, mice were deeply anaesthetized with 5% isoflurane and perfused through the left ventricle with ice‐cold PBS. The cerebral hemispheres (ischaemic side) were then removed and finally homogenized in 50% trichloroacetic acid (1.5 ml·g−1; Macklin, T818876). After centrifugation at 12,000× g for 20 min, the supernatants were diluted with ethanol (1:3), and the concentration of extracted EB dye was determined with a spectrophotometer at 620 nm for absorbance. EB dye leakage of each sample was quantified using a standard curve and expressed as μg·g−1 of brain hemisphere.
2.7. Isolation of mouse BMECs
Mouse BMECs from cerebral hemispheres free of meninges were isolated as reported elsewhere (Ruck, Bittner, Epping, Herrmann, & Meuth, 2014). Briefly, brain tissues were minced and digested with collagenase–DNAse–dispase mixtures as needed in different steps to finally obtain a myelin‐free pellet. Pellets resuspended in 2‐ml DMEM were carefully loaded on top of the Percoll gradient (19 ml of 1× PBS with 1 ml of 10× PBS, 1 ml of fetal calf serum, and 10 ml of Percoll; Sigma‐Aldrich, P1644) and centrifuged (700× g, 4°C, 10 min). The interphase obtained (~12 ml) was centrifuged again (1,000× g, 10 min, 4°C) to collect the final pellet as BMECs for use in real‐time PCR and Western blot assays.
2.8. Flow cytometry analysis of immune cell populations
At 24 hr after eMCAo, mice were deeply anaesthetized with 5% isoflurane and perfused through the left ventricle with ice‐cold PBS. The brains were dissected and homogenized in 7‐ml RPMI without phenol red medium. Percoll (3‐ml) was added to the suspension to bring the final concentration to 30% Percoll. The 10‐ml cell suspension was then slowly layered on top of 2‐ml 70% Percoll. The gradient was centrifuged at 500× g for 30 min at 18°C. Cells were collected from the 70–30% interface and resuspended on 100 μl of 0.1% BSA in PBS with Fc block reagent (BD Pharmingen, San Jose, CA). Cell suspensions were incubated with anti‐CD11b‐FITC (Cat# 11‐0112‐82, RRID:AB_464935; Thermo Fisher Scientific, Waltham, MA), anti‐F4/80‐PE (Cat# 565410, RRID:AB_2687527; BD Biosciences, San Jose, CA), anti‐Ly6G‐Percp‐cy5.5 (BD Biosciences Cat# 560602, RRID:AB_1727563), or anti‐CD45.2‐APC (BD Biosciences Cat# 558702, RRID:AB_1645215) antibodies. Stained cells were washed and resuspended in 400 μl of FACS flow (BD Pharmingen) and analysed immediately using a FACSCalibur flow cytometer with CellQuest software (BD Pharmingen). Isotype controls (BD Pharmingen) were used in parallel.
2.9. Immunofluorescent staining in mouse brain tissue
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Mice were deeply anaesthetized with 5% isoflurane and perfused successively with PBS and 4% PFA. Brains were removed and postfixed overnight in 4% PFA, followed by a 30% sucrose solution for cryoprotection. Serial 40‐μm‐thick coronal sections were prepared using a freezing microtome (Leica, Wetzlar, Germany) and stored in PBS at 4°C as free‐floating sections. After permeabilizing and blocking, sections were incubated with primary antibodies against mouse A2 receptors (5 μg·ml−1, Millipore Cat# 05‐717, RRID:AB_309931) and CD31 (4 μg·ml−1, Cat# DIA‐310, RRID:AB_2631039; Dianova, Hamburg, Germany) overnight at 4°C and then incubated with a mixture of Alexa Fluor 488 goat anti‐mouse IgG (10 μg·ml−1, Cat# ab150077, RRID:AB_2630356; Abcam, Cambridge, MA) and Alexa Fluor 594 goat anti‐rat IgG (10 μg·ml−1, Abcam Cat# ab150160, RRID:AB_2756445) for 1 hr at room temperature. After washing with PBS, the sections were counterstained with DAPI (Invitrogen, Carlsbad, CA) and flat mounted on microscope slides in fluorescence mounting medium. Immunoreactivity was then observed by confocal microscopy (Nikon, Tokyo, Japan).
2.10. Cell culture and treatment
Primary human BMECs (huBMECs; ScienCell Cat# 1000) were cultured in endothelial basal medium (CC‐3156; Lonza, Basel, Switzerland) supplemented with microvascular endothelial cell growth kit (Lonza, CC‐4147). In some experiments, huBMECs were incubated with 10 ng·ml−1 TNF‐α (BD Pharmingen, 554618), and 100 nM KW 6002 was added to the culture medium. THP‐1 cells used for the monocyte adhesion assay were cultured in RPMI 1640 medium (Cat # 11875093, Gibco, Grand Island, NY) containing 10% FBS.
2.11. RNA interference
huBMECs were transfected at 60–70% confluence with 50 nmol·L−1 siRNAs targeting human ADORA2A (siADORA2A; Ribobio, Guangzhou, China) or with a non‐targeting negative control (siCtrl; Ribobio) using Lipofectamine RNAiMAX Reagent (Invitrogen, 13778‐150) according to the manufacturer's protocol. Six hours after transfection, the medium was changed to fresh complete cell culture medium, and cells were cultured for 48 hr.
2.12. Monocyte adhesion assay
THP‐1 cells (ATCC Cat# TIB‐202, RRID:CVCL_0006) were labelled with Calcein AM (0.5 μmol·L−1, Yeasen, Shanghai, China) at 37°C for 15 min in RPMI 1640 and subsequently washed by centrifugation. huBMECs were cultured in 12‐well plates and washed twice with RPMI 1640. The adhesion assay was performed by adding 1 ml of a THP‐1 cell suspension (1 × 106 cells) to each monolayer. After 20 min, non‐adherent cells were removed by gentle washing with RPMI 1640 for three times, and monolayers were fixed with 4% PFA. The number of adherent THP‐1 cells was counted in four random fields per well for quantification.
2.13. Western blotting
Brain tissues or cells were collected and homogenized with ice‐cold RIPA with 1% proteinase inhibitor cocktail (TransGen Biotech, Beijing, China). The amount of total protein was quantified using the BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer's guidelines. Equal amounts of protein per lane (20 μg) were subjected to 8–12% SDS‐PAGE. Antibodies used in this study were as follows: mouse anti‐A2A receptors (0.5 μg·ml−1, Millipore Cat# 05‐717, RRID:AB_309931), mouse anti‐VCAM1 (0.4 μg·ml−1, Cat# sc‐13160, RRID:AB_626846; Santa Cruz Biotechnology, Dallas, TX), rabbit anti‐ ICAM1 (0.4 μg·ml−1, Cat# BA0541, RRID:AB_2797134, Boster Biological Technology, Wuhan, China), mouse anti‐Nlrp3/NLRP3 (1 μg·ml−1, Cat# AG‐20B‐0014, RRID:AB_2490202; AdipoGen, San Diego, CA), rabbit anti‐caspase‐1 (p20; 1:1,000, Cat# 4199, RRID:AB_1903916, Cell Signaling Technology, Danvers, MA), and mouse anti‐IL‐1β (p17; 1:1,000, Cell Signaling Technology Cat# 12242, RRID:AB_2715503). A rabbit anti‐GAPDH antibody (1:2,000, Cell Signaling Technology Cat# 2118, RRID:AB_561053) was used as a loading control. Immunoreactive proteins were detected by the chemiluminescence assay (Millipore) using the ChemiDoc MP system (Bio‐Rad). Quantification was done with ImageJ free software (Version 1.47, RRID:SCR_003070), and each lane was normalized to GAPDH.
2.14. Real‐time PCR analysis
Total RNA was extracted from mouse brain tissues or huBMECs with TRIzol reagent (Invitrogen) following the manufacturer's instructions. A 0.5‐ to 1‐μg sample of RNA was used as a template for reverse transcription using the iScriptTM cDNA synthesis kit (Bio‐Rad). Real‐time PCR was performed via a Bio‐Rad CFX96 instrument (Bio‐Rad). The primers used for PCR are shown in Table S1. The following PCR protocol was used: 10 min at 95°C, followed by 50 cycles of denaturing at 95°C for 15 s, annealing at 60°C for 30 s and extension at 72°C for 60 s. Actb was used as an internal standard. All real‐time PCRs for each sample were performed in triplicate.
2.15. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. The optimum sample sizes and animal numbers were determined by power analysis, prior experience, and our preliminary data to achieve statistical significance. Grouping was performed in a randomized manner. Data were expressed as the mean ± SD in all animal studies and mean ± SEM in cell culture studies. Statistical significance was determined using unpaired Student's t test, one‐way ANOVA followed by Bonferroni's test, or Mann–Whitney U test as appropriate. All statistical analyses were performed using the GraphPad Prism software (Version 5.0, RRID:SCR_002798). *P < .05 was taken to show statistical significance.
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017a,b).
3. RESULTS
3.1. Cerebral ischaemic injury increases expression of Adora2a in brain
To investigate the differential expression of adenosine receptors after thromboembolic stroke, we first examined transcriptional changes of the different subtypes of adenosine receptors in the mouse brain following eMCAo. We found that Adora2a expression was significantly increased, but the Adora1 level was markedly reduced in the ischaemic hemisphere compared with the contralateral, while the other two Adora subtypes remained unchanged (Figure 1a). Western blot assay further confirmed that the transcriptional changes translated into significantly increased protein expression of A2A receptors in the ischaemic hemisphere compared with the contralateral side (Figure 1b). Furthermore, the protein expression of A2A receptors at 24 hr after eMCAo was also significantly increased in the isolated BMECs of ipsilateral hemispheres compared with the contralateral side (Figure 1c). Double immunofluorescence staining showed that cerebral ischaemia enhanced the expression of A2A receptors on endothelial cells as well as other types of cells such as microglia and astrocytes (Figure 1d), suggesting that the endothelial A2A receptors may play a major role in the microvascular dysfunction following stroke.
Figure 1.
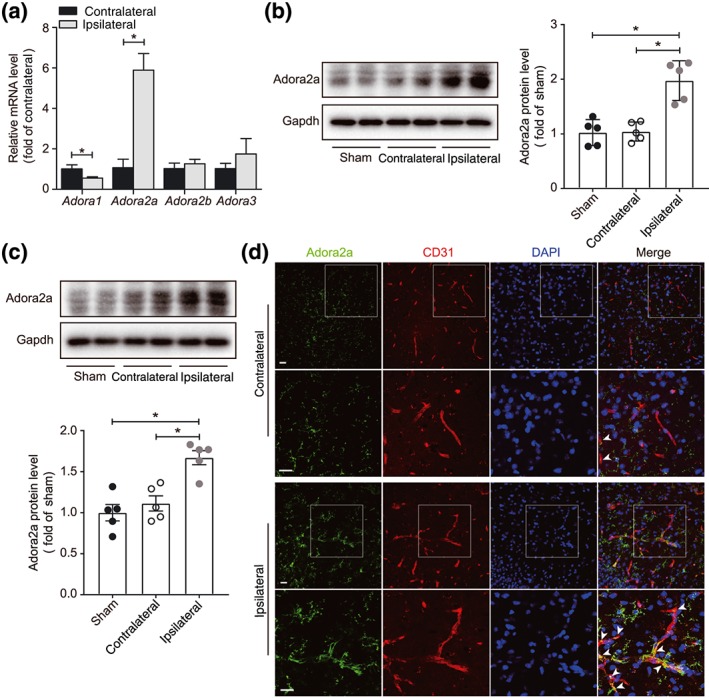
Increased expression of A2A receptors in ischaemic brain. (a) Real‐time PCR analysis of Adora1, Adora2a, Adora2b, and Adora3 mRNA expression in whole brain 24 hr after eMCAo in mice (n = 6 mice per group). (b) Western blot analysis and densitometric quantification of A2A receptor protein expression in whole brain 24 hr after eMCAo (n = 5 mice per group). (c) Western blot analysis and densitometric quantification of A2A receptor protein expression in isolated BMECs, 24 hr after eMCAo (n = 5 per group). (d) Representative images of A2A receptors in the contralateral and ipsilateral cerebral hemispheres in mice with eMCAo. Brain sections were stained for A2A receptors (green), CD31 (red, microvessel), and DAPI (blue, nuclei). Images in the second and fourth rows are magnification of the boxed regions of images in the first and third rows respectively. Scale bar: 20 μm. Data are represented as means ± SD (for a, b) and ± SEM (for c). *P < .05, significantly different as indicated; unpaired Student's t test (for a) and one‐way ANOVA followed by Bonferroni's test (for b, c)
3.2. Adora2a deficiency improves post‐stroke outcomes in mice
To evaluate the effect of A2A receptors on the post‐stroke outcomes, we first examined and compared mice globally deficient in Adora2a (Adora2a −/−) with their WT control, following eMCAo. Infarction volume in Adora2a −/− mice was significantly smaller than that of WT control at 24 hr after eMCAo (Figure 2a), which also translated into a reduced neurological deficit in Adora2a −/− mice (Figure 2b). These data suggest that the deletion of Adora2a is protective in acute thromboembolic stroke.
Figure 2.
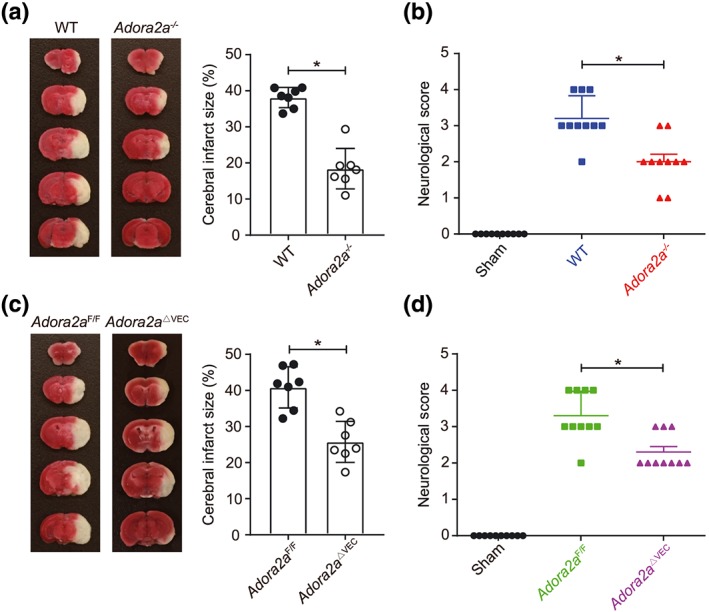
Decreased ischaemic brain injury in Adora2a‐deficient mice. (a) TTC staining of coronal brain sections 24 hr after eMCAo and quantification of infarct size in wild‐type (WT) and Adora2a −/− mice (n = 7 mice per group). (b) Neurological deficit scores of cerebral ischaemic WT and Adora2a −/− mice (n = 10 mice per group). (c) TTC staining of coronal brain sections 24 hr after eMCAo and quantification of infarct size in Adora2a F/F and Adora2a ΔVEC mice (n = 7 mice per group). (d) Neurological deficit scores of cerebral ischaemic Adora2a F/F and Adora2a ΔVEC mice (n = 10 mice per group). Data shown are individual values with means ± SD. *P < .05, significantly different as indicated; unpaired Student's t test (for a, c) and Mann–Whitney U test (for b, d)
In order to investigate the cell‐specific role of Adora2a in ischaemic stroke, we then induced eMCAo in Adora2a ΔVEC and the control Adora2a F/F mice. Interestingly, 24 hr after thromboembolic stroke, Adora2a ΔVEC mice exhibited a noticeable reduction in infarct volume along with improved neurological outcomes compared with the control (Figure 2c,d), indicating that the endothelial A2A receptors may play a prominent role in post‐stroke pathogenesis.
3.3. Deletion of endothelial Adora2a attenuates post‐stroke neuroinflammation and preserves neurovascular integrity
Increased cerebral endothelial inflammation after stroke plays a critical role in leukocyte infiltration in brain and disruption of the BBB, resulting in both acute injury and long‐term adverse prognosis. Therefore, we next investigated the effect of deletion of endothelial Adora2a on post‐stroke adhesion markers, leukocyte infiltration in brain, integrity of the BBB, and oedema. As shown in Figure 3a,b, the Adora2a ΔVEC mice subjected to thromboembolic stroke showed reduced mRNA and protein expression levels of adhesion molecules compared with those in the Adora2a F/F control, indicating that the lack of endothelial Adora2a attenuates post‐stroke neuroinflammation. Moreover, compared with Adora2a F/F mice, Adora2a ΔVEC mice also showed reduced infiltration of leukocytes (CD11b+CD45high) following stroke, including both neutrophils and monocytes (Figure 3c). Significantly reduced BBB leakage and oedema was also observed in Adora2a ΔVEC mice as compared with the control Adora2a F/F mice (Figure 3d,e), demonstrating that reduced neuroinflammation protects the cerebrovascular integrity after stroke.
Figure 3.
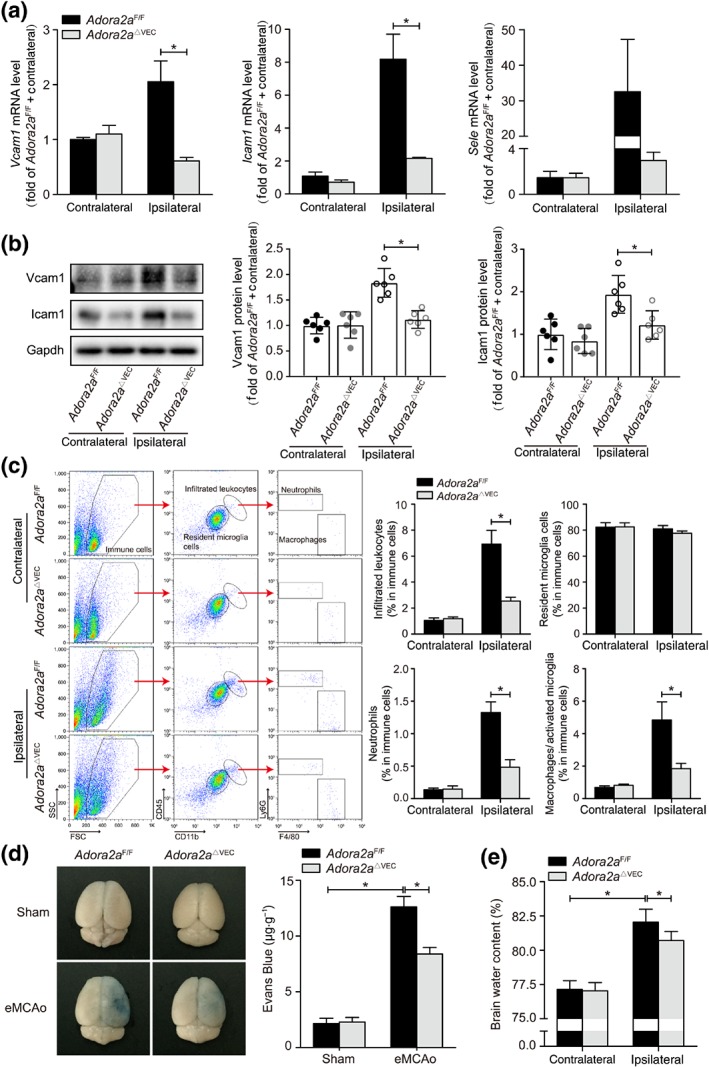
Reduced inflammation in ischaemic brain of endothelial Adora2a‐deficient mice. (a) Real‐time PCR analysis of adhesion molecule expression in brains of Adora2a F/F and Adora2a ΔVEC mice 6 hr after eMCAo (n = 9 mice per group). (b) Western blot analysis and densitometric quantification of adhesion molecule expression in brains of Adora2a F/F and Adora2a ΔVEC mice 24 hr after eMCAo (n = 6 mice per group). (c) The gating strategy and quantification for each inflammatory cell population in the brains of Adora2a F/F and Adora2a ΔVEC mice 24 hr after eMCAo: infiltrated leukocytes (CD11b+CD45high), resident microglia cells (CD11b+CD45low), infiltrated neutrophils (CD11b+CD45highLy6G+), and macrophages/activated microglia cells (CD11b+CD45highF4/80+; n = 6 mice per group). (d) Representative images and quantification of Evans Blue dye extravasation at 24 hr after eMCAo in Adora2a F/F and Adora2a ΔVEC mice (n = 6 mice per group). (e) Brain water content in the contralateral and ipsilateral areas at 24 hr after eMCAo in Adora2a F/F and Adora2a ΔVEC mice (n = 6 mice per group). Data shown are means ± SD. *P < .05, significantly different as indicated; unpaired Student's t test. FSC, forward scatter; SSC, side scatter
3.4. ADORA2A knockdown suppresses endothelial inflammation in vitro
To support our above findings in the mouse model, we next examined whether ADORA2A regulates cerebrovascular dysfunction following inflammatory challenge in huBMECs in an in vitro setting. We used TNF‐α, an inflammatory cytokine that is well known to be increased after stroke and triggers endothelial inflammation and dysfunction (Liu et al., 1994). TNF‐α treatment (10 ng·ml−1) to huBMECs for 2 hr increased the mRNA expression of ADORA2A but not that of the genes of other subtypes of adenosine receptors (Figure 4a), and this effect was dose‐ and time‐dependent (Figure 4b,c). Moreover, TNF‐α treatment of huBMECs increased the mRNA and protein expression of adhesion molecules (VCAM1, ICAM1, and E‐selectin) involved in endothelial‐leukocyte adhesion, which was down‐regulated as a result of silencing the ADORA2A gene using siRNA (Figure 4d,e). Accordingly, ADORA2A gene silencing with siRNA in huBMECs translated into reduced monocyte adhesion to the endothelial monolayer induced by TNF‐α (Figure 4f), thus mechanistically confirming that the products of ADORA2A are involved in the endothelial‐leukocyte interactions following a proinflammatory challenge and that suppression of this gene protects against endothelial inflammation.
Figure 4.
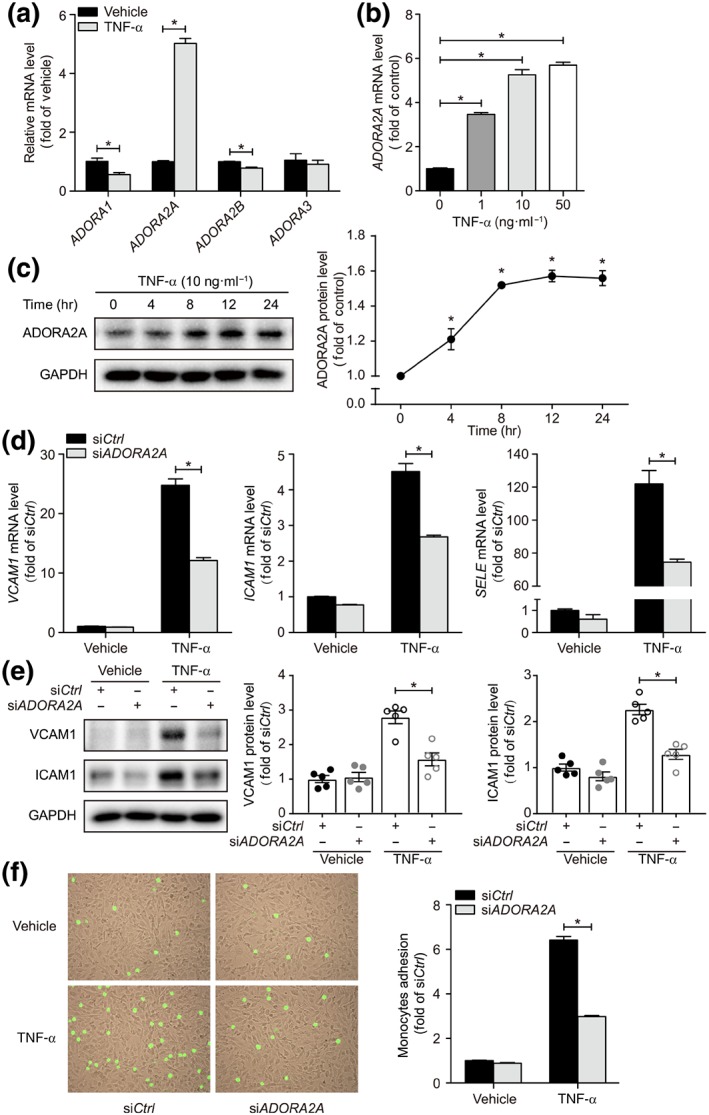
Inhibitory effects of ADORA2A knockdown on huBMEC inflammation. (a) Real‐time PCR analysis of mRNA expression for adenosine receptors in huBMECs treated with TNF‐α at 10 ng·ml−1 for 2 hr (n = 6). (b) Real‐time PCR analysis of ADORA2A mRNA expression in huBMECs treated with TNF‐α for 2 hr at indicated concentrations (n = 6). (c) Western blot analysis and densitometric quantification of A2A receptor protein in huBMECs treated with TNF‐α at 10 ng·ml−1 for indicated times (n = 5). (d) Real‐time PCR analysis of adhesion molecule expression in huBMECs treated with TNF‐α at 10 ng·ml−1 for 2 hr. huBMECs were transfected with siADORA2A or siCtrl (n = 9). (e) Western blot analysis and densitometric quantification of adhesion molecule expression in huBMECs treated with TNF‐α at 10 ng·ml−1 for 4 hr. huBMECs were transfected with siADORA2A or siCtrl (n = 5). (f) Representative images and quantification of monocyte adhesion on huBMECs treated with TNF‐α at 10 ng·ml−1 for 4 hr. huBMECs were transfected with siADORA2A or siCtrl (n = 6). Data shown are means ± SEM. *P < .05, significantly different as indicated; unpaired Student's t test (for a, d, and f) and one‐way ANOVA followed by Bonferroni's test (for b)
3.5. Blockade of A2A receptors protects mice from ischaemic brain injury
In addition to the effect of the inactivation of A2A receptors on ischaemic brain injury using genetically engineered mice, we administered the A2A receptor antagonist KW 6002 to mice to test whether pharmacological inhibition of these receptors benefits mice in our model of acute stroke. The therapeutic treatment with KW6002 (10 mg·kg−1), injected intraperitoneally at 10 min after eMCAo, reduced infarct size (Figure 5a) and improved the neurological outcome (Figure 5b). Along with these observations, KW 6002 treatment suppressed the expression of endothelial adhesion molecules (Figure 5c). This treatment did not affect the BP or heart rate in mice (Figure S1). Furthermore, in the in vitro assay, KW 6002 treatment suppressed the gene and protein expression of adhesion molecules in huBMECs following TNF‐α stimulation (Figure 5d,e). Thus, pharmacological inhibition of A2A receptors, which down‐regulated neuroinflammation in our model of stroke, is likely to be translatable as an acute stroke therapy.
Figure 5.
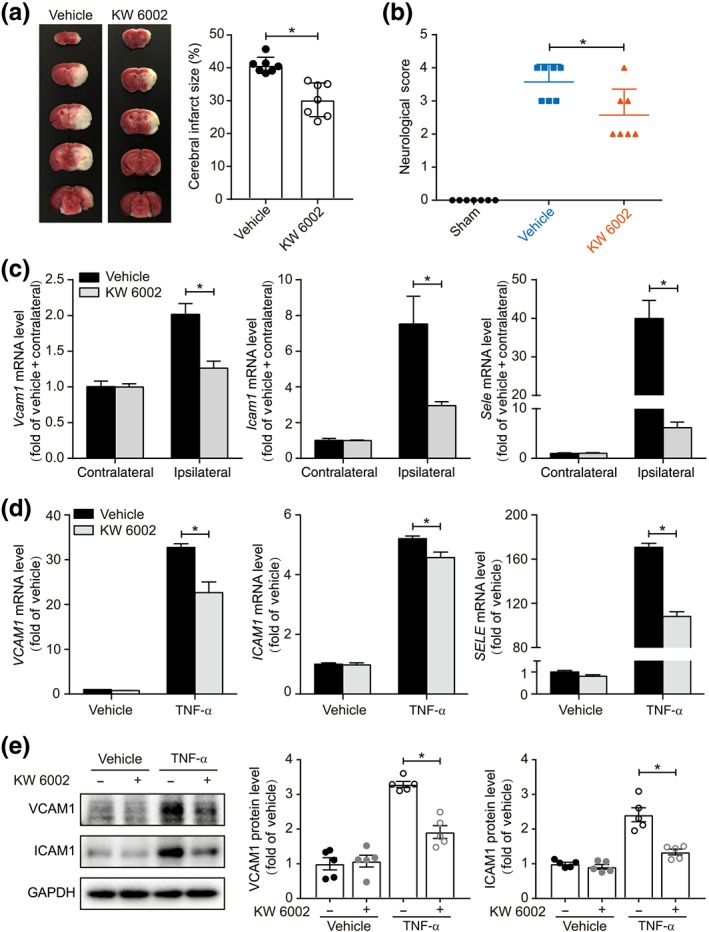
Pharmacological inhibition of A2A receptors prevents cerebral ischaemic injury. (a) TTC staining of coronal brain sections 24 hr after eMCAo and quantification of infarct size in vehicle‐ or KW 6002‐treated mice (n = 7 mice per group). (b) Neurological deficit scores of cerebral ischaemic vehicle‐ or KW 6002‐treated mice (n = 7 mice per group). (c) Real‐time PCR analysis of adhesion molecule expression in brains of vehicle‐ or KW 6002‐treated mice at 6 hr after eMCAo (n = 6 mice per group). (d) Real‐time PCR analysis of adhesion molecule expression in huBMECs treated with TNF‐α. huBMECs were pretreated with KW 6002 at 100 nmol·L−1 for 30 min and then stimulated with 10 ng·ml−1 TNF‐α for 2 hr (n = 6). (e) Western blot analysis and densitometric quantification of adhesion molecule expression in huBMECs treated with TNF‐α. huBMECs were pretreated with KW 6002 at 100 nmol·L−1 for 30 min and then stimulated with TNF‐α at 10 ng·ml−1 for 4 hr (n = 5). Data shown are means ± SD (for a–c) and means ± SEM (for d, e). *P < .05, significantly different as indicated; unpaired Student's t test (for a, c, d, and e) and Mann–Whitney U test (for b)
3.6. NLRP3 is involved in the anti‐inflammatory effect of endothelial Adora2a deficiency
Activation of the NLRP3 inflammasome plays a significant role in the development of post‐stroke neuroinflammation (Qiu et al., 2016; Wang, Li, Wang, Fu, & Ma, 2015). To dissect the mechanisms underlying the protective effect of inactivating A2A receptors, we next investigated alteration of the NLRP3 inflammasome following thromboembolic stroke. Deletion of endothelial Adora2a resulted in reduced activation of NLRP3 following eMCAo in Adora2a ΔVEC mice compared with the control Adora2a F/F mice (Figure 6a). Both knockdown and blockade of A2A receptors reduced the caspase‐1 and IL‐1β activation and cleavage in huBMECs following TNF‐α stimulation (Figure 6b,c). These data demonstrate that the inhibition of A2A receptor‐mediated neuroinflammation is, at least in part, through activation of NLRP3.
Figure 6.
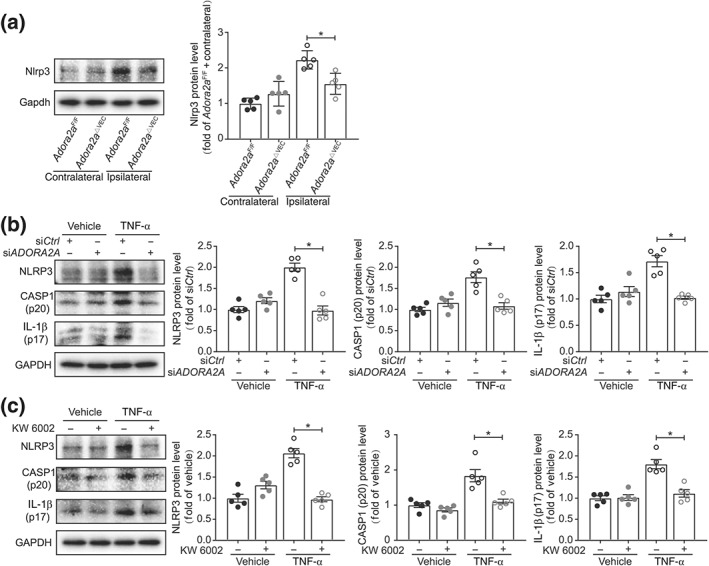
A2A receptors act with NLRP3 to facilitate inflammation in ischaemic brain. (a) Western blot analysis of NLRP3 protein expression in brains of Adora2a F/F and Adora2a ΔVEC mice 24 hr after eMCAo (n = 5 mice per group). (b) Western blot analysis of NLPR3, caspase‐1 (p20), and IL‐1β (p17) expression in huBMECs, treated with TNF‐α at 10 ng·ml−1 for 4 hr. huBMECs were transfected with siADORA2A or siCtrl (n = 5). (c) Western blot analysis of NLPR3, caspase‐1 (p20), and IL‐1β (p17) expression in huBMECs treated with TNF‐α. huBMECs were pretreated with KW 6002 at 100 nmol·L−1 for 30 min and then stimulated with TNF‐α at 10 ng·ml−1 for 4 hr (n = 5). Data shown are individual values with means ± SD (for a) and means ± SEM (for b, c). *P < .05, significantly different as indicated; unpaired Student's t test
4. DISCUSSION
In the present study, our mechanistically oriented experiments using novel genetic tools provide new insights for neurovascular protection following a thromboembolic stroke. Our key finding was that the expression of A2A receptors in endothelium was increased in cerebral microvessels after thromboembolic stroke, and both deletion of the endothelium‐specific Adora2a gene and pharmacological inhibition of its product (A2A receptors) protected against stroke injury, resulting in improved neurological outcomes. Moreover, the inhibition of A2A receptors suppressed the activity of the NLRP3 inflammasome in both mouse brains and huBMECs. Thus, we have delineated a novel mechanism of NLRP3 inflammasome activation by A2A receptors in BMECs.
A significantly large proportion of ischaemic stroke is thromboembolic in nature. Despite Food and Drug Administration approval of two “reperfusion” therapies in stroke, microvascular protection is not essentially guaranteed following reperfusion (Dalkara & Arsava, 2012; Gursoy‐Ozdemir, Yemisci, & Dalkara, 2012). Leukocyte adhesion and infiltration into the ischaemic brain are considered to be two of the earlier events after stroke, causing microvascular inflammation and plugging that disrupts the restoration of microvascular perfusion (Jin et al., 2010). Therefore, therapeutic strategies are urgently needed that can provide microvascular protection in the larger subset of stroke patients.
The A2A receptor is a type of adenosine receptor expressed in many types of cells involved in ischaemic stroke. These receptors are expressed in different regions of the brain and differentially regulates the pathophysiology of ischaemia versus reperfusion during stroke (Chen et al., 1999; Fredholm et al., 2003). In addition, the corresponding gene, Adora2a, in the entire bone marrow‐derived cell population contributed to the production of proinflammatory cytokines and formation of brain infarction in rodent stroke and brain trauma models (Yu et al., 2004). Prior reports demonstrated that inactivation of endothelial A2A receptors inhibited ROS production and preserved endothelial function (Thakur, Du, Hourani, Ledent, & Li, 2010). However, the effect of endothelial A2A receptors remains understudied in a thromboembolic stroke model that closely represents a state of ischaemia–reperfusion in the majority of stroke patients.
Our finding demonstrates that, in comparison with other adenosine receptor subtypes, the expression of A2A receptors in endothelium is elevated after thromboembolic stroke in microvessels, indicating that these receptors are a major regulator of adenosine‐mediated neurovascular inflammation, at least in brain microvessels. Mice genetically deficient in endothelial A2A receptors exhibited reduced endothelial inflammation, suppressed leukocyte infiltration, and decreased thromboembolic stroke injury. Interestingly, in this slowly reperfusing mouse model, pharmacological inhibition of A2A receptors also protected against stroke injury, resulting in reduced oedema/injury volume, improved neurological outcomes, and suppression of inflammatory responses. While others reported the benefits of adenosine receptor agonists in reperfused stroke (Melani, Corti, et al., 2014; Sheardown & Knutsen, 1996; Von Lubitz et al., 1995), our results clearly demonstrate that antagonism of the A2A receptors in BMECs was protective following a thromboembolic stroke. This discrepancy between our findings and those of others may be attributed to differences in the dynamics of cerebral blood flow changes, which significantly affects the post‐stroke pathogenesis and the benefits of a therapeutic intervention (Kim et al., 2008). It should be noted that the majority of thromboembolic stroke patients remain untreated but exhibit spontaneous though slow and partial reperfusion. Therefore, in the present study, we used a partly humanized thromboembolic stroke model in mice that better represents a larger untreated stroke population.
There is evidence that activation of the NLRP3 inflammasome is strongly involved in triggering post‐stroke neuroinflammation, resulting in long‐term adverse prognosis (Qiu et al., 2016; Wang et al., 2015). The NLRP3 inflammasome is predominantly expressed in microvascular endothelial cells in brain and is further induced due to ischaemia. It thus plays a key role in post‐stroke microvascular dysfunction (Yang et al., 2014). Interestingly, our data provide evidence that the deletion of endothelial Adora2a in mice inhibited activation of the NLRP3inflammasome after thromboembolic stroke, leading to reduced production of IL‐1β, an inflammatory cytokine actively involved in the leukocyte‐endothelial adhesion phenomena. Thus, inactivation of NLRP3 inflammasomes protected the integrity of the BBB and leukocyte infiltration following stroke in mice deficient in endothelial Adora2a. While we do not rule out other possible mechanisms in A2A receptor‐mediated, post‐stroke, pathogenesis, the role of A2A receptors in NLRP3‐mediated cerebral endothelial inflammation should be noted and needs in‐depth investigation.
In conclusion, we report, here, a novel finding that A2A receptors in brain endothelial cells mediate inflammation after thromboembolic stroke. Genetic deletion of endothelial A2A receptors or their pharmacological inhibition leads to a reduction in stroke injury and vascular inflammation, and improved post‐stroke outcomes, at least in part via down‐regulation of the endothelial NLRP3 inflammasome. Further studies are warranted to test specific antagonists of A2A receptors, with and without Food and Drug Administration‐approved reperfusion therapies, in order to prevent long‐term microvascular dysfunction and improve prognosis after stroke.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Y.Z., C.W., J.F.‐C., Z.L., M.H., and Y.H. designed the research; Y.Z., X.Z., G.L., Q.Y., J.X., M.Z., X.M., Y.C., L.W., Y.X., Y.W., Y.Z., L.H., Z.H., T.W., and M.N.H. performed the experiments; Y.Z., X.Z., Z.L., and Y.H. analysed the data; Y.Z., M.N.H., Z.L., M.H., and Y.H. wrote and revised the manuscript; and Z.X., C.W., M.H., and Y.H. provided the reagents or materials and participated in the experimental design.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Blood pressure and heart rate of vehicle‐ and KW 6002‐treated mice. KW 6002 was injected intraperitoneally into C57BL/6J mice at 10 mg·kg‐1. Systolic blood pressure, diastolic blood pressure, mean blood pressure and heart rate were monitored at the indicated times after administration (n = 5 mice per group). Data are represented as means ± SD. Statistical significance was determined by unpaired Student's t‐test.
Table S1 Real‐Time PCR primers
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (81870324), the Shenzhen Science and Technology Innovation Committee (JCYJ20170810163238384, JCYJ20160525154531263, JCYJ20160506170316776, JCYJ20170412150405310, and JSGG20160608091824706), Guangdong Natural Science Foundation (2014A030312004), American Heart Association (16GRNT30510010), and the National Institutes of Health (R01HL134934, R01DK095862, and R01 HL142097).
Zhou Y, Zeng X, Li G, et al. Inactivation of endothelial adenosine A2A receptors protects mice from cerebral ischaemia‐induced brain injury. Br J Pharmacol. 2019;176:2250–2263. 10.1111/bph.14673
Contributor Information
Zhiping Liu, Email: zhiping0414@163.com.
Mei Hong, Email: meihong.sz@pku.edu.cn.
Yuqing Huo, Email: yhuo@augusta.edu.
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather, J. , & Iadecola, C. (2016). Inflammation and stroke: An overview. Neurotherapeutics, 13, 661–670. 10.1007/s13311-016-0483-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bederson, J. B. , Pitts, L. H. , Tsuji, M. , Nishimura, M. C. , Davis, R. L. , & Bartkowski, H. (1986). Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke, 17, 472–476. 10.1161/01.STR.17.3.472 [DOI] [PubMed] [Google Scholar]
- Chen, J. F. , Huang, Z. , Ma, J. , Zhu, J. , Moratalla, R. , Standaert, D. , … Schwarzschild, M. A. (1999). A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. The Journal of Neuroscience, 19, 9192–9200. 10.1523/JNEUROSCI.19-21-09192.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. F. , Sonsalla, P. K. , Pedata, F. , Melani, A. , Domenici, M. R. , Popoli, P. , … de Mendonça, A. (2007). Adenosine A2A receptors and brain injury: Broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Progress in Neurobiology, 83, 310–331. 10.1016/j.pneurobio.2007.09.002 [DOI] [PubMed] [Google Scholar]
- Chen, J. F. , Xu, K. , Petzer, J. P. , Staal, R. , Xu, Y. H. , Beilstein, M. , … Schwarzschild, M. A. (2001). Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson's disease. The Journal of Neuroscience, 21, RC143 10.1523/JNEUROSCI.21-10-j0001.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkara, T. , & Arsava, E. M. (2012). Can restoring incomplete microcirculatory reperfusion improve stroke outcome after thrombolysis? Journal of Cerebral Blood Flow and Metabolism, 32, 2091–2099. 10.1038/jcbfm.2012.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm, B. B. , Cunha, R. A. , & Svenningsson, P. (2003). Pharmacology of adenosine A2A receptors and therapeutic applications. Current Topics in Medicinal Chemistry, 3, 413–426. 10.2174/1568026033392200 [DOI] [PubMed] [Google Scholar]
- Gao, Y. , & Phillis, J. W. (1994). CGS 15943, an adenosine A2 receptor antagonist, reduces cerebral ischemic injury in the Mongolian gerbil. Life Sciences, 55, PL61–PL65. [DOI] [PubMed] [Google Scholar]
- Gomes, C. V. , Kaster, M. P. , Tome, A. R. , Agostinho, P. M. , & Cunha, R. A. (2011). Adenosine receptors and brain diseases: Neuroprotection and neurodegeneration. Biochimica et Biophysica Acta, 1808, 1380–1399. 10.1016/j.bbamem.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Gursoy‐Ozdemir, Y. , Yemisci, M. , & Dalkara, T. (2012). Microvascular protection is essential for successful neuroprotection in stroke. Journal of Neurochemistry, 123(Suppl 2), 2–11. 10.1111/j.1471-4159.2012.07938.x [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoda, M. N. , Li, W. , Ahmad, A. , Ogbi, S. , Zemskova, M. A. , Johnson, M. H. , … Sazonova, I. Y. (2011). Sex‐independent neuroprotection with minocycline after experimental thromboembolic stroke. Exp Transl Stroke Med, 3, 16 10.1186/2040-7378-3-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, R. , Yang, G. , & Li, G. (2010). Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. Journal of Leukocyte Biology, 87, 779–789. 10.1189/jlb.1109766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. R. , van Meer, M. P. , Tejima, E. , Murata, Y. , Mandeville, J. B. , Dai, G. , … Lo, E. H. (2008). Functional MRI of delayed chronic lithium treatment in rat focal cerebral ischemia. Stroke, 39, 439–447. 10.1161/STROKEAHA.107.492215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi, M. , & Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell, 157, 1013–1022. 10.1016/j.cell.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Liu, T. , Clark, R. K. , McDonnell, P. C. , Young, P. R. , White, R. F. , Barone, F. C. , & Feuerstein, G. Z. (1994). Tumor necrosis factor‐alpha expression in ischemic neurons. Stroke, 25, 1481–1488. 10.1161/01.STR.25.7.1481 [DOI] [PubMed] [Google Scholar]
- Melani, A. , Corti, F. , Cellai, L. , Vannucchi, M. G. , & Pedata, F. (2014). Low doses of the selective adenosine A2A receptor agonist CGS21680 are protective in a rat model of transient cerebral ischemia. Brain Research, 1551, 59–72. 10.1016/j.brainres.2014.01.014 [DOI] [PubMed] [Google Scholar]
- Melani, A. , Pantoni, L. , Bordoni, F. , Gianfriddo, M. , Bianchi, L. , Vannucchi, M. G. , … Pedata, F. (2003). The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Research, 959, 243–250. 10.1016/S0006-8993(02)03753-8 [DOI] [PubMed] [Google Scholar]
- Melani, A. , Pugliese, A. M. , & Pedata, F. (2014). Adenosine receptors in cerebral ischemia. International Review of Neurobiology, 119, 309–348. 10.1016/B978-0-12-801022-8.00013-1 [DOI] [PubMed] [Google Scholar]
- Owens, B. (2014). Stroke. Nature, 510, S1 10.1038/510S1a [DOI] [PubMed] [Google Scholar]
- Qiu, J. , Wang, M. , Zhang, J. , Cai, Q. , Lu, D. , Li, Y. , … Chen, H. (2016). The neuroprotection of sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. International Immunopharmacology, 40, 492–500. 10.1016/j.intimp.2016.09.024 [DOI] [PubMed] [Google Scholar]
- Ruck, T. , Bittner, S. , Epping, L. , Herrmann, A. M. , & Meuth, S. G. (2014). Isolation of primary murine brain microvascular endothelial cells. Journal of Visualized Experiments, (93), e52204 10.3791/52204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheardown, M. J. , & Knutsen, L. J. S. (1996). Unexpected neuroprotection observed with the adenosine A(2A) receptor agonist CGS 21680. Drug Development Research, 39, 108–114. [DOI] [Google Scholar]
- Thakur, S. , Du, J. , Hourani, S. , Ledent, C. , & Li, J. M. (2010). Inactivation of adenosine A2A receptor attenuates basal and angiotensin II‐induced ROS production by Nox2 in endothelial cells. The Journal of Biological Chemistry, 285, 40104–40113. 10.1074/jbc.M110.184606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz, D. K. , Lin, R. C. , & Jacobson, K. A. (1995). Cerebral ischemia in gerbils: Effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist. European Journal of Pharmacology, 287, 295–302. 10.1016/0014-2999(95)00498-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Tang, X. N. , & Yenari, M. A. (2007). The inflammatory response in stroke. Journal of Neuroimmunology, 184, 53–68. 10.1016/j.jneuroim.2006.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Li, R. , Wang, X. , Fu, Q. , & Ma, S. (2015). Umbelliferone ameliorates cerebral ischemia–reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neuroscience Letters, 600, 182–187. 10.1016/j.neulet.2015.06.016 [DOI] [PubMed] [Google Scholar]
- Yang, F. , Wang, Z. , Wei, X. , Han, H. , Meng, X. , Zhang, Y. , … Yi, F. (2014). NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. Journal of Cerebral Blood Flow and Metabolism, 34, 660–667. 10.1038/jcbfm.2013.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, G. , Chan, P. H. , Chen, J. , Carlson, E. , Chen, S. F. , Weinstein, P. , … Kamii, H. (1994). Human copper–zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke, 25, 165–170. 10.1161/01.STR.25.1.165 [DOI] [PubMed] [Google Scholar]
- Yin, J. , Zhao, F. , Chojnacki, J. E. , Fulp, J. , Klein, W. L. , Zhang, S. , & Zhu, X. (2018). NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer's disease. Molecular Neurobiology, 55, 1977–1987. 10.1007/s12035-017-0467-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Huang, Z. , Mariani, J. , Wang, Y. , Moskowitz, M. , & Chen, J. F. (2004). Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nature Medicine, 10, 1081–1087. 10.1038/nm1103 [DOI] [PubMed] [Google Scholar]
- Zhou, R. , Zhang, S. , Gu, X. , Ge, Y. , Zhong, D. , Zhou, Y. , … Chen, J. F. (2018). Adenosine A2A receptor antagonists act at the hyperoxic phase to confer protection against retinopathy. Molecular Medicine, 24, 41 10.1186/s10020-018-0038-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Blood pressure and heart rate of vehicle‐ and KW 6002‐treated mice. KW 6002 was injected intraperitoneally into C57BL/6J mice at 10 mg·kg‐1. Systolic blood pressure, diastolic blood pressure, mean blood pressure and heart rate were monitored at the indicated times after administration (n = 5 mice per group). Data are represented as means ± SD. Statistical significance was determined by unpaired Student's t‐test.
Table S1 Real‐Time PCR primers