

Abstract
Background and Purpose
L‐DOPA‐induced dyskinesia (LID) remains a major complication of L‐DOPA therapy in Parkinson's disease. LID is believed to result from inhibition of substantia nigra reticulata (SNr) neurons by GABAergic striatal projection neurons that become supersensitive to dopamine receptor stimulation after severe nigrostriatal degeneration. Here, we asked if stimulation of direct medium spiny neuron (dMSN) GABAergic terminals at the SNr can produce a full dyskinetic state similar to that induced by L‐DOPA.
Experimental Approach
Adult C57BL6 mice were lesioned with 6‐hydroxydopamine in the medial forebrain bundle. Channel rhodopsin was expressed in striatonigral terminals by ipsilateral striatal injection of adeno‐associated viral particles under the CaMKII promoter. Optic fibres were implanted on the ipsilateral SNr. Optical stimulation was performed before and 24 hr after three daily doses of L‐DOPA at subthreshold and suprathreshold dyskinetic doses. We also examined the combined effect of light stimulation and an acute L‐DOPA challenge.
Key Results
Optostimulation of striatonigral terminals inhibited SNr neurons and induced all dyskinesia subtypes (optostimulation‐induced dyskinesia [OID]) in 6‐hydroxydopamine animals, but not in sham‐lesioned animals. Additionally, chronic L‐DOPA administration sensitised dyskinetic responses to striatonigral terminal optostimulation, as OIDs were more severe 24 hr after L‐DOPA administration. Furthermore, L‐DOPA combined with light stimulation did not result in higher dyskinesia scores than OID alone, suggesting that optostimulation has a masking effect on LID.
Conclusion and Implications
This work suggests that striatonigral inhibition of basal ganglia output (SNr) is a decisive mechanism mediating LID and identifies the SNr as a target for managing LID.
Abbreviations
- 6‐OHDA
6‐hydroxydopamine
- AAV
adeno‐associated virus
- AIMs
abnormal involuntary movements
- ChR2
channel rhodopsin 2
- dMSN
direct pathway medium spiny neuron
- eYFP
enhanced yellow fluorescent protein
- GPi
internal globus pallidus
- iMSN
indirect pathway medium spiny neuron
- LID
L‐DOPA‐induced dyskinesia
- OID
optostimulation‐induced dyskinesia
- PD
Parkinson's disease
- SNr
substantia nigra reticulata
What is already known
Dyskinesia induced by L‐DOPA involves hyperactivity of striatal medium spiny neurons expressing dopamine D1 receptors.
Severe dopaminergic denervation is a key factor for the development of L‐DOPA induced dyskinesia.
What this study adds
Optostimulation of striatonigral terminals produces a full repertoire of dyskinesia in severely dopamine depleted animals.
Striatonigral inhibition of substantia nigra reticulata is a critical mechanism for the generation of dyskinesia.
What is the clinical significance
The striatonigral synapse could be a novel target for management of dyskinesia in Parkinson's disease.
1. INTRODUCTION
L‐DOPA‐induced dyskinesia (LID) remains a major complication of chronic L‐DOPA therapy in Parkinson's disease (PD). LID arises after chronic pulsatile stimulation of striatal dopamine receptors in patients who, due to severe nigrostriatal terminal degeneration, have lost their capacity to buffer fluctuations of extracellular dopamine (Cenci & Konradi, 2010; Jenner, 2008; Murer & Moratalla, 2011). However, 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐intoxicated patients who quickly develop severe PD symptoms show LID shortly after beginning treatment (Ballard, Tetrud, & Langston, 1985), and similarly, a first challenge with L‐DOPA induces dyskinesia in severely dopamine‐depleted animals. Furthermore, optogenetic or chemogenetic stimulation of severely dopamine‐denervated striatal neurons in L‐DOPA‐naïve rodents can induce dyskinesia resembling LID (Alcacer et al., 2017; F. Hernández, Castela, Ruiz‐DeDiego, Obeso, & Moratalla, 2017; Perez, Zhang, Bordia, & Quik, 2017). Altogether, this work suggests that loss of dopamine input can modify striatal circuit function such that striatal activation induces dyskinesia instead of normal actions (Picconi & Calabresi, 2017).
During dyskinesia induced by dopamine receptor stimulation in patients and animal models of PD, neurons at the internal globus pallidus (GPi) and substantia nigra reticulata (SNr) are markedly inhibited (Aristieta, Ruiz‐Ortega, Miguelez, Morera‐Herreras, & Ugedo, 2016; Boraud, Bezard, Bioulac, & Gross, 2001; Filion, Tremblay, & Bédard, 1991; Lozano, Lang, Levy, Hutchison, & Dostrovsky, 2000; Meissner et al., 2006; Papa, Desimone, Fiorani, & Oldfield, 1999). The main source of SNr inhibition is GABA released by direct pathway medium spiny neurons (dMSNs) from the striatum (Freeze, Kravitz, Hammack, Berke, & Kreitzer, 2013). These neurons express the dopamine D1 receptor, which is crucial and necessary for the development of LID (Darmopil, Martín, De Diego, Ares, & Moratalla, 2009; Halje et al., 2012; Murer & Moratalla, 2011). Whether the indirect pathway contributes to LID is still controversial. Agonists of D2 receptors, expressed in the indirect pathway medium spiny neurons (iMSNs), only induce dyskinesia in animals that have been primed with mixed D1/D2 receptor agonists (Delfino et al., 2004; Luquin, Laguna, & Obeso, 1992) and do not substantially modify LID in patients (Rascol et al., 2006). Interestingly, LID is not modified by inactivation of D2 receptors (Darmopil et al., 2009) but is reduced by chemogenetic activation of iMSNs (Alcacer et al., 2017). Importantly, in previous studies, stimulation of dMSNs produced dyskinesia that was less intense and varied than that observed with L‐DOPA, except when chemogenetic activation of dMSNs was combined with administration of D2 receptor agonists. This suggests that inhibition of the D2 receptors in iMSNs is necessary for full expression of LID (Alcacer et al., 2017), as predicted by classical models and suggests that an activity imbalance between dMSNs and iMSNs drives LID (DeLong, 1990; Jenner, 2008; Suarez, Alberquilla, García‐Montes, & Moratalla, 2018; Suarez, Solis, Aguado, Lujan, & Moratalla, 2016; Suárez et al., 2014). However, recent studies challenge these classical views, as dMSNs and iMSNs are coactivated during movement (Cui et al., 2013; Parker et al., 2018; Ryan, Bair‐Marshall, & Nelson, 2018), and optogenetic co‐stimulation of dMSNs and iMSNs induces dyskinesia in PD rats (F. Hernández et al., 2017). Thus, whether selective dMSN stimulation can induce a full repertoire of dyskinetic movements, comparable in intensity to those induced by L‐DOPA, remains uncertain.
Here, we asked if optogenetically targeting the densely packed dMSN terminals at the SNr rather than the broadly distributed dMSN cell bodies at the striatum would generate dyskinetic movements as intense and varied as those produced with L‐DOPA. We also asked if dyskinesia‐related molecular markers associated with MSN stimulation are expressed during optostimulation of stratonigral axon terminals.
2. METHODS
2.1. Animals
All animal care and experimental procedures conformed to European Community guidelines (2003/65/CE) and were approved by the Cajal Institute's Bioethics Committee (following DC86/609/EU) and, for work carried out in Argentina, protocols were approved by the University of Buenos Aires School of Medicine Institutional Animal Care and Use Committee. Every effort was made to minimise animal discomfort and the overall number of animals used. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. This study was carried out on adult C57BL/6N mice (RRID:MGI:5825016) from Harlan Iberica, Barcelona, Spain. Animals were housed under a 12‐hr light/dark cycle with free access to food and water. Electrophysiological experiments performed in Argentina were also carried out in C57BL/6 mice (RRID:IMSR_CRL:475) from the National Academy of Medicine, Buenos Aires, Argentina.
2.2. Surgical procedures
All surgical procedures were conducted under deep surgical anaesthesia (1% isoflurane). Animals were mounted on a stereotaxic frame (Kopf Instruments, Tujunga, USA) with a mouse adaptor. 6‐Hydroxydopamine (6‐OHDA) injection and viral infection were performed during the same surgery. Fibre optic cannulae were implanted in a second operation, 4–6 weeks later.
2.2.1. 6‐OHDA lesions
6‐OHDA hydrobromide with added ascorbic acid was dissolved in saline, to a final concentration of 5.75‐μg·μl−1 6‐OHDA in 0.02% ascorbic acid. Mice received 0.7‐μl injections of 6‐OHDA or vehicle solution into the left medial forebrain bundle at the following coordinates from bregma (mm): anterior–posterior = −1, medial–lateral = +1.2, and dorso–ventral = −4.8. Injections were performed at 0.5 μl·min−1 using a 300‐μm‐diameter cannula attached to a 1‐μl Hamilton syringe controlled by a motorised pump (Harvard Apparatus, USA). Special care was required after 6‐OHDA lesion to ensure survival. Each mouse was weighed daily and received a subcutaneous injection of saline and an enriched diet. This procedure continued until animals began to regain weight (14–20 days after surgery).
2.2.2. Vector injection
Immediately after 6‐OHDA or vehicle infusion, an adeno‐associated virus (AAV) vector expressing channelrhodopsin 2 (ChR2) tagged with enhanced yellow fluorescent protein (eYFP) and under the CaMKII promoter (AAV‐CaMKIIa‐hChR2(H134R)‐eYFP) was injected into the left dorsolateral striatum. Viral concentration was 2.46 × 1013 viral particles·ml−1; 0.5‐μl AAV was infused in the left striatum at 0.1 μl·min−1 at the following coordinates (mm): anterior–posterior = +0.5, medial–lateral = +2.5, and dorso–ventral = −3. An additional group of 6‐OHDA animals was injected with an AAV vector expressing only eYFP (AAV‐CaMKIIa‐eYFP).
2.2.3. Fibre optic cannula implant
Four to 6 weeks after 6‐OHDA lesion and viral infection, mice were implanted ipsilaterally with a fibre optic cannula over the substantia nigra reticulata (SNr). We used the following coordinates (mm): anterior–posterior = −2.9, medial–lateral = +1.4, and dorso–ventral = −4.25. Fibre optic cannulae had a 200‐μm core fibre, flat cleave, and 0.22 NA (Doric Lenses, Quebec, Canada).
2.3. Behavioural tests
Three weeks after 6‐OHDA injection, behavioural testing was performed on 3 non‐consecutive days during the light phase in order to select only severely lesioned animals. All mazes and apparatus were thoroughly cleaned with 10% ethanol and dried between subjects.
2.3.1. Actimeter
General horizontal and vertical exploratory activity in a novel open field was recorded as described by Ruiz‐DeDiego et al. (2018) using a multi‐cage activity meter (Columbus Instruments, Ohio, USA) with eight individual cages measuring 20 × 20 × 28 cm. Horizontal movement was detected by two arrays of 16 IR beams, with a third array positioned 4 cm above the floor to detect vertical movement. Test sessions lasted 20 min.
2.3.2. Cylinder test
Each mouse was placed in a 10‐cm‐diameter glass cylinder and videotaped for 3 min. Mice responded to the novel environment by standing on their hindlimbs and leaning on the walls of the cylinder with their forelimbs. The number of supporting ipsilateral, contralateral, and simultaneous forepaw placements against the cylinder wall was assessed. Data are expressed as a percentage of contralateral forelimb use (%contralateral forelimb use = (contralateral contacts/(ipsilateral+contralateral+simultaneous contacts/2) × 100).
2.3.3. Accelerating rotarod test
Mice were tested in sets of five, using an accelerating rotarod (from 4 to 40 rpm in 5 min; Ugo Basile, Rome, Italy). The latency to fall from the rod was automatically recorded, and cut‐off time was 5 min as described by García‐Montes et al. (2018). Each animal was assessed in a single day over six trials with 15‐min intertrial intervals.
2.4. Optical stimulation
Striatonigral terminals were activated using a 473‐nm DPSS blue laser (Shanghai Laser, Shanghai, China) with a maximum output power of 100 mW. The laser was controlled by an Optogenetics TTL Pulse Generator (Doric Lenses, Quebec, Canada), and its power was adjusted to be 10 mW at the fibre tip (measured with a PM100D optical power meter with an S120C sensor; Thorlabs Inc, Newton, USA). Two optical stimulation protocols were used in this study. One consisted of a single 15‐s pulse, a 3‐min pause, and a second single 15‐s pulse tested at 1–10 mW. The second stimulation protocol consisted of a 30‐s burst of pulsed light at 20 Hz with a 5‐ or 20‐ms pulse duration at 10 mW, a 3‐min pause, and a second 30‐s burst of light identical to the first. Only one protocol was tested per session, and sessions were separated by at least 72 hr. After eliciting dyskinesias with the two light stimulation protocols, animals were subjected to an L‐DOPA sensitisation protocol using escalating doses of L‐DOPA. L‐DOPA methyl ester was injected i.p. once per day, 20 min after an i.p. benserazide hydrochloride injection. L‐DOPA and benserazide doses were respectively 2 and 6 mg·kg−1; 3 and 6 mg·kg−1; 6 and 6 mg·kg−1; 9 and 12 mg·kg−1; and 20 and 12 mg·kg−1. Animals were light stimulated twice over 1 week. In the first session, abnormal involuntary movements (AIMs) elicited by laser stimulation were assessed 24 hr after three daily doses of L‐DOPA. The second session took place 72 hr later; the simultaneous effect of light stimulation combined with an acute dose of L‐DOPA was assessed. For a graphical explanation of the escalating L‐DOPA treatment protocol, please see Figures 4a and 6a.
Figure 4.
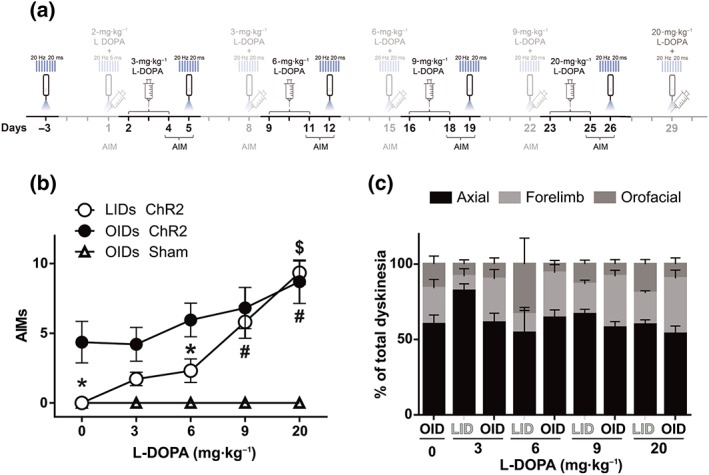
Optostimulation of striatonigral terminals induces greater dyskinesia in primed than in naïve 6‐OHDA mice. (a) Experimental timeline highlighting in black the behavioural manipulation rated in (b) and (c). (b) Total dyskinesia score induced by three consecutive daily doses of L‐DOPA (LID) or by optostimulation (OID) 24 hr after the third day of each escalating dose of L‐DOPA treatment. *P < .05, OID significantly different from LID; $ P < .05 OID at 20 mg·kg−1 significantly different from OID at naïve and OID after 3‐mg·kg−1 L‐DOPA; # P < 0.05, LID at 9 or 20 mg·kg−1 significantly different from LID at 3‐ and 6‐mg·kg−1 L‐DOPA; two‐way repeated measures ANOVA.with significant interaction, Bonferroni post hoc test. (c) Type and intensity of dyskinesia induced by 3 days of each escalating dose of L‐DOPA (LID) or by optostimulation (OID) applied 24 hr after each third day of L‐DOPA treatment. Data shown are means ± SEM from n = 6 Sham and n = 8, 6‐OHDA treated mice. AIMs, axial, oral and forelimb dyskinesia score
Figure 6.
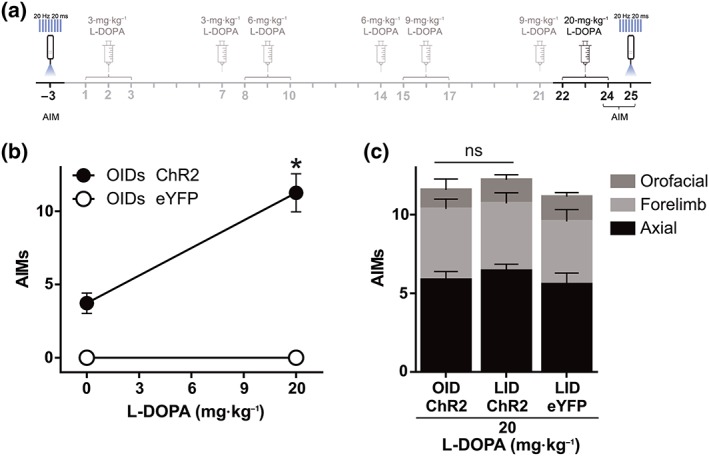
Optostimulation in an independent set of animals confirms the L‐DOPA priming effect on OIDs. (a) Experimental timeline highlighting in black the behavioural manipulation rated in (b) and (c). (b) OID observed in 6‐OHDA‐lesioned animals expressing ChR2‐eYFP in striatonigral terminals (n = 7) before and after chronic L‐DOPA treatment and in an additional group of 6‐OHDA‐lesioned mice expressing eYFP only. Data shown are means ± SEM from n = 7 mice. *P < .05, significant effect of L‐DOPA; paired t test. (c) Composition of total dyskinesia scores for OID after the chronic L‐DOPA treatment shown in (a) and for LID induced by 20‐mg·kg−1 L‐DOPA in the same animals. Total AIM scores did not significantly differ between these conditions (paired t test not significant). Data shown are means ± SEM. AIMs, axial, oral and forelimb dyskinesia score
2.5. Dyskinesia ratings
Dyskinesia was rated by two separate observers, one of whom was blind to treatment (sham or lesion), based on video footage. Lineal regression analysis on raters' scores produced an R2 = 0.84. Therefore, the raters' scores were averaged. Each subtype of dyskinesia (oral, axial, and forelimb) was scored, considering dyskinesia frequency and amplitude, as described in Perez et al. (2017). Briefly, the frequency scale ranged from 0 to 4 (where 0 = no dyskinesia; 1 = occasional dyskinesia displayed for <50% of the observation time; 2 = sustained dyskinesia displayed for >50% of the observation time; 3 = continuous dyskinesia; and 4 = continuous dyskinesia not interruptible by external stimuli). Amplitude scores were subdivided as “A,” which indicates oral dyskinesia without tongue protrusion, forelimb dyskinesia without shoulder engagement, and axial dyskinesia with body twisting <60°, or “B,” indicating oral dyskinesia with tongue protrusion, forelimb dyskinesia with shoulder engagement, or axial dyskinesia with body twisting ≥60°. Overall scores for the frequency and amplitude of dyskinesia used for data analysis were calculated as 1A = 1, 1B = 2, 2A = 2, 2B = 4, 3A = 4, 3B = 6, 4A = 6, and 4B = 8. This entails that scores for any one component (axial, oral, or forelimb) may be rated from 0 to 8. Total dyskinesia scores were calculated as the sum of all components, with a total possible maximum score of 24 per rating point.
2.6. Tissue preparation
After behavioural testing, animals were illuminated for one final session with the 20‐Hz, 20‐ms, and 10‐mW protocol and perfused 50 min later. Mice were anaesthetised (sodium pentobarbital, 100 mg·kg−1) and perfused transcardially with cold saline containing heparin (500 IU·L−1) followed by 4% paraformaldehyde in phosphate buffer (PB). Brains were then dissected, incubated overnight in paraformaldehyde, and then stored in PB. Fixed brains were cut into 30‐μm coronal sections in a vibratome (Leica Microsystems, Wetzlar, Germany), and slices were stored free‐floating in 0.1‐M PB containing 0.1% sodium azide at 4°C until use. 6‐OHDA‐induced nigrostriatal lesions were confirmed by immunohistochemical detection of tyrosine hydroxylase (TH) on free‐floating sections from the striatum (anti‐TH, 1:1,000; Millipore, Temecula, USA). Expression of C‐fos, FosB, and DARPP‐32 were assessed by immunohistochemistry on free‐floating striatal sections (anti‐c‐Fos, 1:15,000; anti‐FosB, 1:7,500; anti‐DARPP‐32, 1:500) as previously described (Granado, Escobedo, O'Shea, Colado, & Moratalla, 2007; Ruiz‐DeDiego, Mellstrom, Vallejo, Naranjo, & Moratalla, 2015).
2.7. In vitro electrophysiology
Experiments were conducted in brain slices prepared from the SNr of 6‐month‐old mice. These mice were sham lesioned and were injected with AAV particles, as described in Section 2.2.2, 16 weeks prior to experiments. Mice were deeply anaesthetised with isoflurane and decapitated. Brains were quickly removed and immersed in ice‐cold slicing solution containing 210‐mM sucrose, 10‐mM NaCl, 1.9‐mM KCl, 1.2‐mM Na2HPO4, 33‐mM NaHCO3, 6‐mM MgCl2, 1‐mM CaCl2, and 10‐mM glucose, pH 7.3–7.4 when bubbled with 95% O2 and 5% CO2. The SNr was then sectioned into 300‐μm coronal slices, using a vibrating microtome (Pelco 1000, Ted Pella, Inc). Slices were immediately placed in an incubation chamber filled with artificial CSF maintained at 36°C and containing 125‐mM NaCl, 2.5‐mM KCl, 1.25‐mM Na2HPO4, 10‐mM glucose, 25‐mM NaHCO3, 0.4‐mM ascorbate, 1‐mM MgCl2, and 2‐mM CaCl2, pH 7.3–7.4 when gassed with 95% O2 and 5% CO2. After 5‐min incubation at 36°C, brain slices were stabilised at room temperature in the same solution for at least 30 min before they were transferred to the recording chamber.
For recording, slices were transferred to a submersion chamber and superfused at 2 ml·min−1 with oxygenated artificial CSF at 30–32°C. Whole‐cell recordings were obtained from visually identified neurons in the SNr using a Nikon microscope equipped with IR‐DIC optics. Pipettes pulled from borosilicate glass capillaries had a resistance of 4–6 MΩ when filled with the following solutions: Solution A (used for current clamp experiments): 120‐mM potassium gluconate, 10‐mM KCl, 10‐mM HEPES, 0.2‐mM EGTA, 4.5‐mM MgATP, 0.3‐mM NaGTP, and 14‐mM sodium phosphocreatine, pH adjusted to 7.2–7.4 with KOH, and Solution B (used for voltage clamp experiments): 130‐mM CsMeSO4, 10‐mM HEPES, 0.2‐mM EGTA, 4.5‐mM MgATP, 0.3‐mM NaGTP, 14‐mM sodium phosphocreatine, 10‐mM TEA‐Cl, and 5‐mM QX‐314‐Br, pH adjusted to 7.2–7.4 with CsOH. Recordings were obtained using Multiclamp 700B amplifiers (Molecular Devices, San Jose, USA). Signals were low‐pass filtered at 6 kHz and digitised at 20 kHz using DigiData 1200 acquisition interfaces (Molecular Devices). Data acquisition and analysis were performed using Clampex 10.2 and CampFit 10.2 software (Molecular Devices). In voltage clamp mode, the pipette capacitance was compensated, and series resistance was continuously monitored but was not compensated. Only recordings with a stable series resistance of <20 MΩ were used for analysis. When current clamp mode was used, series resistance and pipette capacitance were monitored and corrected using bridge and capacitance neutralisation.
2.8. Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Data in figures are expressed as mean ± SEM. Data omission or exclusion criteria were as follows: in some cases, misinjection of AAV caused insufficient AAV‐dependent ChR2 expression, or optic fibres were not placed over the SNr. These animals were excluded, resulting in slightly unbalanced experimental groups. Statistical analysis was performed using SigmaPlot 11.0 (RRID:SCR_003210, Systat Software Inc, UK). The significance level was set at P < .05. Depending on the number of experimental groups and factors to be compared, statistical analyses were performed with t tests or two‐way repeated measures ANOVA followed by post hoc tests (Bonferroni) when appropriate. Before applying parametric tests, data were tested for normality with the Kolmogorov–Smirnov test and homoscedasticity with the Levene median test.
2.9. Materials
6‐OHDA hydrobromide with added ascorbic acid, L‐DOPA, benserazide hydrochloride, NaCl, KCl, Na2HPO4, NaHCO3, MgCl2, CaCl2, ascorbate, HEPES, EGTA, MgATP, NaGTP, sodium phosphocreatine, KOH, CsMeSO4, NaGTP, TEA‐Cl, and CsOH were purchased from Sigma‐Aldrich (St. Louis, USA). QX‐314‐Br was from Tocris (Bristol, UK). AAV was purchased from Vector Core (North Carolina University, Chapel Hill, USA). TH antibody (cat# AB152, RRID:AB_390204) was from Millipore (Burlington, USA). cFos (cat# sc‐7202, RRID:AB_2106765) and FosB (cat# sc‐28213, RRID:AB_2106911) antibodies were from Santa Cruz Biotechnology (Santa Cruz, USA). DARPP‐32 antibody (cat# 611520, RRID:AB_398980) was from Becton Dickinson (Franklin Lakes, USA).
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, S. P. H., Fabbro et al., 2017; Alexander, Peters et al., 2017).
3. RESULTS
3.1. Validation of the optogenetic approach in brain slices
To selectively stimulate striatonigral axon terminals in the SNr, we injected AAV particles expressing the fusion protein ChR2‐EYFP, driven by the CaMKIIa promoter, in the striatum of C57BL6 mice. Histology confirmed the expression of ChR2‐EYFP in the striatum as well as in the synaptic terminal field of striatonigral neurons in the SNr (Figure 1a). Whole‐cell patch clamp recordings of SNr neurons in coronal brainstem slices, 16 weeks after intrastriatal AAV‐ChR2‐EYFP injection, were used to determine if light stimulation of striatonigral terminals induces GABA‐mediated inhibition of SNr neurons (Figure 1b). Exploratory current clamp studies showed that both continuous (1 s) and pulsed train illumination (pulses of 5‐ to 20‐ms duration at 10–20 Hz; 447‐nm light source), at intensities of 1–10 mW, markedly inhibited the firing of SNr neurons (>90% inhibition of firing, four cells from two animals, regardless of light stimulation protocol). Voltage clamp studies showed that 1‐, 5‐, or 20‐ms‐long light pulses evoked inhibitory post‐synaptic currents in SNr neurons, which were almost completely blocked by the non‐competitive GABAA receptor antagonist picrotoxin (10 cells from five animals; data corresponding to 20‐ms pulses are shown in Figure 1c). Additional current clamp experiments were performed to characterise the effect of 20‐Hz trains of light pulses, which in preliminary behavioural experiments provided promising results (see below). On average, it was possible to nearly completely inhibit the spontaneous tonic firing of SNr neurons located near the centre of the light field with 20‐Hz trains of 5‐ or 20‐ms duration pulses of an intensity of ~8 mW (seven cells from five animals; data obtained with 20‐ms pulses are shown in Figure 1d,e). Addition of picrotoxin to the bath completely blocked the effect of optostimulation of striatonigral terminals on SNr firing (Figure 1d,e).
Figure 1.
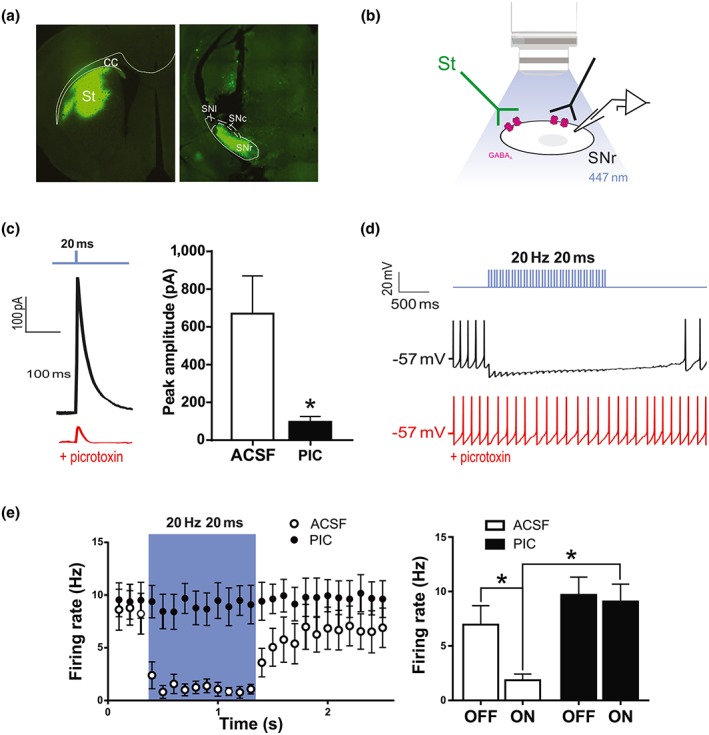
Illumination of ChR2‐transfected striatonigral terminals inhibits SNr neurons. (a) Coronal sections of dorsal striatum (left) and the SNr (right) showing a representative injection site and ChR2‐eYFP expression at striatonigral terminals, respectively. Note the track left by the fibre optic cannula in the SNr section. (b) Schematic of the approach to ex vivo whole‐cell recording and illumination. (c) Representative trace of an inhibitory post‐synaptic current recorded at a holding potential of 0 mV in an SNr neuron, evoked by a 20‐ms light pulse, before and after adding picrotoxin (PIC; 50 μM) to the bath (left), and population data corresponding to 10 SNr neurons recorded from five animals (right). *P < .05, significant effect of picrotoxin; paired t test). (d) Representative current clamp recording of an SNr neuron inhibited during light stimulation of striatonigral terminals. (e) Effect of trains of light pulses (20 ms, 20 Hz, ~8 mW) on the tonic firing of SNr neurons, before and after adding picrotoxin (50 μM) to the bath. Left: Time course of effects. Bin size: 100 ms. Right: Data were integrated along the laser off and laser on periods and subjected to statistical analysis. Values shown are means ± SEM from seven cells from five animals. *P < .05, significantly different as indicated; two‐way ANOVA with Bonferroni post hoc test. cc, corpus callosum; SNl, substantia nigra pars lateralis; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; St, striatum
These results indicate that ChR2 is efficiently expressed and functional in striatonigral terminals.
3.2. Parkinson‐like symptoms and dopamine denervation
Parkinson‐like phenotypes were tested 3 weeks after lesion in order to select animals with marked motor coordination deficits (rotarod) and a reduction of contralateral forelimb use (cylinder test) and vertical activity (multicage activity meter system; Figure 2a–c). Dopaminergic degeneration in these animals was further confirmed post‐mortem by immunohistochemistry, assessing TH fibre loss and determining striatal volume with a complete loss of TH‐immunoreactive fibres (Figure 2d–f). We also verified ChR2‐YFP expression in DARPP32‐positive striatal neurons and in DARPP32‐positive fibres in the SNr—the projection field of dMSNs (Figure 2g).
Figure 2.
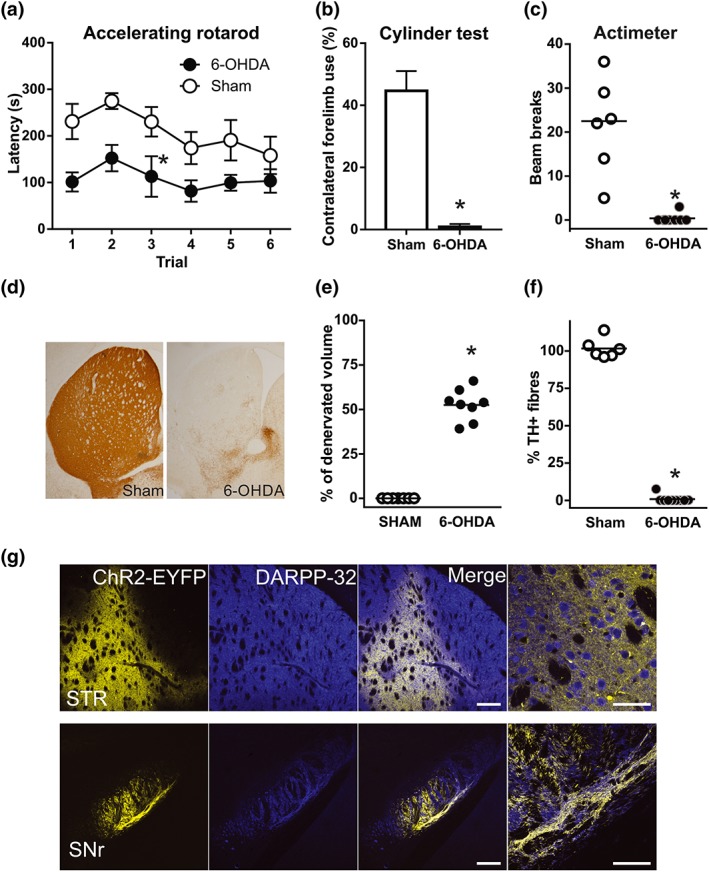
Efficacy of dopamine lesions and channelrhodopsin viral infection. (a–c) 6‐OHDA‐lesioned animals showed motor coordination deficits (a; *P < .05, main effect of treatment, two‐way repeated measures ANOVA), reduced contralateral forelimb use (b;, *P < .05, significant effect of 6‐OHDA; Student's t‐test) and reduced vertical activity (c; *P < .05, significant effect of 6‐OHDA; Mann–Whitney test. (d) Representative photomicrographs of striatal sections illustrating the loss of TH‐positive fibres in 6‐OHDA‐lesioned mice. (e and f) The efficacy of 6‐OHDA lesions was assessed by quantifying ( e) the percentage of striatal volume that was completely denervated and (f) striatal TH‐positive fibre depletion. *P < .05, significant effect of 6‐OHDA; Mann–Whitney test. (g) Representative low‐magnification confocal images obtained from the striatum and SNr illustrating ChR2‐EYFP expression in DARPP‐32 neurons (striatum) and fibres (SNr) in AAV‐ChR2‐infected mice. Right‐most panels show magnifications of the merge panels. Scale bars: 500 and 50 μm. STR, striatum; SNr, substantia nigra reticulata. Sham, n = 6; 6‐OHDA, n = 8. Data are mean ± SEM for (a) and (b); median and individual data points for (c), (e), and (f)
3.3. Optogenetic stimulation of striatonigral terminals induces abnormal involuntary movements in 6‐OHDA‐lesioned mice
To examine the effect of striatonigral terminal optostimulation on dyskinetic behaviour, a fibre optic cannula was implanted on the ipsilateral SNr of lesioned or sham‐operated mice transduced with ChR2‐EYFP. A range of light stimulation protocols were tested in sham and 6‐OHDA mice. To determine the optimal intensity of light pulses, mice were stimulated with a continuous 15‐s blue laser pulse at different intensities (1, 2, 5, 8, 10, and 12 mW). Abnormal movements resembling axial, orofacial, and limb dyskinesia induced by L‐DOPA were readily detected in 6‐OHDA‐lesioned animals from 2 mW and progressively increased to reach a plateau at 10 mW (Figure 3a). We therefore used 10 mW for subsequent experiments. For pulsatile stimulation, we used 30‐s and 10‐mW pulse trains at different frequencies (10–20 Hz) and pulse duration (5–20 ms). No dyskinesia was observed with the 10‐Hz/5‐ms protocol; however, 20 Hz/5 ms elicited weak dyskinetic symptoms (suboptimal protocol), which increased at 20‐Hz/20‐ms bursts (Figure 3b) (Video S1). Dyskinetic symptoms elicited with 20‐Hz/20‐ms light bursts were similar to those elicited with continuous light. Each pulsatile protocol was tested in two different sessions (48 hr apart); each session consisted of two trials, 3 min apart. The scores during intrasession and intersession repetitions of an illumination protocol were similar (Figure 3c). The dyskinesia induced in 6‐OHDA‐lesioned animals was predominantly of the axial type, but abnormal limb and orofacial movements were also observed (Figure 3d,e). Optostimulation of striatonigral terminals also induced contralateral rotations in 6‐OHDA‐lesioned mice (Figure 3e,f). In sham‐lesioned mice, optostimulation at the SNr with continuous light or with trains of light pulses failed to induce abnormal movements and overall produced very subtle behavioural effects (Figure 3a,b,f) (Video S2). Overall, these data show that in animals with severe nigrostriatal terminal degeneration induced by 6‐OHDA, but not in sham‐lesioned mice, optostimulation of striatonigral terminals induces a wide repertoire of abnormal movements resembling LID (henceforth termed optostimulation‐induced dyskinesia [OID]).
Figure 3.
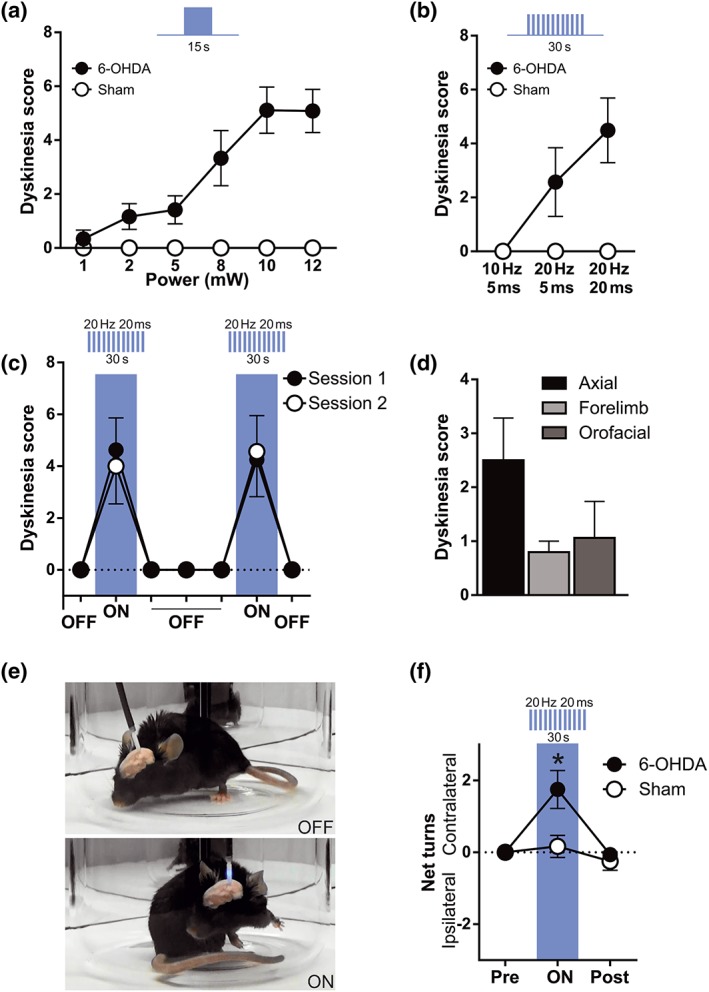
Optostimulation of striatonigral terminals induces dyskinesia in 6‐OHDA mice. Total dyskinesia (axial + forelimb + orofacial) score after continuous (a) or pulsatile (b) illumination of striatonigral terminals in 6‐OHDA‐lesioned mice. Note that illumination in sham‐lesioned mice does not induce dyskinesia. (c) Total dyskinesia score during striatonigral terminal stimulation showed intersession and intrasession stability, two‐way repeated measures ANOVA, intersession main effect not significant, intrasession main effect not significant, and interaction not significant. (d) Types of dyskinesia observed during SNr illumination in 6‐OHDA‐lesioned mice. (e) Photographs of 6‐OHDA‐lesioned mouse with laser OFF and laser ON. Optostimulation of striatonigral terminals induced dyskinesia. (f) Rotational locomotive behaviour induced by the 20‐Hz/20‐ms protocol of optostimulation of striatonigral axon terminals in sham‐ and 6‐OHDA‐lesioned mice. Data shown are means ± SEM from n = 6 Sham and n = 8; 6‐OHDA treated mice. *P < .05, significant effect of 6‐OHDA; two‐way repeated measures ANOVA with significant interaction and Bonferroni post hoc test
3.4. Chronic L‐DOPA administration sensitises dyskinetic responses to striatonigral terminal optostimulation
Optostimulation of MSN cell bodies induces more dyskinetic movements in L‐DOPA‐primed mice than in naïve 6‐OHDA‐lesioned mice (F. Hernández et al., 2017; Perez et al., 2017; Ryan et al., 2018). However, it is not known whether this sensitisation depends only on striatal adaptations induced by L‐DOPA or if it can be partly accounted for by extrastriatal adaptations. To determine if exposure to L‐DOPA sensitises the dyskinetic response to optostimulation of striatonigral terminals, sham‐ and 6‐OHDA‐lesioned mice were treated with escalating doses of L‐DOPA (3, 6, 9, and 20 mg·kg−1 i.p., each dose given for 3 consecutive days) and tested for the effects of optostimulation when they were “off” (L‐DOPA free for 24 hr) as represented in the timeline (Figure 4a).
During the course of L‐DOPA treatment, there was a significant increase of both OID and LID in the 6‐OHDA‐lesioned animals. Stimulation (30 s, 20 Hz, 20‐ms pulses, 10 mW) of striatonigral terminals consistently induced more dyskinesia in L‐DOPA‐primed mice than in naïve 6‐OHDA‐lesioned mice (Figure 4b) (Video S3). Interestingly, the dyskinesia scores during striatonigral terminals optostimulation were consistently higher than those induced by L‐DOPA alone at doses of 3 and 6 mg·kg−1. However, these differences disappeared when animals received higher doses of L‐DOPA. The OID scores measured 24 hr after 9‐ and 20‐mg·kg−1 L‐DOPA were comparable to those induced by L‐DOPA alone (Figure 4b). Sham mice did not show OID at any of the conditions tested (Figure 4b) (Video S4). The significant increase of both the OID and LID scores during L‐DOPA treatment indicates a progressive sensitisation to striatonigral terminal optostimulation.
We also asked if OID and LID involved similar repertoires of dyskinetic movements. Indeed, axial dyskinesia prevailed in both conditions, especially at the beginning of treatment. During treatment, for both conditions, limb and orofacial dyskinesia accounted for ~50% of the dyskinetic scores. Overall, orofacial dyskinesia seemed to be less frequent during OID than in LID (Figure 4c).
To rule out any possible contribution of D1 receptor stimulation in OID, light stimulation (20 Hz, 20‐ms pulses, 10 mW) was applied to striatonigral axon terminals before and 25 min after systemic administration of the prototypical D1 receptor antagonist SCH23390 (0.2 mg·kg−1 i.p.). As expected, SCH23390 increased time of immobility and reduced vertical activity (Figure 5a,b). However, the D1 receptor antagonist had no effect on the intensity or composition of OID (Figure 5c,d).
Figure 5.
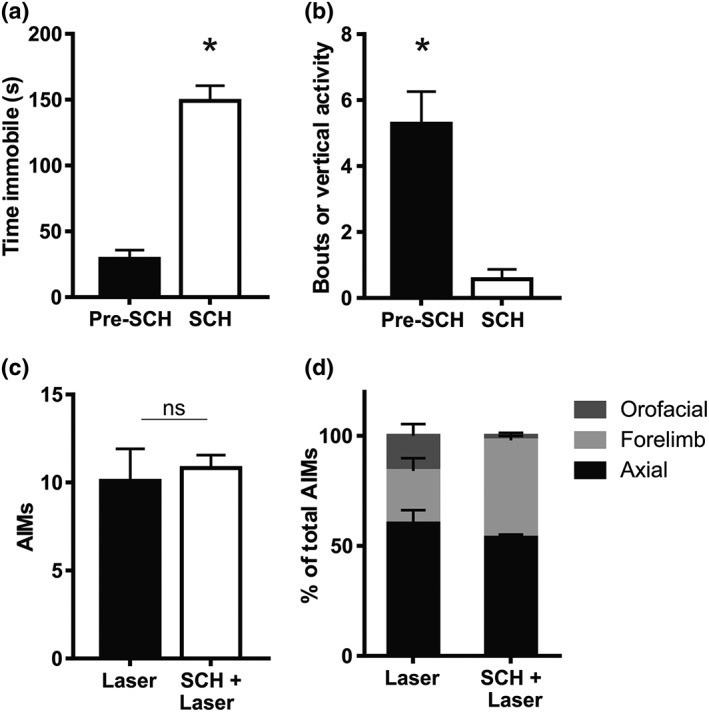
The D1 receptor antagonist SCH23390 does not modify optostimulation‐induced dyskinesia. (a and b) SCH23390 (SCH; 0.25 mg·kg−1, i.p.) increases immobility (a) and reduces vertical activity (b) in 6‐OHDA‐lesioned mice. ; *P < .05, significant effect of SCH23390; paired t test. (c) Total dyskinesia score (axial, orofacial, and limb) induced by optostimulation of striatonigral terminals in 6‐OHDA‐lesioned mice, before and after administration of the D1 receptor antagonist SCH23390 (0.25 mg·kg−1, i.p.). No significant difference was found (paired t test not significant). (d) Composition of OID before and under the effect of SCH23390. Data shown are means ± SEM from n = 8, 6‐OHDA treated mice. AIMs, axial, oral and forelimb dyskinesia score
Although our optostimulation protocols were very brief (1 min of light stimulation during each session), this set of animals was exposed to several illumination sessions before and during L‐DOPA treatment. To exclude the possibility that multiple light sessions would generate plastic changes that could interfere with behavioural results, we prepared an additional set of 6‐OHDA‐lesioned mice (n = 4) that received one illumination session before and one 24 hr after the last escalating dose of L‐DOPA (Figure 6a). In these animals, the OID score was also significantly higher after L‐DOPA treatment. Furthermore, there was no difference between the OID score of the last illumination session and the LID score during the last L‐DOPA challenge (Figure 6b,c). Importantly, there were no statistical differences in OID between the two sets of animals—main set of animals (Figure 4) and additional set of animals (Figure 6)—in either naïve or primed conditions.
Finally, to completely rule out any possible non‐specific effect of light stimulation in our results, a group of 6‐OHDA‐lesioned animals was injected in the striatum with an AAV vector carrying eYFP only and subjected to the treatments displayed in Figure 6a. Light stimulation of striatonigral axon terminals expressing eYFP (20 Hz, 20 ms, 10 mW, for 30 s) did not induce dyskinesia, neither before nor after L‐DOPA administration (Figure 6b). Importantly, these animals showed strong dyskinesia at 20‐mg·kg−1 L‐DOPA (Figure 6c).
3.5. Subthreshold striatonigral terminal optostimulation enhances LID, but suprathreshold stimulation does not
The data described so far show that optostimulation of striatonigral terminals induces more dyskinesia in primed than in naïve 6‐OHDA mice and that OID can be as diverse and severe as LID. If both treatments were recruiting the same neural substrate, additive/synergistic interactions would be expected at subthreshold doses of the treatments (as shown in previous studies [Alcacer et al., 2017; F. Hernández et al., 2017; Perez et al., 2017]). However, if one of the treatments were strong enough to recruit most of the substrate, it would mask the effect of the other treatment. Thus, we next investigated whether there were synergistic or masking interactions between LID and OID.
To test if OID and LID can interact synergistically, 6‐OHDA‐lesioned mice (n = 15) showing almost no dyskinesia with a weak light stimulation protocol (30 s, 20 Hz, 5‐ms pulses, 10 mW) were treated with a subthreshold dose of L‐DOPA (2 mg·kg−1). These mice were then tested again for the effect of the illumination protocol 15 min after the L‐DOPA challenge (Figure 7a). The combined treatment produced higher dyskinesia scores than either treatment alone (Figure 7b; a significant interaction in two‐way repeated measures ANOVA), indicating a synergistic effect.
Figure 7.
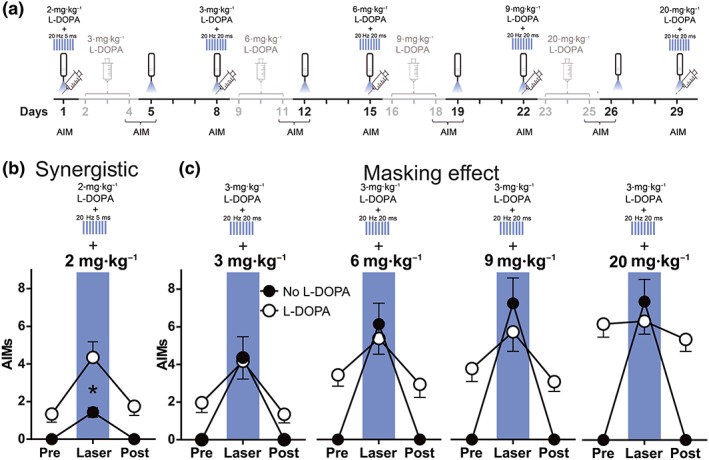
Optical stimulation potentiates the effect of L‐DOPA on dyskinesia. (a) Experimental timeline highlighting in black the behavioural manipulations rated in (b) and (c). (b) Interaction between subthreshold illumination of striatonigral terminals (20 Hz/5 ms, 10 mW) and a low dose of L‐DOPA (2 mg·kg−1; n = 14); *P < .05, significant effect of L‐DOPA; two‐way repeated measures ANOVA.with significant interaction, Bonferroni post hoc test. (c) Interaction between suprathreshold illumination of striatonigral terminals (20 Hz/20 ms, 10 mW) and increasing doses of L‐DOPA (3 to 20 mg·kg−1). Two‐way ANOVA interactions reached significance at 3 (n = 8), 6 (n = 8), 9 (n = 8), and 20 (n = 12) mg·kg−1 L‐DOPA. Light stimulation plus L‐DOPA did not differ from light stimulation alone at any of the conditions tested (Bonferroni post hoc comparisons). Data shown are means ± SEM. AIMs, axial, oral and forelimb dyskinesia score
We speculated that if illumination recruited a large part of the striatonigral terminals, and LID depended mainly on the degree of dMSN activation, additional effects of L‐DOPA on dMSN cell bodies would not further increase the dyskinesia score. Alternatively, if LID were promoted by pathways parallel to dMSNs, such as D2R‐mediated inhibition of the indirect pathway, L‐DOPA combined with striatonigral terminals illumination would yield a higher dyskinesia score than illumination alone. The same animals were therefore further tested for the interaction between our standard illumination protocol (30 s, 20 Hz, 20‐ms‐long pulses, 10 mW) and escalating doses of L‐DOPA (3, 6, 9, and 20 mg·kg−1 i.p.) as shown in Figure 7a.
Importantly, in L‐DOPA‐primed animals, striatonigral terminal illumination plus L‐DOPA did not yield a higher dyskinesia score than illumination alone in any of the conditions tested (Figure 7c). This cannot be attributed to a ceiling effect, since at 3 mg·kg−1 of L‐DOPA, the OID and LID scores were well below those at 20‐mg·kg−1 L‐DOPA. Additionally, the subtypes of dyskinesia composing OID, LID, and dyskinesia induced by combined treatment with light and L‐DOPA were similar.
3.6. Expression of dyskinesia‐related molecular markers in the striatum
Increased phosphorylation of ERK1/2 and increased expression of cFos and FosB in dMSNs in the denervated striatum have been directly associated with LIDs (Andersson, Hilbertson, & Cenci, 1999; Darmopil et al., 2009; Pavón, Martín, Mendialdua, & Moratalla, 2006) and also with dyskinesias induced by optogenetic stimulation of striatal cell bodies (F. Hernández et al., 2017; Perez et al., 2017). We investigated if optostimulation of striatonigral terminals also induces an increase in c‐Fos and FosB in the striatum. Nine days after the last L‐DOPA administration, sham‐ and 6‐OHDA‐lesioned animals were stimulated with the same protocol (20 Hz, 20 ms, 10 mW, 2 × 30 s) and perfused 50 min later. We found a slight but significant increase in the expression of c‐Fos in the ChR2 striatum of the 6‐OHDA‐lesion mice compared with the ChR2 striatum of sham mice (Figure 8a). This increase was much smaller than that expected for animals treated with dyskinetic doses L‐DOPA and perfused 50 min after the last L‐DOPA challenge. For instance, there was a high increase of FosB expression in the 6‐OHDA‐lesioned mice, which probably reflects residual expression after the last L‐DOPA challenge performed 9 days before (Figure 8b). FosB expression was observed in approximately 1,000 striatal cells·mm−2, which is about 60% to 80% the density of FosB‐positive cells observed in the striatum of 6‐OHDA‐lesioned mice shortly after a dyskinetogenic L‐DOPA challenge (Darmopil et al., 2009; Pavón et al., 2006). In contrast, c‐Fos was observed in about 50 striatal cells·mm−2 (Figure 8a). There were no significant differences in the contralateral striatum or in sham mice.
Figure 8.
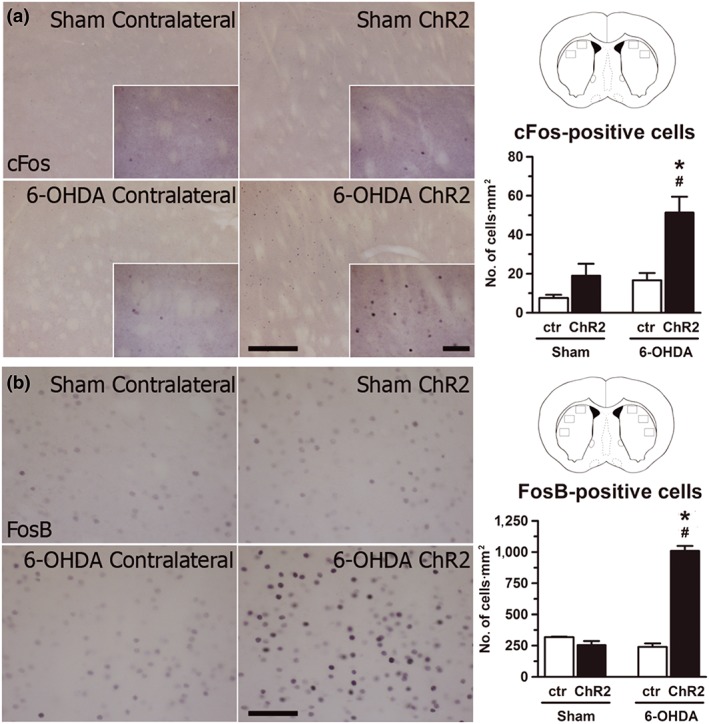
Optostimulation of striatonigral axon terminals in the SNr induced subtle cFos expression in the striatum in 6‐OHDA‐lesioned animals. (a) Left: High‐power photomicrographs of sham‐ and 6‐OHDA‐lesioned mice, representing contralateral and ipsilateral (ChR2) striata immunostaining for cFos. Right: Locations of sampled areas are indicated by black boxes in the modified section from the Paxinos and Franklin (2001) brain atlas at 0.65 mm anterior to bregma. Histograms represent the quantification of cFos‐immunoreactive nuclei. Note the higher expression in the ipsilateral striatum of 6‐OHDA‐lesioned mice. (b) Left: High‐power photomicrographs of sham‐ and 6‐OHDA‐lesioned mice showing contralateral and ChR2 striatal immunostaining for FosB. Right: Sample locations as seen in (a). Histograms represent FosB quantification. Also note the higher expression in ChR2 striatum. Data shown are means ± SEM. *P < .05 significantly different from sham ChR2; # P < .05, significantly different from 6‐OHDA contralateral; two‐way repeated measures ANOVA.with significant interaction, Bonferroni post hoc test. Scale bars: 200 and 50 μm
4. DISCUSSION
Our results showed that selective activation of the striatonigral terminals induced dyskinesia in a mouse model of PD, but not in control animals. Importantly, the repertoire of abnormal movements elicited by this optogenetic approach is similar to that induced by L‐DOPA. Our study also shows that optical stimulation induces more severe dyskinetic behaviour in L‐DOPA‐primed mice, indicating that chronic L‐DOPA administration sensitises the behavioural response to striatonigral terminals optostimulation. Nevertheless, combination of L‐DOPA and optostimulation shows synergy at subthreshold doses of both treatments, yet suprathreshold light stimulation interferes with the dyskinetic effect of higher L‐DOPA doses, suggesting that both treatments act on the same neural substrate (Figure 9). In summary, this work supports that dyskinesia can be triggered by activation of direct pathway axon terminals at the SNr.
Figure 9.
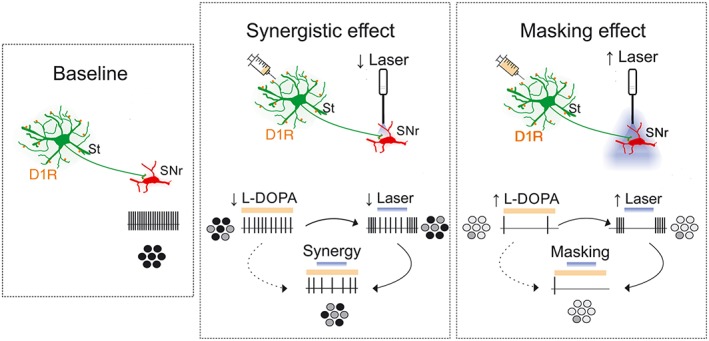
Proposed mechanisms of the interacting effects of optostimulation and L‐DOPA. Without stimulation, the SNr neurons fire action potentials tonically (“baseline”). When L‐DOPA and light are delivered at low doses, their effects add to each other (“synergistic effect”). This could be explained by actions of both treatments on the same pathway (as illustrated) or on parallel pathways converging onto SNr neurons. In contrast, suprathreshold light stimulation masked the effect of even high doses of L‐DOPA (“masking effect”), suggesting that stimulation of striatonigral axon terminals “clamps” basal ganglia output making it insensitive to actions of L‐DOPA on the same or additional pathways. D1R, dopamine D1 receptors; circles in greyscale give out a population perspective of SNr neuronal activity
Pioneering studies in mice have shown that optogenetic stimulation of dMSN cell bodies (Kravitz et al., 2010) and axon terminals (Borgkvist et al., 2015) causes an increase in normal motor activity and an improvement of Parkinson‐like motor symptoms. Improvement of symptoms is linked to inhibition of SNr neurons by GABA released from striatonigral axon terminals (Borgkvist et al., 2015; Freeze et al., 2013), as predicted by classical basal ganglia models (Albin, Young, & Penney, 1989). It is also in line with electrophysiological evidence from studies that find inhibition of GPi and SNr neurons in animal models of PD and patients treated with L‐DOPA and mixed D1/D2 receptor agonists, such as apomorphine, at doses that reduce parkinsonian symptoms (Boraud et al., 2001; Filion et al., 1991; Lozano et al., 2000; Papa et al., 1999). The latter studies also show that the transition from the “on” therapeutic state to the dyskinetic state is related to an escalation of inhibition at the level of the GPi and SNr (Boraud et al., 2001; Filion et al., 1991; Lozano et al., 2000; Papa et al., 1999). Consistent with the view that LID is mediated by dMSN inhibition of basal ganglia output, optogenetic stimulation of dMSN axon terminals at the level of the SNr induced a full repertoire of dyskinetic movements in PD mice. These movements are as severe as those induced by a high dose of L‐DOPA and even mask LID when both treatments are given simultaneously.
Studies in 6‐OHDA‐lesioned rodents not previously treated with L‐DOPA have shown that optogenetic stimulation of striatal neuron cell bodies can induce abnormal movements (F. Hernández et al., 2017; Perez et al., 2017; Ryan et al., 2018). The striatum shows a number of changes after chronic nigrostriatal lesion, including an increase in dMSN somatic excitability (Fieblinger et al., 2014; Suarez et al., 2018; Suarez et al., 2016; Suárez et al., 2014). Because MSNs fire more spikes in PD animals than controls when depolarising current is applied through a somatic recording micropipette, a comparable increase in their firing response would be expected if current were instead applied through somatic ChR2 channels. Therefore, changes in MSN somatodendritic excitability could have contributed to dyskinesia induced by optogenetic stimulation in previous studies. Because stimulation of ChR2 at striatonigral axon terminals could have produced antidromic spike invasion of dMSN cell bodies in our experiments, a contribution of changes in dMSN somatodendritic excitability to our results cannot be ruled out. On the other hand, modification of orthodromic activity by local plasticity at or downstream striatonigral axon terminals is very likely to have contributed to our findings. Borgkvist et al. (2015) have shown that an increased probability of GABA release from striatonigral terminals contributes to a sensitised motor response in 6‐OHDA mice. Additionally, a recent study performed in PD patients suggests that inhibitory transmission in the SNr can be potentiated after repeated stimulation (Milosevic et al., 2018). Further examples of plasticity mechanisms induced by 6‐OHDA lesion in rodents, that could have contributed to OID in our studies, include a reorganisation of functional movement representation maps in the motor cortex (Brown, Hu, Antle, & Teskey, 2009; Viaro, Morari, & Franchi, 2011) and accompanying structural and functional changes occurring in cortical neurons (Lindenbach & Bishop, 2013; Villalba, Mathai, & Smith, 2015).
As with dyskinesia induced by optostimulation of striatal cell bodies (F. Hernández et al., 2017; Perez et al., 2017; Ryan et al., 2018), or chemogenetic stimulation of dMSNs (Alcacer et al., 2017), dyskinesia induced by stimulation of striatonigral axon terminals is enhanced after treating 6‐OHDA animals with L‐DOPA. Interestingly, many of the cellular and circuit adaptations occurring in PD seem to be homeostatic in nature and are partly reversed by L‐DOPA therapy (Fieblinger et al., 2014; Suarez et al., 2018; Suarez et al., 2016). For instance, L‐DOPA treatment does not exacerbate but instead partly normalises the somatic excitability of MSNs in 6‐OHDA mice (Fieblinger et al., 2014; Suarez et al., 2018; Suarez et al., 2016; Suárez et al., 2014). Whether the excitability of striatonigral axon terminals is re‐established or exacerbated after L‐DOPA priming remains unknown, although L‐DOPA administration induces more GABA release in the SNr of L‐DOPA‐treated animals than in naïve 6‐OHDA animals (Yamamoto, Pierce, & Soghomonian, 2006). Further adaptations in movement representations in the motor cortex are seen in L‐DOPA‐treated 6‐OHDA animals, which could also contribute to the abnormal response to striatonigral axon terminals stimulation in our experiments (Viaro et al., 2011).
In previous studies, dyskinesia induced by optogenetic and chemogenetic stimulation of dMSNs was less severe than LID and interacted additively with LID (Alcacer et al., 2017; Perez et al., 2017). These findings were interpreted as suggesting that mechanisms beyond dMSN activation contribute to LID. For instance, Alcacer et al. (2017) found dyskinesia scores resembling those observed in LID when chemogenetic activation of dMSN was combined with a D2 receptor agonist, suggesting that inhibition of iMSN by D2 receptors is necessary for full expression of LID. Furthermore, a genetic model allowing to “trap” active neurons during LID recruited mainly dMSN but also iMSN (Girasole et al., 2018), and recent studies show modulations of activity in both dMSN and iMSN during LID (Parker et al., 2018; Ryan et al., 2018). However, optostimulation of striatonigral axon terminals can cause more powerful dyskinesia than moderate and even high doses of L‐DOPA, which cannot be further increased by combining optostimulation with L‐DOPA. Thus, based on our findings, it is not necessary to invoke mechanisms other than striatonigral inhibition of SNr neurons for full expression of LID. It seems likely that targeting striatonigral axon terminals rather than striatal cell bodies can recruit a wider population of dMSNs than in previous optogenetic studies. It is also likely that optogenetic stimulation of axon terminals might allow a more powerful effect on striatonigral outflow than chemogenetic stimulation of dMSNs. Finally, targeting striatonigral axon terminals could provide a more certain means of selectively activating dMSNs, because even when D1 receptor promoters are used to drive the expression of opto/chemogenetic transgenes in striatal neurons, striatal illumination and systemic administration of CNO could affect D1 receptor‐expressing striatal neurons other than dMSNs and targets of dMSNs other than the SNr.
Regarding the expression of dyskinesia‐related molecular markers, our results show that c‐Fos expression is slightly yet significantly higher only in the 6‐OHDA‐lesioned hemisphere after ChR2 activation in the ipsilateral striatonigral terminals. This result is likely to be linked to the dyskinetic activity displayed by the animals just 50 min before perfusion. FosB expression was also significantly higher in the 6‐OHDA‐lesioned hemisphere, but probably as a consequence of the chronic L‐DOPA treatment stopped 9 days before perfusion (Andersson et al., 1999; Pavón et al., 2006). Interestingly, this residual population of FosB‐positive cells was much higher than the population of cells expressing c‐Fos, suggesting that the full dyskinetic state induced by optostimulation of striatonigral terminals produces very limited transcriptional activity in the striatum. In contrast, dyskinesia induced by light stimulation of striatal cell bodies was accompanied by a marked increase in the expression of dyskinesia‐related molecular markers (F. Hernández et al., 2017; Perez et al., 2017). Whether the observed increase of c‐Fos relates to antidromic activity in dMSN or to the dyskinetic motor activity induced before perfusion remains to be determined. Importantly, sham animals, which showed only a slight increase in motor activity during light stimulation, did not show higher c‐Fos expression in the ChR2 hemisphere. Together with previous studies showing limited effects of backpropagating action potentials in dMSN dendrites (Day, Wokosin, Plotkin, Tian, & Surmeier, 2008), and studies showing plasticity at inhibitory synapses in the SNr in both animal PD models and patients (Borgkvist et al., 2015; Milosevic et al., 2018; Yamamoto et al., 2006), the data favour the view that local plasticity at striatonigral synapses play a role in our findings.
In conclusion, we show that optostimulation of striatonigral axons can produce a full dyskinetic state in chronically and severely dopamine‐depleted animals that cannot be further modulated by simultaneous administration of L‐DOPA. This introduces the striatonigral synapse as a particularly attractive target for future strategies aimed at improving or managing LID in patients.
AUTHOR CONTRIBUTIONS
E.K. performed all experiments, carried out data quantification and analysis, prepared figures, and contributed to the first draft. I.R.‐d.D. performed most of optogenetic behavioural experiments and immunohistochemistry studies, carried out data quantification and analysis, prepared figures and contributed to the first draft. O.S. contributed in data quantification and optogenetic behavioural testing. D.E.P. and R.M.P. performed electrophysiological experiments and reviewed the manuscript. M.G.M. and R.M. conceived the study, provided experimental design, analysed the data, procured funding, interpreted the results, and approved the final version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organisations engaged with supporting research.
Supporting information
Video S1 6‐OHDA after L‐DOPA lo‐fi.
Video S2 6‐OHDA naive to L‐DOPA lo‐fi.
Video S3 Sham after L‐DOPA lo‐fi.
Video S4 Sham naive to L‐DOPA lo‐fi.
ACKNOWLEDGEMENTS
This work was supported by grants from the Spanish Ministries of Economy and Competitiveness (SAF2016‐78207‐R and PCIN‐2015‐098) and Health, Social Services and Equality (PNSD 2016/033 and CIBERNED CB06/05/0055) and from the Fundación Ramón Areces (172275 and OTR02679). Funding was also provided by the EMHE “Enhancing Mobility between Latin American and Caribbean countries and Europe”—CSIC program, by ANPCYT (Agency for the Promotion of Science and Technology, Argentina), PICT 2013 1523 and 2015 3687, and by the University of Buenos Aires (UBACYT 2018). We thank Jesica Unger and Veronica Risso for their technical assistance.
Keifman E, Ruiz‐DeDiego I, Pafundo DE, et al. Optostimulation of striatonigral terminals in substantia nigra induces dyskinesia that increases after L‐DOPA in a mouse model of Parkinson's disease. Br J Pharmacol. 2019;176:2146–2161. 10.1111/bph.14663
Mario Gustavo Murer and Rosario Moratalla have equal contributions.
REFERENCES
- Albin, R. L. , Young, A. B. , & Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends in Neurosciences, 12(10), 366–375. 10.1016/0166-2236(89)90074-X [DOI] [PubMed] [Google Scholar]
- Alcacer, C. , Andreoli, L. , Sebastianutto, I. , Jakobsson, J. , Fieblinger, T. , & Cenci, M. A. (2017). Chemogenetic stimulation of striatal projection neurons modulates responses to Parkinson's disease therapy. Journal of Clinical Investigation, 127(2), 720–734. 10.1172/JCI90132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … Collaborators, C. (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(S1), S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … Collaborators, C. G. T. P. (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … Collaborators, C. G. T. P. (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, M. , Hilbertson, A. , & Cenci, M. A. (1999). Striatal fosB expression is causally linked with l‐DOPA‐induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiology of Disease, 6(6), 461–474. 10.1006/nbdi.1999.0259 [DOI] [PubMed] [Google Scholar]
- Aristieta, A. , Ruiz‐Ortega, J. A. , Miguelez, C. , Morera‐Herreras, T. , & Ugedo, L. (2016). Chronic L‐DOPA administration increases the firing rate but does not reverse enhanced slow frequency oscillatory activity and synchronization in substantia nigra pars reticulata neurons from 6‐hydroxydopamine‐lesioned rats. Neurobiology of Disease, 89, 88–100. 10.1016/j.nbd.2016.02.003 [DOI] [PubMed] [Google Scholar]
- Ballard, P. A. , Tetrud, J. W. , & Langston, J. W. (1985). Permanent human parkinsonism due to 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP): Seven cases. Neurology, 35(7), 949–956. 10.1212/WNL.35.7.949 [DOI] [PubMed] [Google Scholar]
- Boraud, T. , Bezard, E. , Bioulac, B. , & Gross, C. E. (2001). Dopamine agonist‐induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP‐treated monkey. Brain: A Journal of Neurology, 124(Pt 3), 546–557. 10.1093/brain/124.3.546 [DOI] [PubMed] [Google Scholar]
- Borgkvist, A. , Avegno, E. M. , Wong, M. Y. , Kheirbek, M. A. , Sonders, M. S. , Hen, R. , & Sulzer, D. (2015). Loss of striatonigral GABAergic presynaptic inhibition enables motor sensitization in parkinsonian mice. Neuron, 87(5), 976–988. 10.1016/j.neuron.2015.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, A. R. , Hu, B. , Antle, M. C. , & Teskey, G. C. (2009). Neocortical movement representations are reduced and reorganized following bilateral intrastriatal 6‐hydroxydopamine infusion and dopamine type‐2 receptor antagonism. Experimental Neurology, 220(1), 162–170. 10.1016/j.expneurol.2009.08.015 [DOI] [PubMed] [Google Scholar]
- Cenci, M. A. , & Konradi, C. (2010). Maladaptive striatal plasticity in L‐DOPA‐induced dyskinesia. Progress in Brain Research, 183, 209–233. 10.1016/S0079-6123(10)83011-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, G. , Jun, S. B. , Jin, X. , Pham, M. D. , Vogel, S. S. , Lovinger, D. M. , & Costa, R. M. (2013). Concurrent activation of striatal direct and indirect pathways during action initiation. Nature, 494(7436), 238–242. 10.1038/nature11846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmopil, S. , Martín, A. B. , De Diego, I. R. , Ares, S. , & Moratalla, R. (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L‐DOPA–induced dyskinesia and histone activation. Biological Psychiatry, 66(6), 603–613. 10.1016/j.biopsych.2009.04.025 [DOI] [PubMed] [Google Scholar]
- Day, M. , Wokosin, D. , Plotkin, J. L. , Tian, X. , & Surmeier, D. J. (2008). Differential excitability and modulation of striatal medium spiny neuron dendrites. The Journal of Neuroscience, 28(45), 11603 LP–11614. 10.1523/JNEUROSCI.1840-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino, M. A. , Stefano, A. V. , Ferrario, J. E. , Taravini, I. R. E. , Murer, M. G. , & Gershanik, O. S. (2004). Behavioral sensitization to different dopamine agonists in a parkinsonian rodent model of drug‐induced dyskinesias. Behavioural Brain Research, 152(2), 297–306. 10.1016/j.bbr.2003.10.009 [DOI] [PubMed] [Google Scholar]
- DeLong, M. R. (1990). Primate models of movement disorders of basal ganglia origin. Trends in Neurosciences, 13(7), 281–285. 10.1016/0166-2236(90)90110-V [DOI] [PubMed] [Google Scholar]
- F. Hernández, L. , Castela, I. , Ruiz‐DeDiego, I. , Obeso, J. A. , & Moratalla, R. (2017). Striatal activation by optogenetics induces dyskinesias in the 6‐hydroxydopamine rat model of Parkinson disease. Movement Disorders, 32(4), 530–537. 10.1002/mds.26947 [DOI] [PubMed] [Google Scholar]
- Fieblinger, T. , Graves, S. M. , Sebel, L. E. , Alcacer, C. , Plotkin, J. L. , Gertler, T. S. , … Surmeier, D. J. (2014). Cell type‐specific plasticity of striatal projection neurons in parkinsonism and L‐DOPA‐induced dyskinesia. Nature Communications, 5, 5316 10.1038/ncomms6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion, M. , Tremblay, L. , & Bédard, P. J. (1991). Effects of dopamine agonists on the spontaneous activity of globus pallidus neurons in monkeys with MPTP‐induced parkinsonism. Brain Research, 547(1), 152–161. [PubMed] [Google Scholar]
- Freeze, B. S. , Kravitz, A. V. , Hammack, N. , Berke, J. D. , & Kreitzer, A. C. (2013). Control of basal ganglia output by direct and indirect pathway projection neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33(47), 18531–18539. 10.1523/JNEUROSCI.1278-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Montes, J.‐R. , Solís, O. , Enríquez‐Traba, J. , Ruiz‐DeDiego, I. , Drucker‐Colín, R. , & Moratalla, R. (2018). Genetic knockdown of mGluR5 in striatal D1R‐containing neurons attenuates l‐DOPA‐induced dyskinesia in aphakia mice. Molecular Neurobiology. 10.1007/s12035-018-1356-6 [DOI] [PubMed] [Google Scholar]
- Girasole, A. E. , Lum, M. Y. , Nathaniel, D. , Bair‐Marshall, C. J. , Guenthner, C. J. , Luo, L. , … Nelson, A. B. (2018). A subpopulation of striatal neurons mediates levodopa‐induced dyskinesia. Neuron, 97, 787–795.e6. 10.1016/j.neuron.2018.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granado, N. , Escobedo, I. , O'Shea, E. , Colado, M. I. , & Moratalla, R. (2007). Early loss of dopaminergic terminals in striosomes after MDMA administration to mice. Synapse, 62(1), 80–84. 10.1002/syn.20466 [DOI] [PubMed] [Google Scholar]
- Halje, P. , Tamtè, M. , Richter, U. , Mohammed, M. , Cenci, M. A. , & Petersson, P. (2012). Levodopa‐induced dyskinesia is strongly associated with resonant cortical oscillations. The Journal of Neuroscience, 32(47), 16541 LP–16551. Retrieved from http://www.jneurosci.org/content/32/47/16541.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner, P. (2008). Molecular mechanisms of L‐DOPA‐induced dyskinesia. Nature Reviews. Neuroscience, 9(9), 665–677. 10.1038/nrn2471 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz, A. V. , Freeze, B. S. , Parker, P. R. L. , Kay, K. , Thwin, M. T. , Deisseroth, K. , & Kreitzer, A. C. (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature, 466(7306), 622–626. 10.1038/nature09159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach, D. , & Bishop, C. (2013). Critical involvement of the motor cortex in the pathophysiology and treatment of Parkinson's disease. Neuroscience & Biobehavioral Reviews, 37(10, Part 2), 2737–2750. 10.1016/j.neubiorev.2013.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano, A. M. , Lang, A. E. , Levy, R. , Hutchison, W. , & Dostrovsky, J. (2000). Neuronal recordings in Parkinson's disease patients with dyskinesias induced by apomorphine. Annals of Neurology, 47(4 Suppl 1), S141–S146. [PubMed] [Google Scholar]
- Luquin, M. R. , Laguna, J. , & Obeso, J. A. (1992). Selective D2 receptor stimulation induces dyskinesia in parkinsonian monkeys. Annals of Neurology, 31(5), 551–554. 10.1002/ana.410310514 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner, W. , Ravenscroft, P. , Reese, R. , Harnack, D. , Morgenstern, R. , Kupsch, A. , … Boraud, T. (2006). Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6‐OHDA‐lesioned rat in the presence of excessive extracelullar striatal dopamine. Neurobiology of Disease, 22(3), 586–598. 10.1016/j.nbd.2006.01.009 [DOI] [PubMed] [Google Scholar]
- Milosevic, L. , Kalia, S. K. , Hodaie, M. , Lozano, A. M. , Fasano, A. , Popovic, M. R. , & Hutchison, W. D. (2018). Neuronal inhibition and synaptic plasticity of basal ganglia neurons in Parkinson's disease. Brain, 141(1), 177–190. Retrieved from. 10.1093/brain/awx296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murer, M. G. , & Moratalla, R. (2011). Striatal signaling in L‐DOPA‐induced dyskinesia: Common mechanisms with drug abuse and long term memory involving D1 dopamine receptor stimulation. Frontiers in Neuroanatomy, 5, 51 10.3389/fnana.2011.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa, S. M. , Desimone, R. , Fiorani, M. , & Oldfield, E. H. (1999). Internal globus pallidus discharge is nearly suppressed during levodopa‐induced dyskinesias. Annals of Neurology, 46(5), 732–738. [DOI] [PubMed] [Google Scholar]
- Parker, J. G. , Marshall, J. D. , Ahanonu, B. , Wu, Y. W. , Kim, T. H. , Grewe, B. F. , … Schnitzer, M. J. (2018). Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature, 557, 177–182. 10.1038/s41586-018-0090-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavón, N. , Martín, A. B. , Mendialdua, A. , & Moratalla, R. (2006). ERK phosphorylation and FosB expression are associated with L‐DOPA‐induced dyskinesia in hemiparkinsonian mice. Biological Psychiatry, 59(1), 64–74. 10.1016/j.biopsych.2005.05.044 [DOI] [PubMed] [Google Scholar]
- Paxinos, G. , & Franklin, K. B. J. (2001). The Mouse Brain in Stereotaxic Coordinates (2nd Ed.), San Diego: Academic Press. [Google Scholar]
- Perez, X. A. , Zhang, D. , Bordia, T. , & Quik, M. (2017). Striatal D1 medium spiny neuron activation induces dyskinesias in parkinsonian mice. Movement Disorders, 32(4), 538–548. 10.1002/mds.26955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi, B. , & Calabresi, P. (2017). Switching on the lights of dyskinesia: Perspectives and limits of the optogenetic approaches. Movement Disorders, 32(4), 485–486. 10.1002/mds.26999 [DOI] [PubMed] [Google Scholar]
- Rascol, O. , Brooks, D. J. , Korczyn, A. D. , De Deyn, P. P. , Clarke, C. E. , Lang, A. E. , & Abdalla, M. (2006). Development of dyskinesias in a 5‐year trial of ropinirole and L‐dopa. Movement Disorders: Official Journal of the Movement Disorder Society, 21(11), 1844–1850. 10.1002/mds.20988 [DOI] [PubMed] [Google Scholar]
- Ruiz‐DeDiego, I. , Fasano, S. , Solís, O. , Garcia‐Montes, J.‐R. , Brea, J. , Loza, M. I. , … Moratalla, R. (2018). Genetic enhancement of Ras‐ERK pathway does not aggravate L‐DOPA‐induced dyskinesia in mice but prevents the decrease induced by lovastatin. Scientific Reports, 8(1), 15381 10.1038/s41598-018-33713-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐DeDiego, I. , Mellstrom, B. , Vallejo, M. , Naranjo, J. R. , & Moratalla, R. (2015). Activation of DREAM (downstream regulatory element antagonistic modulator), a calcium‐binding protein, reduces L‐DOPA‐induced dyskinesias in mice. Biological Psychiatry, 77(2), 95–105. 10.1016/j.biopsych.2014.03.023 [DOI] [PubMed] [Google Scholar]
- Ryan, M. B. , Bair‐Marshall, C. , & Nelson, A. B. (2018). Aberrant striatal activity in parkinsonism and levodopa‐induced dyskinesia. Cell Reports, 23(12), 3438–3446.e5. 10.1016/j.celrep.2018.05.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez, L. M. , Alberquilla, S. , García‐Montes, J. R. , & Moratalla, R. (2018). Differential synaptic remodeling by dopamine in direct and indirect striatal projection neurons in Pitx3−/− mice, a genetic model of Parkinson's disease. The Journal of Neuroscience, 38(15), 3619 LP–3630. Retrieved from http://www.jneurosci.org/content/38/15/3619.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez, L. M. , Solis, O. , Aguado, C. , Lujan, R. , & Moratalla, R. (2016). L‐DOPA oppositely regulates synaptic strength and spine morphology in D1 and D2 striatal projection neurons in dyskinesia. Cerebral Cortex, 26(11), 4253–4264. Retrieved from. 10.1093/cercor/bhw263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez, L. M. , Solís, O. , Caramés, J. M. , Taravini, I. R. , Solís, J. M. , Murer, M. G. , & Moratalla, R. (2014). L‐DOPA treatment selectively restores spine density in dopamine receptor D2‐expressing projection neurons in dyskinetic mice. Biological Psychiatry, 75(9), 711–722. 10.1016/j.biopsych.2013.05.006 [DOI] [PubMed] [Google Scholar]
- Viaro, R. , Morari, M. , & Franchi, G. (2011). Progressive motor cortex functional reorganization following 6‐hydroxydopamine lesioning in rats. Journal of Neuroscience, 31(12), 4544–4554. 10.1523/JNEUROSCI.5394-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba, R. M. , Mathai, A. , & Smith, Y. (2015). Morphological changes of glutamatergic synapses in animal models of Parkinson's disease. Frontiers in Neuroanatomy, 9, 117 10.3389/fnana.2015.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, N. , Pierce, R. C. , & Soghomonian, J.‐J. (2006). Subchronic administration of l‐DOPA to adult rats with a unilateral 6‐hydroxydopamine lesion of dopamine neurons results in a sensitization of enhanced GABA release in the substantia nigra, pars reticulata. Brain Research, 1123(1), 196–200. 10.1016/j.brainres.2006.09.027 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1 6‐OHDA after L‐DOPA lo‐fi.
Video S2 6‐OHDA naive to L‐DOPA lo‐fi.
Video S3 Sham after L‐DOPA lo‐fi.
Video S4 Sham naive to L‐DOPA lo‐fi.