

Abstract
Background
Chronic obstructive pulmonary disease (COPD) frequent exacerbators (FE) suffer increased morbidity and mortality compared to infrequent exacerbators (IE). The association between the oral and sputum microbiota and exacerbation phenotype is not well defined. The objective of this study was to determine key features that differentiate the oral and sputum microbiota of FEs from the microbiota of IEs during periods of clinical stability.
Methods
We recruited 11 FE and 11 IE who had not used antibiotics or systemic corticosteroids in the last 1 month. Subjects provided oral wash and sputum samples, which underwent 16S V4 MiSeq sequencing and qPCR of 16S rRNA. Data were analyzed using Dada2 and R.
Results
FE and IE were similar in terms of age, FEV1 percent predicted (FEV1pp), pack-years of tobacco exposure, and St. George’s Respiratory Questionnaire score. 16S copy numbers were significantly greater in sputum vs. oral wash (p = 0.01), but phenotype was not associated with copy number. Shannon diversity was significantly greater in oral samples compared to sputum (p = 0.001), and IE samples were more diverse than FE samples (p < 0.001). Sputum samples from FE had more Haemophilus and Moraxella compared to IE sputum samples, due to dominance of these COPD-associated taxa in three FE sputum samples. Amplicon sequencing variant (ASV)-level analysis of sputum samples revealed one ASV (Actinomyces) was significantly more abundant in IE vs. FE sputum (padj = 0.048, Wilcoxon rank-sum test), and this persisted after controlling for FEV1pp. Principal coordinate analysis using Bray-Curtis distance with PERMANOVA analyses demonstrated clustering by anatomic site, phenotype, inhaled corticosteroid use, current tobacco use, COPD severity, and last professional dental cleaning.
Conclusions
FE have less diverse oral and sputum microbiota than IE. Actinomyces was significantly more abundant in IE sputum than FE sputum. The oral and sputum microbiota of COPD subjects cluster based on multiple clinical factors, including exacerbation phenotype. Even during periods of clinical stability, the frequent exacerbator phenotype is associated with decreased alpha diversity, beta-diversity clustering, and changes in taxonomic abundance.
Electronic supplementary material
The online version of this article (10.1186/s12931-019-1080-4) contains supplementary material, which is available to authorized users.
Keywords: Pulmonary disease, chronic obstructive; RNA, ribosomal, 16S; Sputum; Microbiota; Streptococcus; Haemophilus; Moraxella; Phenotype
Background
Chronic obstructive pulmonary disease (COPD) is an inflammatory lung disorder with diverse clinical presentations. Some COPD patients experience severe or frequent exacerbations (episodic worsening of COPD symptoms), while others have a more indolent course. The reasons for this clinical variability are poorly understood. COPD phenotypes have been described in an effort to understand better the clinical heterogeneity of COPD. [1, 2] The frequent exacerbator phenotype [3, 4] (defined as the occurrence of ≥2 COPD exacerbations in a 12-month period) is clinically important because exacerbations accelerate loss of lung function, [5] decrease quality of life, [6] increase health care utilization, [7] and increase morbidity and mortality. [8, 9]
The ECLIPSE study showed that the best predictor of the frequent exacerbator phenotype over the course of a year was a history of exacerbation in the previous year (odds ratio 5.72 for subjects with ≥2 vs. 0 exacerbations). [4] Although the frequent exacerbator phenotype was more common in very severe COPD (by GOLD criteria [10]) than in less severe COPD, 22% of moderate COPD subjects and 47% of very severe COPD subjects were frequent exacerbators. [4, 11] Therefore, the frequent exacerbator phenotype is clinically relevant, easily identified, stable over time, and found at all stages of COPD severity.
Approximately 50% of COPD exacerbations are due to bacterial lung infection. [12] Acquisition of a new strain of Haemophilus influenzae, Moraxella catarrhalis, or Streptococcus pneumoniae in the lung microbiota is associated with an increased risk of exacerbation. [13] These pathogenic bacteria often colonize the lungs of COPD patients between exacerbations and become a component of the lung microbiota. [12] Furthermore, airway colonization with pathogenic bacteria – even in the absence of exacerbation – is associated with increased pulmonary symptoms and systemic inflammation. [14] Therefore, heterogeneity in the COPD lung microbiota and lung inflammation is a potential explanation for the frequent exacerbator vs. infrequent exacerbator phenotype.
Over the last several years, the lung microbiota of individuals with stable, exacerbation-free COPD have been described using 16S rRNA gene sequencing. [15–25] Several authors have also used prospective, observational cohorts to compare the lung microbiota of subjects with COPD both during clinical stability and later during exacerbation, [26–31] the largest studies of which are AERIS [32] and COPDMAP. [33] Several studies note an increase in Proteobacteria during exacerbations compared to periods of clinical stability, [29, 31–33] with some studies specifically citing Moraxella [31–33] and Haemophilus [32] as the genera responsible for this increase. While alpha diversity appears to decrease with a decline in FEV1, there is no consensus on whether or not alpha diversity changes at the time of exacerbation. [31–33] In particular, the COPDMAP study [33] noted differences in alpha diversity patterns across their three study centers, raising the possibility that local practice patterns in the management of COPD exacerbations may influence alpha diversity. The AERIS study [32] also described how alpha diversity and changes in taxonomic abundance are associated with severity of lung obstruction, but did not detect a statistically significant association between alpha diversity and exacerbations status. There were also limited associations between exacerbation status and taxonomic abundance.
Despite these contributions to our understanding of the COPD exacerbation microbiota, several gaps remain in our understanding of how exacerbation phenotype may correlate with the lung microbiota. Firstly, no studies have prospectively identified frequent vs. infrequent exacerbators for further examination. Secondly, we lack a complete understanding of the “stable” lung microbiota (the microbiota present during periods of clinical stability, without recent exposure to microbiota-altering drugs such as systemic corticosteroids or antibiotics) in frequent vs. infrequent exacerbators. Lastly, it is not clear to what extent sputum, which is contaminated by the oral microbiota during expectoration, can adequately reflect the lung microbiota or differentiate between disease phenotypes.
Therefore, we have undertaken the present study to address these three knowledge gaps. This case-control observational study describes the oral and sputum microbiota in relationship to exacerbation phenotype and other relevant clinical characteristics. Our central hypothesis is that the oral and sputum microbiota are important correlates or causes of exacerbations. Any potentially-modifiable clinical factors that correlate with the frequent exacerbator microbiota may suggest future targets for therapeutic intervention.
Methods
Subjects
Twenty-two subjects with COPD (11 frequent and 11 infrequent COPD exacerbators, referred to as FE and IE, respectively) who were over the age of 40, with a minimum of 10 pack-year history of smoking were recruited at the Minneapolis VA Medical Center. FEs were identified based on a history of ≥1 exacerbation in the last 12 months, whereas IEs were free from exacerbations for the last 24 months. All FE study participants met the GOLD criteria for FEs (at least 2 exacerbations in the prior year). [10] All participants had not used antibiotics or oral corticosteroids within the previous month. For full inclusion and exclusion criteria see Additional file 1: Table S1.
Sample acquisition
Oral wash and sputum samples were obtained and processed as described in the supplementary information available online. Three subjects (2 IE and 1 FE) were unable to provide a sputum sample.
DNA extraction and 16S rRNA gene sequencing
Samples and controls were extracted using the MO BIO PowerSoil DNA Isolation Kit as described in the Additional file 1: supplemental information.
Quantitative PCR (qPCR)
To determine 16S rRNA gene copy numbers, qPCR was performed in triplicate for all samples and controls as previously described [20] and detailed in the Additional file 1: Supplemental information. Copy numbers were normalized to sample mass, which accounts for both sample volume and density.
Data processing
Dada2 was used to filter, trim, dereplicate, merge paired reads, and remove primers, phix, and bimeras. [34] Bowtie2 [35] was used to remove human sequences prior to aligning sequences using the Ribosomal Database Project (RDP) Classifier [36] with the SILVA database. [37] The full data set underwent β-diversity analysis with Bray-Curtis dissimilarity to visualize control and sample similarities and determine subsampling depth. Subsampling to 25,955 sequences eliminated all negative control samples and one FE sputum sample. This subsampled data set, which consisted of 398 amplicon sequencing variants (ASVs), was used in subsequent analyses. Alpha diversity indices (Shannon and Simpson indices) were calculated using vegan while β-diversity analyses utilizing the Bray-Curtis dissimilarity were performed using phyloseq. Prior to hierarchical clustering and taxa distribution analyses, additional filtering was performed to remove ASVs that did not have at least 3 reads in 10% of all samples. 169 ASVs remained in this data set. Methods of accounting for and correcting for oral contamination of sputum samples during expectoration were considered but not undertaken to avoid introducing bias (see Additional file 1: Supplemental information for a discussion of this issue). A data processing flow chart is provided in Fig. 1.
Fig. 1.
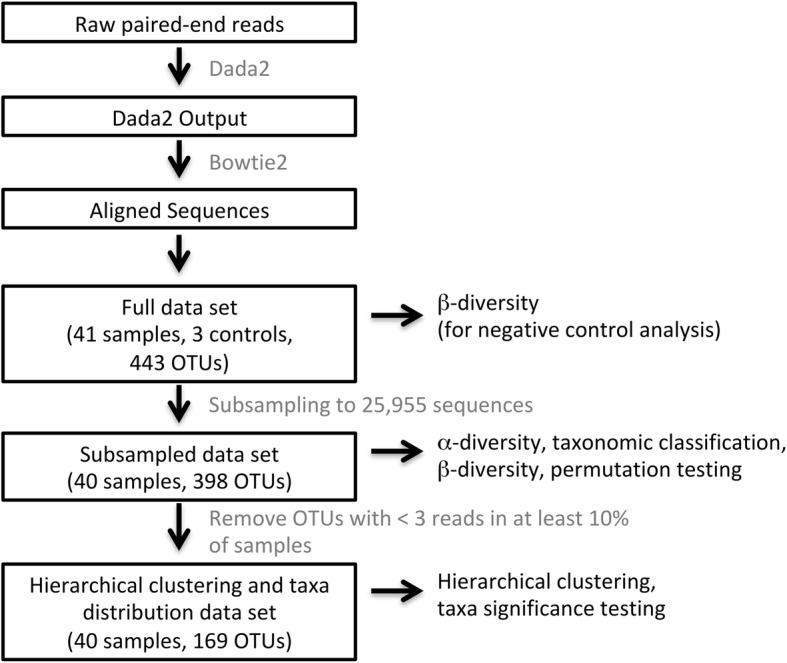
Data processing flow chart. DNA sequences were processed into data sets using the software tools and procedures as described in the text and illustrated here as a pipeline. Boxed black text indicates significant steps in the pipeline or data sets produced for specific analyses. Gray text specifies the software tools or procedures used in the pipeline. Unboxed black text specifies the analyses performed on the indicated data sets
Statistical analysis
Subject characteristics were analyzed with a Fisher-Pitman permutation test for continuous variables and Fisher’s exact test for categorical variables. ANOVA analyses with post-hoc pairwise permutation testing were performed to compare 16S copy numbers across sites. A linear mixed model was used to evaluate for associations between 16S copy numbers, phenotype, and sampling site; p-values were obtained by permutation testing. α-diversity associations were tested using linear mixed models with α-diversity score as a response, patient as a random effect, and site, phenotype and their interaction as fixed effects. To determine clinical factors associated with α-diversity, PERMANOVA testing with default parameters was performed on the Bray-Curtis dissimilarity matrix. Wilcoxon rank-sum permutation tests were used to determine differences in ASV counts between phenotypes. Results were also assessed after controlling for FEV1 percent predicted (FEV1pp), a potential co-linear clinical variable with phenotype. ASV count data was used to cluster samples with complete linkage hierarchical clustering using hclust function in base R. All analyses were performed in R version 3.4.2. Additional statistical analysis methods are found in the Additional file 1: Supplemental information.
Results
Characteristics of the study participants
Subjects were all male and no differences were observed between FEs and IEs based on age, significant medical comorbidities, COPD severity, tobacco exposure, or SGRQ score. FEs, all of whom met the GOLD criteria for frequent exacerbators, [10] experienced a mean of 2.91 exacerbations in the prior year (range: 2–5 exacerbations, median: 3 exacerbations, Table 1). Three subjects were unable to provide sputum samples and one sputum sample was eliminated due to low sequencing yield. Eighteen sputum samples (from 9 FEs and 9 IEs) were available for analysis. Subjects with evaluable sputum samples did not differ significantly from the study population as a whole (Additional file 1: Table S2).
Table 1.
Subject Characteristics
| Infrequent Exacerbator | Frequent Exacerbator | p value* | |
|---|---|---|---|
| N | 11 | 11 | |
| Sex, Male (%) | 11 (100) | 11 (100) | |
| Age, mean (sd) | 68.36 (5.45) | 70.82 (7.12) | 0.39 |
| Race, white (%) | 11 (100) | 10 (90.9) | 1.00 |
| BMI, mean (sd) | 29.25 (6.01) | 26.09 (6.85) | 0.26 |
| Hypertension, Yes (%) | 6 (54.5) | 6 (54.5) | 1.00 |
| Diabetes, Yes (%) | 2 (18.2) | 3 (27.3) | 1.00 |
| COPD Severity (%) | |||
| Moderate | 5 (45.5) | 3 (27.3) | |
| Severe | 4 (36.4) | 3 (27.3) | |
| Very Severe | 2 (18.2) | 5 (45.5) | |
| FEV1% predicted | 46.82 (13.93) | 35.5 (15.18) | 0.088 |
| COPD exacerbations in the last 12 months, mean (sd) | 0 (0) | 2.91 (1.04) | < 0.001 |
| COPD hospitalizations in the last 12 months, mean (sd) | 0 (0) | 0.55 (0.69) | 0.035 |
| Inhaled corticosteroid use, Yes (%) | 6 (54.5) | 7 (63.6) | 1.00 |
| Pack-years of smoking, mean (sd) | 61.18 (31.94) | 69.55 (48.57) | 0.66 |
| Current tobacco use, Yes (%) | 5 (45.5) | 3 (27.3) | 0.66 |
| Current alcohol use, Yes (%) | 7 (63.6) | 9 (81.8) | 0.64 |
| Brush teeth ≥ once daily (%) | 9 (81.8) | 10 (90.9) | 1.00 |
| History of periodontal disease, Yes (%) | 3 (27.3) | 2 (18.2) | 1.00 |
| SGRQ Score, mean (sd) | 49.72 (7.92) | 48.27 (16.20) | 0.792 |
BMI, body mass index; COPD, chronic obstructive pulmonary disease; FEV1% predicted, forced expiratory volume in 1 s, percent of predicted value; sd, standard deviation; SGRQ, St. George’s Respiratory Questionnaire
*A Fisher-Pitman permutation test was conducted for all continuous variables and a Fishers exact test for all categorical variables
16S rRNA copy number is associated with site, but not exacerbation phenotype
16S rRNA copy numbers were determined for each subject and control sample and normalized to sample mass. Sputum samples contained a greater number of 16S rRNA copies (mean 3.04 × 1010) than both oral wash samples (mean 7.17 × 108) and negative control samples (mean 4.21 × 103; ANOVA p < 0.001). Post-hoc pairwise testing for all sample pairs indicated significant differences across all pairs (all p-values < 0.01). Although anatomic site was a significant predictor of 16S copy numbers (p = 0.004), exacerbation phenotype and the interaction of anatomic site and exacerbation phenotype were not significant (Fig. 2).
Fig. 2.
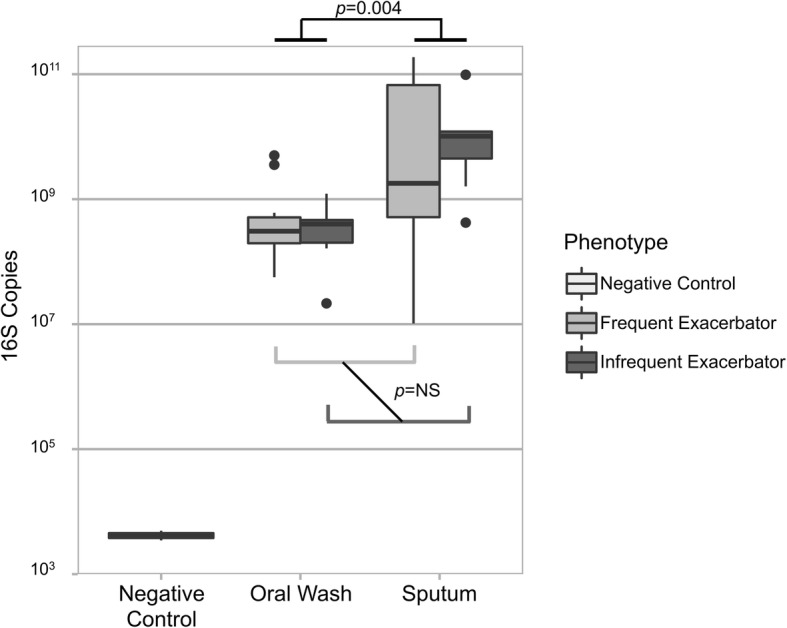
Exacerbation phenotype is not associated with bacterial biomass. 16S rRNA gene copies were determined using quantitative PCR. Negative control samples contained significantly fewer 16S copies than subjects’ oral wash and sputum samples. Sputum contained a greater number of 16S rRNA gene copies than did oral wash samples (linear mixed model with Bonferroni-Holm correction, p = 0.004). Exacerbation phenotype was not associated with 16S rRNA gene copy number changes, although frequent exacerbator sputum samples exhibited greater variability than infrequent exacerbator sputum samples
Samples are distinct from negative controls
Samples contained 17,567–136,275 reads (mean 89,961.7, median 90,420); negative controls contained 222–732 sequences (mean 417.7, median 229). The full data set underwent α-diversity analysis with Bray-Curtis dissimilarity to visualize control and sample similarities and determine subsampling depth (Additional file 1: Figure S1). Negative control samples as well as sequencing center controls clustered together, while the majority of subject samples were distributed elsewhere. Subsampling to 25,955 sequences eliminated all negative control samples and one FE sputum sample.
Alpha diversity is associated with site and exacerbation phenotype
Shannon and Simpson diversity indices, which incorporate both microbial richness and evenness, were determined for each subject sample. Regardless of exacerbation phenotype, alpha diversity indices were lower for sputum samples than for oral wash samples (mean oral wash Shannon diversity 3.05 vs. mean sputum diversity 2.45; mean oral wash Simpson diversity 0.90 vs. mean sputum diversity 0.76). We used a linear mixed model with anatomic site, phenotype, and their interaction as fixed effects (subject as random effect) to determine relationships between alpha diversity, anatomic site, and exacerbation phenotype. Utilizing Shannon diversity, we found that both anatomic site (p = 0.001) and exacerbation phenotype (p < 0.001) were significant predictors of Shannon diversity, with FE exhibiting significantly lower Shannon diversity than IE at both sites. There was no evidence for an interaction between site and phenotype (p = 0.22, Fig. 3a).
Fig. 3.

Alpha diversity is associated with exacerbation phenotype. Linear mixed models were constructed with diversity score as the response variable; site, phenotype, and their interactions as fixed effects; and subject as a random effect. a. Sputum samples had lower Shannon diversity scores than oral wash samples (p = 0.001). Frequent exacerbators had lower Shannon diversity scores than infrequent exacerbators (p < 0.001). The interaction of site and phenotype was 0.22. b. Sputum samples had lower Simpson diversity scores than oral was samples (p = 0.009). Frequent exacerbators had lower Simpson diversity scores than infrequent exacerbators (p = 0.006). The interaction of site and phenotype (p = 0.15) was not statistically significant, although there was some evidence for a difference. These potential associations warrant further study
The linear mixed effects model using Simpson diversity demonstrated that anatomic site was a significant predictor of Simpson diversity (p = 0.009), as was exacerbation phenotype (p = 0.006). The interaction of site and phenotype (p = 0.15) was not statistically significant, although there was some evidence for differences (Fig. 3b). In both Shannon and Simpson linear models of alpha diversity, anatomic site was a more influential predictor of alpha diversity than was exacerbation phenotype or their interactions (Table 2).
Table 2.
Alpha Diversity Effect Estimates Using a Linear Model
| Factor | Shannon Diversity Model | Simpson Diversity Model | ||
|---|---|---|---|---|
| p-value | Effect Estimate | p-value | Effect Estimate | |
| Anatomic Site | 0.001 | −0.86 | 0.009 | −0.20 |
| Exacerbation Phenotype | < 0.001 | 0.31 | 0.006 | 0.038 |
| Interaction of Site and Phenotype | 0.22 | 0.76 | 0.15 | 0.19 |
Our previous work on the COPD lung microbiota identified age as a significant predictor of α-diversity. [17, 38] We therefore included age as a predictor in our alpha diversity linear mixed models. With site, phenotype, and their interactions included in the model, age was not a significant predictor of either Shannon or Simpson diversity (data not shown).
Sputum samples are subject to oral admixture during expectoration, and this may be a particular concern in subjects (such as IE) who may produce less sputum. We sought to understand whether there is a correlation between oral wash and sputum α-diversity in the same subject. In linear models with only oral wash α-diversity as a predictor of sputum α-diversity, both Shannon and Simpson oral wash scores were significant predictors of Shannon (p = 0.0002) and Simpson (p = 0.0003) sputum diversity, respectively (Fig. 4). We expanded these linear models to include exacerbation phenotype and the interaction of oral wash α-diversity and phenotype as predictors of sputum α-diversity. Inclusion of these additional factors in the Shannon diversity model showed that oral wash Shannon diversity alone narrowly missed significance (p = 0.07), while phenotype alone and the interaction of oral wash diversity and phenotype were non-significant. In the Simpson diversity model, oral wash diversity alone remained significant (p = 0.008), while phenotype alone and their interactions were non-significant. Close inspection of the figure, particularly the Simpson diversity model, reveals the presence of a low diversity x-outlier (boxed). Removal of this x-outlier did not significantly change the findings of the Shannon diversity model (oral wash alone remained significant [p = 0.039], while all factors were non-significant in the full model). However, removal of this low diversity x-outlier from the Simpson model did result in all factors becoming non-significant (oral wash alone and in combination with phenotype and their interactions; Additional file 1: Figure S2). This suggests that sputum α-diversity likely is associated with oral wash alpha diversity, but this potential relationship does not appear to be robust.
Fig. 4.

Oral Wash Alpha Diversity Is Associated with Sputum Alpha Diversity. Circles represent FE samples while triangles represent IE samples. Linear models were used to predict sputum alpha diversity based on oral wash alpha diversity. The solid lines represent the aggregated model incorporating both phenotypes while the dotted lines represent the models of frequent or infrequent exacerbator samples alone. a) Oral wash Shannon diversity alone was a significant predictor of sputum Shannon diversity (p = 0.0002), but addition of phenotype and the interaction of oral wash Shannon diversity and phenotype to the model resulted in a near-significant oral wash result (p = 0.07) and non-significant results for phenotype and their interactions. b) Oral wash Simpson diversity alone was also a significant predictor of sputum Simpson diversity (p = 0.0003), and incorporation of phenotype and its interaction resulted in significant results for oral wash diversity (p = 0.008), but non-significant results for phenotype and its interaction with oral wash diversity. The removal of the low diversity x-outlier (boxed in both panels, see text and Additional file 1: Figure S2) resulted in non-significant results for all but oral wash Shannon diversity alone as a predictor of sputum Shannon diversity (p = 0.039)
Upper airway microbiota sites exhibit greater within-subject than between-subject similarity
The subsampled dataset underwent PCoA analysis using Bray-Curtis dissimilarity (Fig. 5). Univariate PERMANOVA analyses were used to identify significant clustering based on anatomic site sampled, exacerbation phenotype, COPD severity (by GOLD category), time since last professional cleaning (in the last 6 months, 6–12 months ago, or more than 12 months ago), current tobacco use, inhaled corticosteroid use, and current alcohol use. All PERMANOVA analyses except current alcohol use were statistically significant, with each variable associated with 4.3–10.4% of the variance on univariate analysis (Table 3). Time since last professional dental cleaning was associated with a larger variance than both anatomic site and exacerbation phenotype. While there was homogeneity of dispersion in beta diversity with respect to anatomic site and COPD severity, exacerbation phenotype (as well as ICS use and current tobacco use) exhibited heterogeneity of dispersion on beta diversity analysis. This suggests that FE and IE microbiotas are not homogeneous with respect to beta diversity variation.
Fig. 5.
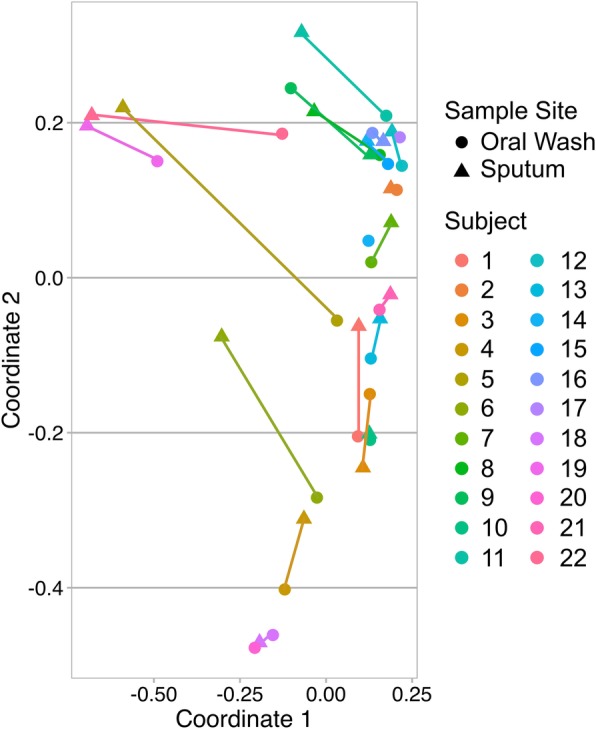
Within-subject similarity is greater than between-subject similarity. Principal coordinate analysis using Bray-Curtis dissimilarity was plotted with color used to define each subject. Each subject’s oral wash (circle) and sputum (triangle) samples are connected by a colored, subject-specific line, which defines the within-subject oral wash-sputum similarity. Four subjects did not have sputum samples available or evaluable. Permutation testing demonstrated greater within-subject than between-subject similarity at these two sites (p < 0.001). Exacerbation phenotype was not significantly associated with changes in within-subject similarity (p = 0.95)
Table 3.
Beta Diversity is Associated with Multiple Clinical Factors on PERMANOVA Analysis
| Factor | Univariate PERMANOVA | ||
|---|---|---|---|
| p-value | r2 | Homogeneity of multivariate dispersions | |
| Anatomic Site | 0.016 | 0.091 | Homogeneous (p = 0.225) |
| Exacerbation Phenotype | 0.025 | 0.050 | Heterogeneous (p = 0.037) |
| Inhaled Corticosteroid Use | 0.045 | 0.043 | Heterogeneous (p = 0.005) |
| Current Tobacco Use | 0.024 | 0.051 | Heterogeneous (p = 0.005) |
| COPD Severity | 0.003 | 0.096 | Homogeneous (p = 0.89) |
| Last Professional Dental Cleaning | 0.006 | 0.104 | Heterogeneous (p = 0.003) |
To understand the degree of similarity between subjects’ paired oral washes and sputum samples, we compared within-subject and between-subject sample similarities. A permutation test comparing the within-subject and between-subject similarities resulted in a p-value of < 0.001, indicating greater within-subject than between-subject similarities at these sites (Fig. 5). We also compared the within-subject similarities for FE to the within-subject similarities of IE using a permutation test. This p-value was 0.95, indicating that within-subject oral and sputum similarities are not significantly modified by exacerbation phenotype.
Hierarchical clustering was used to illustrate similarities between samples. Prior to clustering, taxa with fewer than 3 reads in 10% of samples were removed from the dataset, which left 169 ASVs for analysis. The individual subject, rather than sample type or exacerbation phenotype, was the primary driver of clustering in this analysis as well (Fig. 6). The exceptions to this were FE subjects 5, 19, and 22, whose sputa were dominated by Haemophilus or Moraxella while their oral washes were not (see next section and Fig. 7). We reviewed these subjects’ clinical characteristics to determine if they otherwise differed from the FE group as a whole. All three subjects were using ICSs, which have been associated with microbiota changes, [39] while 7 of the 11 FEs (64%) were using ICSs. These three subjects’ mean FEV1pp was 34%, mean SGRQ score was 47.45, and mean number of exacerbations per year was 3. These values were similar to the group of FEs as a whole, indicating that these subjects with low diversity did not appear to differ from the other FEs with respect to disease severity.
Fig. 6.

Hierarchical clustering illustrates within-subject similarity. Oral samples (circles) and sputum samples (triangles) are represented on this clustering diagram, with subject ID provided at bottom. With the exception of samples from frequent exacerbators 5, 19, and 22, subject ID (rather than sample type or exacerbation phenotype) was the primary driver of clustering. The exceptions to this were FE subjects 5, 19, and 22, whose sputa were dominated by Haemophilus or Moraxella while their oral wash samples were not dominated by these taxa. No sample pairs from IE subjects exhibited such large separations between samples
Fig. 7.

Select frequent exacerbator sputum samples differ markedly from their paired oral wash samples. Oral wash samples are presented in the top row, with sputum samples in the bottom row. Frequent exacerbator samples are present in the left column with infrequent exacerbator samples at right. Samples are labeled by subject ID to facilitate comparisons between sites for the same subject. Class-level taxonomy is provided by color coding (see legend). Oral wash samples are similar across both phenotypes, with the most common taxa being Streptococcus (Bacilli—purple), Veillonella (Negativicutes—orange), and Haemophilus (Gammaproteobacteria—red). Sputum samples differ visually between frequent and infrequent exacerbators, with Haemophilus and Moraxella (both Gammaproteobacteria—red) dominating frequent exacerbator sputum samples 5, 19, and 22. This marked shift in select sputum samples is neither seen across all frequent exacerbator sputum samples nor in any infrequent exacerbator samples. Comparison of frequent exacerbator paired oral wash and sputum samples reveals that this Haemophilus or Moraxella dominance is not reflected in the oral wash samples
Exacerbation phenotype is associated with taxa changes in the sputum microbiota but not in the oral microbiota
To understand taxa shifts associated with exacerbation phenotype, we compared ASVs separately within each site. Of the 169 ASVs in the hierarchical clustering and taxa significance dataset, many were found in only one phenotype. Among oral wash samples, 43 ASVs were found only in FE, while 102 ASVs were found only in IE. Among sputum samples, 34 ASVs were found only in FE, while 101 ASVs were found only in IE. The three most common taxa in oral wash samples did not change based on subject phenotype. In contrast, the most common taxa in sputum samples differed by phenotype. Haemophilus was the most common genus in FE sputum samples, but the 4th most common in IE sputum samples. Prevotella was the 3rd most common genus in IE sputum samples and the 6th most common in FE sputum samples (Fig. 7, Table 4).
Table 4.
Taxa Abundance by Anatomic Site and Exacerbation Phenotype
| Rank* | Oral Wash | Sputum | ||
|---|---|---|---|---|
| Frequent Exacerbator | Infrequent Exacerbator | Frequent Exacerbator | Infrequent Exacerbator | |
| 1 | Streptococcus | Streptococcus | Haemophilus | Streptococcus |
| 2 | Veillonella | Veillonella | Streptococcus | Veillonella |
| 3 | Haemophilus a | Haemophilus | Veillonella | Prevotella |
| 4 | Rothia | Fusobacterium | Moraxella | Haemophilus |
| 5 | Lactobacillus | Rothia | Rothia | Actinomyces |
| 6 | Prevotella | Prevotella | Prevotella | Rothia |
| 7 | Neisseria | Actinomyces | Actinomyces | Fusobacterium |
| 8 | Actinomyces | Neisseria | Megasphaera | Neisseria |
| 9 | Gemella | Lactobacillus | Lactobacillus | Gemella |
| 10 | Granulicatella | Porphyromonas | Neisseria | Leptotrichia |
| 11 | Fusobacterium | Gemella | Porphyromonas | Granulicatella |
| 12 | Leptotrichia | Granulicatella | Fusobacterium | Porphyromonas |
| 13 | Megasphaera | Leptotrichia | Alloprevotella | Megasphaera |
| 14 | Porphyromonas | Alloprevotella | Gemella | Alloprevotella |
| 15 | Alloprevotella | Megasphaera | Leptotrichia | Lactobacillus |
| 16 | Moraxella | Moraxella | Moraxella | |
*Only genera that represent at least 1% of total sequences are shown
aSelect clinically or statistically significant taxa are bolded for emphasis
Wilcoxon rank-sum permutation tests with Benjamini-Hochberg correction were used to test for significant differences in ASV counts between phenotypes. Among oral wash samples with an FDR < 0.10, none of the ASVs differed by exacerbation phenotype. Among sputum samples, we did not find statistically significant differences in Haemophilus, Moraxella, or Streptococcus relative abundance at the ASV or genus level, as only 3 of the 9 available FE sputum samples exhibited striking shifts in abundance of these taxa. We did find 12 less common sputum ASVs that differed by exacerbation phenotype (Table 5). Actinomyces, a gram-positive anaerobic organism capable of causing both oral and pulmonary infections had the most evidence for a difference. It was more common in the sputum of IE compared to FE. Since exacerbation phenotype and FEV1pp may be associated with each other in our dataset, we used FEV1pp as a co-variate in our model. Actinomyces, Capnocytophaga, Gemella, Ruminococcaceae, Candidatus_Saccharimonas, Prevotella, and Bergeyella ASVs all exhibited an FDR < 0.10 with respect to exacerbation phenotype, after correcting for FEV1pp. All ASVs were more abundant in IE sputum compared to FE sputum.
Table 5.
Sputum Taxa Differential Abundance by Exacerbation Phenotype
| Corresponding Genus for Identified ASV | q-value | q-value (corrected for FEV1ppa) | Difference in Group Means (all are more abundant in IE sputum than FE sputum) |
|---|---|---|---|
| Actinomyces b | 0.048 | < 0.001 b | 169.6 |
| Capnocytophaga | 0.087 | < 0.001 | 42.5 |
| Gemella | 0.087 | < 0.001 | 2379.2 |
| Streptococcus | 0.087 | 0.382 | 500.8 |
| Campylobacter | 0.087 | 0.591 | 29.8 |
| Ruminococcaceae | 0.087 | 0.057 | 23.9 |
| Candidatus_Saccharimonas | 0.087 | 0.019 | 63.8 |
| Prevotella | 0.087 | 0.066 | 42.5 |
| Prevotella | 0.087 | 0.443 | 18.4 |
| Lachnospiraceae | 0.087 | 0.289 | 96.3 |
| Bergeyella | 0.087 | 0.057 | 36.6 |
| Treponema | 0.087 | 0.201 | 7.6 |
aFEV1 percent predicted
bASVs with corrected q-values <0.10 are bolded for emphasis
Discussion
Our study of frequent and infrequent COPD exacerbators is among the first comparisons of the oral and sputum microbiota based on exacerbation phenotype. COPD exacerbations are associated with bacterial infection as well as excess morbidity and mortality, and the exacerbation-associated lung microbiota is an area of active study. Few studies have compared the lung microbiota of FE and IE during periods of clinical stability. Here we have shown that the sputum of FE has lower alpha diversity than the sputum of IE even during periods of clinical stability and without recent administration of antibiotics and/or systemic corticosteroids. This finding is consistent with the general observation that lower alpha diversity is associated with worsening lung health. Although our data show significant within-subject similarity of the oral and sputum microbiota, this did not preclude the identification of shifts in sputum abundance of select taxa in association with exacerbation phenotype.
Differences in alpha diversity and taxa abundance between exacerbation phenotypes were primarily driven by the sputum samples. Oral wash samples alone did not appear to distinguish frequent from infrequent exacerbators. Although decreased alpha diversity and changes in taxa abundance correlated with exacerbation phenotype here, observational studies such as ours are unable to determine causation. Other clinical factors associated with exacerbation phenotype (medication use, obstruction severity, etc.) may be responsible for these associations between exacerbation phenotype and alpha diversity or taxa abundance. Although we excluded subjects who had received antibiotics in the last 1 month, it is likely that FEs received a greater number of antibiotic courses in the past year than were received by the IEs over the past year. Prior antibiotic exposure may have been responsible for some of the changes in alpha diversity between phenotypes noted here. Our facility favors the use of doxycycline to treat COPD exacerbations. Doxycycline is a tetracycline antibiotic, whose effects on the oral and gut microbiota were shown to be less persistent than the effects of other antibiotics like ciprofloxacin or clindamycin. [40] However, prior antibiotic exposure should have affected both the oral and sputum microbiota of our subjects. Our study identified greater phenotype-associated changes in the sputum compared to oral microbiota, suggesting that antibiotic-associated changes are unlikely to be responsible for all of the lung-specific changes noted here.
Although it may be hypothesized that FEs harbor a greater number of bacteria in their airways and sputum when compared to IEs, our data do not support this hypothesis. After normalizing 16S copy numbers to sample mass, sputum contained a larger number of 16S copies than did oral wash samples, regardless of phenotype. No differences in 16S copy numbers based on exacerbation phenotype were observed.
Our studies using alpha diversity demonstrate lower diversity in sputum samples compared to oral wash samples, with the lowest alpha diversity observed in the FE sputum samples. This is consistent with studies of other chronic lung diseases such as cystic fibrosis, in which lower alpha diversity is associated with more severe disease. Review of the taxa found in FE sputum samples demonstrates that three subjects (5, 19, and 22) exhibited a high relative abundance of Gammaproteobacteria (specifically Haemophilus and Moraxella, two organisms associated with COPD exacerbations), which corresponded to low alpha diversity scores. The pathogen dominance exhibited by these three FE sputum samples is in contrast to these subjects’ oral wash findings, which had higher diversity scores and did not exhibit pathogen dominance. Conversely, there were no IE sputum samples that demonstrated pathogen dominance and resulted in very low diversity scores. The three FEs with low sputum alpha diversity did not appear to differ from the FE group as a whole with respect to obstruction severity, number of exacerbations in the last year, or symptom severity. Our data are most consistent with the notion that even during exacerbation-free intervals, a minority of FEs have low alpha diversity due to colonization with potentially pathogenic organisms—and they are not readily distinguished from non-colonized FEs based on other clinical factors.
Although the FE sputum samples from subjects 5, 19, and 22 were different from these subjects’ oral wash samples, other FE and IE sputum-oral wash pairs exhibited a high degree of similarity in beta diversity analyses. Clustering analysis demonstrated that for 18 sputum samples, in nine instances the sputum sample’s nearest neighbor in the dendrogram was the oral wash sample from the same subject. Of these nine very similar sputum-oral wash sample pairs from the same subject, six sample pairs were from IE while three sample pairs were from FE. In contrast, sputum and oral washes from FE subjects 5, 19, and 22 did not cluster together in the dendrogram or the beta diversity plots. Despite the overall similarity in oral wash and sputum sample beta diversity within subjects, we did not detect a change in within-subject beta diversity related to exacerbation phenotype (as we may have expected based on the findings from FE subjects 5, 19, and 22). Additionally, oral wash alpha diversity appeared to be a predictor of sputum alpha diversity, further supporting the similarities we saw between subjects’ oral and sputum microbiota. While there is likely a correlation between the sputum and oral wash microbiota from the same subject, additional exploration of the potential role of exacerbation phenotype in this relationship is warranted.
PERMANOVA analyses identified significant clustering based on anatomic site sampled, exacerbation phenotype, COPD severity, time since last professional dental cleaning, current tobacco use, and inhaled corticosteroid use. Due to our sample size of 42, these analyses were performed for all samples simultaneously, rather than separate analyses of oral wash and sputum samples. Although the literature suggests that COPD severity clustering is more likely due to sputum microbiota changes and clustering based on dental habits or tobacco use is more likely driven by the oral microbiota changes, due to our small sample size we did not evaluate which sample site or taxa were the primary drivers of clustering. Time since last professional dental cleaning, COPD severity, and anatomic site appeared to have a greater influence on beta diversity than tobacco use, phenotype, or ICS use.
Our analysis of taxa distribution across anatomic sites and phenotypes identified shifts in the most abundant taxa in FE sputum samples compared to IE sputum samples, although the increases in Haemophilus and Moraxella abundance in FE sputum did not reach statistical significance. The most likely explanation for this lack of statistical significance is that increases in these two taxa were only noted in three of nine FE sputum samples. Actinomyces was significantly more abundant in IE sputum compared to FE sputum, and this result persisted after controlling for obstruction severity (FEV1pp). Increased relative abundance of Actinomyces in the sputum microbiota was also recently associated with survival following hospitalization for COPD exacerbation. [41] While Actinomyces is a common and clinically-relevant oral and lung pathogen, we currently lack robust evidence to assert a protective effect on the microbiome in COPD. It is possible that Actinomyces in the COPD lung provides protective benefits unrelated to other organisms, or that Actinomyces helps eliminate other more pathogenic members of the COPD microbiota, or simply that its relative abundance in the lung appears to increase when harmful and abundant COPD-associated taxa (such as Moraxella or Haemophilus) are not present. Additional studies will be needed to differentiate among these possibilities. Of note, oral wash sample taxa abundance did not appear to be correlated with exacerbation phenotype. This is consistent with exacerbation phenotype primarily influencing the lung (as opposed to oral) microbiota and suggests that phenotype-associated effects are not solely related to differences in antibiotic exposure between phenotypes.
We examined and compared oral wash samples to sputum samples in this study for several reasons. Firstly, we wished to compare the two sites to determine if sputum was or was not too heavily contaminated by oral bacteria during expectoration to identify phenotype-associated factors. Our data show that oral contamination of sputum samples has not prevented us from identifying phenotype-associated sputum microbiota changes. Secondly, we wished to address the possibility that differences in antibiotic exposure between phenotypes are responsible for the associations we identified. While antibiotic use should affect both the oral and sputum microbiota, the changes we observed in the sputum (but not oral) microbiota support the hypothesis that the lung microbiota changes we observed were due to phenotype-associated factors in the lung rather than systemic factors such as antibiotic use. In addition, the oral microbiota is the source of the lung microbiota through microaspiration or direct mucosal dispersion. The two sites are closely related and therefore the inclusion of oral samples in this study enhances our understanding of the COPD lung microbiota.
Our study had several strengths. We were able to study subjects during periods of clinical stability (at least 1 month after exacerbation treatment had ended), when their oral and sputum microbiotas had reached a period of relative clinical stability and were less likely to be significantly influenced by recent antibiotic or systemic corticosteroid exposure. In spite of the similarities between subjects’ oral and sputum microbiotas, we were able to observe differences in sputum taxa abundance and overall beta diversity based on exacerbation phenotype. We suggest that exacerbation phenotype, rather than increased antibiotic exposure among FEs, is responsible for the observed sputum microbiota changes. If increased antibiotic use among FEs had been entirely responsible for the exacerbation phenotype-related changes we observed in the sputum microbiota, we should also have observed similar phenotype-related changes in the oral microbiota. Both sites should be affected by antibiotic exposure.
Although our study successfully achieved our goals, it also had several weaknesses. Most importantly, this study is unable to determine if sputum microbiota changes caused the development of the FE phenotype or if the FE phenotype caused the sputum microbiota changes. Separating cause from effect in this instance would require identifying and sampling subjects prior to development of the FE or IE phenotype or significant antibiotic exposure. As in any non-randomized study, it is possible that factors other than exacerbation phenotype are responsible for the changes observed here. In addition, the degree of similarity between the oral and the sputum microbiota may suggest that sputum is too contaminated by oral taxa to be a reliable indicator of the lung microbiota. This concern is unlikely to have significantly affected our results as we were able to observe several alterations in the sputum microbiota correlated with exacerbation phenotype, and the effect of oral contamination would have been to mask sputum differences between phenotypes. There was a non-significant trend towards more significant lung obstruction in the FE group compared to the IE group. Therefore, some of the observed differences between phenotypes may be related to increased obstruction. However, many of our taxa abundance results remained significant following statistical correction for obstruction severity. Due to our sample size, we were also unable to determine if clustering on beta diversity analyses was primarily the result of differences between oral or sputum samples and we may have been underpowered to detect some differences, particularly between exacerbation phenotypes.
Conclusions
We showed that prospectively-identified, clinically-stable frequent exacerbators have lower upper airway alpha diversity than infrequent exacerbators, and this finding may be driven by a minority of frequent exacerbators whose sputum is dominated by Gammaproteobacteria (Moraxella, Haemophilus). We identified differences in taxa relative abundance between phenotypes in sputum but not oral wash samples, and many of these differences persisted after correction for severity of obstruction. The upper airway microbiota of COPD subjects demonstrated clustering based on COPD exacerbation phenotype, inhaled corticosteroid use, tobacco use, severity of obstruction, and time since last professional dental cleaning. Dental care habits, tobacco cessation, and inhaled corticosteroid use are all modifiable factors and potential future targets for improving lung health via alterations in the upper airway microbiota.
Additional file
Supplementary information. (DOCX 2881 kb)
Acknowledgements
The authors thank Shane Hodgson for assistance with sample processing and figure and table preparation and Susan A. Johnson for assistance with recruitment and sampling and IRB submissions.
Previously presented
Portions of this work were previously presented as oral abstracts at the 2018 American Thoracic Society Annual Meeting and the 2018 Thomas L. Petty Aspen Lung Conference.
Disclaimer
The views expressed in this article are those of the authors and do not reflect the views of the United States Government, the Department of Veterans Affairs, the funders, the sponsors, or any of the authors’ affiliated academic institutions.
Abbreviations
- ANOVA
analysis of variance
- ASV
amplicon sequencing variant
- BMI
body mass index
- COPD
Chronic obstructive pulmonary disease
- FDR
false discovery rate
- FE
frequent exacerbator
- FEV1pp
forced expiratory volume in 1 s percent predicted
- ICS
inhaled corticosteroid use
- IE
infrequent exacerbator
- PERMANOVA
permutational multivariate analysis of variance
- SGRQ
St. George’s Respiratory Questionnaire
- SILVA
(rRNA gene database named after the Latin word for forest)
Authors’ contributions
Conception and design, AAP, REI, CSR, CHW; Sample acquisition, AAP; Sample processing, AAP; Data analysis, AAP, TJG, KAK, CSR. Drafting of manuscript, AAP; Data interpretation, manuscript revision, final approval, all.
Funding
This work was supported in part by a Career Development Award 1IK2CX001095 (A.A.P.) from the United states (U.S.) Department of Veterans Affairs Clinical Sciences Research and Development Service; 5KL2TR113 and the NIH Clinical and Translational Science Award at the University of Minnesota, 8UL1TR000114 (A.A.P.); and NIAID/NIH 5T32AI055433 (A.A.P). The study sponsors did not play a role in the study design; in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. The corresponding author confirms that she had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Availability of data and materials
The datasets generated during the current study are publicly available at NCBI Sequence Read Archive (SRA) under BioProject #543785: http://www.ncbi.nlm.nih.gov/bioproject/543785.
Ethics approval and consent to participate
The institutional review board for human studies at MVAMC approved the protocol (#4541-B) and written informed consent was obtained from the subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alexa A. Pragman, Phone: 612.467.6671, Email: alexa@umn.edu
Katherine A. Knutson, Email: knuts828@umn.edu
Trevor J. Gould, Email: goul0109@umn.edu
Richard E. Isaacson, Email: isaac015@umn.edu
Cavan S. Reilly, Email: cavanr@biostat.umn.edu
Chris H. Wendt, Email: wendt005@umn.edu
References
- 1.Barker BL, Brightling CE. Phenotyping the heterogeneity of chronic obstructive pulmonary disease. Clin Sci (Lond) 2013;124:371–387. doi: 10.1042/CS20120340. [DOI] [PubMed] [Google Scholar]
- 2.Bon J, Liao S, Tseng G, Sciurba FC. Considerations and pitfalls in phenotyping and reclassification of chronic obstructive pulmonary disease. Transl Res. 2013;162:252–257. doi: 10.1016/j.trsl.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Carolan BJ, Sutherland ER. Clinical phenotypes of chronic obstructive pulmonary disease and asthma: recent advances. J Allergy Clin Immunol. 2013;131:627–634. doi: 10.1016/j.jaci.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Hurst JR, Vestbo J, Anzueto A, Locantore N, Mullerova H, Tal-Singer R, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128–1138. doi: 10.1056/NEJMoa0909883. [DOI] [PubMed] [Google Scholar]
- 5.Kanner RE, Anthonisen NR, Connett JE. Lower respiratory illnesses promote FEV(1) decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am J Respir Crit Care Med. 2001;164:358–364. doi: 10.1164/ajrccm.164.3.2010017. [DOI] [PubMed] [Google Scholar]
- 6.Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:1418–1422. doi: 10.1164/ajrccm.157.5.9709032. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan SD, Ramsey SD, Lee TA. The economic burden of COPD. Chest. 2000;117:5S–9S. doi: 10.1378/chest.117.2_suppl.5S. [DOI] [PubMed] [Google Scholar]
- 8.Spencer S, Jones PW. Time course of recovery of health status following an infective exacerbation of chronic bronchitis. Thorax. 2003;58:589–593. doi: 10.1136/thorax.58.7.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:925–931. doi: 10.1136/thx.2005.040527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gold PM. The 2007 GOLD guidelines: a comprehensive care framework. Respir Care. 2009;54:1040–1049. [PubMed] [Google Scholar]
- 11.Vestbo J, Agusti A, Wouters EF, Bakke P, Calverley PM, Celli B, et al. Should we view chronic obstructive pulmonary disease differently after ECLIPSE? A clinical perspective from the study team. Am J Respir Crit Care Med. 2014;189:1022–1030. doi: 10.1164/rccm.201311-2006PP. [DOI] [PubMed] [Google Scholar]
- 12.Sethi S, Murphy TF. Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin Microbiol Rev. 2001;14:336. doi: 10.1128/CMR.14.2.336-363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465–471. doi: 10.1056/NEJMoa012561. [DOI] [PubMed] [Google Scholar]
- 14.Desai H, Eschberger K, Wrona C, Grove L, Agrawal A, Grant B, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2014;11:303–309. doi: 10.1513/AnnalsATS.201310-350OC. [DOI] [PubMed] [Google Scholar]
- 15.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5:372–381. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One. 2012;7:e47305. doi: 10.1371/journal.pone.0047305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cabrera-Rubio R, Garcia-Nunez M, Seto L, Anto JM, Moya A, Monso E, et al. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. 2012;50:3562–3568. doi: 10.1128/JCM.00767-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pragman AA, Lyu T, Baller JA, Gould TJ, Kelly RF, Reilly CS, et al. The lung tissue microbiota of mild and moderate chronic obstructive pulmonary disease. Microbiome. 2018;6:e47305. doi: 10.1186/s40168-017-0381-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diao W, Shen N, Du Y, Erb-Downward JR, Sun X, Guo C, et al. Symptom-related sputum microbiota in stable chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2018;13:2289–2299. doi: 10.2147/COPD.S167618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engel M, Endesfelder D, Schloter-Hai B, Kublik S, Granitsiotis MS, Boschetto P, et al. Influence of lung CT changes in chronic obstructive pulmonary disease (COPD) on the human lung microbiome. PLoS One. 2017;12:e0180859. doi: 10.1371/journal.pone.0180859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Nunez M, Millares L, Pomares X, Ferrari R, Perez-Brocal V, Gallego M, et al. Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:4217–4223. doi: 10.1128/JCM.01967-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HJ, Kim YS, Kim KH, Choi JP, Kim YK, Yun S, et al. The microbiome of the lung and its extracellular vesicles in nonsmokers, healthy smokers and COPD patients. Exp Mol Med. 2017;49:e316. doi: 10.1038/emm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, et al. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:438–445. doi: 10.1164/rccm.201502-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Anna SE, Balbi B, Cappello F, Carone M, Di Stefano A. Bacterial-viral load and the immune response in stable and exacerbated COPD: significance and therapeutic prospects. Int J Chron Obstruct Pulmon Dis. 2016;11:445–453. doi: 10.2147/COPD.S93398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haldar K, Bafadhel M, Lau K, Berg A, Kwambana B, Kebadze T, et al. Microbiome balance in sputum determined by PCR stratifies COPD exacerbations and shows potential for selective use of antibiotics. PLoS One. 2017;12:e0182833. doi: 10.1371/journal.pone.0182833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jubinville E, Veillette M, Milot J, Maltais F, Comeau AM, Levesque RC, et al. Exacerbation induces a microbiota shift in sputa of COPD patients. PLoS One. 2018;13:e0194355. doi: 10.1371/journal.pone.0194355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:2813–2823. doi: 10.1128/JCM.00035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molyneaux PL, Mallia P, Cox MJ, Footitt J, Willis-Owen SA, Homola D, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:1224–1231. doi: 10.1164/rccm.201302-0341OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Bafadhel M, Haldar K, Spivak A, Mayhew D, Miller BE, et al. Lung microbiome dynamics in COPD exacerbations. Eur Respir J. 2016;47:1082–1092. doi: 10.1183/13993003.01406-2015. [DOI] [PubMed] [Google Scholar]
- 32.Mayhew D, Devos N, Lambert C, Brown JR, Clarke SC, Kim VL, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax. 2018;73:422–430. doi: 10.1136/thoraxjnl-2017-210408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Singh R, Miller BE, Tal-Singer R, Van Horn S, Tomsho L, et al. Sputum microbiome temporal variability and dysbiosis in chronic obstructive pulmonary disease exacerbations: an analysis of the COPDMAP study. Thorax. 2017;73:331–338. doi: 10.1136/thoraxjnl-2017-210741. [DOI] [PubMed] [Google Scholar]
- 34.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. Chronic obstructive pulmonary disease lung microbiota diversity may be mediated by age or inhaled corticosteroid use. J Clin Microbiol. 2015;53:1050. doi: 10.1128/JCM.03320-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Contoli M, Pauletti A, Rossi MR, Spanevello A, Casolari P, Marcellini A, et al. Long-term effects of inhaled corticosteroids on sputum bacterial and viral loads in COPD. Eur Respir J. 2017;50:1700451. doi: 10.1183/13993003.00451-2017. [DOI] [PubMed] [Google Scholar]
- 40.Zaura E, Brandt BW, Teixeira de Mattos MJ, Buijs MJ, Caspers MP, Rashid MU, et al. Same exposure but two radically different responses to antibiotics: resilience of the salivary microbiome versus long-term microbial shifts in feces. MBio. 2015;6. [DOI] [PMC free article] [PubMed]
- 41.Leitao Filho FS, Alotaibi NM, Ngan D, Tam S, Yang J, Hollander Z, et al. Sputum microbiome is associated with 1-year mortality following COPD hospitalizations. Am J Respir Crit Care Med. 2018; in press. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information. (DOCX 2881 kb)
Data Availability Statement
The datasets generated during the current study are publicly available at NCBI Sequence Read Archive (SRA) under BioProject #543785: http://www.ncbi.nlm.nih.gov/bioproject/543785.