

Abstract
Numerous studies have demonstrated the importance of altered hyaluronan metabolism to malignant progression of multiple tumor types, including breast carcinomas. Increased hyaluronan (HA) metabolism in the stroma of primary tumors promotes activation of oncogenic signaling pathways that impact tumor initiation, growth, and invasion. Carcinoma cell synthesis and assembly of HA-rich pericellular matricies induces a stromal-independent phenotype, which is associated with cancer progression. Although the pro-tumorigenic role of stromal HA is well established, a novel but unexplored hypothesis is that carcinoma cell-associated HA pericellular matrices promote metastasis of circulating tumor cells. Here, we report the development of an in vitro assay that employs microfluidic techniques to directly measure the importance of an HA-rich pericellular matrix in the entry of carcinoma cells into ectopic sites. This model provides the capability to visualize specific steps in metastasis, which is difficult using animal models. The results show that the presence of a HA-rich pericellular matrix correlates to the invasive and metastatic potential of breast carcinoma cells. Furthermore, enzymatic removal or pharmacologic inhibition of HA synthesis significantly inhibits carcinoma cell extravasation and invasion in this model system. These results implicate pericellular HA-rich carcinoma cell associated pericellular matrices in colonization of ectopic sites by circulating tumor cells and support specific targeting of this matrix to limit metastasis in patients.
Introduction
Changes in the microenvironment at the primary tumor site are key in supporting tumor growth, tumor cell survival, and expansion into surrounding tissues. Changes in both the tumor microenvironment and in tumor cell programming sanction highly metastatic cells to become anchorage independent and escape the primary tumor microenvironment, a critical step in tumor progression and metastasis. Many primary cancers are associated with an increase in hyaluronan (HA) metabolism which functions in a paracrine fashion to enhance carcinoma growth, survival, and invasiveness1-8. As carcinoma cells acquire the capacity to metastasize they develop an autonomous phenotype which can include synthesis and assembly of a pericellular HA-rich matrix. At the cellular level, these HA-rich pericellular matrices function to organize receptors within plasma membrane microdomains, lowering the threshold for activating associated oncogenic signaling pathways, promote cytoskeletal reorganization and impact on an oncogenic transcriptome2. The phenotypic changes associated with these matrices include enhanced therapeutic resistance, increased growth and enhanced motility and invasion. It has been hypothesized that HA-rich tumor cell pericellular matrices function to prevent anoikis, enhance carcinoma cell adhesion to endothelial cells, promote autocrine growth factor sequestration and aid invasion via enhanced HA-receptor signaling cascades3, 7. HA rich pericellular matrices enhance carcinoma cell adhesion to endothelial cellsand transmigration across the endothelium. The tumor cell pericellular matrix may serve as a scaffold for de novo extracellular matrix (ECM) formation in metastatic sites2, 9. Thus, the HA-rich pericellular matrix has potentially critical functions in metastasis and provides a plausible target for better clinical management of cancer patients.
A major barrier to testing this hypothesis and to studying many factors that influence the later stages of metastasis, including extravasation and invasion of the metastatic site, is the lack of appropriate model systems. Traditional in vitro assays lack key components of the tissue microenvironment such as tissue composition, architecture, and physiologically relevant forces, so may not accurately assess tumor cell ability to metastasize. By contrast, mouse models capture many of the salient features of the tissue microenvironment and have been extremely important in confirming findings from in vitro assays; however, they lack the tunability of engineered systems which limits our ability to systematically explore relevant, metastasis-regulating factors. Furthermore, studying the later stages of metastasis in vivo, especially in real time, is challenged by our limited ability to spatially and temporally localize the metastatic event. The introduction of tissue and organ-on-a-chip models has enabled the development of more physiologically relevant in vitro models while maintaining the ability to perform high spatiotemporal resolution imaging of pathologic processes including cancer metastasis10-13. Here, we report the development of an in vitro model of metastatic extravasation that recapitulates critical aspects of the ectopic site, including a perfusion channel with physiologic flow, a functional endothelium, and a three-dimensional (3D) tissue compartment. Using this platform, we can quantify tumor cell-endothelial adhesion, endothelial transmigration, and tissue invasion. Since this platform permits systematic testing and measurement of key variables, it should enable discovery of pathways and microenvironmental factors that regulate metastasis formation in vivo.
In this study, we used our in vitro model of metastasis to systematically investigate how pericellular HA matrices surrounding disseminated tumor cells may impact key stages of metastasis formation in vivo. We tested the hypothesis that limiting the ability of circulating breast carcinoma cells to form a pericellular matrix reduces their ability to adhere to the endothelium, transmigrate across the vessel wall, and invade the metastatic site. By correlating higher surface HA expression of three distinct breast carcinoma cell lines with increased metastatic potential in vivo, we highlight the potential of the pericellular HA coat to influence metastasis. Furthermore, our data demonstrate that the pericellular matrix produced by MDA-MB-231 breast carcinoma cells directly impacts extravasation in our in vitro metastasis model. We also show that pharmacological inhibition of HA synthesis results in reduced pericellular matrix formation and reduced invasiveness of MDA-MB-231 breast carcinoma cells, suggesting that the HA pericellular matrix may be a potential therapeutic target.
Materials and Methods
Cell Culture and Reagents
GFP expressing MDA-MB-231 cells, stable infection with lentivirus, generously provided by Dr. Paolo Provenzano (University of Minnesota) were cultured in DMEM (Gibco, catalog # 11995–065) supplemented with 10% FBS (Sigma Aldrich, catalog # F9423) and 1 μg/ml puromycin (Invivogen, catalog # ant-pr-1). HS578T cells, generously provided by Dr. Kaylee Schwertfeger (University of Minnesota, Twin Cities), were cultured in DMEM supplemented with 10% FBS and 0.01 mg/ml human insulin (Sigma Aldrich, catalog # 11061–680). MCF7 cells were purchased from Lonza and cultured in in DMEM supplemented with 10% FBS and 1X non-essential amino acids (Gibco, catalog # 11140–050) and 1mM sodium pyruvate (Gibco, catalog # 11360–070). All cell lines were grown to 80% confluency in at 37°C and 5% CO2 before passaging or 60% confluency before treatment with 4-methylumbelliferone (4MU) (Sigma Aldrich, catalog # M1508). Primary human umbilical vein cells (HUVECs) (Lonza) and cultured in Endothelial Cell Growth Media (EGM)-2 bullet kit media (Lonza, catalog # cc-3162). HUVECs were grown in tissue treated culture flasks coated with 1% gelatin (Sigma Aldrich, catalog # G1383) and grown to 80% confluence before passaging or device seeding.
Measurement and Visualization of Pericellular HA
Preexisting HA matrices were enzymatically removed from the cell surface via hyaluronidase treatment. Breast cancer cells were washed with PBS (Corning, catalog # 21–031-CV) and released with 5mM EDTA (Corning, catalog # 46–034-CI). To measure pericellular HA, 1×106 cancer cells were incubated in in suspension of 2.5 mg/ml type I hyaluronidase (bovine, Sigma Aldrich, catalog # H3506) in PBS or suspended in PBS alone for 30 minutes at 37°C. Pericellular HA was quantified by flow cytometry using biotinylated HA binding protein (HABP) (Millipore, catalog # 80502–722), which has a high binding specificity for HA, combined with fluorophore labelled streptavidin14. Cells treated with hyaluronidase and control cells were washed and incubated with 0.5 μg/ml HABP overnight at 4°C. Cells were then washed to remove unbound HABP and stained with streptavidin-Cy5 (Invitrogen, catalog # S21374)for 30 minutes on ice. Cell surface HA was then measured using a BD Acurri flow cytometer (Becton Dickson).
Additionally, a particle exclusion assay was used to visualize cell surface HA6, 15. Cancer cells were plated at a concentration of 1×104 cells/ml into 96 well plates and incubated overnight at 37°C with 5% CO2. Cells were then either treated with 2.5 mg/ml hyaluronidase in PBS or PBS alone for 30 minutes at 37°C. They were subsequently washed and incubated with 2 mg/ml aggrecan (Sigma Aldrich, catalog # A1960) for 90 minutes. Glutaraldehyde fixed sheep red blood cells (RBCs) (VWR, catalog # 102768–578) were added to each well at a concentration of 1×107cells/ml. The pericellular HA matrix of cells physically excludes the cells, leaving a cell free region around untreated cells, visualized using phase contrast microscopy. Alternatively, to directly stain cell surface HA, MDA-MB-231 cells were plated at a density of 1.5×105 cells/ml on a tissue treated well plate. Cells were incubated overnight to allow cells to adhere to the plate. Cells were either treated with 2.5 mg/ml hyaluronidase in PBS or with PBS alone for 30 minutes at 37°C and washed with PBS. Cells were incubated with biotinylated HABP for 2 hours at 37°C, followed by washing and a 30 minute incubation with streptavidin-Cy5. Cell nuclei were stained using 0.01 mg/ml Hoechst (Invitrogen, catalog # H3569) and epi-fluorescent images were captured on an inverted microscope.
Fabrication of Microfluidic Extravasation Model
Microfluidic models of tumor cell adhesion and extravasation were fabricated using standard soft lithography of polydimethylsiloxane (PDMS)16-19 (Ellsworth, catalog # 4019862). A 10:1 mixture of PDMS to curing agent was cast onto photolithographically-defined molds (SU-8 on silicon) with 100 μm feature height. After heat curing, the PDMS was cut from the mold, punched with biopsy punches to make access ports, and bonded to PDMS coated glass slides using oxygen plasma, ultimately creating two adjacent, 100 um tall channels with posts separating the larger, 3D tissue compartment (700 μm width) from the smaller, perfusion channel (400 μm width). To create a 3D tissue, a 6 mg/ml solution of rat tail collagen I (Corning, catalog # CD354249) buffered with pH8 HEPES (Boston Bioproducts, catalog # BBH-80) was first allowed to nucleate for at least 30 minutes on ice and then brought to a pH of 7.4 – 7.6. This solution of unpolymerized collagen was perfused into the tissue compartment and polymerized for at least 30 minutes at 37°C. Collagen did not enter the endothelial compartment because the capillary burst pressure of the valves was not exceeded20. For experiments that required an endothelial monolayer to model transmigration, the perfusion channel was coated with 1% gelatin for 30 minutes and rinsed with EGM (HUVECs were released with trypsin and (HUVECs; Lonza) were suspended in a 2% w/v dextran (Sigma Aldrich, catalog # 31392) in EGM at 106 cells/mL and 200 uL of cell suspension was added to the inlet of the perfusion channel. Endothelial cells were grown to confluency creating an endothelial monolayeralong the walls of the perfusion channel. Breast carcinoma cells were delivered into the perfusion channel in a 10,000 or 50,000 cell bolus under constant perfusion, depending on the experiment. Devices were imaged every 24 hours for 5 days and adhesion, transmigration, and invasion were measured as described below
Characterization of Microfluidic Model System
Shear within the perfusion channel was measured by flowing 1 μm diameter fluorescent beads (Thermo Scientific) through the perfusion channel and recording distance traveled over time with a high-speed camera (Hamamatso). These measurements were used with the dimensions of the channel to calculate the shear on the endothelial monolayer. To confirm a continuous endothelial layer around the perfusion channel, we stained endothelial-lined devices with CD31-RPE (Bio-Rad) to highlight the tight junctions between endothelial cells. To determine the permeability of the endothelium, a mixture of 48 μg/ml 150 kD TRITC dextran (Sigma Aldrich, catalog # T1287) and 10 μg/ml FITC (Sigma Aldrich, catalog # F2502) in EGM was added to the perfusion channel and the endothelial barrier along the collagen tissue was imaged every 3 minutes. Linear regression of the change in concentration over time for a quasi-steady state was used to calculate the permeability of the endothelial layer10. Permeability was measured and averaged across devices from 3 experiments.
Analysis of Adhesion, Transmigration, and Invasion of Cancer Cells
Following seeding and proliferation of HUVECs, 50,000 breast cancer cells either treated with hyaluronidase, as previously described, or untreated were delivered to the perfusion channel (Figure 3a). Cancer cell adhesion to the endothelium was measured by counting the number of cells within the perfusion channel 24 hours after cancer cell introduction into the system. The number of cells within the tissue compartment was counted for every 24-hour time point over a 5-day period to quantify cell transmigration. To measure invasion distance (Figure 3a, lower right), ImageJ21 was used to draw a line from the edge of the endothelium to the center of each invaded cell body within the tissue compartment. The average distance of all cells in each device within a trial was calculated. The mean invasion distance for each treatment group in each trial was averaged and these means were normalized to the untreated group for ease of comparison. The data were normalized within each experiment trial containing a minimum of n=3 devices per treatment group and experiments were repeated at least 3 times. The means of the normalized data from each experiment were averaged and cell transmigration, adhesion, or average invasion distance was compared using an unpaired t-test across treatment types or an ANOVA with Tukey’s t-test for comparison across cell lines to determine statistical significance. Experimental trials containing an n of less than 30 were checked for normality using the Shapiro-Wilk normality test. For data sets with an n larger than 30 the sampling distribution of the mean is assume to be normal per the central limit theorem.
Figure 3. Removal of pericellular HA reduces tumor cell metastatic potential in vitro.
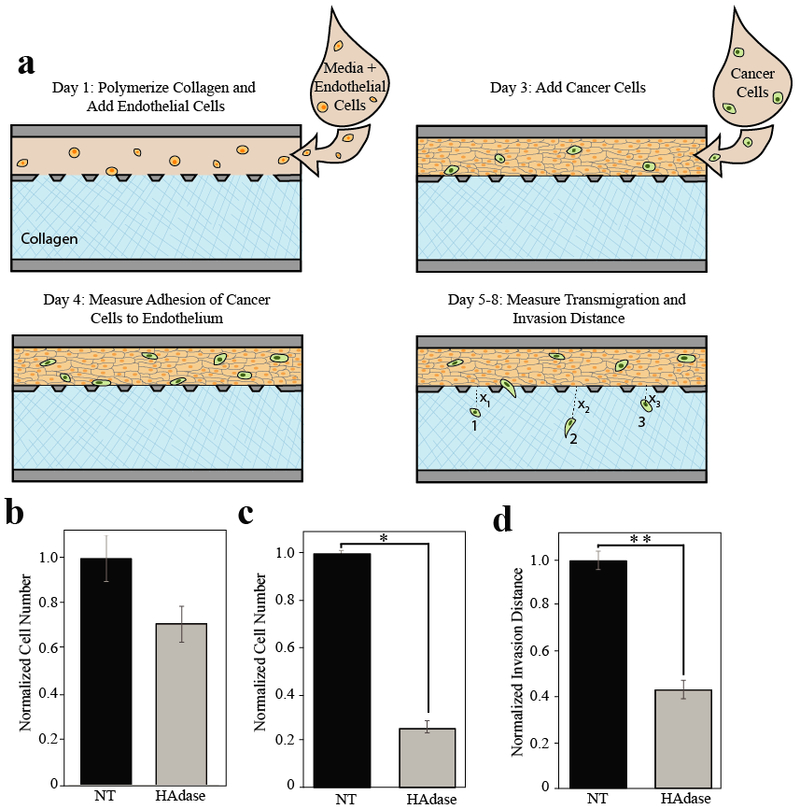
a) Timeline of experimental process. On day one unpolymerized collagen solution (blue) was perfused into larger of two adjacent channels. After collagen was polymerized, media and endothelial cells (tan) were added to the perfusion channel. Devices were then placed in a 37°C incubator for 24-48 hours until endothelial cells formed a confluent monolayer. On day three, cancer cells (green) were added to the perfusion channel under constant flow. Media was perfused for a total of 5 days after cancer cells were added, and each device was imaged every 24 hours. Adhesion was quantified by counting cells adhered in the endothelial channel 24 hours after cancer cells were added. The number of cells that transmigrated and the average distance of migration was measured (ie. Average distance = (Σ X1+ X2+ X3+… Xn)/n) using ImageJ to obtain the distance from the edge of the endothelium to the middle of the cell body. b-d) Quantification of metastatic potential of MDA-MB-231 breast carcinoma cells with and without hyaluronidase treatment. c) Adhesion of cancer epithelial cells to endothelium. There is no significant difference in endothelial adhesion between hyaluronidase (HAdase) treatedand untreated (NT) tumor cells. d) Transmigration between treatment groups was statistically significant, p < 0.0005. e) The average invasion distance also showed a statistiaclly significant difference across treatment groups (*p < 0.0005 and **p < 0.007). All experiments were performed at least three separate times with at least n=3 in each treatment group. Data was then normalized within each experiment and experiments were averaged before comparison.
Treatment of Cancer Cells with 4-methylumbelliferone
4-methylumbellerferone (4MU) (Sigma Aldrich, catalog # M1508) was used to reduce hyaluronan synthase (HAS) activity in breast cancer epithelial cells. To determine the appropriate working concentration of 4MU that would decrease cellular HA production without causing a significant decrease in cell growth, we measured both the population doubling time and the level of HA synthesis of cells treated with a range of HA inhibitor concentrations (data not shown). For these studies, MDA-MB-231eGFP cells were plated in a 12 well plate and subjected to varying concentration of 4MU ranging from 0–800 μM. HA output of MDA-MB-231 cells treated with varying concentrations of 4MUwas measured by ELISA (R&D Systems, catalog # DHYAL0) and compared to untreated cultures. HA output was used to define changes in HA synthesis in cells subjected to 4MU treatment. The level of HA synthesis identified by ELISA experiments was used to determine the concentration of 4MU necessary to inhibit HA synthesis without causing a change in growth or survival of carcinoma cells. This concentration (400μM) was then used to treat cells overnight, at which time they were released, washed, and counted in an automated hemocytometer. To measure metastatic potential of 4MU treated cancer cells, 10,000 cells were added to the perfusion channel of each device without endothelium (Figure 4a). Devices were imaged every 24 hours to measure tumor cell invasion by counting the number of cells in the tissue at each time point, as previously described. The level of cellular HA synthesis within the perfusion channel of these chambers was measured by collecting media output from devices and performing an ELISA. To obtain normalized HA output on a per cell basis, MDA-MB-231 cells were plated in microwells and treated with or without 400 μM 4MU overnight. Media was collected for ELISA analysis of HA concentration and cells were realeased and counted. HA concentration in each well was divided by the cell number from the corresponding well.
Figure 4. Pharmacologic inhibition of HA synthesis reduces invasiveness of tumor cells in vitro.
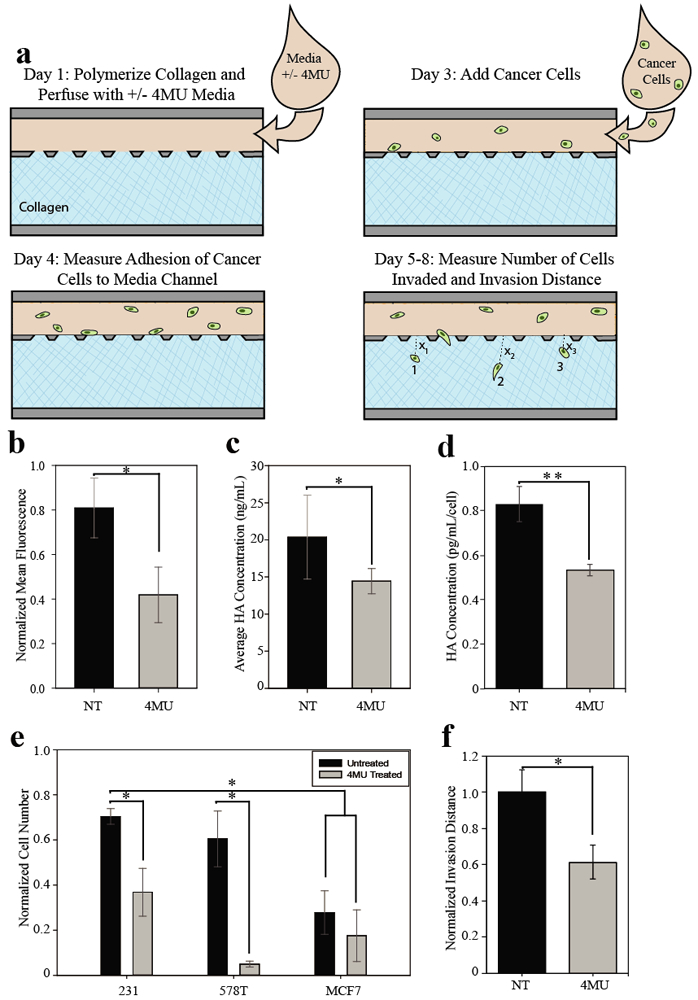
a) Schematic of experimental set up. After collagen polymerization, breast cancer epithelial cells that were pretreated with 400 μM 4MU or untreated were loaded with media containing 400 μM 4MU or no 4MU respectively. Devices were imaged for 5 days following addition of carcinoma cells. On day two, the number of cells adhered in the media channels was measured (data not shown). The number of cells that invaded the collagen was measured and used to calculate invasion potential. b) Cell surface HA of MDA-MB-231eGFP cells was measured using flow cytometry as previously described. Measurement of cell surface HA of MDA-MB-231eGFP cells showed a significant decrease when cells were treated with 4MU. c) HA output from microfluidic devices. Media was collected from the outlets of devices, media was pooled among treatment groups and the HA in the output was measured by ELISA. Three trials of experiments were averaged. Cells treated with 4MU had less free HA in the media collected. d) HA output of MDA-MB-231 cells in multiwell plates, treated with 4MU or untreated. MDA-MB-231 cells were plated and treated with our without 400 μM 4MU overnight. Media was collected for ELISA analysis of HA concentration, and cells were released and counted in an automated hemocytometer. 4MU significantly reduced free HA in treated samples. e) Invasion potential of breast carcinoma cells treated with 400 μM 4MU. MDA-MB-231 cells were the most invasive of the cells tested. This invasion is significantly reduced when cells were treated with 4MU. HS578T cells were most effected by 4MU treatment with a more than five-fold decrease in invasion potential. MCF7s showed no significant difference in invasion potential when treated with 4MU (*p < 0.05 and **p ≤ 0.005). Data was collected and normalized in a similar fashion to extravasation data (Figure 3), where data was normalized within each experiment and experiments were averaged before comparison. f) Effect of 4MU on MDA-MB-231 invasion distance. Invasion disstance of each cell was measured using ImageJ to find the distance from the collagen-endothelium interface to the center of each cell. The average invasion distance was calculated for each device, this data was then averaged on a per trial basis for each treatment group. Four independent trials of 4MU treatment were compared and results were normalized to find 4MU had a significant effect on the invasion distance of the highly metastatic MDA-MB-231 breast carcinoma cells.
Results
Presence of HA in Pericellular Matrix Correlates with Metastatic Potential
The goal of this project was to determine if limiting the ability of circulating breast carcinoma cells to form an HA-rich pericellular matrix reduced their ability to adhere to endothelium, transmigrate, and invade the underlying stroma. The HA pericellular matrix can be directly visualized using a particle exclusion assay (Figure 1a, top panel), where red blood cells are physically excluded from the pericellular region by the presence of the HA-rich matrix. The presence of the pericellular matrix is further confirmed by removing it using exogenously introduced hyaluronidase and showing that RBCs are not excluded from the pericellular region (Figure 1a, bottom panel). Since type 1 hyaluronaidase, as with all other vertebrate hyaluronidases, is an endo-β-N-acetyl-hexosaminidase that degrades hyaluronan through a non-processive endolytic process, it should reduce HA surface expression on breast cancer cells without altering other functions22. The HA pericellular matrix can also be visualized using biotinylated HABP followed by fluorescently labelled streptavidin (Figure 1b, top), and removal of the matrix with hyaluronidase is evidenced by the absence of fluorescent labeling (Figure 1b, bottom). Additionally, we used this fluorescent labeling procedure to directly measure cell surface HA on populations of tumor cells with flow cytometry (Figure 1c, d). MDA-MB-231 cells show high levels of surface HA with mean fluorescence well above background. Further, treatment of MDA-MB-231 with hyaluronidase reduced the mean fluorescence of tagged HA on the cell surface by an order of magnitude, producing a significant change in fluorescence between untreated controls (mean values of 220,000) versus hyaluronidase treated cells (mean values of 20,000) (Figure 1c). Using this assay, we also measured surface HA for two other genetically unrelated breast cancer cell lines and found that surface HA correlates with the metastatic potential of these cell lines in vivo (Figure 1d; MDA-MB-231 ≥ HS578T > MCF7). Both MDA-MB-231 and HS578T cells have significantly more surface HA than MCF7 cells (Figure 1d), and these cells are also more invasive and mesenchymal-like than MCF7s23. Moreover, when hyaluronidase treatment was used to remove the pericellular matrix from these cells, MDA-MB-231 cells show an order of magnitude decrease in mean fluorescence of cell surface HA across three trials (p = 0.0020), and HS578T cells showed a 5-fold decrease (p = 0.0213), while MCF7 cells showed no significant change (p = 0.3244) which confirms the difference in pericellular HA among these cell lines (Figure 1d). These results indicate that differences in HA expression between MDA-MB-231 cells and MCF7s are correlated to known differences in their metastatic potential in vivo. MDA-MB-231 are highly metastatic in nude mice, while MCF7s do not form tumors after mammary fat pad injection in nude mice unless supplemented with some form of estrogen23-25. HS578T cells contain many of the same genetic and phenotypic metastasis markers as MDA-MB-231 cells; however, in vivo they do not form tumors when injected intra-dermally, unlike the highly metastatic MDA-MB-231 cells23. HS578T cells do form tumors when injected into the mammary fat pad, but do not form as many metastatic colonies as MDA-MB-231 cells and do not metastasize to the lung23, 26.
Figure 1. Tumor cell pericellular matrix correlates with in vivo metastatic potential.
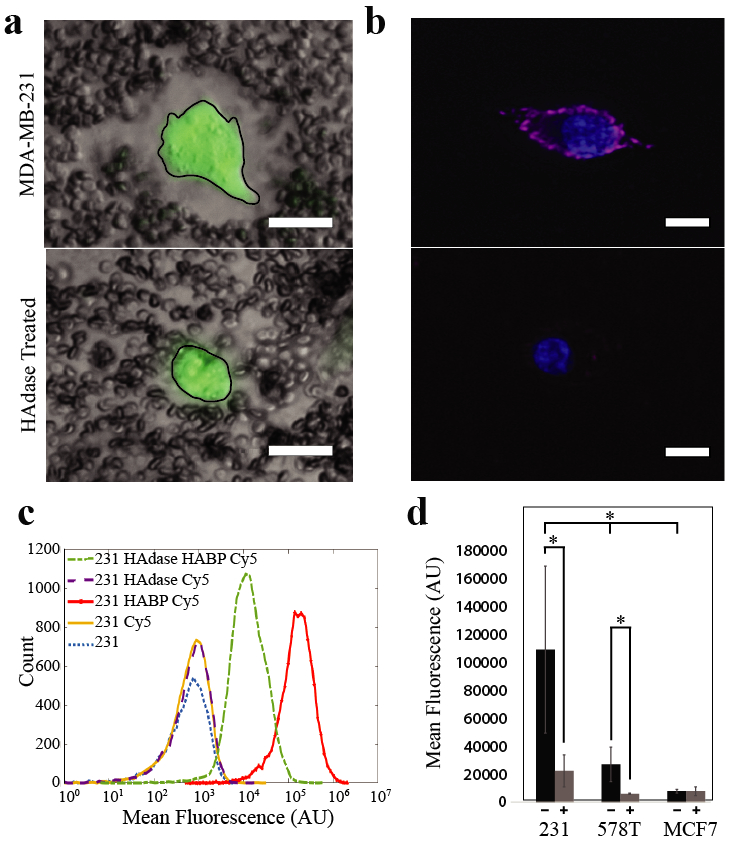
a) Particle Exclusion Assay. The negative charge of aggrecan bound to HA in the pericellular matrix repels negatively charged fixed sheep red blood cells creating space around the cell (outlined in black), top. After treatment with hyaluronidase (HAdase), the sheep red blood cells are close to the cell membrane demonstrating the removal of HA from the cell surface, bottom. Scale bar 20 μm. b) Pericellular HA on live MDA-MB-231 cells with and without hyaluronidase treatment was stained with biotinylated HABP and streptavidin-Cy5 (pink). Nuclei are shown in blue. Scale bars denote 10 μm. c) Histogram of HABP-Cy5 labled MDA-MB-231 breast cancer epithelial cells with and without hyaluronidase treatment. Treating MDA-MB-231 cells with hyaluronidase reduced fluorescence, denoting a change in HA on the cell surface by an order of magnitude. This is visualized as a shift from the peak on the far right representing untreated cells (red) to the peak in the middle representing hyaluronidase treated cells (green). d) Comparison of cell surface HA across cell types. MDA-MB-231 cells have highest mean fluorescence of cell surface HA before hyaluronidase treatment and the biggest change in mean fluorescence of cell surface HA after treatment. The less metastatic HS578Ts have significantly less cell surface HA in untreated cells than MDA-MB-231 cells. Hyaluronidase treatment of HS578Ts significantly reduced cell surface HA. Cell surface HA levels in the least-metastatic MCF7 cells was lowest of all untreated cell types and there was no significant change in the cell surface HA after hyaluronidase treatment. *p < 0.05 and **p < 0.01.
Microfluidic Model of Extravasation and Invasion
To quantify the progression of a cancer cell through the steps of extravasation, we devised an in vitro system that recapitulates critical aspects of the metastatic microenvironment, including physiologic shear at the vessel wall, a functional endothelium, and a 3D ECM-based tissue environment to represent the metastatic site. The model comprises two adjacent chambers: a 3D tissue composed of collagen type I and a perfusion channel that can be endothelialized to model transmigration (Figures 2a and 2b). As shown in Figure 2b (left inset), the collagen matrix can be visualized with second harmonic generation microscopy. To qualitatively assess endothelial structure and function, we visualized endothelial morphology and tight junctions by immunofluorescence. A cross section of the device is projected to the right of the top down view to show that the endothelial layer is contiguous creating a lumen surrounded by endothelial cells within the microchannel (Figure 2c). We also quantitatively assessed the endothelial barrier function in the devices using a high molecular weight fluorescent tracer (150 kDa fluorescent dextran). When the tracer is present in the perfusion channel, a sharp drop in fluorescence intensity across the endothelium (Figure 2d) demonstrates the endothelial barrier to passive transport of macromolecules. An average permeability of 3.77 ± 0.31 × 10−7 cm/s was calculated form the intensity profiles, which is comparable to similar in vitro model systems that have reported endothelial permeability on the order of 10−7-10−6 cm/s for molecules above the molecular weight cutoff for passive transport10, 11. During device operation, continuous media flow is maintained through the perfusion channel to maintain a physiologically relevant shear in the range of 1–6 dynes/cm2 (data not shown), which has shown to be important for maintaining endothelial function11, 27, 28. Our model recapitulates many of the salient physical and biological features of the metastatic microenvironment and should permit investigation of factors that regulate metastatic adhesion, transmigration, and invasion.
Figure 2. An in vitro microfluidic model of metastatic extravasation and invasion.
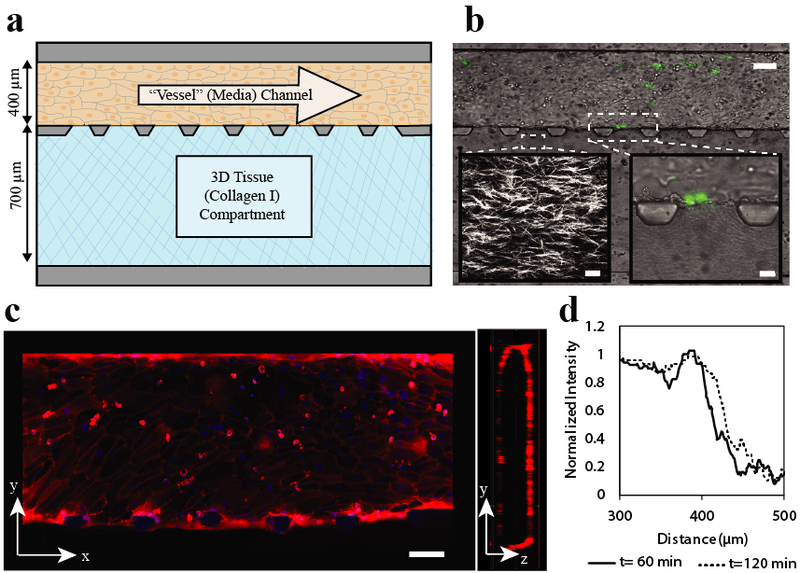
a) Device design. Device consists of two adjacent channels separated by trapezoid-shaped posts. The top perfusion channel (dimensions 100 μm high × 400 μm wide × 10 mm long) is attached to a resistor that slows flow to create physiologically relevant shear (resitor not shown). Endothelial cells are seeded in this perfusion channel and form an endothelial monolayer around the channel. The bottom tissue compartment (100 μm high × 700 μm wide × 10 cm long) is filled with collagen that is polymerized to create a 3D tissue adjacent to the endothelial monolayer. b) Brightfield image of experimental imaging area with MDA-MB-231 eGFP cells adhered to the endothelium and beginning to transmigrate. Scale bar 100 μm. Left inset: Second harmonic generation microscopy image of collagen fibers after polymerizaion. Scale bar 20 μm. Right inset: Brightfield image of MDA-MB-231 eGFP cells beginning transmigration into the tissue compartment. c) Walls of endothelial perfusion channel. Tight junctions of endothelial cells are stained with CD-31 (red), nuclei with hoescht (blue). These images show that there is a coherent layer of endothelial cells on all four sides of the channel walls. Scale Bar 100 μm d) Diffusion profile across the endothelial barrier. To measure endothelial permeability to protein sized molecules, 48 μg/ml 150 kDa TRITC dextran was added to the endothelial cell coated perfusion channel. A line scan of the normalized fluorescent intensity, shown here, illustrates the behavior of the endothelial barrier over time. The endothelial barrier is evident as a sharp drop in fluoresence intensity and shows a slight increase at two hours as fluorescent tracer slowly passes through.
Removal of Pericellular Matrix from Breast Cancer Cells Decreases Metastatic Potential in vitro
To test the hypothesis that tumor cell pericellular matrix influences metastatic extravasation, we used our microfluidic model to compare the metastatic potential of MDA-MB-231 cells with or without hyaluronidase treatment. Briefly, tumor cells were introduced into the perfusion channel under constant shear, and we quantified adhesion and transmigration by counting the number of cells in the vascular and tissue compartments, respectively, at multiple time points (Figures 3a). Adhesion measurements were taken 24 hours after cancer cells were added to the perfusion channel, and transmigration and invasion was assessed over several days. Tissue invasion was determined by measuring average distance of cells which had invaded the adjacent collagen compartment. Notably, we observed no significant difference in tumor cell adhesion to the endothelium when cells were treated with hyaluronidase (Figure 3b). However, we observed significant decreases in the number of breast carcinoma cells that transmigrated after hyaluronidase treatment (Figure 3c) and in the invasion distance of the cells within the 3D tissue microenvironment after removal of pericellular HA (Figures 3d). Hyaluronidase treatment resulted in a roughly 70% decrease (p < 0.0005) in the number of invaded cancer cells and a roughly 50% decrease (p = 0.0070) in the average distance of invasion compared to cells with intact pericellular matrix. Since we observed no significant proliferation of treated or untreated tumor cells within the collagen matrix in additional experiments (data not shown), we concluded that differences in the number of invaded cells were due solely to differences in transmigration, not proliferation. These results support the hypothesis that the HA-rich pericellular matrix promotes metastatic breast cancer extravasation and invasion.
Inhibition of Hyaluronic Acid Synthesis Decreases Invasiveness of Breast Cancer Cells in vitro
As an alternative method to reduce pericellular matrix formation, we also treated cells with 4-methylumbelliferone (4MU) to inhibit HA synthesis in carcinoma cells5, 7. To avoid potential off-target effects of 4MU on tumor cell metabolism, we treated cells with 400μM 4MU after determining this concentration inhibited HA production without inhibiting cell viability or growth. Pretreatment of carcinoma cells with 4MU decreased the mean fluorescence of HA on the surface of MDA-MB-231 cells by half (p = 0.0057) (Figure 4b) and was associated with a significant decrease in HA output, as measured by ELISA, for cells in the device (Figure 4c, p = 0.0133) and cells cultured in multiwell plates (Figure 4d, p = 0.0111). In experiments where cells were treated with hyaluronidase, we observed that the primary effect was on cell motility and invasiveness (Figure 3c and 3d). To reproduce this effect pharmacologically, we quantified invasive potential of tumor cells using a similar protocol to that shown in Figure 3a, but we used a version of our device without endothelium to avoid any confounding effects of 4MU treatment on the endothelial cells (Figure 4a). We compared metastatic potential of highly metastatic MDA-MB-231 cells, less metastatic HS578T cells, and poorly metastatic MCF7 cells. We observed that MDA-MB-231 cells treated with 4MU showed significantly less invasion into the collagen compared to controls (Figure 4e). HS578T cells also showed significantly (p = 0.0357) reduced invasion potential when treated with 4MU. By contrast, we observed no significant inhibition of the invasion of poorly metastatic MCF7 cells. We also observed that the average invasion distance of highly metastatic MDA-MB-231 cells was significantly reduced with 4MU treatment (Figure 4f). These results confirm our findings that the absence of the HA pericellular matrix leads to a less migratory and invasive phenotype and suggest that pharmacologically inhibiting formation of the pericellular matrix may function to limit metastatic carcinoma cell extravasation and invasion.
Discussion
In this study, we have developed an in vitro model of tumor cell extravasation at the metastatic site and have used this platform to study the role of tumor cell pericellular matrix on the metastatic potential of breast carcinoma cells. We showed that pericellular HA correlates with the metastatic potential of breast carcinoma cells in vivo and that removal of the pericellular matrix significantly reduces metastatic potential in our microfluidic model. Additionally, we tested a putative therapeutic that targets HA synthesis, and measured reduced pericellular matrix formation that correlates with a reduction in metastatic potential in our in vitro model system. These findings highlight a potentially significant role for the pericellular matrix in metastatic extravasation.
We observed significant decreases in the number of breast carcinoma cells that transmigrated the endothelium and in the distance these cells migrated into the 3D tissue after enzymatic removal of pericellular HA. This decrease in extravasation potential may be linked to HA-dependent increases in survival and motility of cancer cells via several mechanisms including activation of downstream signaling from HA binding to receptors such as CD44 and RHAMM2, 29 and by trapping free growth factors and cytokines in close proximity to their respective cell surface receptors2. We had initially hypothesized that the presence of the pericellular coat would also mediate cell adhesion before integrins and focal adhesions, but we did not observe a significant difference in adhesion of hyaluronidase treated tumor cells to endothelium in our devices. We expect that this may be because the HA-endothelial interaction is transient and the steady state differences in adhesion, as represented in our measurement, are not substantial. Thus, although we cannot rule out an effect of the pericellular matrix on endothelial adhesion, we have clear evidence that the pericellular matrix promotes endothelial transmigration and an invasive phenotype in our microfluidic model.
The receptors CD44 and RHAMM are likely candidates as HA-mediated drivers of the observed differences in invasiveness and transmigration after removal of the pericellular matrix or inhibition of pericellular matrix formation. While both CD44 and RHAMM bind HA, they differ in their primary and secondary structures of their HA binding sites and/or their response to different size HA polymers and fragments. High molecular weight HA polymers (105 to >106 Da) function to cluster cell surface CD44 within lipid raft signaling complexes. This helps carcinoma cells maintain an autonomous phenotype by lowering the activation threshold of invasion-regulating receptor tyrosine kinases (e.g. c-MET, ERBB2, EGFR, IGFR1-β) activated by autocrine growth factors30. In addition to activating multiple survival mechanisms, these CD44 clusters also reorganize the cytoskeleton which impacts cellular motility30, 31. High levels of low molecular weight HA fragments released into the microenvironment can reverse the protumorigenic influence of high molecular weight HA-induced CD44 clustering and improve sensitivity to therapeutic agents1. In contrast, RHAMM has a more complex subcellular distribution, since it is localized both to the cell surface and within cells31. Cell surface RHAMM can bind to HA fragments to promote motility and regulate the duration of activated oncogenic pathways such as MAPK30, 32. Additional work is needed to identify the most important signaling pathways that link the pericellular matrix to increased metastatic potential, but the microfluidic platform we have developed will also be useful in systematically probing these pathways and may help uncover new therapeutic targets.
Although more investigation is necessary to pinpoint specific HA-dependent mechanisms that influence metastatic potential, previous studies have shown that the pericellular matrix is a key influence in the increased malignancy and resistance to therapy observed in highly metastatic subpopulations3-6, making HA synthesis reduction a prime therapeutic target. Our results using 4MU to inhibit HA synthesis suggest that a decrease in pericellular matrix formation is correlated with decreased invasiveness, adding more evidence that inhibiting pericellular matrix formation by reducing HA synthesis may be a viable strategy for blocking metastatic progression. Notably, 4MU has been shown to be extremely effective without toxicity in pre-clinical trials for the treatment of prostate cancer33, 34, which is a glandular epithelial carcinoma like breast cancer, so may also be a potential treatment for breast carcinoma. In addition to targeting HA synthesis, other potential targets related to the pericellular matrix include the HA-CD44 complex. There are many attempts to disrupt this signaling complex using competitive binding substrates to both CD44 (i.e., antibodies, soluble CD44, BiKes, etc.) and HA (soluble hyaluronan binding proteins, HA oligomers bound to larger molecules) 3-4. These techniques have also been used to disrupt RHAMM which has been linked to the ERK and SRC signaling pathways and is instrumental in maintaining spindle integrity3. Although these treatments have shown promise and often cause tumor growth arrest in mice, these treatments have not transitioned to the clinic, suggesting a need for better tools to assess their potential efficacy before clinical trials.
An important outcome of this study is the development of several tools for testing new therapies and exploring mechanisms of metastasis. The flow cytometric analysis we present can be used to measure HA pericellular matrix expression in live cells and thus demonstrate the efficacy of potential treatments that inhibit pericellular matrix formation or that remove an established matrix. Additionally, the hyaluronidase treatment demonstrated here could serve as a positive control for therapies intended to inhibit pericellular matrix formation, as it appears to remove the matrix to a basal level. The microfluidic model of extravasation we developed could also be used to test potential therapies, obviating some of the challenges of traditional in vitro and in vivo models. Other microfluidic in vitro models have already contributed significantly to our understanding of metastatic progression10, 11. Although our model is a simplified representation of conditions in vivo, our results show that we can measure phenotypic response to removal of cell surface HA and reduction of HA synthesis through the relevant stages of the extravasation process. Moreover, the biological and biochemical components of this platform are highly modular, allowing application of this model to study other mechanisms of tumor progression and metastasis, including the roles of somatic and immune cells in the pre-metastatic niche and the composition of the ECM in the pre-metastatic microenvironment. For example, metastasis to bone could be modeled using fenestrated bone marrow endothelial cells grown on an underlying ‘bone marrow stroma’ by including appropriate stromal cells within the ECM. As a tunable model system of the metastatic microenvironment, this in vitro model may serve as an ideal platform for testing many types of potential therapeutics as well as uncovering their mechanisms of action before proceeding to in vivo trials.
Conclusion
In this study, we report the development of an in vitro model of tumor cell metastatic extravasation and show that the presence of an HA-rich pericellular matrix correlates with metastatic potential of breast carcinoma cells in our in vitro model. Additionally, we show removal of this pericellular matrix limits carcinoma extravasation and invasion and that a pharmacological inhibitor of HA synthesis reduces pericellular matrix formation and invasiveness in breast carcinoma cells. This study highlights a potentially key role for carcinoma associated HA in metastatic extravasation, and it demonstrates the utility of engineered in vitro models to discover mechanisms of metastasis and to test potential therapies.
Acknowledgements
This work was supported by the NCI under grants CA205455 and CA210190. We would like to thank the Minnesota Nanofabrication Center for support with device fabrication and the University Imaging Centers for microscopy support. We also would like to thank Ali Khammanivong and Erin Dickerson for their input on measuring cell surface HA, Paolo Provenzano for his generous gift of MDA-MB-231eGFP cells, and Kaylee Schwertfeger for her generous gift of HS578T cells.
References
- 1.Toole BP and Hascall VC, The American Journal of Pathology, 2002, 161, 745–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turley EA, Wood DK and McCarthy JB, Cancer Res, 2016, 76, 2507–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toole BP, Nat Rev Cancer, 2004, 4, 528–539. [DOI] [PubMed] [Google Scholar]
- 4.Monslow J, Govindaraju P and Pure E, Front Immunol, 2015, 6, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McAtee CO, Barycki JJ and Simpson MA, Adv Cancer Res, 2014, 123, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson MA, The American Journal of Pathology, 2006, 169, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avnet S and Cortini M, Stem Cell Rev, 2016, 12, 464–475. [DOI] [PubMed] [Google Scholar]
- 8.Dai L, Guinea MC, Slomiany MG, Bratoeva M, Grass GD, Tolliver LB, Maria BL and Toole BP, Am J Pathol, 2013, 182, 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grass GD, Tolliver LB, Bratoeva M and Toole BP, J Biol Chem, 2013, 288, 26089–26104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB and Kamm RD, Proceedings of the National Academy of Sciences, 2012, 109, 13515–13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M and Kamm RD, Proc Natl Acad Sci U S A, 2015, 112, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen MB, Whisler JA, Jeon JS and Kamm RD, Integr Biol, 2013, 5, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, Farach-Carson MC and Jia X, Biotechnol Adv, 2014, 32, 1256–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de la Motte CA and Drazba JA, J Histochem Cytochem, 2011, 59, 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fotia C, Messina GML, Marletta G, Baldini N and Ciapetti G, Eur Cells Mater, 2013, 26, 133–149. [DOI] [PubMed] [Google Scholar]
- 16.Brett M-E, Deflorio R, Stone DE and Eddington DT, Lab on a Chip, 2012, 12, 3127–3134. [DOI] [PubMed] [Google Scholar]
- 17.Li CY, Wood DK, Hsu CM and Bhatia SN, Lab Chip, 2011, 11, 2967–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love JC, Wolfe DB, Jacobs HO and Whitesides GM, Langmuir, 2001, 17, 6005–6012. [Google Scholar]
- 19.Qin D, Xia Y and Whitesides GM, Nat Protoc, 2010, 5, 491–502. [DOI] [PubMed] [Google Scholar]
- 20.Hsu Y-H, Moya ML, Abiri P, Hughes CCW, George SC and Lee AP, Lab on a Chip, 2013, DOI: papers3://publication/doi/10.1039/c2lc40787f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abramoff MD, Magalhaes PJ and Ram SJ, Biophotonics International, 2004, 11, 36–42. [Google Scholar]
- 22.Stern R and Jedrzejas MJ, Chem Rev, 2006, 106, 818–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacroix M and Leclercq G, Breast Cancer Res Tr, 2004, 83, 249–289. [DOI] [PubMed] [Google Scholar]
- 24.Su YR, Pogash TJ, Nguyen TD and Russo J, Cancer Med-Us, 2016, 5, 558–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang RD, Fidler IJ and Price JE, Invas Metast, 1991, 11, 204–215. [PubMed] [Google Scholar]
- 26.Koh M, Yong HY, Kim ES, Son H, Jeon YR, Hwang JS, Kim MO, Cha Y, Choi WS, Noh DY, Lee KM, Kim KB, Lee JS, Kim HJ, Kim H, Kim HH, Kim EJ, Park SY, Kim HS, Moon WK, Kim HRC and Moon A, Int J Cancer, 2016, 138, 1232–1245. [DOI] [PubMed] [Google Scholar]
- 27.Koutsiaris AG, Tachmitzi SV, Batis N, Kotoula MG, Karabatsas CH, Tsironi E and Chatzoulis DZ, Biorheology, 2007, 44, 375–386. [PubMed] [Google Scholar]
- 28.Tarbell JM, Cardiovascular Research, 2010, 87, 320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Agostino A, Stellavato A, Corsuto L, Diana P, Filosa R, La Gatta A, De Rosa M and Schiraldi C, Carbohyd Polym, 2017, 157, 21–30. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton SR, Fard SF, Paiwand FF, Tolg C, Veiseh M, Wang C, McCarthy JB, Bissell MJ, Koropatnick J and Turley EA, J. Biol. Chem, 2007, 282, 16667–16680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Misra S, Hascall VC, Markwald RR and Ghatak S, Frontiers in Immunology, 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Telmer PG, Tolg C, McCarthy JB and Turley EA, Communicative & integrative biology, 2011, 4, 182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yates TJ, Lopez LE, Lokeshwar SD, Ortiz N, Kallifatidis G, Jordan A, Hoye K, Altman N and Lokeshwar VB, Jnci-J Natl Cancer I, 2015, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagy N, Kuipers HF, Frymoyer AR, Ishak HD, Bollyky JB, Wight TN and Bollyky PL, Front Immunol, 2015, 6, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]