

Abstract
Wound healing is a complex process that consists of multiple phases, each of which are indispensable for adequate repair. Timely initiation and resolution of each of these phases namely, hemostasis, inflammation, proliferation and tissue remodeling, is critical for promoting healing and avoiding excess scar formation. While platelets have long been known to influence the healing process, other components of blood particularly coagulation factors and the fibrinolytic system also contribute to efficient wound repair. This review aims to summarize our current understanding of the role of platelets, the coagulation and fibrinolytic systems in cutaneous wound healing, with a focus on how these components communicate with immune and non-immune cells in the wound microenvironment. We also outline current and potential therapeutic strategies to improve the management of chronic, non-healing wounds.
Keywords: cutaneous wound healing, coagulation factors, platelets, neutrophils, macrophages, chronic wounds
Introduction
The skin is a vital organ that serves as a barrier to external agents. A wound results from a break in continuity of the surface epithelium or underlying connective tissue secondary to either mechanical, thermal or chemical injuries. Wound healing is a homeostatic process that restores skin integrity following injury or tissue damage and helps prevent the entry of infectious pathogens.
Wound healing is a delicate and dynamic process that involves four distinct, sequential phases that overlap in time: hemostasis, inflammation, proliferation and tissue remodeling (Figures 1 and 2).1–3 Timely initiation, effective progression and resolution of each phase is key to timely tissue regeneration and wound closure.
Figure 1. Phases of wound healing.
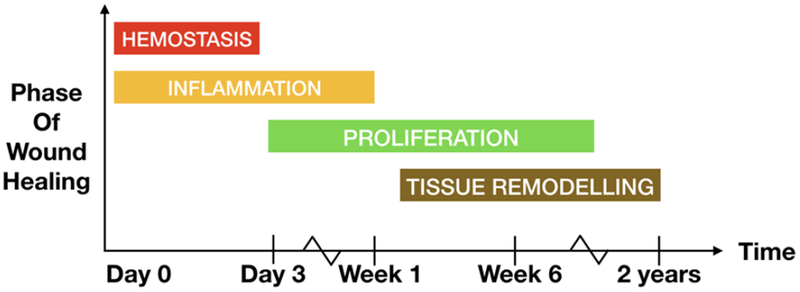
Timeline depicting the sequential yet overlapping phases of wound healing namely, hemostasis (red), inflammation (yellow), keratinocyte proliferation, angiogenesis (green) and tissue remodeling (brown).
Figure 2. Contribution of hematopoietic cells to wound healing.
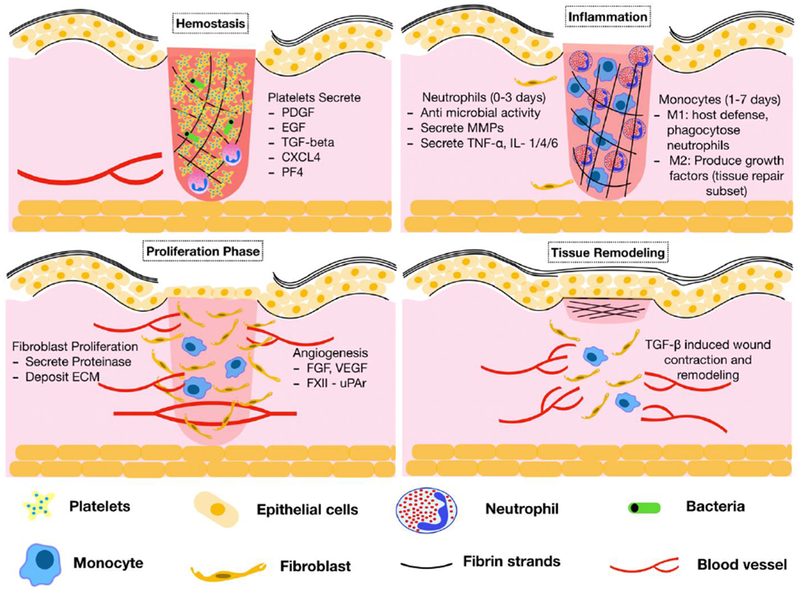
The phases of wound healing are pictorially described here. During the hemostatic phase of wound healing, platelets secrete growth factors and chemokines that facilitate recruitment of inflammatory cells. The inflammatory phase is characterized by early neutrophil responses followed subsequently, by monocyte and macrophage recruitment. MMPs from neutrophils and the fibrinolytic system clear the extracellular matrix and fibrin clot leading to keratinocyte migration and proliferation for wound closure. TGF-β from platelets and macrophages facilitates Myofibroblast differentiation and proliferation thus enabling wound contraction and scar formation. CXCL4=CXC chemokine ligand 4; ECM=extracellular matrix; EGF=epidermal growth factor; FGF=fibroblast growth factor; IL=interleukin; MMP=matrix metalloproteinase; PDGF=platelet derived growth factor; PF4=platelet factor 4; TGF-β= transforming growth factor-β; TNF-α=tumor necrosis factor-α; uPAR=urokinase plasminogen activator receptor; VEGF=vascular endothelial growth factor.
In this review, we focus on the role and significance of platelets, the coagulation and fibrinolytic systems in cutaneous wound healing, their interactions with the wound microenvironment, their contribution to the development of chronic wounds and therapeutic interventions that can improve wound healing.
Physiologic wound healing: phases, components, functions and mediators Hemostasis
Most wounds to the skin will cause leakage of blood from damaged blood vessels and result in rapid platelet recruitment. The formation of a clot then serves as a temporary shield protecting the denuded wound tissues and provides a provisional matrix over and through which cells can migrate during the repair process. The clot consists of platelets embedded in a mesh of cross-linked fibrin fibers derived by thrombin cleavage of fibrinogen, together with smaller amounts of plasma fibronectin, vitronectin, and thrombospondin.3,4
At injury sites, the clot serves as a reservoir of cytokines and growth factors that are released as activated platelets degranulate. Dense granule contents (ADP, serotonin and polyphosphate) promote additional platelet recruitment, aggregation and fibrin formation whereas alpha granules produce and release multiple chemokines, growth factors, pro-and anti-inflammatory mediators.5 Among them, platelet-derived growth factor (PDGF) is chemotactic for macrophages and promotes fibroblast proliferation.6,7 Epidermal growth factor (EGF) is also released from platelets and primarily serves as a keratinocyte proliferation signal.3 Other growth factors and chemokines secreted from platelets include CXC chemokine ligand-4 (CXCL-4), platelet factor 4 (PF4) and transforming growth factor-β (TGF-β) that recruit inflammatory cells, promote keratinocyte migration, fibroblast matrix synthesis and remodeling.8,9 In addition to platelet-derived soluble mediators, platelets themselves establish heterotypic interactions with inflammatory cells recruited at wound sites. These neutrophil-platelet and macrophage-platelet interactions, mediated by integrin receptors and P-selectin on the surface of activated cells, upregulate inflammatory cell recruitment and facilitate progression into the inflammatory phase of wound healing.10,11 The hemostasis phase leads to the formation of fibrin which is cross-linked at injury sites by factor XIII (FXIII).12,13 In addition to preventing bleeding, the fibrin clot serves an important role in cell adhesion, endothelial cell (EC) migration, angiogenesis and acts as a temporary extracellular matrix.14 The fibrin clot also serves as a host defense by containing bacterial spread.15,16 Recently, in a murine model of dermal infection, the fibrin biofilm retained blood cells and protected against transcutaneous microbial dissemination.17
Mechanistic insight from murine wound models
Genetically modified mouse models have proven extremely helpful at dissecting the contribution of specific coagulation factors in tissue repair (Table 1). Monroe et al. examined wound healing in mice with defects in the initiation (low tissue factor) and propagation (low factor IX, hemophilia B) phases of coagulation. In hemophilia B mice, dermal wound healing was delayed and bleeding into the granulation tissue was noted. Pretreating hemophilic mice with replacement therapy (factor IX or bypass agents) just prior to wounding, did not restore wound healing potential.18 In another mouse model of hemophilia B, macrophage infiltration in skin wounds was delayed compared to controls (1-5 days versus 10 days), suggesting a role for FIX in macrophage-related inflammation.19 Similar findings were found in factor IX knockout mice which demonstrated defective joint healing after episodes of hemarthrosis.20 Use of glycopegylated factor IX preparation, resulted in improved synovial healing and preserved osteochondral architecture.20 Low tissue factor mice also displayed defective wound healing.18,21 Gao et al. evaluated cutaneous wound healing in Factor VIII deleted mice and found that they exhibited decreased wound contraction and delayed rate of wound healing compared to wild type and heterozygous mice.22 While no studies have addressed the contribution of Factor XI (FXI) to cutaneous wound healing, combined deficiency in FXI and Plasminogen (Pg), resulted in progressively worse fibrin deposition and increased lung inflammation compared to plasminogen deficient only mice.23 It will be important to investigate if increased fibrin deposition, itself accounts for this proinflammatory phenotype or whether FXI directly influences the inflammatory response. To test directly the effect of fibrinogen deficiency on cutaneous tissue repair, fibrinogen deficient mice and control mice were investigated using incisional and excisional wounds.24 The time required to overtly heal wounds was similar in fibrinogen deficient and control mice, but histologic evaluation revealed distinct differences in the repair process, including an altered pattern of epithelial cell migration and increased epithelial hyperplasia.24 Furthermore, granulation tissue in fibrinogen deficient mice failed to adequately close the wound gap, resulting in persistent open wounds or partially covered sinus tracts. The tensile strength of these wounds was also reduced compared with control mice. These studies show that reepithelialization, granulation tissue formation, including the establishment of neovasculature, and the formation of fibrotic scar tissue can proceed in the absence of fibrin(ogen) and all of its proteolytic derivatives. However, fibrin (ogen) is important for appropriate cellular migration and organization within wound fields and in initially establishing wound strength and stability.25 Similarly, in a murine model of Factor XIII deficiency, wound healing was delayed and histologic analysis of wounds showed decreased reepithelialization and necrotic fissures. Recombinant factor XIII replacement, restored wound healing potential.26
Table 1. Hematopoietic cells, their functions and mediators released in tissue repair. This table also summarizes genetic mouse models of inflammation and cutaneous wound healing.
CXCL-4=CXC chemokine ligand 4; ECM=extracellular matrix; EGF=epidermal growth factor; MMP=matrix metalloproteinase; NETs=neutrophil extracellular traps; PDGF=platelet derived growth factor; PF4=platelet factor 4; Pg=plasminogen; TGF-β= transforming growth factor-β, uPA= urokinase plasminogen activator, uPAR= urokinase plasminogen activator receptor.
| Phase | Coagulation factor/Blood component | Growth factor, Mediator | Effect | Mechanistic insight from murine knockouts (reference) |
|---|---|---|---|---|
| Hemostasis | Platelets | PDGF | Chemotaxis of macrophages, fibroblast mitogen | |
| EGF | Keratinocyte proliferation | |||
| CXCL-4 PF4 | Recruitment of inflammatory cells | |||
| Factor IX | Early and late wound healing (cutaneous and joint), macrophage inflammation | Hemophilia B mice: Delayed dermal wound healing with bleeding into granulation tissue (63); delayed macrophage infiltration (64); defective joint healing (65) | ||
| Factor XIII | Re-epithelialization and wound closure | FXIII deficient mice: Delayed re-epithelialization compared to controls (67) | ||
| Inflammation | Neutrophils | Serine proteases; NETs | Antimicrobial activity | Cathepsin G deficient mice: increased neutrophilic inflammation, delayed wound healing (76) |
| MMPs | ECM degradation, cell migration | PU.1 null mouse: normal wound healing as wild type mice (77) | ||
| Macrophages | Inflammation, angiogenesis M1: host defense M2: wound repair | Prevention of macrophage infiltration: defective wound healing (78) | ||
| Transgenic mice with macrophage elimination: Impaired re-epithelialization, collagen deposition and angiogenesis (79) | ||||
| Protease expression, re-epithelialization and angiogenesis | Plasminogen | Extracellular proteolysis | Pg deficient mice: Delayed wound healing and prolonged inflammation (69, 70) | |
| uPA | Extracellular proteolysis (via Pg activation) | uPA knockout mice: Impaired wound healing (73) | ||
| uPAR | Extracellular proteolysis, inflammation (via FXII) | uPAR knockout (Plaur−/−)mice: no defects in wound healing (73) | ||
| Plau(GFDhu/GFDhu) mice: selective abrogation of uPA-uPAR interaction: no disruption of wound healing (74) | ||||
| Tissue remodeling | Platelets | TGF-β | Wound contraction | CD18 deficient mice: defective wound contraction (80) |
| Macrophages Macrophages | Wound contraction | |||
In summary, platelets and the formation of a fibrin clot serve as important pools of cytokines and growth factors that “jump start” the wound closure process: they provide chemotactic cues to inflammatory cells, initiate cell movements of re-epithelialization and connective tissue contraction, and stimulate the wound angiogenic response.
Recruitment of inflammatory cells to the wound site
The inflammatory phase is the second phase of wound healing. It is characterized by migration of neutrophils and monocytes into wound sites. The presence of inflammatory cells into the wound microenvironment, primarily helps in clearing bacteria and debris. It also lays the background for keratinocyte proliferation to restore skin architecture. Neutrophils arrive into wounds within minutes of injury and form the major inflammatory cell type during the first day of wound healing.27 Neutrophils are attracted into wounds by a variety of mediators including fibrinopeptides, fibrin degradation products, neutrophil activating peptide-2 (NAP-2), growth-related oncogene a (GROα), and interleukin-8 (IL-8).27–30 Neutrophil interactions with endothelial cells and platelets occur via P- and E- selectins and facilitate further inflammatory cell recruitment.31 On arrival, neutrophils release chemoattractants [leukotriene B4 (LTB4) and CXCL8] and begin the process of clearing microbes and devitalized tissues.32 The armamentarium of neutrophils is rich and diverse. Antimicrobial activity is executed by free radical-dependent and -independent mechanisms.33,34 The former include intracellular killing of pathogens (phagocytosis) and the latter includes formation of neutrophil extracellular traps (NETs).35,36 NETs are web-like structures composed of DNA material (citrullinated histones) decorated with antimicrobial granular contents such as neutrophil elastase (NE) and myeloperoxidase (MPO).35,36 The process of releasing NETs is termed NETosis and can result in cell death (suicidal NETosis) or preservation of neutrophils (vital NETosis).37 Whereas suicidal NETosis requires hours of stimulation and oxidant production, vital NETosis takes place within minutes of stimulation of neutrophils with bacteria or bacterial products, Toll-Like Receptor (TLR) 4-activated platelets, or complement proteins.38 NETosis was initially described as an additional mechanism through which neutrophils help catch and kill bacteria. However, increasing evidence suggests that this process might also occur in noninfectious, sterile inflammation. In such settings, aberrant suicidal NETosis over time can be harmful to the host and lead to the development of chronic, non-healing wounds. This will be discussed in detail elsewhere in this review. In addition to their antimicrobial role, neutrophils are also a source of pro-inflammatory cytokines that serve some of the earliest signals to activate local fibroblasts and keratinocytes.39,40,41,42 Among them are the members of the matrix metalloproteinases (MMP) family, each of which cleaves a specific subset of matrix proteins.39 Matrix metalloproteinase 8 (MMP-8) is a collagenase secreted early from neutrophils. Its primary role is to remove damaged type I collagen and wound debris.40,41 MMP-9 is another important gelatinase secreted at later stages of wound healing, usually by days 2-4 post-wounding.41 MMP-9 can cut basal lamina collagen (type IV) and fibril collagen (type VII) and is thought to be responsible for releasing keratinocytes from their tethers to the basal lamina.42 Ultimately, neutrophils pave the way for subsequent macrophage entry into wound sites through release of interleukin (IL) – 4, IL-1, IL-6 and tumor necrosis factor α (TNF-α).43,44 Macrophage influx starts approximately 24 – 48 hours from the time of injury.45
Macrophages are the most abundant hematopoietic cells in intact skin but the majority of macrophages involved in wound healing are derived from blood monocytes.46 Thrombin and PF4 produced by platelets act as chemokines for monocytes, enhance monocyte recruitment,47 promote monocyte differentiation into macrophages and help in the differentiation of monocytes into dendritic cells.9,48,47,49,50,51 On arrival at the site of injury, macrophages develop into two distinct phenotypes namely, the M1 and M2 subtypes.52 The M1 subtype promotes inflammation and plays an important part in host defense against infection. In contrast, the M2 subtype suppresses inflammation and helps in wound repair by producing growth factors. In the early phase, under the influence of TNF-alpha, macrophages are mainly of the M1 subtype.53 Later in wound healing, M1 macrophages induce apoptosis of neutrophils and phagocytose expended neutrophils.54 The process of neutrophil phagocytosis initiates a cascade of events during which cytokine production is turned off and macrophages transition to the M2 or tissue reparative phenotype where they release transforming growth factor - β1 (TGF-β1), a key regulator of myofibroblast differentiation and wound contraction.55 This process ensures timely resolution of inflammation and transition to the proliferative phase of wound healing.
Timely ‘phasing out’ of the neutrophil component in wounds, is key to efficient wound healing. In fact, it has previously been reported that depletion of neutrophils accelerates the rate of re-epithelialization in sterile, non-diabetic and diabetic murine wounds.56 Similar studies with the PU.1 null mouse, which lacks cells of the myeloid lineage (neutrophils, macrophages, mast cells, eosinophils) and B cells showed that despite little inflammation at the wound site, healing occurred at a similar rate to wild-type siblings and repair progressed normally.57 In a study of cathepsin G deficient mice, a higher neutrophil recruitment was noted and impaired wound healing was observed, suggesting that persistent inflammation can be deleterious to the healing wound.58
In contrast to the aforementioned studies, macrophages are indispensable to the healing process; if macrophage infiltration is prevented, then healing is severely impaired.59 Studies using transgenic mice where macrophages could be eliminated during wound healing showed that macrophage depletion leads to impaired reepithelization, decreased collagen deposition and impaired angiogenesis.60,61 Similarly, eliminating all neutrophil entry into wound sites such as reported in CD18 deficient mice, has the potential to deprive macrophages of their main stimulus to release TGF-β1 and was shown to lead to impaired wound contraction.61
Fibrinolytic protease expression, reepithelialization and angiogenesis
Once the inflammatory phase subsides, the reepithelialization phase is geared towards closing the excisional skin wound. This stage overlaps with angiogenesis leading to the formation of healthy and vascularized granulation tissue. In this phase, keratinocytes at the wound edge and epithelial cells from hair follicles in the vicinity, migrate and proliferate. Signals that promote keratinocyte proliferation include EGF, TGF-α, heparin binding epidermal growth factor and fibroblast growth factor secreted from platelets, macrophages and dermal fibroblasts.3 Activated protein C (APC) also facilitates proliferation via cleavage of protease activated receptor-1 (PAR-1) and by binding to PAR-2, both of which are expressed on keratinocytes.62,63 Similar to TGF-β, PAR2 agonists inhibit proliferation or differentiation of human neonatal keratinocytes, whereas PAR-1 agonists stimulate proliferation.64 In contrast, agonists of both PAR-2 and PAR-1 are mitogenic for endothelial cells.65 Moreover, PAR-2 regulates expression of cell adhesion molecules in primary cultures of human endothelial cells and cell lines.66,67 Finally, PAR-2 appears to play a role in the regulation of leukocyte-endothelial interactions in humans and mice in vivo, as shown for atopic dermatitis or experimentally induced contact dermatitis.67,68
Keratinocyte migration is triggered by the loss of physical tension at points of cell attachment to the basal lamina. In order to pass through the fibrin clot, leading–edge keratinocytes have to dissolve the fibrin barrier ahead of them. Plasmin, the chief fibrinolytic enzyme, is derived from plasminogen (Pg) within the clot and can be activated by tissue-type plasminogen activator (tPA) or urokinase-type plasminogen activator (uPA).69 While tPA is found in the circulation and plays a key role in intravascular fibrinolysis, uPA is essential in extracellular proteolysis, wound healing and tissue remodeling. Both uPA and its receptor uPAR are up-regulated in migrating edge keratinocytes.70 In these cells, uPA binding to uPAR and other transmembrane receptors such as integrins and epidermal growth factor receptor (EGFR) affects cell migration, adhesion, differentiation and proliferation via intracellular signaling.71 Generated plasmin degrades fibrin and other matrix glycoproteins (peri-cellular proteolysis), facilitates keratinocyte migration during reepithelialization, regulates growth factor production and release, and activates matrix metalloproteinases which assist in ECM degradation and tissue remodeling.72,73,74 Plasmin also mediates a negative feedback loop by activating transforming growth factor-β (TGF-β) which in turn, stimulates plasminogen activator inhibitor-1 (PAI-1) to turn off fibrinolysis.75,76 Expression of MMPs namely MMP-1 (collagenase) and MMP-9 (gelatinase B) is also increased in wound edge keratinocytes, both of which cut through matrix proteins and aid in cell migration.3
It is important to note that uPA and uPAR have differential roles in the wound microenvironment. Both uPA and its receptor are not only expressed in keratinocytes but they are also abundantly upregulated in inflammatory cells (neutrophils and macrophages) where they promote cell influx into inflamed areas.77,78 Moreover, recent data show that uPAR on the neutrophil surface serves as the receptor for zymogen factor XII (FXII). The FXII-uPAR interaction upregulates neutrophil functions including adhesion, migration, chemotaxis and NET formation. The sum of these activities results in persistent inflammation and delayed wound healing.79
Preclinical studies reinforce the importance of components of the fibrinolytic system in wound repair. In a study of plasminogen deficient mice, wound healing was disrupted when compared to wild type mice.80 In another study, wounds of plasminogen deficient mice exhibited increased fibrin deposition and sustained inflammation several months after re-epithelialization, suggesting a role for plasminogen in inflammation modulation.81 Plasminogen treatment of wild type mice with burn wounds or diabetic mice, accelerated wound healing.82 There have been conflicting reports on the wound healing potential of uPAR knockout mice.83,85 Bugge and Connolly show that loss of uPA, but not uPAR, delays wound healing.84,85 Importantly, abrogating the uPA-uPAR interaction has no effect on wound healing.85 The authors conclude that uPA promotes wound repair independent of binding to its receptor.85 It was previously shown that uPA not only binds uPAR but also extracellular matrix proteins.86 This may well explain why uPA is not functionally redundant in wound repair.
Angiogenesis is induced by Fibroblast Growth Factor (FGF) secreted by macrophages and damaged endothelial cells whereas, VEGF is primarily produced by keratinocytes and wound-resident macrophages.87 These pro-angiogenic factors allow endothelial cells from exiting blood vessels to establish sprouts within the wound. In endothelial cells, zymogen FXII binds to a multi-receptor complex that consists of uPAR, EGFR and β1 integrin to promote phosphor-AktS473 leading to cell proliferation and post-natal angiogenesis.88,89 A direct role for von Willebrand factor (vWF) in wound healing has not been clearly established but a recent observational study found that various angiogenic mediators including angiopoietin 1 and 2, VEGF and galectin-3, are significantly different between types of vW disease.90
Tissue remodeling
The tissue remodeling phase is characterized by wound contraction to ease epithelialization and scar formation. Fibroblasts migrate into wounds on day 3 to 4 and start depositing a collagen-rich matrix.3 Fibroblast proliferation and migration occurs in response to cytokines and growth factors released from platelets and macrophages [PDGF, TGF-β and FGF].91,92 TGF-β also induces differentiation of fibroblasts to myofibroblasts, a critical step for wound contraction.92,93 Scar formation occurs with replacement of collagen III by collagen I that exhibits higher tensile strength. Besides the role of macrophages, no other blood cells or coagulation system components are implicated in this phase of wound healing.
Chronic non-healing wounds, current therapeutic targets
With increasing age, both the morphology and functions of the skin change, due to intrinsic (e.g., hormone levels) and extrinsic factors (e.g., sun exposure). The age-related alterations in the skin result in delayed, but not defective wound healing.94 In the hemostasis phase, aggregation and degranulation of platelets are enhanced in the elderly.95 During the inflammatory phase, increased neutrophil response and delayed monocyte infiltration in wounds have been observed in the aged compared with young controls.96 Moreover, phagocytic activity of wound macrophages in aged mice is decreased compared to young mice, which may account for the increased production of proinflammatory cytokines including IL-1, IL-6, TNF-α, and decreased secretion of VEGF.95,97,98 These responses lead to delayed reepithelialization, angiogenesis and granulation tissue formation in the elderly. In addition to these age-related alterations in the skin, factors associated with aging, e.g., reduced sex steroid hormones, immobilization, malnutrition, medications and comorbidities (diabetes, venous insufficiency, peripheral artery disease), increase susceptibility to chronic wounds. Chronic, non-healing wounds represent a major health care burden, costing 25 billion dollars annually in US health care costs and are associated with high mortality.99 Current treatments for impaired wound healing focus mainly on optimization of controllable healing factors, e.g., mechanical protection, nutritional support and clearance of infections.100 Several approaches have also been studied for local delivery of therapeutic agents to wound sites. Sub-atmospheric pressure dressings are very costly.101 Similarly, hyperbaric oxygen therapy was shown to be efficacious in the treatment of burn wounds but its use is limited by low availability and high cost.102 Tissue-engineered human skin equivalents are ultimately rejected, so their primary task appears to be a transient restoration of the dermis.103 Autologous skin transplantation requires a viable and well-perfused wound site to be successful.104,105 Alternative strategies include the topical use of recombinant growth factors such as platelet-derived growth factor (rPDGF). Clinically, rPDGF has demonstrated only modest improvements in healing diabetic and pressure ulcers and recent data reported an increased cancer risk in connection with its use.106,107 Platelet rich plasma (PRP) has also been used for difficult-to-treat wounds. Driver et al. carried out the first reported prospective, randomized, controlled multicenter trial in the U.S. regarding the use of autologous PRP for the treatment of diabetic foot ulcers.108 In this study, investigators compared the effectiveness of autologous PRP gel to that of normal saline gel for 12 weeks. The primary objective of this study was to evaluate the safety of PRP and the incidence of complete wound closure, defined as 100 percent re-epithelialization when compared to control treatment, a secondary objective was rate of wound closure. The study found that 68.4 percent (13/19) of patients in the PRP group and 42.9 percent (9/21) in the control group had wounds that healed. Wounds in the PRP group healed after a mean of 42.9 days (SD 18.3) vs. 47.4 days (SD 22.0) in the control group. This study was limited somewhat because it excluded ulcers with “challenging presentations” such as mild to moderate vascular disease and exposed tendon or bone, in addition to patients with hyperglycemia and/or inadequate nutritional status.108 In contrast to findings by Driver et al., another randomized prospective double-blind placebo-controlled study by Krupski et al. investigated the use of autologous platelet-derived wound healing formula (PDWHF), a mixture of growth factors including PDGF, PF-4, TGF-β, platelet-derived epidermal growth factor (PDEGF), and platelet-derived angiogenesis factor (PDAF).109 PDWHF was investigated in 18 patients with 26 lower extremity wounds of at least eight weeks duration. Only 78 percent of the patients were diabetic and all were men ranging from 57 to 75 years old. Over the 12-week study period, the investigators did not find any improvement in wound healing with the use of PDWHF. Three (33 percent) wounds healed in two patients in the control group, and four (24 percent) wounds healed in three patients in the PDWHF group (p > 0.05). While this study was limited by a small sample size, its results suggested that treatment of chronic wounds with PDWHF is no better than traditional therapy. Human studies also failed to confirm a beneficial role for aspirin in the management of chronic venous leg ulcers.110,111 Lastly, tranexamic acid has been shown to improve the tensile strength of wounds in murine models however, this effect was thought to be independent of its antifibrinolytic activity.112 Currently, there are no human studies examining the use of tranexamic acid for treatment of chronic wounds.
Future prospects for the management of chronic wounds
Despite their heterogeneity, most non-healing wounds fail to progress through the normal phases of wound repair, but instead remain in a chronic inflammatory state.113 Indeed, continued recruitment, or buildup of active neutrophils, can prolong inflammation and contribute to the development of chronic wounds. Animal models show that excess neutrophil influx into wound sites impairs keratinocyte migration and proliferation.114 The persistence of neutrophils in wounds leads to unrestricted proteolytic activity mediated by neutrophil granular enzymes that are considered the final executor of a pathogenic chain leading to matrix disruption and proteolysis of growth factors and their receptors.115 Neutrophil elastase was previously shown to be markedly increased in the exudate of non-healing human wounds and is thought to reflect a chronic, inflammatory, tissue-destructive microenvironment.116 In contrast, high levels of alpha1-antitrypsin, an in vivo neutrophil elastase inhibitor, are a biomarker of successful wound healing.115 Neutrophil Elastase associates with NETs and is critical to their function.35 Recent studies show that circulating neutrophils from diabetic humans are primed to produce NETs and NETosis delayed diabetic wound healing in mice and humans.117,118 The involvement of NETs was corroborated by showing that DNase I treatment enhanced wound healing in wild type diabetic mice.117 Altogether, these data support that limiting the activity of neutrophils may be beneficial for the treatment of recalcitrant wounds. Future studies are needed to establish the benefit from an array of compounds designed to specifically inhibit peptidylarginine deiminase 4 (PAD4), an essential enzyme in the formation of NETs. Interestingly, the first generation PAD inhibitor, Cl-amidine, does not effectively block NETosis in human neutrophils,119 but new specific PAD4 inhibitors have been developed to inhibit both NET formation and histone citrullination.120
Concluding remarks
In summary, the healing of an adult skin wound is a complex process requiring the collaborative efforts of different tissues, cell lineages and soluble pro- and anti-inflammatory mediators. Components of the hemostatic and fibrinolytic systems play an indispensable role in the wound healing process. Besides their immediate contribution to the formation of a barrier ‘clot’ against blood loss and pathogens, their cross talk with inflammatory cells lays the ground for antimicrobial activity, ECM degradation, keratinocyte migration and proliferation and wound contraction. Our understanding of wound healing mechanisms has progressed considerably in recent years. Part of the difficulty in unraveling tissue repair mechanisms is a consequence of redundancy and cross-talk in the system. Most wound signals probably control more than one cell activity, and most cell activities are responses to cocktails of signals. Experimental mouse models have been particularly useful in answering open questions, because of our ability to manipulate the genetic, systemic, and wound environment. Although only a handful of knockout mice have been wounded so far, there have been some surprisingly normal healing phenotypes reported. Reports have raised questions on the validity of the essential prerequisite of inflammation for efficient tissue repair. Indeed, in experimental models of repair, inflammation has been shown to delay healing and to result in increased scarring. In this framework, the next few years in wound healing research will be exciting as we improve on our current understanding of the mechanisms controlling wound repair and test novel therapeutic targets to improve pathological would healing.
Highlights.
Platelets, coagulation and fibrinolytic factors influence cutaneous wound healing.
There is extensive crosstalk between the hemostatic system and the wound milieu.
Timely resolution of each phase of wound healing is critical for wound repair.
Buildup of active neutrophils, contributes to the development of chronic wounds.
Acknowledgments
FINANCIAL SUPPORT and SPONSORHIP
Work in Dr. Stavrou’s laboratory is supported by a grant from the National Institute of Health (HL137695) and the Oscar D. Ratnoff Endowed Professorship. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Abbreviations:
- ADP
adenosine diphosphate
- CXCL
CXC chemokine ligand
- EC
endothelial cell
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FGF
fibroblast growth factor
- GRO-α
growth related oncogene-α
- IL
interleukin
- LTB4
leukotriene B4
- MMP
matrix metalloproteinase
- MPO
myeloperoxidase
- NAP-2
neutrophil activating peptide-2
- NE
neutrophil elastase
- NETs
neutrophil extracellular traps
- PAR
Protease Activated Receptor
- PAI-1
plasminogen activator inhibitor-1
- PDGF
platelet derived growth factor
- PDWHF
platelet-derived wound healing formula
- PF4
platelet factor 4
- Pg
plasminogen
- PRP
platelet rich plasma
- TGF
transforming growth factor
- TNF
tumor necrosis factor
- tPA
tissue plasminogen activator
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
- VEGF
vascular endothelial growth factor
- vWF
von Willebrand factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341(10):738–746. [DOI] [PubMed] [Google Scholar]
- 2.Witte MB, Barbul A. General principles of wound healing. Surg Clin North Am. 1997;77(3):509–528. [DOI] [PubMed] [Google Scholar]
- 3.Martin P Wound healing--aiming for perfect skin regeneration. Science. 1997;276(5309):75–81. [DOI] [PubMed] [Google Scholar]
- 4.Clark RA. Regulation of fibroplasia in cutaneous wound repair. Am J Med Sci. 1993;306(1):42–48. [DOI] [PubMed] [Google Scholar]
- 5.Ross R, Glomset J, Kariya B, Harker L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(4):1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heldin CH, Westermark B. Platelet-derived growth factor: three isoforms and two receptor types. Trends in genetics : TIG. 1989;5(4): 108–111. [DOI] [PubMed] [Google Scholar]
- 7.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79(4):1283–1316. [DOI] [PubMed] [Google Scholar]
- 8.Gleissner CA, von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. 2008;28(11):1920–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia CQ, Kao KJ. Effect of CXC chemokine platelet factor 4 on differentiation and function of monocyte-derived dendritic cells. IntImmunol. 2003;15(8):1007–1015. [DOI] [PubMed] [Google Scholar]
- 10.Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev. 2007;21(2):99–111. [DOI] [PubMed] [Google Scholar]
- 11.Nurden AT. Platelets, inflammation and tissue regeneration. Thromb Haemost. 2011;105 Suppl 1:S13–33. [DOI] [PubMed] [Google Scholar]
- 12.Fang J, Hodivala-Dilke K, Johnson BD, et al. Therapeutic expression of the platelet-specific integrin, alphaIIbbeta3, in a murine model for Glanzmann thrombasthenia. Blood. 2005;106(8):2671–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ariens RA, Lai TS, Weisel JW, Greenberg CS, Grant PJ. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood. 2002;100(3):743–754. [DOI] [PubMed] [Google Scholar]
- 14.Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost. 2006;4(5):932–939. [DOI] [PubMed] [Google Scholar]
- 15.Dunn DL, Simmons RL. Fibrin in peritonitis. III. The mechanism of bacterial trapping by polymerizing fibrin. Surgery. 1982;92(3):513–519. [PubMed] [Google Scholar]
- 16.Mullarky IK, Szaba FM, Berggren KN, et al. Infection-stimulated fibrin deposition controls hemorrhage and limits hepatic bacterial growth during listeriosis. Infection and immunity. 2005;73(7):3888–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macrae FL, Duval C, Papareddy P, et al. A fibrin biofilm covers the blood clot and protects from microbial invasion. J Clin Invest. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monroe DM, Mackman N, Hoffman M. Wound healing in hemophilia B mice and low tissue factor mice. Thrombosis research. 2010;125 Suppl 1:S74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman M, Harger A, Lenkowski A, Hedner U, Roberts HR, Monroe DM. Cutaneous wound healing is impaired in hemophilia B. Blood. 2006;108(9):3053–3060. [DOI] [PubMed] [Google Scholar]
- 20.Sun J, Hua B, Livingston EW, et al. Abnormal joint and bone wound healing in hemophilia mice is improved by extending factor IX activity after hemarthrosis. Blood. 2017;129(15):2161–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Z, Xu H, Ploplis VA, Castellino FJ. Factor VII deficiency impairs cutaneous wound healing in mice. Molecular medicine (Cambridge, Mass.). 2010;16(5-6):167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao G, Mashausi DS, Negi H, Li D, Li D. A new mouse model for wound healing in hemophilia A. Int JClin Exp Pathol. 2015;8(3):3015–3021. [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng Q, Zhao Y, Lawson WE, et al. The effects of intrinsic pathway protease deficiencies on plasminogen-deficient mice. Blood. 2005;106(9):3055–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drew AF, Liu H, Davidson JM, Daugherty CC, Degen JL. Wound-healing defects in mice lacking fibrinogen. Blood. 2001;97(12):3691–3698. [DOI] [PubMed] [Google Scholar]
- 25.Barinka C, Parry G, Callahan J, et al. Structural basis of interaction between urokinase-type plasminogen activator and its receptor. JMolBiol. 2006;363(2):482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inbal A, Lubetsky A, Krapp T, et al. Impaired wound healing in factor XIII deficient mice. Thromb Haemost. 2005;94(2):432–437. [DOI] [PubMed] [Google Scholar]
- 27.Engelhardt E, Toksoy A, Goebeler M, Debus S, Brocker E- B, Gillitzer R. Chemokines IL-8, GROα, MCP-1, IP-10, and Mig Are Sequentially and Differentially Expressed During Phase-Specific Infiltration of Leukocyte Subsets in Human Wound Healing. The American journal of pathology. 1998;153(6):1849–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richardson DL, Pepper DS, Kay AB. Chemotaxis for human monocytes by fibrinogen-derived peptides. Br J Haematol. 1976;32(4):507–513. [DOI] [PubMed] [Google Scholar]
- 29.Brandt E, Petersen F, Ludwig A, Ehlert JE, Bock L, Flad HD. The beta-thromboglobulins and platelet factor 4: blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. Journal of leukocyte biology. 2000;67(4):471–478. [DOI] [PubMed] [Google Scholar]
- 30.Gross TJ, Leavell KJ, Peterson MW. CD1 lb/CD18 mediates the neutrophil chemotactic activity of fibrin degradation product D domain. Thromb Haemost. 1997;77(5):894–900. [PubMed] [Google Scholar]
- 31.Carvalho-Tavares J, Hickey MJ, Hutchison J, Michaud J, Sutcliffe IT, Kubes P. A role for platelets and endothelial selectins in tumor necrosis factor-alpha-induced leukocyte recruitment in the brain microvasculature. Circ Res. 2000;87(12): 1141–1148. [DOI] [PubMed] [Google Scholar]
- 32.de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nature reviews. Immunology. 2016;16(6):378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehrer RI, Ganz T. Antimicrobial peptides in mammalian and insect host defence. Curr Opin Immunol. 1999; 1 l(l):23–27. [DOI] [PubMed] [Google Scholar]
- 34.Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92(9):3007–3017. [PubMed] [Google Scholar]
- 35.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663): 1532–1535. [DOI] [PubMed] [Google Scholar]
- 36.Kawasaki H, Iwamuro S. Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets. 2008;8(3): 195–205. [DOI] [PubMed] [Google Scholar]
- 37.Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–2794. [DOI] [PubMed] [Google Scholar]
- 38.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature medicine. 2007;13(4):463–469. [DOI] [PubMed] [Google Scholar]
- 39.Parks WC. Matrix metalloproteinases in repair. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 1999;7(6):423–432. [DOI] [PubMed] [Google Scholar]
- 40.Nwomeh BC, Liang HX, Cohen IK, Yager DR. MMP-8 is the predominant collagenase in healing wounds and nonhealing ulcers. The Journal of surgical research. 1999;81(2): 189–195. [DOI] [PubMed] [Google Scholar]
- 41.Armstrong DG, Jude EB. The role of matrix metalloproteinases in wound healing. Journal of the American Podiatric Medical Association. 2002;92(1): 12–18. [DOI] [PubMed] [Google Scholar]
- 42.Salo T, Makela M, Kylmaniemi M, Autio-Harmainen H, Larjava H. Expression of matrix metalloproteinase-2 and −9 during early human wound healing. Lab Invest. 1994;70(2): 176–182. [PubMed] [Google Scholar]
- 43.Brandt E, Woerly G, Younes AB, Loiseau S, Capron M. IL-4 production by human polymorphonuclear neutrophils. Journal of leukocyte biology. 2000;68(1): 125–130. [PubMed] [Google Scholar]
- 44.Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. JInvest Dermatol. 2007;127(3):514–525. [DOI] [PubMed] [Google Scholar]
- 45.Arumugam S, Jang YC, Chen-Jensen C, Gibran NS, Isik FF. Temporal activity of plasminogen activators and matrix metalloproteinases during cutaneous wound repair. Surgery. 1999;125(6):587–593. [PubMed] [Google Scholar]
- 46.Dupasquier M, Stoitzner P, van Oudenaren A, Romani N, Leenen PJ. Macrophages and dendritic cells constitute a major subpopulation of cells in the mouse dermis. The Journal of investigative dermatology. 2004;123(5):876–879. [DOI] [PubMed] [Google Scholar]
- 47.von Hundelshausen P, Koenen RR, Sack M, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105(3):924–930. [DOI] [PubMed] [Google Scholar]
- 48.Deuel TF, Senior RM, Chang D, Griffin GL, Heinrikson RL, Kaiser ET. Platelet factor 4 is chemotactic for neutrophils and monocytes. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(7):4584–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheuerer B, Ernst M, Durrbaum-Landmann I, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood. 2000;95(4):1158–1166. [PubMed] [Google Scholar]
- 50.Bar-Shavit R, Kahn A, Wilner GD, Fenton JW, 2nd. Monocyte chemotaxis: stimulation by specific exosite region in thrombin. Science. 1983;220(4598):728–731. [DOI] [PubMed] [Google Scholar]
- 51.Joseph S, MacDermot J. Thrombin promotes actin polymerization in U937 human monocyte-macrophage cells. Analysis of the signalling mechanisms mediating actin polymerization. Biochemical Journal. 1992;286(Pt 3):945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–6173. [DOI] [PubMed] [Google Scholar]
- 53.Mosser DM, Zhang X. Activation of murine macrophages. Carr Protoc Immunol. 2008;Chapter 14:Unit 14 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meszaros AJ, Reichner JS, Albina JE. Macrophage-induced neutrophil apoptosis. J Immunol. 2000; 165( 1 ):435–441. [DOI] [PubMed] [Google Scholar]
- 55.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. The Journal of clinical investigation. 1998;101(4):890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dovi JV, He LK, DiPietro LA. Accelerated wound closure in neutrophil-depleted mice. J Leukoc Biol. 2003;73(4):448–455. [DOI] [PubMed] [Google Scholar]
- 57.Martin P, D’Souza D, Martin J, et al. Wound Healing in the Pu.1 Null Mouse—Tissue Repair Is Not Dependent on Inflammatory Cells. Current Biology. 2003; 13(13): 1122–1128. [DOI] [PubMed] [Google Scholar]
- 58.Abbott RE, Corral CJ, MacIvor DM, Lin X, Ley TJ, Mustoe TA. Augmented inflammatory responses and altered wound healing in cathepsin G-deficient mice. Archives of surgery (Chicago, Ill. : 1960). 1998;133(9):1002–1006. [DOI] [PubMed] [Google Scholar]
- 59.Leibovich SJ, Ross R. A macrophage-dependent factor that stimulates the proliferation of fibroblasts in vitro. The American journal of pathology. 1976;84(3):501–514. [PMC free article] [PubMed] [Google Scholar]
- 60.Mirza R, DiPietro LA, Koh TJ. Selective and Specific Macrophage Ablation Is Detrimental to Wound Healing in Mice. The American journal of pathology. 2009;175(6):2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters T, Sindrilaru A, Hinz B, et al. Wound-healing defect of CD18(−/−) mice due to a decrease in TGF-beta1 and myofibroblast differentiation. EMBO J. 2005;24(19):3400–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xue M, Campbell D, Sambrook PN, Fukudome K, Jackson CJ. Endothelial protein C receptor and protease-activated receptor-1 mediate induction of a wound-healing phenotype in human keratinocytes by activated protein C. J Invest Dermatol. 2005;125(6):1279–1285. [DOI] [PubMed] [Google Scholar]
- 63.Julovi SM, Xue M, Dervish S, Sambrook PN, March L, Jackson CJ. Protease activated receptor-2 mediates activated protein C-induced cutaneous wound healing via inhibition of p38. Am J Pathol. 2011;179(5):2233–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Derian CK, Eckardt AJ, Andrade-Gordon P. Differential regulation of human keratinocyte growth and differentiation by a novel family of protease-activated receptors. Cell Growth Differ. 1997;8(7):743–749. [PubMed] [Google Scholar]
- 65.Hwa JJ, Ghibaudi L, Williams P, et al. Evidence for the presence of a proteinase-activated receptor distinct from the thrombin receptor in vascular endothelial cells. Circ Res. 1996;78(4):581–588. [DOI] [PubMed] [Google Scholar]
- 66.Lindner JR, Kahn ML, Coughlin SR, et al. Delayed onset of inflammation in protease-activated receptor-2-deficient mice. J Immunol. 2000;165(11):6504–6510. [DOI] [PubMed] [Google Scholar]
- 67.Steinhoff M, Buddenkotte J, Shpacovitch V, et al. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26(1):1–43. [DOI] [PubMed] [Google Scholar]
- 68.Seeliger S, Derian CK, Vergnolle N, et al. Proinflammatory role of proteinase-activated receptor-2 in humans and mice during cutaneous inflammation in vivo. FASEB J. 2003;17(13):1871–1885. [DOI] [PubMed] [Google Scholar]
- 69.Saksela O, Rifkin DB. Cell-Associated Plasminogen Activation: Regulation and Physiological Functions. Annual Review of Cell Biology. 1988;4(1):93–120. [DOI] [PubMed] [Google Scholar]
- 70.Grondahl-Hansen J, Lund LR, Ralfkiaer E, Ottevanger V, Dano K. Urokinase- and tissue-type plasminogen activators in keratinocytes during wound reepithelialization in vivo. J Invest Dermatol. 1988;90(6):790–795. [DOI] [PubMed] [Google Scholar]
- 71.Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: coordination of signaling for cell adhesion, migration and growth. Curr Opin Cell Biol. 2000;12(5):613–620. [DOI] [PubMed] [Google Scholar]
- 72.Rifkin DB, Gleizes PE, Harpel J, et al. Plasminogen/plasminogen activator and growth factor activation. Ciba Found Symp. 1997;212:105–115; discussion 116-108. [DOI] [PubMed] [Google Scholar]
- 73.Ra H-J, Parks WC. Control of Matrix Metalloproteinase Catalytic Activity. Matrix biology : journal of the International Society for Matrix Biology. 2007;26(8):587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chapman HA. Plasminogen activators, integrins, and the coordinated regulation of cell adhesion and migration. Curr Opin Cell Biol. 1997;9(5):714–724. [DOI] [PubMed] [Google Scholar]
- 75.Keski-Oja J, Raghow R, Sawdey M, et al. Regulation of mRNAs for type-1 plasminogen activator inhibitor, fibronectin, and type I procollagen by transforming growth factor-beta. Divergent responses in lung fibroblasts and carcinoma cells. J Biol Chem. 1988;263(7):3111–3115. [PubMed] [Google Scholar]
- 76.Overall CM, Wrana JL, Sodek J. Transforming growth factor-beta regulation of collagenase, 72 kDa-progelatinase, TIMP and PAI-1 expression in rat bone cell populations and human fibroblasts. Connect Tissue Res. 1989;20(1-4):289–294. [DOI] [PubMed] [Google Scholar]
- 77.Sitrin RG, Pan PM, Harper HA, Todd RF 3rd, Harsh DM, Blackwood RA. Clustering of urokinase receptors (uPAR; CD87) induces proinflammatory signaling in human polymorphonuclear neutrophils. J Immunol. 2000;165(6):3341–3349. [DOI] [PubMed] [Google Scholar]
- 78.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med. 1998;188(6):1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stavrou EX, Fang C, Bane KL, et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. 2018;128(3):944–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romer J, Bugge TH, Pyke C, et al. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2(3):287–292. [DOI] [PubMed] [Google Scholar]
- 81.Sulniute R, Shen Y, Guo YZ, et al. Plasminogen is a critical regulator of cutaneous wound healing. Thromb Haemost. 2016;115(5):1001–1009. [DOI] [PubMed] [Google Scholar]
- 82.Shen Y, Guo Y, Mikus P, et al. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood. 2012;119(24):5879–5887. [DOI] [PubMed] [Google Scholar]
- 83.D’Alessio S, Gerasi L, Blasi F. uPAR-deficient mouse keratinocytes fail to produce EGFR-dependent laminin-5, affecting migration in vivo and in vitro. J Cell Sci. 2008;121(Pt 23):3922–3932. [DOI] [PubMed] [Google Scholar]
- 84.Bugge TH, Flick MJ, Danton MJ, et al. Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci U S A. 1996;93(12):5899–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Connolly BM, Choi EY, Gardsvoll H, et al. Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood. 2010;116(9):1593–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carmeliet P, Moons L, Dewerchin M, et al. Receptor-independent role of urokinase-type plasminogen activator in pericellular plasmin and matrix metalloproteinase proteolysis during vascular wound healing in mice. J Cell Biol. 1998;140(1):233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lucas T, Waisman A, Ranjan R, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184(7):3964–3977. [DOI] [PubMed] [Google Scholar]
- 88.Mahdi F, Madar ZS, Figueroa CD, Schmaier AH. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood. 2002;99(10):3585–3596. [DOI] [PubMed] [Google Scholar]
- 89.LaRusch GA, Mahdi F, Shariat-Madar Z, et al. Factor XII stimulates ERK1/2; and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood. 2010;115(24):5111–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Groeneveld DJ, Sanders YV, Adelmeijer J, et al. Circulating Angiogenic Mediators in Patients with Moderate and Severe von Willebrand Disease: A Multicentre Cross-Sectional Study. Thromb Haemost. 2018;118(1):152–160. [DOI] [PubMed] [Google Scholar]
- 91.Schultz GS, Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009;17(2):153–162. [DOI] [PubMed] [Google Scholar]
- 92.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fadok VA, McDonald PP, Bratton DL, Henson PM. Regulation of macrophage cytokine production by phagocytosis of apoptotic and post-apoptotic cells. Biochem Soc Trans. 1998;26(4):653–656. [DOI] [PubMed] [Google Scholar]
- 94.Sgonc R, Gruber J. Age-related aspects of cutaneous wound healing: a mini-review. Gerontology. 2013;59(2):159–164. [DOI] [PubMed] [Google Scholar]
- 95.Ashcroft GS, Mills SJ, Ashworth JJ. Ageing and wound healing. Biogerontology. 2002;3(6):337–345. [DOI] [PubMed] [Google Scholar]
- 96.Ashcroft GS, Horan MA, Ferguson MW. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab Invest. 1998;78(1):47–58. [PubMed] [Google Scholar]
- 97.Swift ME, Burns AL, Gray KL, DiPietro LA. Age-related alterations in the inflammatory response to dermal injury. J Invest Dermatol. 2001;117(5):1027–1035. [DOI] [PubMed] [Google Scholar]
- 98.Swift ME, Kleinman HK, DiPietro LA. Impaired wound repair and delayed angiogenesis in aged mice. Lab Invest. 1999;79(12):1479–1487. [PubMed] [Google Scholar]
- 99.Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17(6):763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leaper DJ, Schultz G, Carville K, Fletcher J, Swanson T, Drake R. Extending the TIME concept: what have we learned in the past 10 years?(*). Int Wound J. 2012;9 Suppl 2:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ubbink DT, Westerbos SJ, Evans D, Land L, Vermeulen H. Topical negative pressure for treating chronic wounds. Cochrane Database Syst Rev. 2008(3):CD001898. [DOI] [PubMed] [Google Scholar]
- 102.Eskes AM, Ubbink DT, Lubbers MJ, Lucas C, Vermeulen H. Hyperbaric oxygen therapy: solution for difficult to heal acute wounds? Systematic review. World journal of surgery. 2011;35(3):535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Veves A, Falanga V, Armstrong DG, Sabolinski ML, Apligraf Diabetic Foot Ulcer S. Graftskin, a human skin equivalent, is effective in the management of noninfected neuropathic diabetic foot ulcers: a prospective randomized multicenter clinical trial. Diabetes Care. 2001;24(2):290–295. [DOI] [PubMed] [Google Scholar]
- 104.Zgonis T, Stapleton JJ, Roukis TS. Advanced plastic surgery techniques for soft tissue coverage of the diabetic foot. Clin Podiatr Med Surg. 2007;24(3):547–568, x. [DOI] [PubMed] [Google Scholar]
- 105.Mahmoud SM, Mohamed AA, Mahdi SE, Ahmed ME. Split-skin graft in the management of diabetic foot ulcers. J Wound Care. 2008;17(7):303–306. [DOI] [PubMed] [Google Scholar]
- 106.Gilligan AM, Waycaster CR, Motley TA. Cost-effectiveness of becaplermin gel on wound healing of diabetic foot ulcers. Wound Repair Regen. 2015;23(3):353–360. [DOI] [PubMed] [Google Scholar]
- 107.Papanas N, Maltezos E. Benefit-risk assessment of becaplermin in the treatment of diabetic foot ulcers. Drug Saf. 2010;33(6):455–461. [DOI] [PubMed] [Google Scholar]
- 108.Driver VR, Hanft J, Fylling CP, Beriou JM. A prospective, randomized, controlled trial of autologous platelet-rich plasma gel for the treatment of diabetic foot ulcers. Ostomy/wound management. 2006;52(6):68–70, 72, 74 passim. [PubMed] [Google Scholar]
- 109.Ganio C, Tenewitz FE, Wilson RC, Moyles BG. The treatment of chronic nonhealing wounds using autologous platelet-derived growth factors. The Journal of foot and ankle surgery : official publication of the American College of Foot and Ankle Surgeons. 1993;32(3):263–268. [PubMed] [Google Scholar]
- 110.Darby IA, Weller CD. Aspirin treatment for chronic wounds: Potential beneficial and inhibitory effects. Wound Repair and Regeneration. 2017;25(1):7–12. [DOI] [PubMed] [Google Scholar]
- 111.Jull A, Wadham A, Bullen C, Parag V, Kerse N, Waters J. Low dose aspirin as adjuvant treatment for venous leg ulceration: pragmatic, randomised, double blind, placebo controlled trial (Aspirin4VLU). BMJ. 2017;359:j5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bjorlin G, Nilsson IM. The effect of antifibrinolytic agents on wound healing. Int J Oral Maxillofac Surg. 1988;17(4):275–276. [DOI] [PubMed] [Google Scholar]
- 113.Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol. 1998;111(5):850–857. [DOI] [PubMed] [Google Scholar]
- 114.Mori R, Power KT, Wang CM, Martin P, Becker DL. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J Cell Sci. 2006;119(Pt 24):5193–5203. [DOI] [PubMed] [Google Scholar]
- 115.Buchstein N, Hoffmann D, Smola H, et al. Alternative proteolytic processing of hepatocyte growth factor during wound repair. Am J Pathol. 2009;174(6):2116–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eming SA, Koch M, Krieger A, et al. Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. JProteome Res. 2010;9(9):4758–4766. [DOI] [PubMed] [Google Scholar]
- 117.Wong SL, Demers M, Martinod K, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fadini GP, Menegazzo L, Rigato M, et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes. 2016;65(4):1061–1071. [DOI] [PubMed] [Google Scholar]
- 119.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349(6245):316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lewis HD, Liddle J, Coote JE, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11(3):189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]