

Abstract
Tea oil camellia (Camellia oleifera) is an important woody oil tree in southern China. However, little is known regarding the molecular mechanisms that contribute to high oleic acid accumulation in tea oil camellia. Herein, we measured the oil content and fatty acid compositions of high- and low-oil tea oil camellia seeds and investigated the global gene expression profiles by RNA-seq. The results showed that at the early, second and third seed developmental stages, a total of 64, 253, and 124 genes, respectively, were significantly differentially expressed between the high- and low-oil cultivars. Gene ontology (GO) enrichment analysis of the identified differentially expressed transcription factors (TFs; ABI3, FUS3, LEC1, WRI1, TTG2 and DOF4.6) revealed some critical GO terms associated with oil biosynthesis and fatty acid accumulation, including glycolysis, zinc ion binding, positive regulation of fatty acid biosynthetic process, triglyceride biosynthetic process, seed coat development, abscisic acid-mediated signaling pathway and embryo development. Comprehensive comparisons of transcriptomic profiles and expression analysis of multigenes based on qRT-PCR showed that coordinated high expression of the upstream genes HAD, EAR and KASI directly increased the relative levels of C16:0-ACP, which provided enough precursor resources for oleic acid biosynthesis. Continuous high expression of the SAD gene accelerated oleic acid synthesis and accumulation, and coordinated low expression of the downstream genes FAD2, FAD3, FAD7, FAD8 and FAE1 decreased the consumption of oleic acid for conversion. The coordinated regulation of these multigenes ensures the high accumulation of oleic acid in the seeds of tea oil camellia. Our data represent a comprehensive transcriptomic study of high- and low-oil tea oil camellia, not only increasing the number of sequences associated with lipid biosynthesis and fatty acid accumulation in public resource databases but also providing a scientific basis for genetic improvement of the oleic acid content in woody oil trees.
Keyword: Camellia oleifera, RNA-seq, Seed oil content, Fatty acid accumulation, Coordinated regulation of upstream and downstream multigenes
Introduction
Tea oil camellia (Camellia oleifera Abel.) is an important woody oil tree in southern China. The seed oil of this tree contains rich unsaturated fatty acids and numerous nutrients, such as vitamins, camelliaside, and tea polyphenols. The oleic acid content in tea oil camellia seed oil, a high-grade edible vegetable oil, accounts for over 75% of all fatty acids (Yu et al. 1999; Yao et al. 2011). Oleic acid is not easily oxidized, exhibits high chemical stability (Chen 2007) and is resistant to storage (Ohlrogge 1994), with special effects in the prevention of cardiovascular diseases and cancer and in reducing cholesterol and blood lipid levels (Liao et al. 2005). One of the important aims for the genetic improvement of this species is the breeding of cultivars of tea oil camellia with high oleic acid content.
The use of Illumina technology for high-throughput transcriptome sequencing of seeds at different developmental stages could lead to the identification of stage-specific expression patterns of genes associated with seed development and lipid biosynthesis (Libault et al. 2010). To date, high-throughput next-generation sequencing (NGS) technologies have been utilized widely to study many oil crops (Liu et al. 2016; Qun et al. 2017; Zhang et al. 2017). Based on the transcriptome sequencing analyses of soybean embryos, Li et al. (2015a) identified 37,593 transcripts from seeds collected at five different developmental stages, 2879 of which were differentially expressed and played crucial roles in some metabolic pathways, such as fatty acid biosynthesis, glycolysis, and sucrose catabolism. Xu et al. (2015) investigated candidate genes associated with oil-related biological processes by transcriptome sequencing of pod tissues of Brassica napus cultivars at different developmental stages and found that a total of 33 differentially expressed genes (DEGs) were potentially involved in lipid metabolism. By performing transcriptome sequencing analyses of soybean embryos, Collakova et al. (2013) identified 73,320 transcripts and revealed the expression of 41,619 genes at one time point during 10 different seed development stages. These expressed genes were involved in cellular metabolic processes, including embryonic programs and general cell maintenance, cell growth, molecular trafficking, signaling, and regulation. Furthermore, Severin et al. (2010) found 27,945 genes that were differentially expressed in the developing seeds of soybean 28 days after flowering relative to the expression in flowers; these genes played important roles in lipid biosynthesis, sucrose transport and seed coat development. Based on the transcriptome sequencing data, O’Rourke et al. (2014) identified and evaluated the expression patterns of key genes associated with lipid biosynthesis in different soybean seed developmental stages and found that the FAD2 (fatty acid desaturase 2)-2B and FAD2-2C genes were highly expressed in the early stages of seed formation and that the FAD2-1B and FAD2-1A genes were highly expressed at later seed developmental stages. To explore the differences in seed oil content and composition among multiple soybean genotypes, transcriptome sequencing analyses for mid-maturation seeds identified the most abundantly expressed seed transcripts to be those of lipid metabolism genes, SSPs (seed storage proteins) and oil body proteins (Goettel et al. 2014), indicating that differential expressions of genes involved in lipid biosynthesis may be responsible for the levels of different fatty acids.
Lipid biosynthesis-related genes exhibit programmed expression during seed development (Gupta et al. 2017). Studies have shown that overexpression of some genes encoding key enzymes of lipid biosynthesis metabolic pathways, such as acetyl-CoA carboxylase (ACC) and 3-ketoacyl-ACP synthase (KAS), could successfully increase oil biosynthesis and accumulation (Baud et al. 2010; González-Mellado and Martínez-Force 2010). The high expression level of the glycerol-3-phosphate dehydrogenase 1 (GPD1) gene in Saccharomyces cerevisiae promoted G3P (glycerol-3-phosphate) synthesis, increasing the seed oil content (Remize et al. 2001). Overexpression of the Tropaeolum majus DGAT1 (diacylglycerol acyltransferase 1) gene in wild-type Arabidopsis and high-erucic-acid rapeseed (HEAR) and canola B. napus resulted in an increase in oil content (3.5–10% on a dry weight basis, or a net increase of 11–30%) (Xu et al. 2010). Overexpression of the soil fungus Umbelopsis ramanniana DGAT2 (diacylglycerol acyltransferase 2) gene in soybean resulted in a 1.5% increase in the oil content of mature seeds (Lardizabal et al. 2008).
Multiple enzymes are involved in fatty acid biosynthetic pathways. Expression of the Lupinus micranthus SAD (stearoyl-ACP desaturase) gene in tobacco rapidly increased the oleic acid content, indicating a close correlation between SAD expression and fatty acid accumulation (Zaborowska et al. 2002). Overexpression of the acyl carrier protein thioesterase B1 gene (FATB1) from Arabidopsis in Arabidopsis increased the palmitic acid content fourfold and increased the saturated fatty acid content in seed oil (Dörmann et al. 2000). Antisense expression of the FAD2 gene of Brassica rapa in Brassica juncea showed that the oleic acid content reached 73% in the transgenic lines, which was 20% higher than the level observed in the receptor parent (Sivaraman et al. 2004).
To improve yield and quality, different genes involved in seed oil biosynthesis and fatty acid accumulation in tea oil camellia have been cloned and functionally characterized, including ACCase (Wang et al. 2018), SAD (Zhang et al. 2008), DGAT1 (Xia et al. 2014) and FAD2 (Zeng et al. 2014). Recently, Xia et al. (2014) identified 2300 simple sequence repeats (SSRs) and 20,200 single-nucleotide polymorphisms (SNPs) of tea oil camellia based on high-throughput RNA-seq data for four tissues—tender shoots, young leaves, flower buds and flowers, and identified the major unigenes associated with lipid metabolism. High-throughput transcriptomic data of drought-tolerant and drought-sensitive cultivars showed that many DEGs are involved in metabolic pathways associated with drought stress in tea oil camellia (Dong et al. 2017). Chen et al. (2017) tested variations in the transcriptome of wild tea oil camellia growing at different latitudes and elevations and discovered candidate genes for cold acclimation. However, there are several limitations associated with studies on seed oil biosynthesis and fatty acid accumulation in tea oil camellia: (1) only one gene or a single gene family was examined in most studies, which does not correctly reflect the coordinated regulatory mechanism of multigenes involved in lipid biosynthesis; (2) there has been no report on the regulatory mechanism of multigenes involved in lipid biosynthesis based on high- and low-oil materials of tea oil camellia. Although Lin et al. (2018) reported that the increased expression levels of the SAD gene and decreased activity of the FAD2 gene might be a response to the increase in oleic acid levels during the late stage of seed development in tea oil camellia, this study was performed based only on materials collected at different seed developmental stages. This method could not determine the contribution of the differential expression of related genes to seed oil biosynthesis and fatty acid accumulation.
In this study, we first used high-throughput RNA-seq technology to obtain the transcriptomic profiles of high- and low-oil seeds of tea oil camellia at different developmental stages. Next, the DEGs associated with seed development and lipid biosynthesis were identified based on the RNA-seq data. Finally, the contributions of the DEGs were determined based on qRT-PCR analyses and RNA-seq data, which included the contributions of the differential expression of GPD1 and DGAT to seed oil biosynthesis and the effects of changes in the expression of upstream and downstream multigenes on oleic acid synthesis and accumulation. Our datasets and results provide a scientific resource and basis for the improvement of tea oil camellia with high-oil seeds, with high oleic acid content, in the future.
Materials and methods
Plant materials
Two tea oil camellia cultivars—‘M3’ with high seed oil content and ‘M8’ with low seed oil content—were selected as sample trees from fields in Yuping Dong Autonomous County, Guizhou Province, China. These two cultivars were selected and bred from the natural population, with the same age of 12 years. Fruits of two cultivars were harvested on June 2, July 4, August 5, and September 3 in 2016. The fruits were collected from different parts of each cultivar. Seeds obtained by stripping the fruit shells were immediately wrapped in tin foil and placed in liquid nitrogen. After being transported to the laboratory, the seeds were stored at − 80 °C for subsequent experiments.
Tea oil camellia has been widely cultivated and utilized as oil tree in Yuping Dong Autonomous County of Guizhou province of China, for over 600 years. This county was named the ‘hometown of tea oil camellia’ by the Chinese first Prime Minister Enlai Zhou in 1958, because the seed yields of tea oil camellia were so high in that year. Tea oil camellia cultivar, ‘M3’ and ‘M8’ grew at the Chahuaquan garden, Yuping Dong Autonomous County. The orchard had a mean annual rainfall of 1200 mm, mean annual temperature of 16.4 °C, effective accumulative temperature of 2460.4 °C, maximum temperature of 39.7 °C, minimum temperature of − 10.7 °C, and 1293.1-h sunshine a year.
Determination of seed oil content and fatty acid compositions
The seed oil content of the two cultivars at four different developmental stages was determined by the method of Ding et al. (2016). Fatty acid compositions were determined by GC-TOF/MS (gas chromatography time-of-flight mass spectrometry) (Sánchez-Salcedo et al. 2016). Each sample analysis was performed in triplicate.
Seed oil from each sample was isolated using a methanol–chloroform extraction procedure. Seed sample powder (300 mg) was homogenized in methanol (2 ml) for 1 min, and after adding chloroform (4 ml), homogenization was continued for an additional 2 min. The mixtures were sonicated in an ultrasonic bath for 30 min, centrifuged and filtered. The solid residues were resuspended in chloroform/methanol (2:1, v/v, 4 ml) and then homogenized again for 3 min and filtered. Then, 1 ml of 0.88% KCl solution was added to the combined filtrates, and the mixtures were mixed thoroughly by vortexing and then centrifuged. The lower phase containing the purified oils was collected and evaporated to dryness under nitrogen.
The fatty acid composition was determined as fatty acid methyl esters (FAMEs) by boron trifluoride-methanol catalysis. GC-TOF/MS analysis of the FAMEs was performed on a Clarus 680 GC coupled with an AxION iQT TOF/MS system (PerkinElmer, Shelton, USA). The system was equipped with an Agilent J&W DB-23 capillary column (60 m × 0.25 mm × 0.25 µm). The fatty acid composition was measured and expressed as the weight percentage of each fatty acid compared to the total fatty acid content as described in our previous study.
Total RNA extraction and high-throughput sequencing
Total RNA from each sample was extracted according to the manufacturer’s instructions for the Total RNA Purification Kit (TRK1001, LC Sciences, Houston, TX). The quality and quantity of the total RNA were analyzed by 1% agarose gel electrophoresis and a Thermo NanoDrop 2000 spectrophotometer (Thermo Scientific, CA, USA) and with a Bioanalyzer 2100 (Agilent, CA, USA), respectively. Six micrograms of total RNA from each sample was used for library preparation. mRNA purification was performed by oligo-dT-attached magnetic beads, and the purified mRNA was randomly fragmented into small fragments using fragmentation buffer (Illumina, San Diego, USA), which was used to construct a cDNA library and for sequencing. Library construction and sequencing were performed by LC Biotech (Hangzhou, China) on an Illumina HiSeq 2000 platform. The Illumina HiSeq 2000 platform generated 150-bp reads via paired-end sequencing.
Quality control analysis
To obtain accurate and reliable transcriptome sequencing data, the raw data were preprocessed using the Illumina paired-end RNA-seq approach to remove the adapter sequences, low-quality sequences (average quality score < 20), short sequences (length < 100 bp) and poly-A tails before assembly. High-quality sequencing data for each sample were generated based on the clean reads.
Assembly and functional annotation
Trinity Software was used to carry out sequencing data stitching for de novo assembly of the global transcriptome profiles. After splicing to obtain genes, the functional annotation of all the assembled distinct unigenes was compared by BLASTX search with a threshold E-value less than 1e-10 against five public databases of manually annotated and curated protein sequences: Swiss-Prot, the NCBI nonredundant protein database (NR), the Kyoto Encyclopedia of Genes and Genomes database (KEGG), the eukaryotic orthologous groups (KOG) database, and the protein family (Pfam) database. Gene ontology (GO) annotation of all the unigenes was performed by the hypergeometric distribution algorithm based on molecular function, biological process and cellular component (http://www.geneontology.org), and KEGG annotation was performed using the online KEGG automatic annotation server (KAAS; http://www.genome.jp/kegg/kaas/) (Moriya et al. 2007).
Gene expression level analysis
Gene expression level analysis was performed using RSEM software. The RPKM (reads per kilobase per million mapped reads) value was used to represent the gene expression abundance in different samples (Mortazavi et al. 2008). DEGs from different developmental stages of one cultivar and high- and low-oil samples were analyzed using the DESeq method described by Anders and Huber (2010). The FDR (false discovery rate) was characterized by the method described by Benjamini and Hochberg (1995), with p value correction, and genes with the default threshold of |log2foldchange| ≥ 1, P < 0.05 were defined as significant DEGs.
KEGG and GO enrichment analyses
To further analyze the main biological functions of the DEGs, GO and KEGG enrichments were performed by the hypergeometric test, and significant terms had a default threshold corrected p value ≤ 0.05.
Validation of qRT-PCR for gene expression
The unigenes associated with lipid metabolism were selected for validation of gene expression using qRT-PCR analysis. The total RNA samples were the same as those used in the above sequencing experiments. First-strand cDNA was synthesized using PrimeScript™ RT Master Mix (TaKaRa Biotech, Dalian, China) according to the manufacturer’s instructions. All the specific primer pairs for the selected target genes (Table 1) were designed using PrimerQuest online software (http://sg.idtdna.com/PrimerQuest/Home/Index). Elongation factor 1-alpha (CoEF1α) was selected as the reference gene, and qRT-PCR experiments were performed using the SYBR Premix Ex Taq™ II Kit (Tli RNaseH Plus; TaKaRa Biotech, Dalian, China) with a 20-μL reaction system: SYBR Premix Ex Taq II (Tli RNaseH Plus): 10 μL, ROX reference dye II (50×): 0.4 μL, FP (10 μΜ): 1.0 μL, RP (10 μΜ): 1.0 μL, template cDNA: 2.0 μL, RNA-free water: 5.6 μL. The qRT-PCR procedure was conducted using the recommended amplification procedure on an ABI7500 real-time PCR instrument (Applied Biosystems, Foster, USA): 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 34 s. The melting curve was analyzed to confirm the specificity of the amplification reaction, and the qRT-PCR experiment for each sample was performed in triplicate. The relative gene expression level was calculated using the 2−ΔΔCt method (Schmittgen and Livak 2008).
Table 1.
Gene names and real time fluorescent quantitative PCR primer used in this study
| Gene name | Protein name | Primer sequence (5′–3′) | Fragment length (bp) |
|---|---|---|---|
| CoEF1α | Elongation factor 1-alpha |
F:CATGATCACTGGTACCTCACAG R: CAAGGGTAAAGGCAAGCAAAG |
128.0 |
| KAR | 3-Ketoacyl-ACP-reductase |
F: TGCAAAAGCAGGAGTGATTG R: GGTGAAAACCTGTCCGGTAA |
83.4 |
| HAD | 3-Hydroxyacyl-ACP dehydrase |
F: CAGATTCCGGAAGCCAGTTA R: GGTTGCCTCAGGTTTTGCTA |
83.1 |
| EAR | Enoyl-ACP reductase |
F: TGGGCCAGAGGTCACTAAAC R: GCCTCAAAAGCAAGCACTTT |
83.7 |
| KASI | β-Ketoacyl-ACP-synthase I |
F: GCTCTGTCTCAGAGGAATGATG R: CTCCAGAACCTTCACCCATAAC |
91.0 |
| KASII | β-Ketoacyl-ACP-synthase II |
F: GGGAGAAGGAGCTGGAGTTT R: CTGGTGTGGATGTAGCATGG |
82.4 |
| SAD | Stearoyl-ACP-desaturase |
F: TTGGAGGATTGGGCTGAT R: AAGGGCTTCTTCTGTGAT |
198.0 |
| FATB | Palmitoyl-ACP thioesterase B |
F: AGTCCGATCAGCCCCTATCT R: GAGGATGATTCGCCATCACT |
84.6 |
| FAE1 | Fatty acid elongase 1 |
F: GACACTTCTGGTGTTCCTATCC R: TGACGAGCATCTTCTGGTTTAT |
99.0 |
| FAD2 | Fatty acid desaturase 2 |
F: CCCATTGTTTTCGTCGATCT R: TGTGTCATCAAGCCATTGGT |
84.3 |
| FAD3 | Fatty acid desaturase 3 |
F: GCCAAAAAAATCGGGTC R: ATCGCAAAGAATCACTCC |
83.0 |
| FAD7 | Fatty acid desaturase 7 |
F: ATTACCCGAACTCTGCG R: AGGACTTCTACCCCACA |
75.0 |
| FAD8 | Fatty acid desaturase 8 |
F: ATGACAAAGGACAGGCCAAC R: TCATGGCACCCATTATCTGA |
85.9 |
| GPD1 | Glycerol-3-phosphate dehydrogenase 1 |
F:ACGGGTTGGAAATGGGAAATA R:CCTTGACAGACGAGAACAGAAG |
99 |
| DGAT1 | Diacylglycerol acyltransferase 1 |
F:GACGATGTAACCGTTCTTGCG R:TTGGCTGCGGCTGCTGT |
178 |
| DGAT2 | Diacylglycerol acyltransferase 2 |
F:TGGAAGCCTGATGGGAAAC R:ATGCATTGGACGGTGATAGG |
111 |
Results
Dynamic changes in seed oil content in high- and low-oil cultivars of tea oil camellia during seed development
From June 2 to September 3, there were distinct changes in the colors and sizes of the fruits and seeds of the two cultivars ‘M3’ and ‘M8’ of tea oil camellia (Fig. 1a). From June 2 to July 4, the sizes of the fruits gradually increased, and the seed color changed from white to yellow. After July 4, the seed color gradually changed from yellow to brown, and the seed coats became increasingly hard (Fig. 1a). The seed oil content of the two cultivars of tea oil camellia showed gradual upward trends, with an increase from 0.16 to 24.1% observed for ‘M3’ during seed development (Fig. 1b). After July 4, a high rate of increase in seed oil content appeared between August 5 and September 3 for the two cultivars ‘M3’ and ‘M8’. Throughout seed development, the seed oil content in each developmental stage of the ‘M3’ cultivar was consistently higher than in that of the ‘M8’ cultivar.
Fig. 1.

a Morphology and color of fruits and seeds of two cultivars, namely, “M3” and “M8”, of tea oil camellia at four developmental stages; b dynamic changes in the oil content of two cultivars, namely, “M3” and “M8” during seed development. *Indicates significant differences in oil content between the two cultivars at the same developmental stage at the 0.05 level
Dynamic changes in fatty acid compositions in high- and low-oil tea oil camellia during seed development
Based on GC-TOF/MS analysis, six main fatty acids were detected in the seed oils of the two cultivars ‘M3’ and ‘M8’ of tea oil camellia, including the two saturated fatty acids palmitic acid (C16:0) and stearic acid (C18:0), two monounsaturated fatty acids oleic acid (C18:1) and arachidonic acid (C20:1) and two polyunsaturated fatty acids linoleic acid (C18:2) and linolenic acid (18:3) (Fig. 2a, b). Oleic acid accounted for a majority of the total fatty acids, and the relative percentage of oleic acid was 82.51% and 76.64% in oils extracted from mature seeds of the ‘M3’ and ‘M8’ cultivars, respectively (Fig. 2c, d). During early seed development, the palmitic acid content in the seed oils of the two cultivars ‘M3’ and ‘M8’ was relatively high, increasing to 26.33% and 28.1%, respectively, from June 2 to July 4; then, the levels rapidly decreased to 5.62% for ‘M3’ and 9.42% for ‘M8’ by September 3.
Fig. 2.

Total ion chromatogram of fatty acids in the seed oils of two cultivars, namely, ‘M3’ (a) and ‘M8’ (b), and dynamic changes in fatty acid compositions in the seed oils of the cultivars ‘M3’ (c) and ‘M8’ (d) during seed development. C16:0 palmitic acid, C18:0 stearic acid, C18:1 oleic acid, C18:2 linoleic acid, C18:3 linolenic acid, C20:1 arachidonic acid
During seed development, the oleic acid content in the seed oils of both cultivars exhibited distinct upward trends. On June 2, the oleic acid content was 25.65% for the cultivar ‘M3’ and 18.82% for ‘M8’; the oleic acid content rapidly increased to 68.21% for the cultivar ‘M3’ and 59.24% for ‘M8’ over only 32 days from June 4 to August 5. After August 5, the oleic acid content continued to gradually increase, reaching peak values of 82.51% for the cultivar ‘M3’ and 76.64% for ‘M8’ by September 3. The linoleic acid content in the two cultivars of tea oil camellia was highest during early seed development, with peak values of 35.81% for the cultivar ‘M3’ and 43.19% for the cultivar ‘M8’ on July 4. Then, the linoleic acid content rapidly decreased and exhibited minimum values of 3.51% for the cultivar ‘M3’ and 6.47% for the cultivar ‘M8’ on September 3. During seed development, the linolenic acid content of the two cultivars was very low and gradually decreased from June 2 to September 3, with minimum values of 0.71% for the cultivar ‘M3’ and 0.87% for the cultivar ‘M8’ observed on September 3 (Fig. 2c, d).
RNA-seq and de novo assembly
To comprehensively identify key genetic pathways associated with lipid biosynthesis and fatty acid formation and accumulation in seeds of high- and low-oil cultivars of tea oil camellia during seed development, the sixteen cDNA libraries were sequenced using the Illumina HiSeq 2000 platform, with two biological replicates for each sample. A total of 872,341,300 raw reads were generated. After preprocessing of the raw reads, a total of 851,725,244 clean reads (97.61% of the valid ratio, ~ 124.6 G bases) were obtained and were used for assembly (94.98% Q ≥ 30, 45.03 ~ 50.07% GC content). Utilizing Trinity Software, all the clean reads of each sample cDNA library were assembled de novo to obtain 89,049 transcripts, with an average length and N50 of 996 bp and 1749 bp, respectively. Based on further analysis, a total of 53,507 unigenes were obtained, with a mean length of 678 bp and an N50 of 1,142 bp (Table 2).
Table 2.
Statistics of de novo assembly and transcriptome annotation in tea oil camellia
| Items | Genes | Transcripts |
|---|---|---|
| All | 53,507 | 89049 |
| Median GC % | 42.30 | 42.10 |
| Mean GC % | 43.35 | 42.88 |
| Min length | 201 | 201 |
| Median length | 319 | 415 |
| Mean length | 678 | 996 |
| Max length | 69,317 | 69,317 |
| Total assembled bases | 363,06013 | 88,749,500 |
| N50 | 1142 | 1749 |
N50: sorting genes from long to short, increasing the base number of genes successively, the total base number of genes 50% of the genes length, transcript as above described
Functional annotation of unigenes from the transcriptomes of developing seeds
Functional annotation of all the unigenes from the transcriptomes of developing high- and low-oil seeds was performed by similarity comparisons with five public protein databases—the Swiss-Prot, NR, KEGG, KOG and Pfam databases—using the BLASTX method with a default threshold corrected e value ≤ 1e−10. The results showed that a total of 19,082 unigenes (35.66%) were annotated in the Swiss-Prot database, 32,080 unigenes (59.95%) in the NR database, 22,795 unigenes (42.60%) in the Pfam database, 10,175 unigenes (19.02%) in the KEGG database, 25,058 unigenes (46.83%) in the KOG database and 16,834 unigenes (31.46%) in the GO database (Table 3).
Table 3.
Statistics of the BLAST annotation results
| Annotated databases | Gene number | Percentage (%) |
|---|---|---|
| Swiss-Prot | 19,082 | 35.66 |
| NR | 32,080 | 59.95 |
| Pfam | 22,795 | 42.60 |
| KEGG | 10,175 | 19.02 |
| KOG | 25,058 | 46.83 |
| GO | 16,843 | 31.46 |
Transcriptomic changes in high- and low-oil cultivars during seed development
To thoroughly analyze the differences in global transcriptome profiles between high- and low-oil cultivars of tea oil camellia during seed development, the expression levels of all the unigenes were compared, and DEGs were identified using RESM software with the default threshold: genes with |log2foldchange| ≥ 1, P < 0.05 were defined as significant DEGs. The results showed that there were 2,647 DEGs between the ‘M3’ and ‘M8’ libraries at the third stage (Aug. 5), including 883 upregulated genes and 1,764 downregulated genes (Fig. 3); in the GO enrichment analysis, a total of 155 significantly enriched GO terms were identified, which were mainly associated with plasmodesma, extracellular region, response to defense, cellular cell wall organization, and the xenobiotic-transporting ATPase activity pathway (Fig. 4c). In the second stage (July 4), a total of 253 DEGs were identified, including 88 upregulated genes and 165 downregulated genes (Fig. 3); in the GO enrichment analysis, a total of 73 significantly enriched GO terms were identified, which were involved in secondary cell wall biogenesis, lignin biosynthetic process, and the cell wall, hydroquinone:oxygen oxidoreductase activity pathway (Fig. 4b). At the first stage (June 2), only 64 DEGs were identified, including 18 upregulated genes and 46 downregulated genes (Fig. 3); 30 significantly enriched GO terms were identified, which were mainly involved in peroxidase activity, heme binding, the ATP biosynthetic process and the peroxidase activity pathway (Fig. 4a). There was no DEG between the high- and low-oil cultivars at the fourth stage (September 3).
Fig. 3.
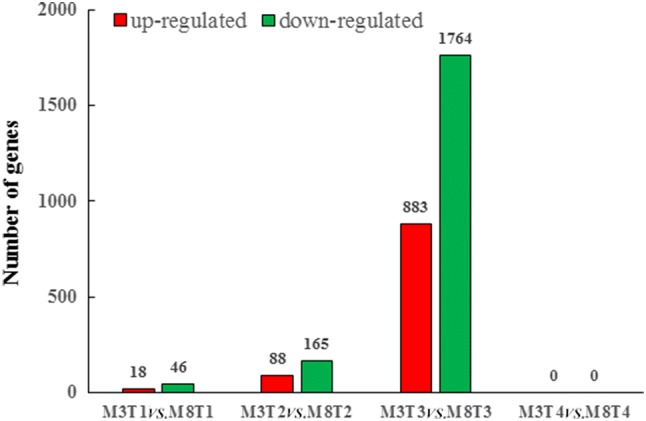
Differentially expressed genes among different groups in ‘M3’ and ‘M8’ at four seed developmental stages. The number of upregulated (red) and downregulated (green) DEGs in ‘M3’ relative to M8’ are shown at different seed developmental stages
Fig. 4.


Analysis of significant GO enrichment scatterplot distribution maps based on identified DEGs. The top 20 enriched GO terms for the early stage (a), second stage (b) and third stage (c)
Venn diagram analysis could visually depict the number of DEGs and the number of common DEGs in comparisons among samples of different seed developmental stages of each cultivar. There was no common DEG in the cultivar ‘M3’ in all the stages (Fig. 5a). In contrast, there were some common DEGs in ‘M8’ (Fig. 5b), indicating that these common DEGs could provide a favorable foundation for genetic improvement of tea oil camellia.
Fig. 5.
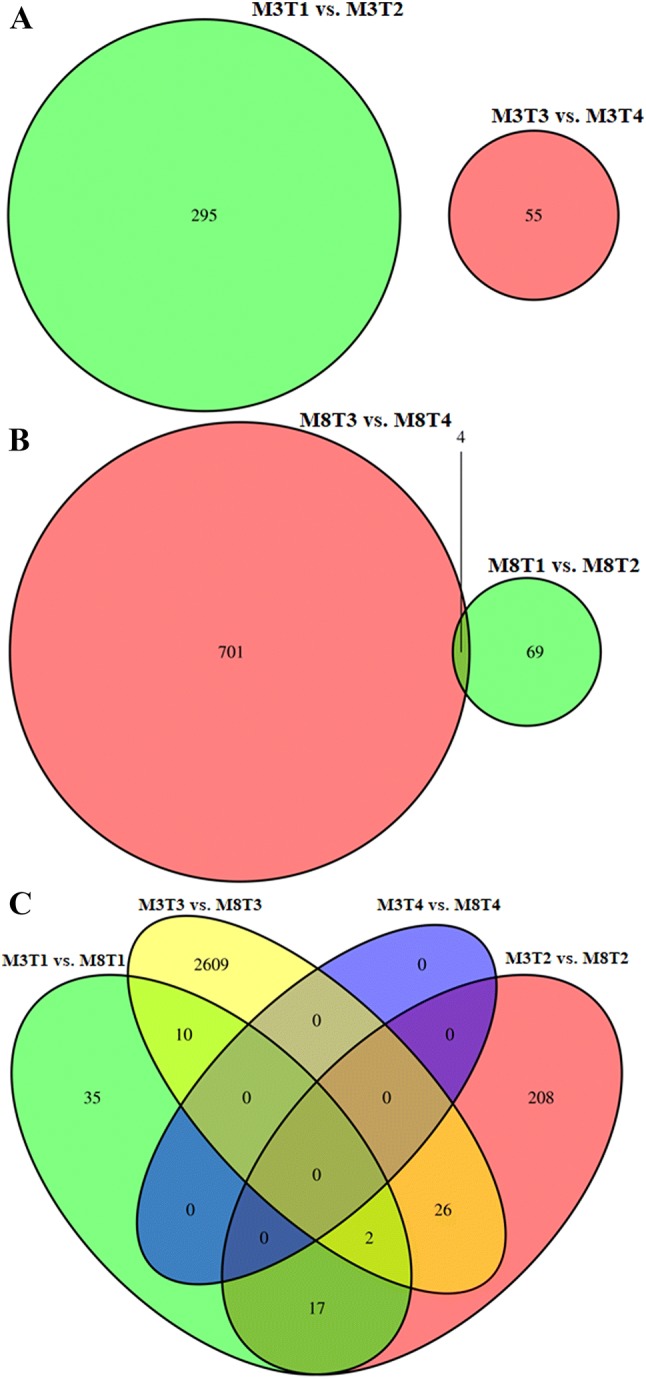
Venn diagram of the distribution of significant DEGs among different seed developmental stages in ‘M3’ (a) and ‘M8’ (b) and between ‘M3’ and ‘M8’ (c)
To further understand the biological functions of genes in the developing seeds of tea oil camellia, eight DEGs in KEGG pathways were identified by a mining analysis of the KEGG pathways for the M3T1 and M8T1 libraries, which mainly exhibited three KEGG pathways. The genes that were downregulated in M3T1 relative to M8T1 were associated with methane metabolism (37%), phenylpropanoid biosynthesis (37%) and phenylalanine metabolism (37%) (Fig. 6a). Compared to M8T2, the upregulated genes in M3T2 were associated ribosome (33%), gap junction (33%) and ABC transporters (33%) (Fig. 6b), and the downregulated genes in M3T2 were involved in naphthalene and anthracene degradation (10%), starch and sucrose metabolism (15%) and nitrogen metabolism (15%). Compared to M8T3, the upregulated genes in M3T3 were mainly involved in phenylalanine metabolism (7%), starch and sucrose metabolism (9%), and phenylpropanoid biosynthesis (14%), and the downregulated genes were associated with glycolysis/gluconeogenesis (5%), starch and sucrose metabolism (7%) and cysteine and methionine metabolism (5%) (Fig. 6c). During seed development, the number of DEGs that changed markedly varied from seed embryogenesis to maturation, and there were diverse numbers of DEGs that increased gradually and participated in related metabolic processes. In addition, compared to the early seed developmental stage, the expression levels of DEGs at the third stage increased substantially. Thus, the key-related DEGs and GO terms may play important roles in providing reservoir activity and may be involved in the seed filling process, functioning throughout seed development. Mining for these key DEGs associated with lipid biosynthesis could provide a scientific basis for improvement of the seed oil quality and lipid composition of tea oil camellia in the future.
Fig. 6.

KEGG pathway enrichment analysis based on identified DEGs. KEGG pathway enrichment analysis for the early stage (a), second stage (b) and third stage (c)
Differentially expressed genes (DEGs) involved in fatty acid metabolism
Eleven DEGs associated with fatty acid metabolism were identified between the high- and low-oil cultivars of tea oil camellia at four different seed developmental stages. Two DEGs, namely, FAD3 (omega-3 fatty acid desaturase, delta-15 desaturase, comp50825_c0) and FAD7A-1 (omega-3 fatty acid desaturase, delta-15 desaturase, and comp54259_c0), were involved in the KO01040 metabolic pathway of unsaturated fatty acid biosynthesis. Two ACCase genes (acetyl-CoA carboxylase, comp59939_c0 and comp50840_c0) and KASI (β-ketoacyl-ACP-synthase I, comp40069_c0) were involved in the KO00061 metabolic pathway of fatty acid biosynthesis. Four long-chain acyl-CoA synthetases, namely, LACS2 (comp60841_c0), LACS4 (comp51115_c0), LACS6 (comp49262_c0) and AAE15 (comp67596_c0), CHY1 (enoyl-CoA hydratase, comp65605_c0) and AAT1 (acetyl-CoA C-acetyltransferase, comp66752_c0), were involved in the KO00071 metabolic pathway of fatty acid biosynthesis.
Interestingly, significantly different expression levels of 11 DEGs were observed in the third stage of seed development, but there was no significant expression in the other stages (Fig. 7a). During early seed development (June 2), most of the DEGs were upregulated in ‘M3’ relative to ‘M8’, but the expression of KSAI was downregulated in ‘M3’ relative to ‘M8’ during the second stage of seed development (July 4). During the third stage of seed development, the expression of only two DEGs (AAE15 and LACS2) was upregulated, and the other DEGs were significantly downregulated in ‘M3’ relative to ‘M8’.
Fig. 7.

Heatmap analysis of significant DEGs associated with fatty acid biosynthesis from four developmental stages in ‘M3’ and ‘M8’ (a), key transcription factors (b) and significant DEGs associated with TAG biosynthesis (c)
Differentially expressed key transcription factors (TFs) in high- and low-oil cultivars during seed development
Transcription factors (TFs) play important roles in various biological pathways during seed development. Six key differentially expressed TFs were identified in high- and low-oil cultivars of tea oil camellia during different stages of seed development, namely, ABI3 (abscisic acid insensitive 3), FUS3 (fusca 3), LEC1 (leafy cotyledon1), WRI1 (wrinkled1), TTG2 (wrky44) and DOF4.6 (DNA binding with one finger 4.6). The GO terms of these TFs were mainly involved in regulation of glycolysis (GO: 0006110), zinc ion binding (GO: 0008270), positive regulation of the fatty acid biosynthetic process (GO: 0045723), triglyceride biosynthetic process (GO: 0019432), seed coat development (GO: 0010214), abscisic acid-mediated signaling pathway (GO: 0009738) and embryo development (GO: 0009790). Among these six TFs, ABI3 and FUS3 play important roles in the regulation of embryo maturation (Suzuki and Mccarty 2008). The two TFs LEC1 and WRI1 (AP2-like factor, ANT lineage) could increase the oil content during seed oil accumulation (Kwong et al. 2003; Focks and Benning 1998; Tan et al. 2011). TTG2 is involved in positive regulation of epidermal hair growth and negatively regulates the auxin-mediated response to salt stress (Li et al. 2015b). DOF4.6 plays an important role in cell development (Gardiner et al. 2010). During early seed development, the six TFs ABI3, WRI1, FUS3, LEC1, DOF4.6 and TTG2 were upregulated in ‘M3’ relative to ‘M8’. During the fourth stage of seed development (September 3), with the exception of three downregulated TFs (WRI1, comp68282_c0; DOF4.6, comp60186_c0; FUS3, comp57500_c0), the TFs were upregulated in ‘M3’ relative to ‘M8’. During the third stage of seed development (August 5), ABI3 (comp68374_c1) and FUS3 were significantly upregulated in ‘M3’ relative to ‘M8’ but downregulated during late seed development (September 3). The expression of WRI1 in ‘M3’ was first upregulated and then downregulated, and peak expression was observed on August 5. The expression of LEC1 in ‘M3’ was first upregulated and then downregulated; maximum expression was observed on July 4, and expression of this gene promoted rapid seed oil accumulation. In contrast, the relative expression levels of LEC1 in ‘M8’ were low during seed development, and only slight changes were observed in the expression levels of this gene (Fig. 7b).
DEGs involved in triacylglycerol (TAG) biosynthesis
Based on the functional annotation of the transcriptomes of developing seeds of tea oil camellia, ten key DEGs involved in triacylglycerol (TAG) biosynthesis were identified (Fig. 7c), namely, GPD1, GPAT3 (glycerol-3-phosphate acyltransferase 3), GPAT8, LPAT1 (lysophosphatidic acid acyltransferase), LAPT2, LPP2 (lipoma-preferred partner), PDAT1 (phospholipid:diacylglycerol acyltransferase), PDAT2, DGAT1 and DGAT1. Most of the DEGs were upregulated in ‘M3’ compared to ‘M8’, such as the GPD1 (comp66955_c0) gene encoding glycerol-3-phosphate, which was distinctly upregulated after the second seed developmental stage. The GPAT3 gene (comp53016_c0) was upregulated at the third stage of seed development in ‘M8’ compared to ‘M3’ but downregulated at the fourth stage. The GPAT8 (comp60752_c0) gene was upregulated at the second stage of seed development in ‘M3’ compared to ‘M8’, while LPP2 (comp61892_c0), LPAT1 (comp56565_c0) and LPAT2 (comp64140_c0) were upregulated during early seed development. During late seed development, PDAT1 (comp62924_c0) and PDAT2 (comp67513_c0) were downregulated in ‘M3’ relative to ‘M8’. In addition, DGAT1 (comp67933_c0) and DGAT2 (comp53935_c0) were upregulated in ‘M3’ relative to ‘M8’ throughout seed development.
qRT-PCR validation of the expression of genes involved in lipid biosynthesis
Based on the qRT-PCR analyses, the expression of the GPD1 gene in ‘M3’ showed a slight downregulation from June 2 to July 4 and then exhibited upregulation from July 4 to September 3, with peak expression observed on September 3. The expression of the GPD1 gene in ‘M8’ first showed slight downregulation from June 2 to July 4; after July 4, the expression was first upregulated and then downregulated after August 5, which was consistent with the increase in seed oil content observed after July 4. The expression levels of the GPD1 gene in ‘M3’ were significantly higher than those in ‘M8’ after July 4 (Fig. 8a). The expression of the DGAT1 gene in ‘M3’ first showed upregulation from June 2 to July 4 and then downregulation from July 4 to August 5, after which, the expression was upregulated. However, the expression of the DGAT1 gene in ‘M8’ first exhibited distinct downregulation from June 2 to August 5 and then exhibited slight upregulation from August 5 to September 3 (Fig. 8b). The expression levels of the DGAT1 gene in ‘M3’ were consistently higher than those in ‘M8’ throughout seed development. The expression of the DGAT2 gene in ‘M3’ first appeared upregulated from June 2 to July 4 and then downregulated from July 4 to August 5, exhibiting upregulation again after August 5 (Fig. 8c). The expression of the DGAT2 gene were always higher in ‘M3’ than in ‘M8’, and the difference was significant between July 4 and September 3, which was consistent with the seed oil content in ‘M3’ being significantly higher than that in ‘M8’ during the relatively late developmental stage.
Fig. 8.

Differences in the expression of GPD1 (a), DGAT1 (b) and DGAT2 (c) in the seeds of the ‘M3’ and ‘M8’ cultivars of tea oil camellia. *Indicates significant differences in relative gene expression levels between two cultivars at the same developmental stage at the 0.05 level
The genes KAR (β-ketoacyl-ACP reductase), EAR (enoyl-ACP reductase), HAD (3-hydroxyacyl-ACP dehydrase) and KASI are key genes that regulate the biosynthesis of C16:0-ACP, which is a precursor of C18 fatty acids in the seeds of tea oil camellia. During different seed developmental stages of tea oil camellia, the expression patterns of these four genes were similar in ‘M3’ and ‘M8’ (Fig. 9). From June 2 to July 4, the expression levels of the HAD and KASI genes were very similar (Fig. 9c, d); the expression levels of the KAR gene increased substantially; and the expression level of the EAR gene decreased considerably. From July 4 to September 3, the KASI, EAR, and HAD genes were upregulated in ‘M3’ and ‘M8’, and peak expression was observed on September 3; however, the expression levels of the KAR gene first decreased and then increased slightly during these stages (Fig. 9a). The expression levels of KASI, KAR, HAD and EAR genes in ‘M3’ were significantly higher than those in ‘M8’ throughout seed development, providing high levels of C16:0-ACP, the precursor of C18 fatty acids.
Fig. 9.

Expression patterns of genes involved in fatty acid biosynthesis in the seeds of two cultivars, namely, ‘M3’ and ‘M8’. KAR (a), EAR (b), HAD (c), KASI (d), FATB (e), KASII (f), SAD (g), FAD2 (h), FAE1 (i), FAD3 (j), FAD7 (k), FAD8 (l). *Indicates significant differences in relative gene expression levels between two cultivars at the same developmental stage at the 0.05 level
FATB (palmitoyl-acyl-ACP thioesterase) is a key enzyme-encoding gene that mainly controls the conversion of the 16-carbon palmitoyl-ACP into palmitic acid. The FATB gene appeared to be upregulated in ‘M3 and M8’ from June 2 to July 4 (Fig. 9e), and the expression levels of this gene increased, which was consistent with the high palmitic acid content observed at related seed developmental stages (Fig. 2c). Subsequently, the expression levels of the FATB gene gradually decreased, which was consistent with the downward trend of the palmitic acid content, which reached peak value on July 4 and then decreased. The KASII gene encodes a key enzyme that catalyzes the formation of stearic acid, and KASII gene expression levels are closely associated with changes in the stearic acid content. From June 2 to July 4, the expression levels of the KASII gene appeared to decrease slightly, which was consistent with the slight decrease in stearic acid content observed in related seed developmental stages (Figs. 2c, 9f); subsequently, the expression levels of the KASII gene showed a rapid upward trend from July 4 to September 3, which contributed to the rapid synthesis of stearic acid, and the stearic acid content increased steadily at these related seed developmental stages (Figs. 2c, 9f).
SAD mainly catalyzes the desaturation of stearic acid to form oleic acid in the fatty acid biosynthetic pathway, which is the first step in the desaturation of C18 fatty acids to produce C18:1. During seed development of tea oil camellia, the expression levels of the SAD gene showed an upward trend (Fig. 9g), increasing slightly from June 2 and July 4; these values were similar, with the proportion of oleic acids increasing slightly during related seed developmental stages. From July 4 to September 3, the expression levels of the SAD gene increased rapidly and reached peak values on September 3. The relative expression levels of the SAD gene in ‘M3’ and ‘M8’ increased 12.68 and 8.75 from July 4 to September 3, respectively; while the oleic acid content increased from 28.53 to 82.51% in ‘M3’ and from 20.18 to 76.64% in ‘M8’. In addition, the expression levels of the SAD gene in ‘M3’ were significantly higher than those in ‘M8’ throughout seed development of tea oil camellia, which resulted in the oleic acid content in M3 being higher than those in ‘M8’ (Figs. 2c, 9g), with a significant difference observed on September 3 (82.51% vs. 76.64%).
FAD2 mainly regulates the desaturation of oleic acid to linoleic acid in the fatty acid biosynthetic pathway. The expression levels of the FAD2 gene first increased from June 2 to July 4 (Fig. 9h), which was consistent with the upward trend for the proportion of linoleic acid in seed oil. After July 4, the expression level of the FAD2 gene rapidly decreased, which led to a rapid decrease in the percentage of linoleic acid in these related seed developmental stages (Figs. 2c, 9h). Subsequently, the linoleic acid content in ‘M3’ and ‘M8’ decreased to the lowest values of 3.51% and 6.47%, respectively. The expression levels of the FAD2 gene in ‘M8’ were always higher than those in ‘M3’, which was the main reason behind the linoleic acid content in ‘M8’ being higher than that in ‘M3’. FAE1 (fatty acid elongase 1) is a key enzyme for the synthesis of arachidonic acid. From June 2 to August 5, the expression level of the FAE1 gene increased slightly and then decreased slightly in the seeds of tea oil camellia, which was consistent with the level of arachidonic acid first exhibiting an increase, followed by a decrease (Figs. 2c, 9i).
The genes FAD3, FAD7 and FAD8 are key genes that regulate the conversion of linoleic acid into linolenic acid. The expression levels of these three genes were low throughout the seed development of tea oil camellia (Fig. 9j–l), which was consistent with the low linolenic acid content in the seed oil of tea oil camellia. The expression levels of FAD3 and FAD7 first increased and then decreased at different seed developmental stages of the two cultivars ‘M3’ and ‘M8’. FAD8 expression showed a downward trend between June 2 and August 5, after which, the expression increased slightly but remained relatively low. These results showed that the selected genes involved in lipid metabolism exhibited different expression patterns and differences in expression between the high- and low-oil cultivars of tea oil camellia during seed development.
Discussion
Transcriptomic differences between high- and low-oil cultivars of tea oil camellia during seed development
In this study, a total of 851,725,244 clean reads were generated from RNA-seq data obtained from seeds of high- and low-oil cultivars of tea oil camellia at different developmental stages; these reads were assembled de novo to obtain 89,049 transcripts and further obtain 53,507 unigenes. Interestingly, a large number of DEGs were obtained by pairwise comparisons of the high- and low-oil cultivars at same developmental stage (M3T1 vs. M8T1, M3T2 vs. M8T2, M3T3 vs. M8T3 and M3T4 vs. M8T4), and the number of DEGs gradually increased from early to late seed development, with a peak value of 2647 observed at the third stage. However, no DEG was detected at the fourth stage of seed development, possibly because there was little difference between the two cultivars during the seed maturation stage. In addition, comparative GO enrichment analysis of the high- and low-oil cultivars at the same developmental stage showed that the number of GO terms gradually increased, and only 30 significantly enriched GO terms were identified during early seed development, reaching 155 during the third stage of seed development. On the other hand, based on the Venn diagram analyses of the number of common DEGs in comparisons among samples of different seed developmental stages of each cultivar, there was no common DEG in ‘M3’ throughout seed development; in contrast, there were some common DEGs in ‘M8’. Mining for these common DEGs in ‘M8’ could provide some interesting clues for improvement of tea oil camellia.
Furthermore, these DEGs, which increased in number, were involved in various metabolic processes from seed embryogenesis to maturation. During early seed development, the DEGs associated with KEGG pathways were involved in only three pathways: methane metabolism, phenylpropanoid biosynthesis and phenylalanine metabolism. However, from seed embryogenesis to maturation, the DEGs that participated in related metabolic processes switched to five other pathways—glycolysis/gluconeogenesis, starch metabolism, sucrose metabolism, cysteine and methionine metabolism. Analysis of the transcripts and GO terms involved in different relevant biological pathways for functional transcriptomic studies of tea oil camellia in the future will be an important breakthrough.
Coordinated expression of source and sink genes that regulate seed oil biosynthesis
The GPD1 gene catalyzes the conversion of dihydroxyacetone phosphate into G3P, positively regulating G3P synthesis and directly affecting the levels of G3P and TAG (Remize et al. 2001). Overexpression of the GPD1 gene could enhance the Kennedy metabolic pathway and increase the TAG content in plants (Gibon et al. 2002; Vigeolas and Geigenberger 2004). The GPD1 gene of yeast was overexpressed in transgenic oilseed rape, resulting in a 40% increase in seed oil content (Vigeolas et al. 2007). The DGAT gene mainly catalyzes the conversion of DAG into TAG and has two nonhomologous transcripts, namely, DGAT1 and DGAT2, which play important roles in TAG biosynthesis (Wang et al. 2006; Lung and Weselake 2006). Overexpression of the DGAT gene of Arabidopsis in wild-type Arabidopsis could significantly increase the seed oil content and seed weight (Jako and Taylor 2001). The DGAT1 gene of T. majus was transferred into wild-type Arabidopsis, which resulted in an increase of 3.5–10% in dry seed oil content (Xu et al. 2010).
The GPD1 gene, which is the rate-limiting gene in the glycerol anabolic pathway, is also called the “source gene” of the TAG synthesis pathway (Ruan and Li 2008), which is responsible for regulating G3P synthesis in the TAG biosynthetic pathway; the DGAT gene, which is the most critical gene that catalyzes the conversion of DAG into TAG in the final step of the Kennedy pathway, is called the “sink gene” of the TAG synthesis pathway (Ruan and Li 2008). Vanhercke et al. (2013) defined the increase in fatty acid biosynthesis as “Push” and participation in TAG synthesis and assembly as “Pull”. Coexpression of “Push” and “Pull” could effectively increase the seed oil content. Thus, it is important to study the coordinated expression of the source gene GPD1 and the sink gene DGAT to understand seed oil biosynthesis in oil crops.
During seed development of tea oil camellia, the seed oil content of the two cultivars ‘M3’ and ‘M8’ showed gradual upward trends, increasing rapidly after the second seed developmental stage. The seed oil content in ‘M3’ was higher than that in ‘M8’ throughout seed development; interestingly, the expression levels of the GPD1 gene in the seeds of ‘M3’ were significantly higher than those in the seeds of ‘M8’. After the second stage of seed development, the expression of the GPD1 gene in ‘M3’ was rapidly upregulated, which was consistent with the seed oil content increasing gradually at this stage. The expression levels of the DGAT1 and DGAT2 genes in seeds of ‘M3’ were consistently higher than those in the seeds of ‘M8’ at all the seed developmental stages, leading to increased conversion of DAG to TAGs, resulting in higher seed oil levels in ‘M3’ than in ‘M8’.
The high expression levels of the source gene GPD1 in the seeds of the high-oil ‘M3’ cultivar at all the seed developmental stages provided a high amount of the primary substrate G3P for TAG synthesis, with high coexpression of the sink genes DGAT1 and DGAT2 accelerating TAG synthesis and assembly. Peak expression of the GPD1 gene was observed during third seed development, and peak expression of the DGAT1 and DGAT2 genes was observed during late seed development. There was positive correlation of expression levels among the GPD1, DGAT1 and DGAT2 genes. Thus, the coordinated high expression of the source gene GPD1 and sink genes DGAT1 and DGAT2 promoted greater lipid biosynthesis and accumulation in the seeds of ‘M3’ than in the seeds of ‘M8’. Detailed demonstration of the coordinated regulatory mechanism of source and sink genes will be another important breakthrough in the improvement of lipid metabolism in the seeds of tea oil camellia and other woody oil trees.
Coordinated high expression of multigenes directly increases C16:0-ACP biosynthesis
The coordinated regulation of multigenes directly affects fatty acid metabolism. Overexpression of the FATB1 gene in Arabidopsis seeds resulted in a fourfold increase in the palmitic acid content in the seed oil; alteration of the expression of FATB1 would lead to decreased palmitic acid content and saturated fatty acid content (Dörmann et al. 2000). The KASII gene determines the synthesis ratio of C16 to C18 fatty acids. Suppression of the expression of the KASII gene in the leaves of Jatropha curcas by gene silencing techniques led to a 149.69% increase in the palmitic acid content; moreover, the stearic acid content decreased, and the ratio between the levels of these two types of fatty acids increased 2.87-fold relative to the control group (Ye et al. 2009).
During seed development of tea oil camellia, the expression of the KASI, HAD and EAR genes showed an upward trend between July 4 and September 3. The coordinated high expression of these three genes provided sufficient C16:0-ACP for the synthesis of stearic acid and palmitic acid. While the expression levels of the FATB gene showed a rapid decrease, which weakened the conversion of C16:0-ACP into palmitic acid during these stages, the rapidly increased expression levels of the KASII gene promoted the conversion of C16:0-ACP into C18:0-ACP, which provided an increased amount of C18:0-ACP for the conversion of C18 unsaturated fatty acids. Therefore, the coordinated high expression of the KASI, HAD, and EAR genes directly increased the relative level of C16:0-ACP, which provided enough precursor resources for the synthesis of oleic acid in the seeds of tea oil camellia. Downregulation of FATB weakened the conversion of C16:0-ACP into palmitic acid, and the high expression of KASII contributed to the promotion of stearic acid synthesis. The coordinated regulation of upstream and downstream multigenes contributed enough C18:0 for rapid oleic acid synthesis in the seeds of tea oil camellia.
High expression levels of key SAD genes and low expression levels of downstream multigenes contributing to high oleic acid accumulation
Oleic acid accounts for a major proportion of the seed oil of tea oil camellia. The SAD gene is a key gene and encodes an enzyme that catalyzes the first-stage desaturation of saturated fatty acids to oleic acid. This enzyme determines the total levels of unsaturated fatty acids over C18 and the ratio of saturated and unsaturated fatty acids in seed oil. Overexpression of the yellow lupine SAD gene in tobacco resulted in an 11% increase in the oleic acid content of tobacco leaves (Zaborowska et al. 2002). Antisense expression of the SAD gene in rapeseed led to a significant increase in the stearic acid content from 2 to 40% (Knutzon et al. 1992). The expression of the SAD gene appeared to be upregulated in ‘M3’ compared to ‘M8’ at all the seed developmental stages, which was consistent with the finding that the oleic acid content in ‘M3’ was consistently higher than that in ‘M8’.
The FAD2 gene is responsible for the regulation of the desaturation of oleic acid to produce linoleic acid. Heterologous expression of the Vernicia fordii FAD2 gene in Arabidopsis thaliana showed that the relationship between the expression level of VfFAD2 and the increase in linoleic acid content was statistically linear (Chen et al. 2015). The expression of the FAD2 gene was first upregulated and then continuously downregulated in the seeds of tea oil camellia. The expression level of the FAD2 gene during early seed development was relatively higher than the levels observed during the other three seed developmental stages, corresponding to the relatively high proportion of linoleic acid. Then, the expression level of the FAD2 gene decreased significantly, which resulted in a continuous decrease in the linoleic acid content.
During early seed development, the expression levels of the SAD gene in the seeds of the two cultivars of tea oil camellia were relatively low compared with the levels in other three stages, and the expression levels of the FAD2 gene were relatively high. The expression levels of the FAD2 gene in ‘M8’ were consistently higher than those in ‘M3’ at all the seed developmental stages, which was consistent with the fact that the linoleic acid content in the cultivar ‘M8’ was higher than that in ‘M3’. This effect may have led to a relatively low accumulation rate of oleic acid and acceleration of the conversion rate, which resulted in the oleic acid content in the cultivar ‘M8’ being significantly lower than that in ‘M3’. During late seed development, the expression levels of the SAD gene increased rapidly to high levels, while the expression levels of the downstream FAD2 gene decreased rapidly to low levels, which further explained the coordinated regulatory mechanism of the upstream and downstream genes for high oleic acid accumulation.
The FAE1 gene is involved in the biosynthesis of super-long-chain fatty acids in seeds and regulates the conversion of oleic acid to arachidonic acid (Wu et al. 2015). During early seed development of tea oil camellia, the expression level of the FAE1 gene did not change significantly. The expression level of this gene showed a slight increase and then a slight decrease in both ‘M3’ and ‘M8’, resulting in little change in the arachidonic acid content, which was relatively stable and low. This finding indicated that the metabolic pathways involved in the conversion of C18:1 into C20:1 were weak in seeds of tea oil camellia. During early seed development, arachidonic acid was not detected in the seed oil of tea oil camellia, which may be associated with a rapid increase in fruit volume and low dry matter accumulation.
The genes FAD3, FAD7, and FAD8 are key genes that regulate the conversion of linoleic acid into linolenic acid. During seed development of tea oil camellia, the expression of the FAD3 and FAD7 genes showed slight upregulation during early seed development, and the linolenic acid content in the seed oil was higher than that observed during late seed development. This finding was consistent with the upregulated expression of the FAD2 gene and with the linoleic acid content being relatively high in seed oil at this stage. Subsequently, the expression of the FAD3 and FAD7 genes was clearly downregulated, resulting in a corresponding decrease in the linolenic acid content, which was consistent with the significantly downregulated expression of the FAD2 gene resulting in a rapid decrease in linoleic acid content at this stage. During the different seed developmental stages of tea oil camellia, the expression levels of the FAD8 gene remained at a relatively low level, with no significant effects on the linolenic acid content. The continuous downregulation of FAD2 gene expression resulted in a distinct decrease in the expression levels of the FAD3 and FAD7 genes. The coordinated regulation of these upstream and downstream genes affects the conversion of linoleic acid into linolenic acid.
The continuous high expression of the SAD gene accelerated the synthesis and accumulation of oleic acid, and the coordinated low expression of FAD2, FAD3, FAD7, FAD8 and FAE1 reduced the desaturation of oleic acid. The coordinated regulation of these upstream and downstream multigenes contributes to ensuring the high accumulation of oleic acid in seeds of tea oil camellia.
Conclusions
In the transcriptomic profiles of seeds of high- and low-oil tea oil camellia at different developmental stages, a total of 53,507 unigenes and 89,049 transcripts were detected, and many DEGs were identified. Further functional enrichment of DEGs visibly revealed that the DEGs were mainly associated with biological processes and that some key DEGs were associated with lipid metabolism and fatty acid biosynthesis. The seed oil content increased dramatically after the second seed developmental stage, and rapid accumulation of oleic acid was observed during late seed development. In addition, qRT-PCR validation of the expression levels of key genes that regulate lipid biosynthesis showed that coordinated expression of the source gene GPD1 and the sink genes DGAT1 and DGAT2 resulted in rapid lipid biosynthesis and accumulation in seeds of tea oil camellia. The coordinated high expression of the HAD, EAR and KASI genes directly increased the relative levels of C16:0-ACP, which provided enough precursor resources for the synthesis of oleic acid in the seeds of tea oil camellia. Downregulation of the expression of FATB weakened the conversion of C16:0-ACP to palmitic acid, and the high expression levels of the KASII gene contributed to the promotion of stearic acid synthesis, which provided enough resources for the synthesis of oleic acid. The continuous high expression of the SAD gene accelerated the synthesis and accumulation of oleic acid, and the coordinated low expression of FAD2, FAD3, FAD7, FAD8 and FAE1 reduced the desaturation of oleic acid. The coordinated mechanism of regulation of these upstream and downstream multigenes contributed to ensure the high accumulation of oleic acid in seeds of tea oil camellia. In summary, our data provide a scientific basis for improvement of the seed oil content and oleic acid content in tea oil camellia in the future.
Acknowledgements
This research was supported by the Key Technology Research and Development Program of Guizhou Province ([2019]2404, [2018]5252, [2018]4016, [2017]2521, [2016]2517-1, [2016]2519) and the Fundamental Research Funds for the Central Universities (0918-110071). The original mRNA-seq data of this study was uploaded to NCBI’s database and released, and the data path is /fasp/ruanchengjiang/CleanData.
Compliance with ethical standards
Conflict of interest
All the authors declare that there is no conflict of interest.
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud S, Guyon V, Kronenberger J, et al. Multifunctional acetyl-CoA carb-oxylase 1 is essential for very long chain fatty acid elongation and embryo development in Arabidopsis. Plant J. 2010;33(1):75–86. doi: 10.1046/j.1365-313X.2003.016010.x. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc. 1995;57(1):289–300. [Google Scholar]
- Chen YH (2007) Physiochemical properties and bioactivities of tea seed (Camellia oleifera) oil. Clemson University
- Chen Y, Cui Q, Xu Y, et al. Effects of tung oilseed FAD2 and DGAT2 genes on unsaturated fatty acid accumulation in Rhodotorula glutinis and Arabidopsis thaliana. Mol Genet Genom. 2015;290(4):1605. doi: 10.1007/s00438-015-1011-0. [DOI] [PubMed] [Google Scholar]
- Chen J, Yang X, Huang X, et al. Leaf transcriptome analysis of a subtropical evergreen broadleaf plant, wild oil-tea camellia (Camellia oleifera), revealing candidate genes for cold acclimation. BMC Genom. 2017;18(1):211. doi: 10.1186/s12864-017-3570-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collakova E, Aghamirzaie D, Fang Y, et al. Metabolic and transcriptional reprogramming in developing soybean (Glycine max) embryos. Metabolites. 2013;3(2):347–372. doi: 10.3390/metabo3020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Ruan CJ, Shan JY, et al. Expression of key genes involved in lipid biosynthesis and accumulation during seeds formation and development in Hippophae rhamnoides. Acta Botanica Boreali-occidentalia Sinica. 2016;36(8):1642–1647. [Google Scholar]
- Dong B, Wu B, Hong W, et al. Transcriptome analysis of the tea oil camellia (Camellia oleifera) reveals candidate drought stress genes. PLoS One. 2017;12(7):e0181835. doi: 10.1371/journal.pone.0181835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörmann P, Voelker TA, Ohlrogge JB. Accumulation of palmitate in Arabidopsis mediated by the acyl-acyl carrier protein thioesterase FATB1. Plant Physiology. 2000;123(2):637–644. doi: 10.1104/pp.123.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focks N, Benning C. Wrinkled1: a novel, low-seed-oil mutant of Arabidopsis with a deficiency in the seed-specific regulation of carbohydrat-e metabolism. Plant Physiol. 1998;118(1):91–101. doi: 10.1104/pp.118.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner J, Sherr I, Scarpella E. Expression of DOF genes identifies early stages of vascular development in Arabidopsis leaves. Int J Dev Biol. 2010;54(8–9):1389–1396. doi: 10.1387/ijdb.093006jg. [DOI] [PubMed] [Google Scholar]
- Gibon Y, Vigeolas H, Tiessen A, et al. Sensitive and high throughput metabolite assays for inorganic pyrophosphate, ADPGlc, nucleotide phosphates, and glycolytic intermediates based on a novel enzymic cycling system. Plant J. 2002;30(2):221–235. doi: 10.1046/j.1365-313X.2001.01278.x. [DOI] [PubMed] [Google Scholar]
- Goettel W, Xia E, Upchurch R, et al. Identification and characterization of transcript polymorphisms in soybean lines varying in oil composition and content. BMC Genom. 2014;15(1):299. doi: 10.1186/1471-2164-15-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Mellado D, Martínez-Force E. The role of beta-ketoacyl-acyl carrier protein synthase III in the condensation steps of fatty acid biosynthesis in sunflower. Planta. 2010;231(6):1277–1289. doi: 10.1007/s00425-010-1131-z. [DOI] [PubMed] [Google Scholar]
- Gupta M, Bhaskar PB, Sriram S, et al. Integration of omics approaches to understand oil/protein content during seed development in oilseed crops. Plant Cell Rep. 2017;36(5):637–652. doi: 10.1007/s00299-016-2064-1. [DOI] [PubMed] [Google Scholar]
- Jako C, Taylor DC. Seed-specific over-expression of an Arabidopsis cDNA encoding a diacylglycerol acyltransferase enhances seed oil content and seed weight. Plant Physiol. 2001;126(2):861–874. doi: 10.1104/pp.126.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutzon DS, Thompson GA, Radke SE, et al. Modification of Brassica seed oil by antisense expression of a stearoyl-acyl carrier protein desaturase gene. Proc Natl Acad Sci USA. 1992;89(7):2624–2628. doi: 10.1073/pnas.89.7.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong RW, Bui AQ, Lee H, et al. LEAFY COTYLEDON1-LIKE defines a class of regulators essential for embryo development. Plant Cell. 2003;15(1):5–18. doi: 10.1105/tpc.006973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardizabal K, Effertz R, Levering C, et al. Expression of Umbelopsis ramanniana DGAT2A in seed increases oil in soybean. Plant Physiol. 2008;148(1):89–96. doi: 10.1104/pp.108.123042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Hur M, Lee JY, et al. A systems biology approach toward understanding seed composition in soybean. BMC Genom. 2015;16(S3):S9. doi: 10.1186/1471-2164-16-S3-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Mei Y, Li Y, et al. Expression of Brassica napus TTG2, a regulator of trichome development, increases plant sensitivity to salt stress by suppressing the expression of auxin biosynthesis genes. J Exp Bot. 2015;66(19):5821–5836. doi: 10.1093/jxb/erv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao SJ, Ji DL, Tong HR. Study on fatty acid composition and nutrition health protection function of the oiltea Camellia seed oil. J Cereals Oils. 2005;6:7–9. [Google Scholar]
- Libault M, Farmer A, Joshi T, et al. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J. 2010;63(1):86–99. doi: 10.1007/s00299-016-2064-1. [DOI] [PubMed] [Google Scholar]
- Lin P, Wang K, Zhou C, et al. Seed transcriptomics analysis in Camellia oleifera uncovers genes associated with oil content and fatty acid composition. Int J Mol Sci. 2018;19(1):118. doi: 10.3390/ijms19010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SC, Jin JQ, Ma JQ, et al. Transcriptomic analysis of tea plant responding to drought stress and recovery. PLoS One. 2016;11(1):e0147306. doi: 10.1371/journal.pone.0147306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lung SC, Weselake RJ. Diacylglycerol acyltransferase: a key mediator of plant triacylglycerol synthesis. Lipids. 2006;41(12):1073–1088. doi: 10.1007/s11745-006-5057-y. [DOI] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, et al. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:182–185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, Mccue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- O’Rourke JA, Bolon YT, Bucciarelli B, et al. Legume genomics: understanding biology through DNA and RNA sequencing. Ann Bot. 2014;113(7):1107–1120. doi: 10.1093/aob/mcu072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlrogge JB. Design of new plant products: engineering of fatty acid metabolism. Plant Physiol. 1994;104(3):821–826. doi: 10.1104/pp.104.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qun W, Shao HB, Xu ZL. Salinity tolerance mechanism of osmotin and osmotin-like proteins: a promising candidate for enhancing plant salt tolerance. Curr Genom. 2017;18:4. doi: 10.2174/1389202918666170705153045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remize F, Barnavon L, Dequin S. Glycerol export and glycerol-3-phosphate dehydrogenase, but not glycerol phosphatase, are rate limiting for glycerol production in Saccharomyces cerevisiae. Metab Eng. 2001;3(4):301–312. doi: 10.1006/mben.2001.0197. [DOI] [PubMed] [Google Scholar]
- Ruan CJ, Li Q. Status of oil content control in seed by genetic regulation and the strategy for improving the gene of Kosteletzkya for biodiesel production. Renew Energy Resour. 2008;26(4):35–40. [Google Scholar]
- Sánchez-Salcedo EM, Sendra E, Carbonell-Barrachina ÁA, et al. Fatty acids composition of Spanish black (Morus nigra L.) and white (Morus alba L.) mulberries. Food Chem. 2016;190:566–571. doi: 10.1016/j.foodchem.2015.06.008. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Severin AJ, Woody JL, Bolon YT, et al. RNA-Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 2010;10(1):160. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman I, Arumugam N, Sodhi YS, et al. Development of high oleic and low linoleic acid transgenics in a zero erucic acid Brassica juncea L. (Indian mustard) line by antisense suppression of the fad2 gene. Molecular Breeding. 2004;13(4):365–375. doi: 10.1023/B:MOLB.0000034092.47934.d6. [DOI] [Google Scholar]
- Suzuki M, Mccarty DR. Functional symmetry of the B3 network controlling seed development. Curr Opin Plant Biol. 2008;11(5):548–553. doi: 10.1016/j.pbi.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Tan H, Yang X, Zhang F, et al. Enhanced seed oil production in canola by conditional expression of Brassica napus LEAFY COTYLEDON1 and LEC1-LIKE in developing seeds. Plant Physiol. 2011;156(3):1577–1588. doi: 10.1104/pp.111.175000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhercke T, Ei TA, Shrestha P, et al. Synergistic effect of WRI1 and DGAT1 coexpression on triacylglycerol biosynthesis in plants. FEBS Lett. 2013;587(4):364–369. doi: 10.1016/j.febslet.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Vigeolas H, Geigenberger P. Increased levels of glycerol-3-phosphate lead to a stimulation of flux into triacylglycerol synthesis after supplying glycerol to developing seeds of Brassica napus L. in planta. Planta. 2004;219(5):827–835. doi: 10.1007/s00425-004-1273-y. [DOI] [PubMed] [Google Scholar]
- Vigeolas H, Waldeck P, Zank T, et al. Increasing seed oil content in oil-seed rape (Brassica napus L.) by over-expression of a yeast glycerol-3-phosphate dehydrogenase under the control of a seed-specific promoter. Plant Biotechnology Journal. 2007;5(3):431–441. doi: 10.1111/j.1467-7652.2007.00252.x. [DOI] [PubMed] [Google Scholar]
- Wang HW, Zhang JS, Gai JY, et al. Cloning and comparative analysis of the gene encoding diacylglycerol acyltransferase from wild type and cultivated soybean. Theor Appl Genet. 2006;112(6):1086–1097. doi: 10.1007/s00122-006-0210-9. [DOI] [PubMed] [Google Scholar]
- Wang B, Tan X, Jiang J, et al. Molecular cloning and expression of two genes encoding accase subunits of Camellia Oleifera (Theaceae) Pak J Bot. 2018;50(1):103–110. [Google Scholar]
- Wu L, Jia YL, Wu G, et al. Molecular evidence for blocking erucic acid synthesis in rapeseed (Brassica napus L.) by a two-base-pair deletion in FAE1 (fatty acid elongase 1) J Integr Agric. 2015;14(7):1251–1260. doi: 10.1016/S2095-3119(14)60853-4. [DOI] [Google Scholar]
- Xia EH, Jiang JJ, Huang H, Zhang LP, Zhang HB, Gao LZ. Transcriptome analysis of the oil-rich tea plant, Camellia oleifera, reveals candid-ate genes related to lipid metabolism. PLoS One. 2014;9(8):e104150. doi: 10.1371/journal.pone.0104150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Francis T, Mietkiewska E, et al. Cloning and characterization of an acyl-CoA-dependent diacylglycerol acyltransferase 1 (DGAT1) gene from Tropaeolum majus, and a study of the functional motifs of the DGAT protein using site-directed mutagenesis to modify enzyme activity and oil content. Plant Biotechnol J. 2010;6(8):799–818. doi: 10.1111/j.1467-7652.2008.00358.x. [DOI] [PubMed] [Google Scholar]
- Xu HM, Kong XD, Chen F, et al. Transcriptome analysis of Brassica napus pod using RNA-Seq and identification of lipid-related candidate genes. BMC Genom. 2015;16(1):858. doi: 10.1186/s12864-015-2062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Wang Y, Wang K, Ren H. Effects of geographic latitude and longitude on fat and its fatty acid composition of oil tea Camellia seeds. China Oils Fats. 2011;36(4):31–34. [Google Scholar]
- Ye J, Qu J, Bui HT, Chua NH. Rapid analysis of Jatropha curcas gene functions by virus-induced gene silencing. Plant Biotechnol J. 2009;7(9):964–976. doi: 10.1111/j.1467-7652.2009.00457.x. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ren S, Tan K. Study on climatic regionalization and layer and belt distribution of oil tea camellia quality in China. J Nat Res. 1999;14:123–127. [Google Scholar]
- Zaborowska Z, Starzycki M, Femiak I, et al. Yellow lupine gene encoding stearoyl-ACP desaturase–organization, expression and potential application. Acta Biochim Pol. 2002;49(1):29–42. [PubMed] [Google Scholar]
- Zeng Y, Tan X, Zhang L, et al. Identification and expression of fructose-1,6-bisphosphate aldolase genes and their relations to oil content in developing seeds of tea oil tree (Camellia oleifera) PLoS One. 2014;9(9):e107422. doi: 10.1371/journal.pone.0107422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DQ, Tan XF, Hu FM. The cDNA cloning and characteristic of stearoyl-ACP desaturase gene of Camellia Oleifera. Acta Horticulturae. 2008;769(769):55–61. doi: 10.17660/ActaHortic.2008.769.5. [DOI] [Google Scholar]
- Zhang DY, Kumar M, Xu L, et al. Genome-wide identification of Major Intrinsic Proteins in Glycine soja and characterization of GmTIP2;1 function under salt and water stress. Sci Rep. 2017;7:4106. doi: 10.1038/s41598-017-04253-z. [DOI] [PMC free article] [PubMed] [Google Scholar]