

Abstract
Increasing negative charge density at the surfaces of CdSe quantum dots (QDs) effects a bathochromic shift of their ground state optical spectra with increasing pH due to electrostatic and chemical modifications at the QD surface. These modifications are enabled by weakly-bound ligands that expose the surface to the aqueous environment.
TOC Figure:
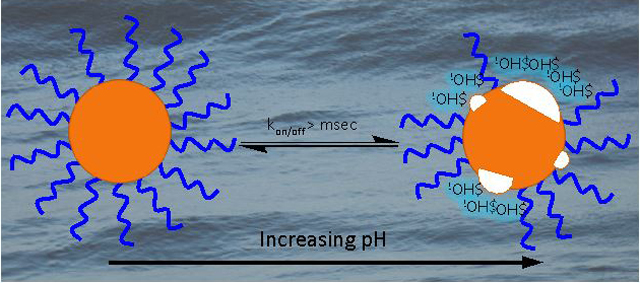
Weakly-bound ligands in dynamic exchange expose the surface of CdSe quantum dots to pH-dependent modification.
Water-soluble semiconductor quantum dots (QDs) are an important class of material for bio-labeling and biosensing1–3 and photocatalysis.4–7 Water-solubility of these particles is generally achieved by exchanging the native lipophilic ligands with thiolates functionalized with a hydrophilic tail group,6, 8–10 or by encapsulation of the QD in a hydrophilic layer.10, 11 Thiolate exchange however introduces mid band-gap trap states for photogenerated holes4 that inhibit catalytic turnover and decrease the quantum yield of emission, and encapsulation of the QDs blocks the access of either catalytic substrates or biological analytes to the QD surface.10, 12, 13 In bio-labeling applications, the quantum yield of emission can be recovered by shelling the QDs with a higher bandgap semiconductor, but an inorganic shell decreases the sensitivity of some sensing mechanisms, and presents a tunneling barrier for the excitonic carriers that makes their extraction prohibitively slow in photocatalytic applications.
We previously demonstrated the use of 3-phosphonopropionic acid (PPA) as an alternative to thiolate ligands to provide colloidal stability of Cd-chalcogenide QDs in water.14 Core-only CdS and CdSe QDs with PPA surfactant have higher oxidation potentials and higher photoluminescence (PL) quantum yields than QDs coated with mercaptopropionic acid (MPA). The QDs within CdS/PPA mixtures also exhibit a reversible bathochromic shift of their absorbance and emission spectra with increasing pH, due to changes in the electrostatic potential within Angstroms of their surfaces. We show here that, unlike the CdS/PPA system, the CdSe QDs are susceptible to pH-dependent surface modifications in water, similar to those observed at bulk semiconductor/liquid interfaces, which render the response of their optical spectra to pH only partially reversible.15 Here, we describe this response and its chemical mechanism. For QDs used as colloidal photo-redox catalysts where the majority of reactions require multiple outer sphere charge transfers, the formation of hydroxide or oxide layers at some pH values, as we see in this system, can slow extraction of photocarriers, but has also been shown to inhibit photocorrosion upon continuous illumination.16
Scheme 1 outlines the simultaneous ligand-exchange/phase-transfer procedure that we use to remove the native oleate ligands from the QD surface and disperse the QDs in water. The transfer of the particles into water requires addition of both acid, to promote proton exchange with the oleate ligands, and DMF, to further strip oleate from the QD surface17 and stabilize the QDs after ligand removal. The addition of 300 equivalents of PPA per QD from an isopropanolic solution results in the precipitation of the QDs from hexanes and displaces 9% – 25% of the oleate monolayer from the QD surface (ESI,† Figure S10). We resuspend the QDs in DMF, a good intermediate solvent probably because the amide acts as a dative ligand which acts to strip oleate from the QD surface and stabilizes the system in the final aqueous solution. We remove the hexanes layer and add aqueous KOH under a N2 atmosphere to achieve a pH, depending on the acid used, of 6 – 7.5 post-exchange. We dilute the solution with water to achieve a ratio of water-to-DMF of 2:1 (v/v), mix the solution rapidly under a N2 atmosphere, and extract the DMF with CHCl3. We do not detect any oleate by NMR (ESI,† Figure S11), but 10 mM DMF is retained in the final aqueous solution of QDs (ESI,† Figure S12).
Scheme 1.
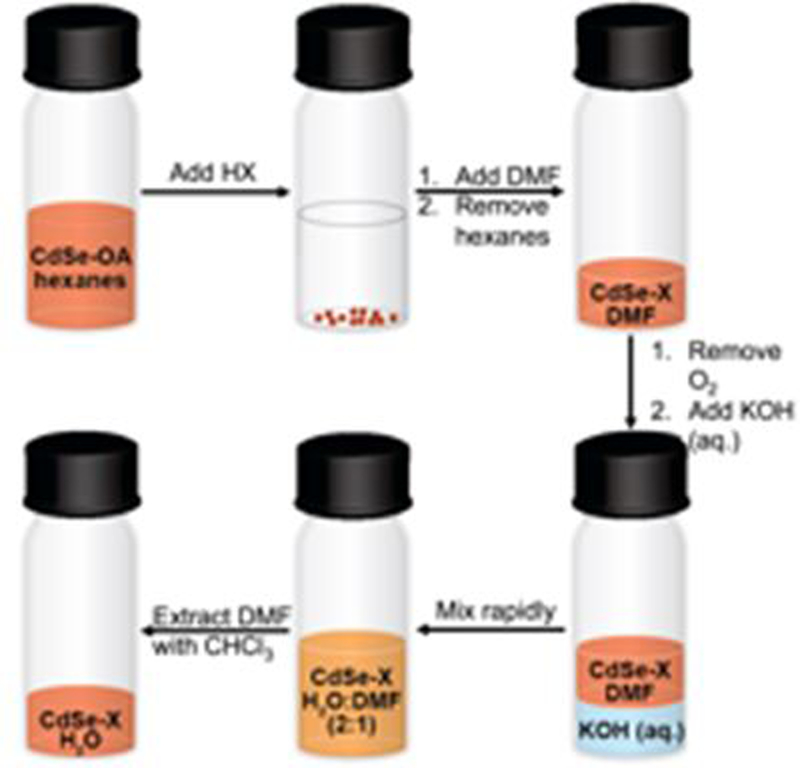
The ligand-exchange/phase-transfer procedure. We add one of the three acids to as-synthesized oleate-capped QDs, followed by DMF addition and removal of hexanes. We add KOH (aq.) to the QDs under N2 slowly, then mix rapidly and extract DMF with CHCl3.
The 1H NMR spectra of PPA within PPA/QD mixtures are identical in peak position, peak width, and integration to those of PPA in similarly-prepared solutions without added QDs over the entire pH range studied (5 – 12) (ESI,† Figure S12A and Figure S13A). These data indicate that PPA is in fast exchange, relative to the NMR timescale, on and off the surfaces of CdSe QDs, as they are on CdS QDs.14 We nonetheless know that PPA is acting as a surfactant for the QDs, as opposed to not interacting with them at all, because the use of fewer than 300 equivalents of PPA per QD during the exchange results in significant aggregation and accelerated precipitation of the QDs from water (ESI,† Figure S14), and because the QD/PPA system has measureable PL, whereas the QDs exchanged through HCl addition do not (ESI,† Figure S15). Based upon a generalized Poisson-Boltzmann model that explicitly includes (i) the acid-base equilibrium of the various protonation states of PPA on the QD surface and free in solution, and (ii) the exchange of PPA on and off the surface of the QD, we estimate the binding constant of PPA to have a lower bound of and an upper bound of . For details, see the ESI.†
We performed the analogous NMR experiments with MPA and we see evidence of binding to the QD surface in a slow exchange regime (ESI,† Figure S13B) as shown previously for thiolates.18
The measured pH values of the initial water dispersions of CdSe/PPA and CdSe/Cl QDs are one to two pH units lower than the theoretical pH values given the amount of KOH we add (ESI,†). The use of negatively charged surfactant during the QD synthesis results in cadmium ion surface enrichment,19 here, a measured 133 excess Cd2+ ions per particle (ESI ,†), which sequester some of the OH− to the QD surface upon exchange. This sequestration is enabled by the weak adsorption of both PPA and Cl- ligands in the moderately basic conditions under which the QDs are brought into water. Neutralization of excess surface Cd2+ and localization of an additional estimated 26 OH− per QD and PPA2- at each particle surface also explains the moderately negative ζ potential of the QDs with PPA surfactant upon exchange into water (−50 ± 1 mV, where −30 mV is approximately the minimum needed for colloidal stability).20
Figure 1 shows the change in the optical bandgap of the QDs (calculated as the average of the absorption and emission peak energies), relative to its value at pH 5, as a function of pH for all three types of water-solubilized CdSe QDs. Separate plots of absorption and emission energies are in the ESI (ESI,† Figure S16). The bandgap of CdSe QDs with PPA surfactant shifts to lower energy by ~30 meV with increasing pH. Dilution (ESI,† Figure S17) and changes in ionic strength (ESI,† Figure S18) do not account for the optical shift. We observe similar behaviour of the absorption energy with HCl-exchanged QDs (these QDs are non-emissive), Fig. 1, dark gray, though the magnitude of the shift is greater by about 10 meV than for the PPA/QD system. The MPA-coated QDs do not show any change in optical bandgap over the full pH range studied, Fig. 1, light gray. Both the bathochromic shift of the QD bandgap in the QD/PPA system and the lack of such a shift in the QD/MPA system are consistent with our previous results for CdS QDs.21 Based on the shape of the response and on simulations, we interpreted the shift in the CdS/PPA case as a Stark shift of the exciton by the pH-dependent charge density within a few Angstroms of the QD surface (primarily through the protonation equilibrium of the phosphonate group in PPA, pKa = 8.22 ± 0.01, ESI,†).21 The results in Fig. 1 with the HCl-exchanged QDs suggest that, even in the absence of PPA, if the surface of the QD is sufficiently unpassivated (as is the case for the weak binding ligand, Cl−, but not for the strong binding ligand, MPA), the changing local concentration of OH− can modulate the energy of the exciton. MPA-coated QDs do not experience this shift because MPA prevents exposure of the QD surface to proximate ions and because the carboxylate in MPA is not close enough to the QD surface to influence the energy of the core exciton upon protonation/deprotonation.
Figure 1.
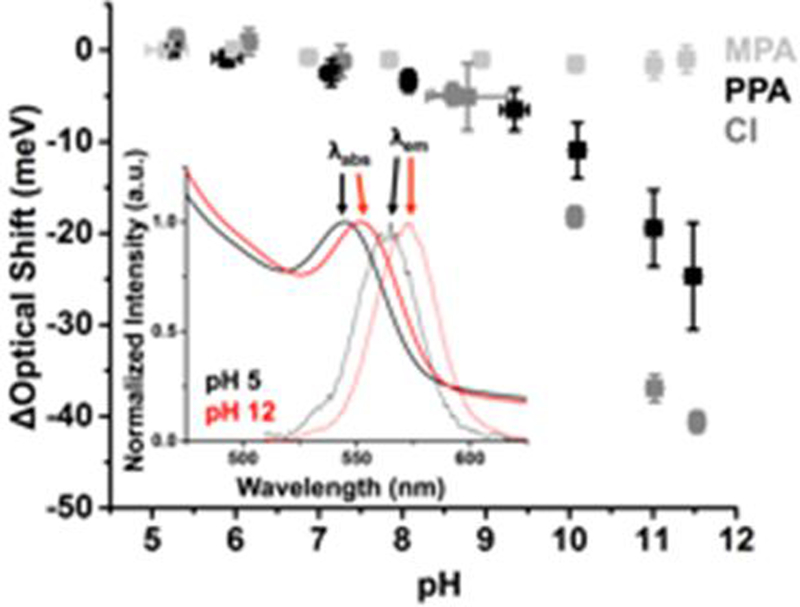
Shift of the excitonic energies of 0.7 μM CdSe QDs with 300 equivalents of MPA (light gray), 0.6 μM CdSe QDs with 300 equivalents of PPA (black) (see inset), or 0.6 μM CdSe QDs with 150 equivalents of HCl (dark gray) as a function of pH. Each data point is the the average of the shifts of the absorbance (λabs) and emission (λem) peaks within the optical spectra of at least three separately prepared samples of the QDs. The HCl-capped QDs were non-emissive (ESI†), so data for those samples is collected only from absorbance spectra. The data at each pH value is from a separate sample.
The important difference between the CdS and CdSe cases is the reversibility of the optical response to changes in pH. The shift in the optical spectra of CdS/PPA system is hysteretic but completely reversible between pH 6 and 12, which indicates that the charge density is controlled by a reversible process (such as protonation of PPA or electrostatic ion adsorption).21, 22 In contrast, the response of the optical bandgap of CdSe/PPA to pH is only ~50% reversible (Figure 2A), which indicates that an irreversible chemical process contributes to the bathochromic shift of the exciton energy. This chemical process is likely entirely responsible for the response of the optical bandgap of HCl-exchanged QDs to increasing pH, which is completely irreversible, and is accompanied by a decrease in colloidal stability (ESI,† Figure S19).
Figure 2.
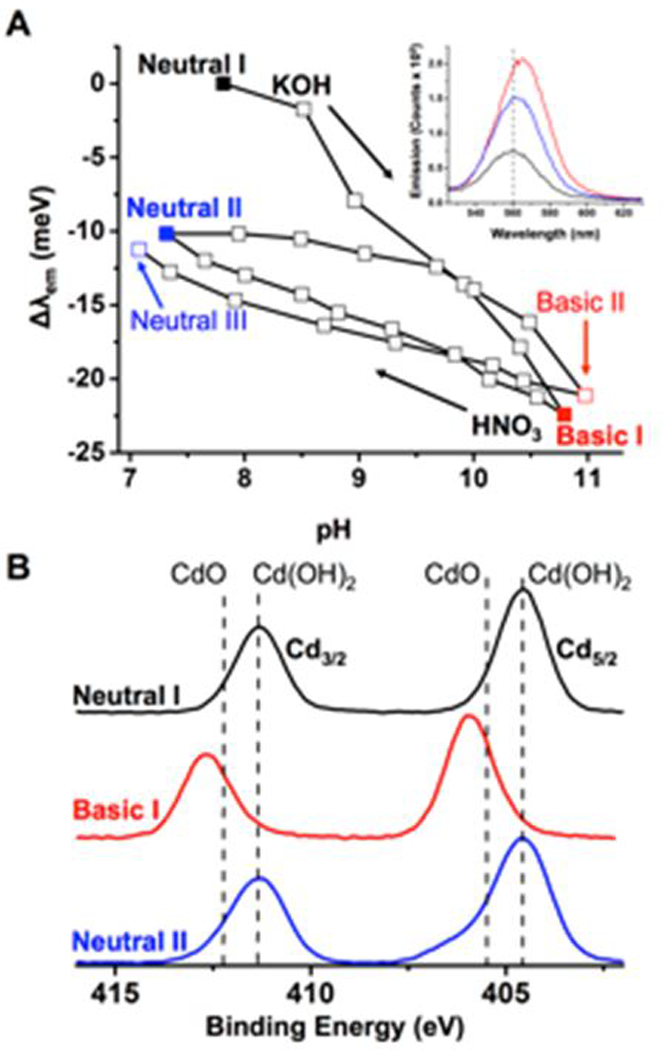
A) The Δλem of 1.0 μM CdSe with 300 equivalents of PPA during a pH cycling experiment in water using 0.1 M KOH and 0.1 M HNO3 to adjust the pH. The inset shows the emission spectra at pH 8 (Neutral I), pH 11 (Basic I), and upon return to pH 7 (Neutral II). B) The average of three scans of the Cd5/2 and Cd3/2 regions of the X-ray photoelectron spectra of the CdSe/PPA system, deposited from a pH 7 solution (Neutral I, black), a pH 12 solution (Basic I, red), and upon bringing a solution back to pH 7 from pH 12 (Neutral II, blue). The dashed lines indicate the peak maxima for bulk CdO and Cd(OH)2 (ESI†).
We propose that the reversible portion of the pH response of the optical bandgap of CdSe QDs within the CdSe/PPA system is due to changes in local charge density through net adsorption and desorption of OH− and PPA, which has a pH-dependent adsorption equilibrium constant due to a second deprotonation of the phosphonate at and above pH 8.22 ± 0.01 (ESI†). This deprotonation encourages displacement of PPA by OH− with increasing pH due to (i) a change from higher-affinity binding mode of PPA2- to lower affinity binding mode of PPA3-,23 (ii) increased intermolecular repulsion of the more highly charged ligands on the QD surface, and (iii) increased competition for binding sites as the [OH−] increases from ~10 OH− per QD at pH 9 to ~10,000 at pH 12. Above pH ~9.5 the bandgap continues to decrease in energy due to increasing OH− localization to the QD surface. The smaller hydrodynamic radius of OH− (compared to PPA) allows it to accumulate in higher density than PPA at the QD surface resulting in a degree of charge compensation not available with PPA.
The contribution of an irreversible chemical process to the bathochromic shift of the optical bandgap of CdSe QDs within the CdSe/PPA system is supported by the Cd regions of X-ray Photoelectron Spectra (XPS) of this system at the “Neutral I”, “Basic I”, and “Neutral II” points of the pH titration in Fig. 2A. For QDs, changes in peak binding energies and line shapes are primarily indicative of changes in the degree of passivation and/or oxidation of the population of these atoms on the surfaces of the particles in different environments.24 As shown in Figure 2B, at Neutral I, the Cd3/2 peak is at 411.3 eV and the Cd5/2 peak is at 404.6 eV, identical to those of bulk Cd(OH)2 (dashed line). This result supports our above assertion that ligand exchange results in the formation of Cd(OH)xy (where x = 1 or 2 and y = +1 or 0) from excess Cd2+ on the particles.25, 26 Upon bringing the dispersion to pH ~11 (Basic I in Fig. 2A), both Cd peaks shift to higher binding energy by more than 1 eV, indicative of both decreased passivation of the QD surface by PPA, which increases the surface’s exposure to OH− and further forms Cd(OH)xy,27, 28 and oxidation of the exposed surface to CdO.25 Increased local density and sequestration of OH− results in a shift in the ζ potential of CdSe QDs within the CdSe/PPA system from −50 ± 1 mV at Neutral I to −57 ± 2 at Basic I, Figure 3. Upon returning to neutral pH (Neutral II), a portion of Cd ions remain in the more oxidized state due to the irreversible formation of CdO and Cd(OH)xy at basic pH, as shown by the broader linewidths of the XPS signals than at Neutral I, Fig. 2B (see fits in the ESI,† Table S1). At Neutral II, the ζ potential of the particles is −43 ± 1 mV, less negative than Neutral I. The differences in the XPS linewidths and ζ potential values observed at Neutral I and Neutral II suggest that neutralization of excess positive charge at the QD surface by formation of CdO and Cd(OH)xy leads to decreased localization of mobile negative counterions around the particle upon the return to Neutral II.29 These chemical changes, and the resultant fewer proximate negative charges in the Neutral II vs. Neutral I states, are responsible for the irreversibility of the response of the optical shift to pH cycling, Fig. 2A. Further pH cycling results in a basic endpoint (Basic II, Fig. 2A) similar to Basic I and a third neutral endpoint (Neutral III, Fig. 2A) similar to Neutral II, which suggests that irreversible formation of CdO and Cd(OH)xy occurs primarily over the first pH cycle and that subsequent cycles are reversible.
Figure 3.
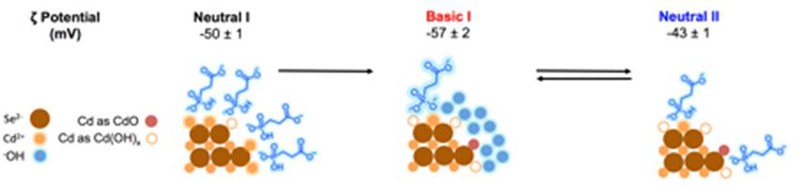
Top: ζ potentials for a sample of 0.8 μM of CdSe QDs brought into water with 300 equivalents of PPA, at the three points in the titration labelled in Figure 2A. Bottom: schematic of the chemical processes at the surface of the QD as a function of pH consistent with optical, XPS, and ζ-potential data on the CdSe/PPA aqueous system. Co-ions in the electrostatic double layer have been omitted for clarity.
We believe that the formation of Cd(OH)xy at basic pH, and the resultant irreversibility of the response of the optical bandgap of CdSe QDs to increasing pH, is related to the inability of CdSe to photo-oxidize OH− to OH• in room light. In the CdS QD case, where the pH response is reversible, we see formation of OH• above pH 9.521 upon addition of terephthalic acid to the CdS/PPA dispersion;30, 31 this result suggests that CdS QDs oxidize OH− such that it is less available to form a Cd(OH)xy layer. In the CdSe case, we see no evidence of OH• even at pH 12 (ESI,† Figure S20), probably because CdSe is ~0.6 V less oxidizing than CdS. We therefore conclude that the reversibility of the hydroxide interaction at the QD surface depends at least in part on the oxidation potential of the QD at a given pH.
In summary, the pH dependence of the excitonic energy of CdSe QDs, along with supporting analytical data, indicates that, above neutral pH, labile electrostatically-bound ligands expose QDs to OH−, which increasingly neutralizes excess positive charge at the CdSe QD surface with increasing pH through both a reversible electrostatic interaction and an irreversible chemical modification. These ions may be beneficial in QD-based photocatalysis by stabilizing reactive intermediates through hydrogen bonding or, at neutral-to-weakly basic pH, providing a reservoir of surface-localized protons for use in proton-coupled electron transfer reactions at QD surfaces.
Supplementary Material
Acknowledgments
We kindly acknowledge the National Science Foundation (CHE-1664184) and the National Institutes of Health (R21GM127919) for funding, and Dr. Kédy Edmé, Dr. Shichen Lian, and Dr. Cameron Rogers for helpful discussion. Research supported as part of the Center for Bio-Inspired Energy Science, funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award #DE-SC0000989. This work made use of the IMSERC at Northwestern University, which has receives support from the NIH (1S10OD012016–01/ 1S10RR019071–01A1); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the State of Illinois and International Institute for Nanotechnology (IIN). This work made use of the Keck-II facility of Northwestern’s NUANCE Center, which has received support from SHyNE; the MRSEC program (NSF DMR-1720139); IIN; the Keck Foundation; and the State of Illinois. Metal analysis was performed at the Northwestern University Quantitative Bio-element Imaging Center.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
References
- 1.Chen Y, Cordero JM, Wang H, Franke D, Achorn OB, Freyria FS, Coropceanu I, Wei H, Chen O, Mooney DJ and Bawendi MG, Angew. Chem. Int. Ed., 2018, 57, 4652–4656. [DOI] [PubMed] [Google Scholar]
- 2.Sajwan RK, Bagbi Y, Sharma P and Solanki PR, J. Lumin., 2017, 187, 126–132. [Google Scholar]
- 3.Mordvinova N, Vinokurov A, Kuznetsova T, Lebedev OI and Dorofeev S, Dalton Trans., 2017, 46, 1297–1303. [DOI] [PubMed] [Google Scholar]
- 4.McClelland KP and Weiss EA, ACS Appl. Energy Mater., 2018, DOI: 10.1021/acsaem.8b01652. [DOI] [Google Scholar]
- 5.Zhang Z, Edme K, Lian S and Weiss EA, J. Am. Chem. Soc., 2017, 139, 4246–4249. [DOI] [PubMed] [Google Scholar]
- 6.Jensen SC, Bettis Homan S and Weiss EA, J. Am. Chem. Soc., 2016, 138, 1591–1600. [DOI] [PubMed] [Google Scholar]
- 7.Caputo JA, Frenette LC, Zhao N, Sowers KL, Krauss TD and Weix DJ, J. Am. Chem. Soc., 2017, 139, 4250–4253. [DOI] [PubMed] [Google Scholar]
- 8.Brown KA, Wilker MB, Boehm M, Dukovic G and King PW, J. Am. Chem. Soc., 2012, 134, 5627–5636. [DOI] [PubMed] [Google Scholar]
- 9.Zhan N, Palui G and Mattoussi H, Nat. Protoc., 2015, 10, 859. [DOI] [PubMed] [Google Scholar]
- 10.Palui G, Aldeek F, Wang W and Mattoussi H, Chem. Soc. Rev., 2015, 44, 193–227. [DOI] [PubMed] [Google Scholar]
- 11.Montaseri H and Forbes PBC, Mat. Today Comm., 2018, 17, 480–492. [Google Scholar]
- 12.Bruchez M, Moronne M, Gin P, Weiss S and Alivisatos AP, Science, 1998, 281, 2013–2016. [DOI] [PubMed] [Google Scholar]
- 13.Gerion D, Pinaud F, Williams SC, Parak WJ, Zanchet D, Weiss S and Alivisatos AP, J. Phys. Chem. B, 2001, 105, 8861–8871. [Google Scholar]
- 14.Calzada R, Thompson CM, Westmoreland DE, Edme K and Weiss EA, Chem. Mater., 2016, 28, 6716–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allongue P and Tenne R, J. Electrochem. Soc., 1991, 138, 261–268. [Google Scholar]
- 16.Wakerley DW, Kuehnel MF, Orchard KL, Ly KH, Rosser TE and Reisner E, Nat. Energy, 2017, 2, 17021. [Google Scholar]
- 17.Anderson NC, Hendricks MP, Choi JJ and Owen JS, J. Am. Chem. Soc., 2013, 135, 18536–18548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldana J, Lavelle N, Wang Y and Peng X, J. Am. Chem. Soc., 2005, 127, 2496–2504. [DOI] [PubMed] [Google Scholar]
- 19.Morris-Cohen AJ, Frederick MT, Lilly GD, McArthur EA and Weiss EA, J. Phys. Chem. Lett., 2010, 1, 1078–1081. [Google Scholar]
- 20.Kuznetsova YV and Rempel AA, Inorg. Mater., 2015, 51, 215–219. [Google Scholar]
- 21.Thompson CM, Kodaimati M, Westmoreland D, Calzada R and Weiss EA, J. Phys. Chem. Lett., 2016, 7, 3954–3960. [DOI] [PubMed] [Google Scholar]
- 22.Chamousis RL and Osterloh FE, Energy Environ. Sci., 2014, 7, 736–743. [Google Scholar]
- 23.Gomes R, Hassinen A, Szczygiel A, Zhao Q, Vantomme A, Martins JC and Hens Z, J. Phys. Chem. Lett., 2011, 2, 145–152. [Google Scholar]
- 24.Peterson MD, Jensen SC, Weinberg DJ and Weiss EA, ACS Nano, 2014, 8, 2826–2837. [DOI] [PubMed] [Google Scholar]
- 25.Ismail KM, J. Appl. Electrochem., 2001, 31, 1333–1338. [Google Scholar]
- 26.Will FG and Hess HJ, J. Electrochem. Soc., 1973, 120, 1–11. [Google Scholar]
- 27.Brookins DG, Chem. Geol., 1986, 54, 271–278. [Google Scholar]
- 28.Wardle W. J. T. a. N., Corros. Sci., 1975, 15, 663–665. [Google Scholar]
- 29.Walker DA, Kowalczyk B, de la Cruz MO and Grzybowski BA, Nanoscale, 2011, 3, 1316–1344. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Su S, Liu B, Cao M and Hu C, Chem. Commun, 2013, 49, 7830–7832. [DOI] [PubMed] [Google Scholar]
- 31.Simon T, Bouchonville N, Berr MJ, Vaneski A, Adrović A, Volbers D, Wyrwich R, Döblinger M, Susha AS, Rogach AL, Jäckel F, Stolarczyk JK and Feldmann J, Nat. Mat., 2014, 13, 1013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.