

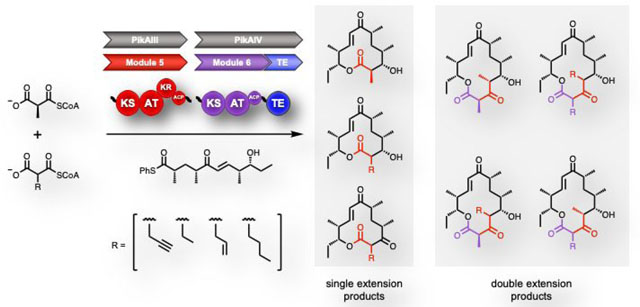
Abstract
There is significant interest in diversifying the structures of polyketides to create new analogues of these bioactive molecules. This has traditionally been done by focusing on engineering the acyltransferase (AT) domains of polyketide synthases (PKSs) responsible for the incorporation of malonyl-CoA extender units. Non-natural extender units have been utilized by engineered PKSs previously; however, most of the work to date has been accomplished with ATs that are either naturally promiscuous and/or located in terminal modules lacking downstream bottlenecks. These limitations have prevented the engineering of ATs with low native promiscuity and the study of any potential gatekeeping effects by domains downstream of an engineered AT. In an effort to address this gap in PKS engineering knowledge, the substrate preferences of the final two modules of the pikromycin PKS were compared for several non-natural extender units and through active site mutagenesis. This led to engineering of the methylmalonyl-CoA specificity of both modules and inversion of their selectivity to prefer consecutive non-natural derivatives. Analysis of the product distributions of these bimodular reactions revealed unexpected metabolites resulting from gatekeeping by the downstream ketoreductase and ketosynthase domains. Despite these new bottlenecks, AT engineering provided the first full-length polyketide products incorporating two non-natural extender units. Together, this combination of tandem AT engineering and the identification of previously poorly characterized bottlenecks provides a platform for future advancements in the field.
Graphical Abstract
INTRODUCTION
Type I polyketide synthases (PKSs) are responsible for the biosynthesis of some of the most clinically important bioactive compounds in Nature, including the blockbuster drugs erythromycin A (antibiotic), rapamycin (immunosuppressant/anti-cancer), and avermectin (anthelminthic).1 These PKSs are giant assembly line pathways that can be broken down into individual modules (Figure 1), each of which is responsible for incorporation of a single extender unit, often a coenzyme A (CoA)-linked malonate derivative. The acyltransferase (AT) within each module acts as the “gatekeeper” domain due to its innate ability to select a specific extender unit for priming of its cognate acyl carrier protein (ACP). Despite the structural diversity of polyketides, the AT domains responsible for selecting the extender units for each module typically include only three substrates: malonyl-CoA, methylmalonyl-CoA, and to a lesser degree, ethylmalonyl-CoA.2 Thus, except in a few rare cases, the selected substrates account for relatively narrow chemical diversity.3–7 Instead, polyketide diversity in nature comes from varying oxidations, cyclization patterns, or post-PKS modifications. This is represented by the four final products that result from the pikromycin (Pik) PKS (Figure 1). The development of chemo-enzymatic approaches that employ non-natural malonyl-CoA analogues affords the opportunity to increase the structural diversity of polyketides by engineering PKSs.8–10
Figure 1.

The pikromycin polyketide synthase and its products. ACP = acyl carrier protein; AT = acyltransferase; DH = dehydratase; ER = enoylreductase; KR = ketoreductase; KS = ketosynthase; KSQ = ketosynthase-like decarboxylase; TE = thioesterase.
Traditionally, PKS engineering has focused on exchanging modules or domains to alter the final product structure, but there are three critical limitations: (1) most PKS modules incorporate natural extenders that lack useful chemical handles, (2) non-native protein-protein interactions often result in chimeras with poor catalytic efficiencies,11–12 and (3) to achieve site-selective installation of a given non-natural extender unit into a polyketide, the specificity of the domain/module chimera must be orthogonal to that of the native, intact extension modules. In order to produce non-natural extender units, we and others have utilized and engineered malonyl-CoA synthetases or similar enzymes to create a panel of PKS substrates bearing a variety of useful chemical moieties.8–10, 13–15 The second issue has been approached through introduction of AT active site mutations, with varying levels of success.9, 16–19 For example, replacing the conserved YASH motif that is found in methylmalonyl-specific ATs with motifs from other natural ATs (e.g., HAFH from malonyl-CoA specific ATs) can lead to changes in AT specificity. However, these changes alone have not completely inverted AT specificity between natural substrates and therefore do not provide the requisite orthogonality for site-selective modification of the polyketide structure.20 In contrast, we and others have demonstrated that inherent extender unit promiscuity of some ATs provides a platform for creating new substrate specificities via site-directed mutagenesis. For example, the methylmalonyl-CoA-utilizing EryAT6 and corresponding terminal extension module (Ery6) of the 6-deoxyerythronolide B synthase (DEBS) from erythromycin A biosynthesis has significant promiscuity towards larger non-natural extender units.21 These non-natural substrates were utilized by an engineered Ery6 module with the YASH to RASH variant resulting in a switch from 92% methylmalonyl-CoA incorporation (wild-type enzyme) to 88% propargylmalonyl-CoA (non-natural) incorporation into the polyketide chain.18 The ability to manipulate the substrate preference of another AT from DEBS (EryAT2) towards longer alkyl chains via a VASH motif (found in some natural ethylmalonyl-CoA-specific ATs) was also demonstrated, albeit to a lesser extent.9 These shifts in substrate selectivity are notable and rely on the inherent promiscuity of the AT as an opportunity for redesigning substrate specificity in PKSs. Additionally, most AT engineering is accomplished with terminal extension modules that are at the end of the assembly line lacking downstream bottlenecks, and involve installation of only one non-natural extender unit into the final product structure.
Herein, the ability of site-specific mutagenesis to manipulate the extender unit specificity of ATs that do not display inherent promiscuity was explored. To this end, the Pik PKS, responsible for the biosynthesis of two core macrolactones, a 12-membered 10-deoxymethynolide (10-dML, 1) and a 14-membered narbonolide (2) in Streptomyces venezuelae ATCC 15439, was selected as a target for mutagenesis.22 We proposed that the extension modules of this pathway would be less promiscuous towards larger extenders than the prototypical DEBS modules due to its evolution in a host that also produces ethylmalonyl-CoA and to hydrolytic proof-reading by the AT and PikAV (TEII).23–25 Using the final two modules from this PKS, the native promiscuity of each module was first compared with a panel of natural and non-natural extender units and via a series of domain exchanges. Next, the substrate selectivity of each module was successfully engineered towards non- natural extender units via site-directed mutagenesis. Finally, a hitherto unrecognized bottleneck in PKS engineering was highlighted. To our knowledge, this is the first example of successful substrate selectivity inversion in an AT that does not belong to the prototypical DEBS assembly line. Moreover, and to the best of our knowledge, it represents the first example of two non-natural extender units being incorporated into a single full-length polyketide product.
RESULTS
Characterization of the PikAIII/PikAIV system
To date, the majority of AT substrate selectivity engineering work has been conducted in DEBS Ery6, a terminal module chosen at least in part because the fully-extended non-natural chains do not need to be passed through other modules.16–18, 21, 26 The Pik PKS provides a unique opportunity to probe the specificity of two adjacent monomodules that control formation of a 12- or 14- membered ring macrolactone, 10-dML (1) and narbonolide (2), respectively. (Figure 1). These two enzymes are evolutionarily related, with 74% amino acid identity over two-thirds of their sequences (PikAIV lacks a KR domain). Both AT domains (88% identical) utilize methylmalonyl-CoA. In contrast, the two ATs are quite dissimilar to the highly promiscuous EryAT2 (48% PikAT5, 47% PikAT6) and EryAT6 (46% PikAT5, 45% PikAT6) (Supplementary Figure S1).9, 21 To better understand substrate promiscuity beyond the DEBS ATs and to determine a baseline level of promiscuity for the Pik ATs, the modules were tested as lysates in vitro with 10, a thiophenol- activated form of the natural pentaketide chain elongation intermediate and equimolar amounts of a natural (8) and a non-natural extender unit (e.g., 9a, Scheme 1). The modules were prepared as lysates given that in our hands,27 and consistent with previous reports,28–30 module stability and reaction consistency were negatively affected by protein purification (data not shown). In this assay, pentaketide 10 is loaded onto the Cys active site of Pik KS5 for extension by one or both modules. As observed in the wild-type system (Scheme 1), after extension by PikAIII in the presence of two competing extender units, the hexaketide is transferred directly to the PikAIV TE domain resulting in cyclization as one of two 10-dML analogues, thus ‘skipping’ PikAIV (PikAIV does not accept the pentaketide substrate.31 In this way, the 10-dML products must be derived from extension by PikAIII and terminated by the PikAIV TE in the absence of the second extension. Concurrently, a series of narbonolide analogues bearing variant extender units are generated by sequential extensions by PikAIII and PikAIV. Notably, a third 12-membered product (4a-d) is also possible if the non-natural elongated intermediate bypasses the PikAIII KR. These features collectively enable a precise assessment of the ability of each module to process non-natural extender units on the basis of the corresponding product distribution, which can then be readily detected and quantified by high-resolution LC-MS.28, 32
Scheme 1.

Bimodular extender unit competition assay. The two final Pik modules are incubated with the synthetic pentaketide chain mimic 10 and a mixture of the native extender 8 and an equimolar amount of one of 9a-d in vitro. Products 1, 3a-d, and 4a-d are produced when the PikAIII-extended chain bypasses module 6 and is cyclized by the TE. Products 4a-d bypass the KR domain in PikAIII. Non-reduced products derived from the native extender 8 were not observed. NADPH was produced in situ with an NADPH regeneration system. Product distributions shown are for the wild-type system and are calculated separately for one- and two-extension products. Error was ± 5% of indicated value.
In the native system, both modules only accept methylmalonyl-CoA (8), but upon incubation of the lysate with 10 and each of 9a-d, the propargyl- (9a), ethyl- (9b), and allyl- (9c) malonyl-CoA extender units were accepted in the presence of 8 (Scheme 1 and Supplementary Tables S1 and S2). Notably though, in all cases, the non-natural single-extension products (3a-b and 4a-b) made up 5% or less of the total 10-dML products while no more than 31% of the total narbonolide product was derived from the non-natural extender unit (e.g., 5a+6a+7a; Scheme 1). Of the three non- natural extender units that were utilized, the allyl (9c) was the poorest substrate, accounting for only 4.3% of the double extension products. Regardless of the distribution of non-natural incorporation between PikAIII and PikAIV, together both modules are far less promiscuous than Ery6.21 This narrow substrate scope provides an opportunity for expansion through mutagenesis and to explore whether inherent and extensive promiscuity is initially required for expansion via single amino acid mutations. Additionally, the discrepancy between the proportion of single and double extension products derived from the non-natural extender unit posed questions about promiscuity of individual modules within the same PKS. Furthermore, detection of new keto-10-dML products (4a-4b, vide infra) raised the issue of processing efficiency by downstream domains.
Comparison of the pikromycin PKS acyltransferases
There are three gatekeeping domains potentially responsible for the low production of 3a-d/4a-d by the modular Pik system: (1) the Pik AT5, (2) the downstream ketosynthase, Pik KS6, or (3) the Pik TE. Several chimeric modules were subsequently designed to probe the role of the AT domains in determining the observed product profile. The native AT of PikAIV was substituted with PikAT5 (R2, Figure 2), and the native AT of PikAIII was substituted with PikAT6 (R3, Figure 2). In both of these chimeras, newly defined boundaries were used33 but nevertheless resulted in less total product compared to the wild-type system, with the R2 chimera less than a tenth as active as the wild-type system (Table 1, Supplementary Table S3, and Supplementary Figure 2). As expected, based upon the assumption that the terminal AT (Pik AT6) was more promiscuous than those upstream, the chimera that contained two Pik AT5 domains, R2, produced almost 25% less propargyl 5a/6a products than the wild-type system (R2, Table 1). Consistent with these results, the system containing two Pik AT6 domains, R3, produced more than 15-fold higher levels of propargyl 3a/4a and 1.6-fold more 5a/6a products than the wild-type system (R3, Table 1). Together, these data indicate that Pik AT6 is significantly more promiscuous than Pik AT5 and that this feature may contribute to the low production of 3a-d/4a-d by the wild-type system.
Figure 2.

Wild-type and chimeric module systems designed to probe the specificity of each AT from PikAIII and PikAIV. R1 is the wild-type PikAIII (red) and wild-type PikAIV (purple) with the native PikAIV TE (blue). AT swaps were used to create R2 and R3 from R1. R4 is PikAIII fused to the PikAIV TE. Each Pik AT was swapped into Ery6TE (R6, grey) to generate R7 and R8.
Table 1.
Product distributions catalyzed by domain swapped chimeras.
| Enzyme | 10-dMLa | Narbonolideb | Relative Activityc | |
|---|---|---|---|---|
| 3a + 4a | 5a + 6a | 7a | ||
| R1 | 4.2%d | 23.6%d | 0.3%d | 100.0d |
| R2 | 11.8%d | 17.9%d | 0.2%d | 9.0d |
| R3 | 66.7%d | 37.2%d | 0.6%d | 47.5d |
| R4 | 0.8%d | -f | -f | 100.0d |
| R5 | N.D.e | -f | -f | N.D.e |
| R6 | 7.4%d | -f | -f | 586.0d |
| R7 | 7.2%d | -f | -f | 180.3d |
| R8 | N.D.e | -f | -f | N.D.e |
Percent of 3a + 4a out of all 10-dML products (1, 3a, and 4a). See Scheme 1 for structures of products.
Percent of (5a + 6a) or 7a out of all narbonolide products (2, 5a, 6a, and 7a). See Scheme 1 for structures of products.
Total activity of each system relative to R1 (for bimodular systems) or R4 (for monomodular systems) is based on total amount of products for each reaction (sum of 1, 2, 3a, 4a, 5a, 6a, and 7a).
Error was ± 5% of indicated value
N.D. = Not detected
Not applicable
Interestingly, only 0.6% of the double-extension product from R3 contained two propargyls (7a) even though 66% of the single-extension products (3a+4a) are derived from the propargyl extender (Table 1). However, upon closer examination, 99% of the 10-dML single-extension products was the unreduced propargyl 10-dML analogue 4a (Supplementary Table S3). The discrepancy between the portion of single and double propargyl products could be due to the native screening ability of Pik KR534 and/or to perturbations in the KR due to the impaired function of the PKS AT domain chimera. However, with the natural extender unit 8, only 10-dML product (1; bearing C-3 OH) is observed (Scheme 1), suggesting that the native processing ability of Pik KR5 is at least dependent on the extender unit C2 side-chain. As mass ions consistent with a keto-narbonolide product were not found (Scheme 1), it is likely that Pik KS6 fails to process C-3 keto hexaketide chains.
To account for any substrate bias by domains within PikAIV that impact product distributions, particularly the KS, PikAIII and PikAIV were decoupled by fusing PikAIII to the Pik TE. In addition to substrate competition assays with both modules (Scheme 1), this now enables competition assays with PikAIII-TE alone (Scheme 2)—though not PikAIV alone as it does not accept the pentaketide substrate 10. As expected for a single extension module, PikAIII-TE produced only 10-dML-like products, and narbonolides were not detected. Notably though, of these products, only 0.8% were derived from the propargyl extender unit, with half of these comprising the corresponding keto-10-dML product 4a (Scheme 2 and Table 1, R4). By comparison, 4.2% of the 10-dML products were derived from the propargyl extender unit when the wild-type bimodular system was used (Table 1, R1). Given that the ratio of propargyl product from R4 was no higher than that with the wild-type bimodular system, this result suggests that the lack of extender unit promiscuity displayed by PikAIII is likely due to PikAIII itself and not due to downstream gatekeeping by components of PikAIV—with the possible exception of the TE.27, 35
Scheme 2.

Single module extender unit competition assay. The fusion protein PikAIIITE or the final module of the DEBS PKS, Ery6TE, are incubated with the synthetic pentaketide chain mimic 10 and a mixture of the native extender 8 and an equimolar amount of 9a in vitro. NADPH was produced in situ with an NADPH regeneration system. Error was ± 5% of indicated value. Percentages are shown for wild-type systems.
Finally, to determine the possible effect of the domains surrounding the AT within each module, each of the two Pik ATs were introduced into Ery6-TE (R6, Figure 2), a module and TE pair known to be capable of producing high yields of non-natural 10-dML products,18 yielding the chimeras R7 and R8 (Figure 2). Once again, issues with chimera stability prevented a direct comparison of the two ATs in these chimeras, as Ery6TE_PikAT6 (R8) was completely inactive (Table 1). However, the Ery6-TE_PikAT5 (R7) construct had a significantly more relaxed substrate selectivity (7.2% 3a/4a) than did PikAIII-TE (0.8% 3a/4a) and nearly equal to that of wild-type Ery6-TE (7.4% 3a/4a) (Table 1). This 9-fold increase in propargyl incorporation by PikAT5 when substituted into Ery6-TE from its native module, along with the results of the other three domain swaps establishes both the importance of AT substrate selection and of the substrate promiscuity of the other post- AT domains, especially the KS and the KR.
Effects of active site mutations on extender unit incorporation
In addition to the insight into substrate incorporation, the domain swaps also highlighted two other aspects important for PKS engineering. First, even with the recently updated AT boundaries,33 domain exchanging often results in inactive or poorly active enzymes. Second, some modules, regardless of AT selectivity, are naturally more substrate-permissive.9, 15, 32 To circumvent the first issue and take advantage of the second, AT active site mutagenesis could be utilized for maximum engineering efficiency.
By inspection of homology models created for PikAT5 and PikAT6, there appears to be no significant structural differences between the two active sites that could otherwise explain the difference in extender unit promiscuity and guide mutagenesis efforts (Supplementary Figure S4). Indeed, as indicated by amino acid sequence alignments and the homology models, both ATs also share many of the same active site residues as EryAT6, including the YASH motif (Supplementary Figures S1 and S4). Given that the structural features that explain the observed difference in extender unit promiscuity of PikAT5 and PikAT6 might be subtle and could not be detected by homology modelling, we set out to identify mutations that could impact the specificity. Twenty mutants, spanning mutations at nine active site residues in AT5 of PikAIIITE were first tested with the pentaketide 10 and a mixture of extender units, 8/9a (Supplementary Table S4). This preliminary set, including the two best characterized mutations from EryAT6, V742A and Y744R (Ery6TE numbering), were introduced into PikAIII-TE17–18 Analysis of the distribution of 1/3a supported by each PikAIIITE mutant revealed that surprisingly, Y755R resulted in a completely inactive enzyme in contrast to the flip in selectivity observed in Ery6TE (Supplementary Table S4).18 The V753A mutation on the other hand, did support improved production of 3a compared to the wild-type enzyme. Accordingly, the product distributions of V753A and Y755R were analyzed in more detail. In this single module system, 3a/4a production increased 26-fold upon introduction of the mutation V753A, as judged by the relative amount of 3a/4a vs. the methyl product, 1 (R10, Table 2). As anticipated, no other products were detected with Y755R (R11, Table 2), while the apo version of the wild-type enzyme was also completely inactive (R13, Table 2).
Table 2.
Product distributions catalyzed by engineered mono and bimodular Pik systems.
| Enzyme | 10-dMLa | Narbonolideb | Relative | ||||
|---|---|---|---|---|---|---|---|
| Entry | PikAIIITE | PikAIII | PikAIV | 3a + 4a | 5a + 6a | 7a | Activityc |
| R9 | WT | -d | -d | 0.8%e | -d | -d | 100.0e |
| R10 | V753A | -d | -d | 21.1%e | -d | -d | 4.2e |
| R11 | Y755R | -d | -d | N.D.f | -d | -d | N.D.f |
| R12 | Y755V | -d | -d | 42.2%e | -d | -d | 81.5e |
| R13 | WT (apo) | -d | -d | N.D.f | -d | -d | N.D.f |
| R14 | -d | WT | WT | 4.2%e | 23.6%e | 0.3%e | 100.0e |
| R15 | -d | V753A | WT | 7.7%e | 21.3%e | 0.8%e | 92.5e |
| R16 | -d | Y755V | WT | 55.9%e | 61.9%e | 16.7%e | 82.1e |
| R17 | -d | WT | Y753V | 4.4%e | 77.2%e | 1.3%e | 86.8e |
| R18 | -d | Y755V | Y753V | 44.6%e | 59.6%e | 14.7%e | 51.7e |
| R19 | -d | WT (apo) | WT (apo) | N.D.f | N.D.f | N.D.f | N.D.f |
Percent of 3a + 4a out of all 10-dML products (1, 3a, and 4a). See Scheme 1 for structures of products.
Percent of (5a + 6a) or 7a out of all narbonolide products (2, 5a, 6a, and 7a). See Scheme 1 for structures of products.
Total activity of each system relative to R9 (for monomodular systems) or R14 (for bimodular systems) is based on total amount of products (sum of 1, 2, 3a, 4a, 5a, 6a, and 7a).
Not applicable
Error was ± 5% of indicated value
N.D. = Not detected
To explore why the Tyr→Arg mutation is effective in one methyl-specific AT (EryAT6) but not in the Pik ATs, molecular dynamics (MD) simulations were utilized with EryAT6 and PikAT6 homology models (Supplementary Figure S3). In EryAT6, Arg744 forms a salt bridge with the neighboring Asp743, which in turn competes in another salt bridge with Arg674 (Figure 3A). According to the MD simulations, both arginines also interact with Asp613, facilitating distribution of the positive charge and also maintaining integrity of the active site. In the MD simulation of PikAT6 Y753R, Arg753 appears to extend into the active site rather than forming salt bridges as seen in DEBS. Arg674 in EryAT6 has been replaced with Ala683 in PikAT6, which in turn interacts with the hydrophobic Val621 in place of Asp613 in EryAT6 (Figure 3B). Accordingly, compared to EryAT6, the predicted nonpolar environment in the PikAT6 active site is not as suitable for accepting the substrate of the engineered Arg753, and the distance between the two catalytic residues widens commensurately (8.4 Å in PikAT6 Y753R vs 4.8 Å in EryAT6 Y744R). This distance would be too far to maintain the interactions between Ser653 and His756 necessary for catalytic activity.
Figure 3.

Models of (A) EryAT6 Y744R and (B) PikAT6 Y753R ATs after undergoing MD simulations. Distances between catalytic residues in the mutant EryAT6 (4.8 Å) are long but catalytically-competent. The same distance in the mutant PikAT6 (8.4 Å) is characteristic of an inactive AT domain.
Given the MD simulations and that Y755R resulted in an inactive PikAIII-TE, the corresponding Tyr→Val sequence variation shared between many ethylmalonyl-CoA-specific ATs was introduced into PikAIII-TE (R12, Table 2).36 This mutation, which has also recently been introduced into EryAT2,9 resulted in a significantly more promiscuous module with nearly half of the products derived from 9a. Next, both functional mutations were introduced into PikAIII and PikAIV and assayed as part of a bimodular system (R14-R19, Table 2). The Val→Ala mutation in PikAIII resulted in a more modest formation of the propargyl products (3a, 4a, and 7a) compared to the wild-type bimodular system (R15, Table 2); however, the Tyr→Val mutation in either module flipped the selectivity of the bimodular system, preferring the non-natural substrate 9a over the natural substrate 8, as judged by the ratio of narbonolide products 2, 5a/6a, and 7a, e.g., 21.4% 2, 61.9% 5a/6a, and 16.7% 7a (R16, Table 2). With PikAIII Y755V, a 13-fold increase in preference for propargyl was observed in the single extension product compared with the wild-type bimodular system, but unlike with the unstable chimera R3, 90% of the product was reduced (R16 and R14, respectively, Supplementary Table S3). Subsequently, whereas the wild-type bimodular system only supports production of 0.3% di-propargyl narbonolides, nearly 17% of the narbonolides formed by PikAIII Y755V are derived from two propargyl extender units (7a). This successful selectivity shift demonstrates that even with other domains potentially impacting substrate selection (e.g., KS, KR, and/or TE), even the most stringent of systems can be engineered to accept a desired new substrate.
The PikAIII Y755V/PikAIV Y753V system was also probed for substrate tolerance beyond methyl and propargyl by including either ethyl-, allyl-, or butylmalonyl-CoA in equimolar concentrations with methylmalonyl-CoA (Table 3, Supplementary Table S2, and Supplementary Figure 4). As desired, the double mutant system showed improved activity with all three additional non-natural substrates tested. For ethyl, the non-natural narbonolide products were also more abundant than the native product 2. Gratifyingly, the allyl and butyl extenders that were not accepted to any degree by wild-type PikAIII (or even by the more promiscuous PikAIV for butyl) were accepted by both mutant modules, even resulting in the double allyl product 7c, albeit in reduced percentage yields as compared to the propargyl and ethyl products. This is particularly interesting given that non- natural narbonolide products were not produced with an engineered bimodular DEBS system.18
Table 3.
Product distributions catalyzed by the double mutant bimodular Pik system with non-natural extender units.a
| PikAIII Y755V + PikAIV Y753V | ||||
|---|---|---|---|---|
| 10-dMLa | Narbonolideb | |||
| Extender unit | 3a-d | 4a-d | 5a-d + 6a-d | 7a-d |
| Propargyl (a) | 41.0%c | 3.6%c | 59.6%c | 14.7%c |
| (10.6)d | (2.5)d | (49.0)d | ||
| Ethyl (b) | 8.0%c | 1.8%c | 49.1%c | 17.4%c |
| (61)d | (16)d | (24.9)d | ||
| Allyl (c) | 3.7%c | 4.3%c | 16.7%c | 3.3%c |
| (>8,000)d | (3.9)d | (>3,300)d | ||
| Butyl (d) | N.D. | 11.3%c | 5.4%c | N.D.e |
| (>11,300)d | (>5,400)d | -f | ||
Percent of 3a-d or 4a-d out of all 10-dML products (1, 3a-d, and 4a-d)
Percent of (5a-d + 6a-d) or 7a-d out of all narbonolide products (2, 5a-d, 6a-d, and 7a-d).
Error was ± 5% of indicated value.
Fold increase calculated based on values from the wild-type system in Scheme 1. In the case of a product that was not detected with the wild-type enzyme, the fold increase was calculated based on the limit of detection, 0.001% of total products.
N.D. = Not detected
Not applicable
The differences in activities between substrates is notable, especially with the two C3 substrates, propargyl (9a, robust) and allyl (9c, poor), in both the wild-type (Scheme 1) and mutant (Table 3) systems. The primary difference between the two substrates that might explain their disparate activities with PikAIII/PikAIV is their relative flexibilities, whereby the rigid propargyl side chain can orient towards the rear of the active site, while the more flexible allyl side chain could clash more with the YASH motif, even with residues that lie beyond Tyr755 targeted in this study.
As with the propargyl extender, each non-natural substrate resulted in significant production of the unreduced 10-dML products, especially in the cases of allyl and butyl where greater than half of the single-extension products were the unreduced 4c and 4d products (Table 3 and Supplementary Table S3). In fact, none of the single-extension butyl product was reduced by the KR, preventing production of any double-butyl narbonolide product likely due to the downstream PikKS6 discriminating against chains with the non-native oxidation pattern. This additional gatekeeping by the KR and KS sets the stage for further engineering in non-terminal PKS modules.
DISCUSSION
PKSs provide the ability to template a series of reactions in order to achieve a desired product, with each module responsible for recruitment of a single extender unit into the polyketide scaffold. However, the envisioned goal of being able to stitch together any domain or module seamlessly still cannot be attained without significant loss in enzyme function,11, 33, 37 likely due to delicate protein:protein interactions that are faulty in chimeric PKSs and prevent proper vectoral biosynthesis.38–39 In this study, even with two ATs sharing 88% identity, the AT exchanges resulted in vastly different enzyme activity levels. Rather than directly addressing the impact of AT exchanges, which has been well documented previously,11, 40–42 the chimeras were used to identify the source of extender unit selectivity in the pikromycin PKS. Instead, to realize the potential of PKS engineering, site-specific mutations can be introduced to shift selectivity towards or away from a given substrate.
Herein, two very similar ATs from the pikromycin PKS were compared with each other and with the well-characterized EryAT6. Despite these three ATs sharing all residues known or predicted to influence substrate selectivity, they exhibit very different levels of substrate promiscuity. Additionally, mutations that cause a substrate selectivity flip in EryAT6 (Y744R) result in inactive enzymes in the Pik ATs. In other cases, such as with the Tyr→Val mutation explored here, the mutation results in a shift towards larger extenders, demonstrating that an AT with as little native promiscuity as PikAT5 can be engineered to prefer non-natural substrates (4.2% 3a/4a to 55.9% 3a/4a, R14 and R16, Table 2).
In its native setting, PikAIV accounted for the clear majority of non-natural extender unit incorporation by the bimodular system. Through AT exchanges and mutagenesis, much of this discrepancy in promiscuity between PikAIII and PikAIV was shown to locate with the ATs themselves; however, as demonstrated by the 9-fold change in percent propargyl product for two PikAT5-containing modules (R4 and R7, Table 1) and the significant discrimination by PikKR5 against larger extenders (Table 3), the surrounding domains play a significant role in the distribution of natural vs non-natural products. The effect of the KR is especially notable regarding the incorporation of non-natural extender units—especially as the extenders become larger (e.g., butyl). As the selectivity of the AT is altered, the ketoreductase (and potentially other domains) will need to be exchanged or engineered to be compatible with the desired product. As engineering of these domains progresses, larger and more exotic extenders can be used to further push the limits of these modular assembly line-like enzymes.
Cumulatively, site-specific mutagenesis of PikAT5 and PikAT6 has led to robust yields of the first full-length polyketide with two non-natural extenders and to the first non-terminal methyl-specific module to take and process a non-natural extender unit. Subsequently, this work significantly expands the synthetic potential of engineered PKSs and opens up new avenues for engineering other systems. Additionally, the new 10-dML and narbonolide analogues reported herein can be further derivatized through semi-synthetic chemistry, especially ‘click chemistry’.
MATERIALS AND METHODS
General
Materials and reagents were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise noted. Isopropyl β-D-thioglactoside (IPTG) was purchased from Calbiochem (Gibbstown, NJ). The E. coli BAP1 strain was provided by Dr. Blaine Pfeifer at the University at Buffalo.43 Construction of the Ery6TE-pET28 plasmid was described previously.18 All module sequences are listed in Supplementary Table S5. Primers were purchased from Integrated DNA Technologies (Coralville, IA). All holo proteins were expressed in BAP1 cells and all apo proteins were expressed in E. coli BL21 (DE3) cells. All primers are listed in Supplementary Table S6.
Lysate preparation
Modules (wild-type and mutant PikAIII(TE), wild-type and mutant PikAIV, and wild-type and mutant Ery6TE) were expressed overnight at 16 °C in 300 mL cultures of LB media with the appropriate antibiotics. Protein production was induced with 1 mM IPTG at OD600 of 0.6. After overnight expression, the culture was centrifuged at 4,700 rpm for 20 min, and the supernatant was discarded. The cells were resuspended in 1 mL module storage buffer (100 mM sodium phosphate, pH 7.4, 1 mM EDTA, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 20% v/v glycerol, 0.1 μL Benzonase (NEB), and EDTA-free protease inhibitor cocktail (Roche, Basel, CH)) and sonicated using 51% amplitude, 10 sec on, 20 sec off for 10 min. After sonication, the lysed cells were centrifuged at 18,000 rpm for 1 h. The lysates were aliquoted and stored at −80 °C. Protein purity was verified by SDS-PAGE. Protein quantification was carried out using the Bradford Protein Assay Kit from Bio-Rad.
MatB reactions and acyl-CoA preparation
Wild-type and mutant T207G/M306I MatB were purified and 8 mM malonyl-CoA (8, 9a-d) stocks were set up as previously disclosed10 and described in the Supplemental Methods.
Pentaketide assay
The pentaketide assay was set up with a total volume of 80 μL in 100 mM sodium phosphate, pH 7.0, and 2 mM MgCl2. The reaction conditions included 1 mM TP-pentaketide (10), 1.75 mM of each competing malonyl-CoA from MatB reactions (8, 9a-d), an NADPH regeneration system (5 mM glucose-6-phosphate, 500 μM NADP+, and 0.008 U/mL glucose-6-phosphate dehydrogenase), and lysate containing the module or modules. For the bimodular reactions, the proteins were included as a total of 29.4 μL lysate, at a final concentration of 1–3 μM. Module concentrations were calculated by Bradford assay and SDS-PAGE gel analysis, and results were normalized based on protein content for each reaction. Negative controls (R13 and R19) were run with apo modules lacking the phosphopantetheine modifications on the ACP domains. Reactions proceeded at room temperature for 16 h and were quenched with an equal volume of −20 °C methanol. After quenching, all reactions were centrifuged at 13,300 rpm three times for 3 h total, and the supernatant was filtered through a nylon 0.2 μm filter. Analysis was carried out on a high-resolution mass spectrometer (ThermoFisher Scientific Exactive Plus MS, a benchtop full-scan Orbitrap™ mass spectrometer) using Heated Electrospray Ionization (HESI). The sample was analyzed via LC-MS injection into the mass spectrometer at a flow rate of 225 μL/min. The mobile phase B was acetonitrile with 0.1% formic acid and mobile phase A was water with 0.1% formic acid (see Table S7 for gradient and scan parameters). The mass spectrometer was operated in positive ion mode. The LC column was a Thermo Hypersil Gold 50 × 2.1 mm, 1.9 μm particle size. This assay produces 10-dML and narbonolide products that can be seen as their [M+H]+, [M+H-H2O]+, and [M+Na]+ ions and keto-10-dML products that can be seen as their [M+H]+ and [M+Na]+ ions. Extracted ions for each listed ion were summed for comparison purposes. For retention times, calculated masses, observed masses, representative extracted ion counts, and representative chromatograms, see Tables S1, S2, and S4 and Figures S2 and S5.
Homology models and molecular dynamics
Wild-type homology models for EryAT6, PikAT5, and PikAT6 were created using the I-TASSER online server.44–46 Mutations were introduced into structurally-converged wild-type models with Discovery Studio 4.1 from Accelrys Software, Inc. (San Diego, CA). Molecular graphics and analyses of MD trajectories and PDB snapshots were performed with VMD 1.9.2 and UCSF Chimera 1.10.1.47–49 Further analysis was performed with CPPTRAJ.50 Images were rendered with POV-Ray.51
Using the AMBER14 software package, individual models’ charges were neutralized with sodium ions in Xleap.52 All models were then solvated with a 15 Å buffer of TIP3P water, also within Xleap. The enzymes and substrates were parameterized with ff12SB and GAFF force fields from the AMBER14 software package. Prior to production MD simulations, solvated systems were treated with four heating and seven minimization steps. Steps 2, 3, 5, and 11 heated the system to 300 K over times of 20–100 ps each. The first nine steps held the protein fixed, with the restraint constant being lowered each step. Steps 10 and 11 used no restraints. Minimization steps were completed when the change in the root mean square was below 0.01 kcal/mol•Å for the first two minimization steps and below 0.001 kcal/mol•Å for the remaining minimizations. Production simulations lasted between 10 ns and 60 ns for each model. Step times were 2 fs. The non-bonded interaction cut-off was imposed at 9.0 Å. For the two mutants, models were based on the simulated wild-type enzymes (Supplementary Figure S3).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by National Institutes of Health grants GM104258 (G.J.W.) and GM118101 (D.H.S.), and the Hans W. Vahlteich Professorship (D.H.S.). All high resolution LC- MS measurements were made in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University.
Footnotes
SUPPORTING INFORMATION
Includes supplementary tables, supplemental figures, and detailed methods that describe site- directed mutagenesis of modules, construction of chimeras, expression and purification of wild- type and mutant MatB, and synthesis of acyl-CoA’s by MatB.
REFERENCES
- 1.Wang H; Fewer DP; Holm L; Rouhiainen L; Sivonen K, Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. USA 2014, 111 (25), 9259–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan YA; Thomas MG, Formation and characterization of acyl carrier protein-linked polyketide synthase extender units. Methods Enzymol. 2009, 459, 143–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan YA; Boyne MT 2nd; Podevels AM; Klimowicz AK; Handelsman J; Kelleher NL; Thomas MG, Hydroxymalonyl-acyl carrier protein (ACP) and aminomalonyl-ACP are two additional type I polyketide synthase extender units. Proc. Natl. Acad. Sci. USA 2006, 103 (39), 14349–14354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mo S; Kim DH; Lee JH; Park JW; Basnet DB; Ban YH; Yoo YJ; Chen SW; Park SR; Choi EA; Kim E; Jin YY; Lee SK; Park JY; Liu Y; Lee MO; Lee KS; Kim SJ; Kim D; Park BC; Lee SG; Kwon HJ; Suh JW; Moore BS; Lim SK; Yoon YJ, Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues. J. Am. Chem. Soc 2011, 133 (4), 976–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang C; Huang R; Yan Y; Ma H; Dai Z; Zhang B; Deng Z; Liu W; Qu X, Uncovering the formation and selection of benzylmalonyl-CoA from the biosynthesis of splenocin and enterocin reveals a versatile way to introduce amino acids into polyketide carbon scaffolds. J. Am. Chem. Soc 2015, 137 (12), 4183–90. [DOI] [PubMed] [Google Scholar]
- 6.Yoo HG; Kwon SY; Kim S; Karki S; Park ZY; Kwon HJ, Characterization of 2-octenoyl-CoA carboxylase/reductase utilizing pteB from Streptomyce avermitilis. Biosci Biotechnol Biochem 2011, 75 (6), 1191–3. [DOI] [PubMed] [Google Scholar]
- 7.Eustáquio AS; McGlinchey RP; Liu Y; Hazzard C; Beer LL; Florova G; Alhamadsheh MM; Lechner A; Kale AJ; Kobayashi Y; Reynolds KA; Moore BS, Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethylmalonyl-coenzyme A from S-adenosyl-L-methionine. Proc. Natl. Acad. Sci. USA 2009, 106 (30), 12295–12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koryakina I; Williams GJ, Mutant malonyl-CoA synthetases with altered specificity for polyketide synthase extender unit generation. ChemBioChem 2011, 12 (15), 2289–2293. [DOI] [PubMed] [Google Scholar]
- 9.Vogeli B; Geyer K; Gerlinger PD; Benkstein S; Cortina NS; Erb TJ, Combining promiscuous acyl-CoA oxidase and enoyl-CoA carboxylase/reductases for atypical polyketide extender unit biosynthesis. Cell Chem Biol 2018, 25 (7), 833–839 e4. [DOI] [PubMed] [Google Scholar]
- 10.Koryakina I; McArthur J; Randall S; Draelos MM; Musiol EM; Muddiman DC; Weber T; Williams GJ, Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem. Biol 2013, 8, 200–208. [DOI] [PubMed] [Google Scholar]
- 11.McDaniel R; Thamchaipenet A; Gustafsson C; Fu H; Betlach M; Ashley G, Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc. Natl. Acad. Sci. USA 1999, 96 (5), 1846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan YA; Podevels AM; Kevany BM; Thomas MG, Biosynthesis of polyketide synthase extender units. Nat. Prod. Rep 2009, 26 (1), 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ad O; Thuronyi BW; Chang MCY, Elucidating the mechanism of fluorinated extender unit loading for improved production of fluorine-containing polyketides. Proc. Natl. Acad. Sci. USA 2017, 114 (5), E660–E668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musiol-Kroll EM; Zubeil F; Schafhauser T; Hartner T; Kulik A; McArthur J; Koryakina I; Wohlleben W; Grond S; Williams GJ; Lee SY; Weber T, Polyketide bioderivatization using the promiscuous acyltransferase KirCII. ACS Synth. Biol 2017, 6 (3), 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moller D; Kushnir S; Grote M; Ismail-Ali A; Koopmans KRM; Calo F; Heinrich S; Diehl B; Schulz F, Flexible enzymatic activation of artificial polyketide extender units by Streptomyces cinnamonensis into the monensin biosynthetic pathway. Lett Appl Microbiol 2018, 67 (3), 226–234. [DOI] [PubMed] [Google Scholar]
- 16.Bravo-Rodriguez K; Klopries S; Koopmans KR; Sundermann U; Yahiaoui S; Arens J; Kushnir S; Schulz F; Sanchez-Garcia E, Substrate flexibility of a mutated acyltransferase domain and implications for polyketide biosynthesis. Chem. Biol 2015, 22 (11), 1425–30. [DOI] [PubMed] [Google Scholar]
- 17.Sundermann U; Bravo-Rodriguez K; Klopries S; Kushnir S; Gomez H; Sanchez-Garcia E; Schulz F, Enzyme-directed mutasynthesis: a combined experimental and theoretical approach to substrate recognition of a polyketide synthase. ACS Chem. Biol 2013, 8, 443–450. [DOI] [PubMed] [Google Scholar]
- 18.Koryakina I; Kasey C; McArthur JB; Lowell AN; Chemler JA; Li S; Hansen DA; Sherman DH; Williams GJ, Inversion of extender unit selectivity in the erythromycin polyketide synthase by acyltransferase domain engineering. ACS Chem. Biol 2017, 12 (1), 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y; Zhang W; Zhang H; Tian W; Wu L; Wang S; Zheng M; Zhang J; Sun C; Deng Z; Sun Y; Qu X; Zhou J, Structural basis of a broadly selective acyltransferase from the polyketide synthase of splenocin. Angew. Chem. Int. Ed. Engl 2018, 57 (20), 5823–5827. [DOI] [PubMed] [Google Scholar]
- 20.Del Vecchio F; Petkovic H; Kendrew SG; Low L; Wilkinson B; Lill R; Cortes J; Rudd BAM; Staunton J; Leadlay PF, Active-site residue, domain and module swaps in modular polyketide synthases. J Ind Microbiol Biotechnol 2003, 30 (8), 489–494. [DOI] [PubMed] [Google Scholar]
- 21.Koryakina I; McArthur JB; Draelos MM; Williams GJ, Promiscuity of a modular polyketide synthase towards natural and non-natural extender units. Org. Biomol. Chem 2013, 11, 4449–4458. [DOI] [PubMed] [Google Scholar]
- 22.Xue Y; Zhao L; Liu HW; Sherman DH, A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: architecture of metabolic diversity. Proc. Natl. Acad. Sci. USA 1998, 95 (21), 12111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung WS; Kim E; Yoo YJ; Ban YH; Kim EJ; Yoon YJ, Characterization and engineering of the ethylmalonyl-CoA pathway towards the improved heterologous production of polyketides in Streptomyces venezuelae. Appl. Microbiol. Biotechnol 2014, 98 (8), 3701–13. [DOI] [PubMed] [Google Scholar]
- 24.Bonnett SA; Rath CM; Shareef AR; Joels JR; Chemler JA; Hakansson K; Reynolds K; Sherman DH, Acyl-CoA subunit selectivity in the pikromycin polyketide synthase PikAIV: steady-state kinetics and active-site occupancy analysis by FTICR-MS. Chem. Biol 2011, 18 (9), 1075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim BS; Cropp TA; Beck BJ; Sherman DH; Reynolds KA, Biochemical evidence for an editing role of thioesterase II in the biosynthesis of the polyketide pikromycin. J. Biol. Chem 2002, 277 (50), 48028–34. [DOI] [PubMed] [Google Scholar]
- 26.Kalkreuter E; Williams GJ, Engineering enzymatic assembly lines for the production of new antimicrobials. Curr. Opin. Microbiol 2018, 45, 140–148. [DOI] [PubMed] [Google Scholar]
- 27.Hansen DA; Koch AA; Sherman DH, Identification of a thioesterase bottleneck in the pikromycin pathway through full-module processing of unnatural pentaketides. J. Am. Chem. Soc 2017, 139 (38), 13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chemler JA; Tripathi A; Hansen DA; O’Neil-Johnson M; Williams RB; Starks C; Park SR; Sherman DH, Evolution of efficient modular polyketide synthases by homologous recombination. J. Am. Chem. Soc 2015, 137 (33), 10603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pieper R; Gokhale RS; Luo G; Cane DE; Khosla C, Purification and characterization of bimodular and trimodular derivatives of the erythromycin polyketide synthase. Biochemistry 1997, 36 (7), 1846–51. [DOI] [PubMed] [Google Scholar]
- 30.Bycroft M; Weissman KJ; Staunton J; Leadlay PF, Efficient purification and kinetic characterization of a bimodular derivative of the erythromycin polyketide synthase. Eur. J. Biochem 2000, 267 (2), 520–6. [DOI] [PubMed] [Google Scholar]
- 31.Aldrich CC; Beck BJ; Fecik RA; Sherman DH, Biochemical investigation of pikromycin biosynthesis employing native penta- and hexaketide chain elongation intermediates. J. Am. Chem. Soc 2005, 127 (23), 8441–52. [DOI] [PubMed] [Google Scholar]
- 32.Mortison JD; Kittendorf JD; Sherman DH, Synthesis and biochemical analysis of complex chain-elongation intermediates for interrogation of molecular specificity in the erythromycin and pikromycin polyketide synthases. J. Am. Chem. Soc 2009, 131 (43), 15784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuzawa S; Deng K; Wang G; Baidoo EE; Northen TR; Adams PD; Katz L; Keasling JD, Comprehensive in vitro analysis of acyltransferase domain exchanges in modular polyketide synthases and its application for short-chain ketone production. ACS Synth. Biol 2017, 6 (1), 139–147. [DOI] [PubMed] [Google Scholar]
- 34.Holzbaur IE; Ranganathan A; Thomas IP; Kearney DJ; Reather JA; Rudd BA; Staunton J; Leadlay PF, Molecular basis of Celmer’s rules: role of the ketosynthase domain in epimerisation and demonstration that ketoreductase domains can have altered product specificity with unnatural substrates. Chem. Biol 2001, 8 (4), 329–340. [DOI] [PubMed] [Google Scholar]
- 35.Koch AA; Hansen DA; Shende VV; Furan LR; Houk KN; Jimenez-Oses G; Sherman DH, A single active site mutation in the pikromycin thioesterase generates a more effective macrocyclization catalyst. J. Am. Chem. Soc 2017, 139 (38), 13456–13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith L; Hong H; Spencer JB; Leadlay PF, Analysis of specific mutants in the lasalocid gene cluster: evidence for enzymatic catalysis of a disfavoured polyether ring closure. ChemBioChem 2008, 9 (18), 2967–2975. [DOI] [PubMed] [Google Scholar]
- 37.Menzella HG; Reid R; Carney JR; Chandran SS; Reisinger SJ; Patel KG; Hopwood DA; Santi DV, Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol 2005, 23 (9), 1171–1176. [DOI] [PubMed] [Google Scholar]
- 38.Lowry B; Li X; Robbins T; Cane DE; Khosla C, A Turnstile Mechanism for the Controlled Growth of Biosynthetic Intermediates on Assembly Line Polyketide Synthases. ACS Cent Sci 2016, 2 (1), 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klaus M; Ostrowski MP; Austerjost J; Robbins T; Lowry B; Cane DE; Khosla C, Protein-protein interactions, not substrate recognition, dominate the turnover of chimeric assembly line polyketide synthases. J. Biol. Chem 2016, 291 (31), 16404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruan X; Pereda A; Stassi DL; Zeidner D; Summers RG; Jackson M; Shivakumar A; Kakavas S; Staver MJ; Donadio S; Katz L, Acyltransferase domain substitutions in erythromycin polyketide synthase yield novel erythromycin derivatives. J. Bacteriol 1997, 179 (20), 6416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hans M; Hornung A; Dziarnowski A; Cane DE; Khosla C, Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J. Am. Chem. Soc 2003, 125 (18), 5366–5374. [DOI] [PubMed] [Google Scholar]
- 42.Patel K; Piagentini M; Rascher A; Tian ZQ; Buchanan GO; Regentin R; Hu Z; Hutchinson CR; McDaniel R, Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem. Biol 2004, 11 (12), 1625–33. [DOI] [PubMed] [Google Scholar]
- 43.Pfeifer BA; Admiraal SJ; Gramajo H; Cane DE; Khosla C, Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 2001, 291 (5509), 1790–2. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy A; Kucukural A; Zhang Y, I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protocols 2010, 5 (4), 725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J; Yan R; Roy A; Xu D; Poisson J; Zhang Y, The I-TASSER Suite: protein structure and function prediction. Nat. Methods 2015, 12 (1), 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Humphrey W, D. A, and Schulten K, VMD - Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 48.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- 49.Sanner MF; Olson AJ; Spehner JC, Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 1996, 38 (3), 305–320. [DOI] [PubMed] [Google Scholar]
- 50.Roe DR; Cheatham TE 3rd, PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9 (7), 3084–95. [DOI] [PubMed] [Google Scholar]
- 51.Persistence of Vision Pty Ltd Persistence of Vision Raytracer (Version 3.6), 2004.
- 52.Case DA, B. JT, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Salomon-Ferrer R, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, York DM, and Kollman PA AMBER 2015, University of California, San Francisco, 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.