

Abstract
Although emerging evidence suggests a potential role of calcium/calmodulin-dependent kinase II (CaMKII) in prostate cancer (PCa), its role in PCa tumorigenesis is largely unknown. Here we examine whether the acetyl CoA-CaMKII pathway, first described in frog oocytes, promotes PCa tumorigenesis. In human PCa specimens, metastatic PCa expressed higher levels of active CaMKII compared to localized PCa. Correspondingly, basal CaMKII activity was significantly higher in the more tumorigenic PC3 and PC3-mm2 cells relative to the less tumorigenic LNCaP and C4–2B4 cells. Deletion of CaMKII by CRISPR/Cas9 in PC3-mm2 cells abrogated cell survival under low-serum conditions, anchorage-independent growth and cell migration; overexpression of constitutively active CaMKII in C4–2B4 cells promoted these phenotypes. In an animal model of PCa metastasis, genetic ablation of CaMKII reduced PC3-mm2 cell metastasis from the prostate to the lymph nodes. Knockdown of the acetyl-CoA transporter carnitine acetyltransferase (CRAT) abolished CaMKII activation, providing evidence that acetyl-CoA generated from organelles is a major activator of CaMKII. Genetic deletion of the β-oxidation rate-limiting enzyme ACOX family proteins decreased CaMKII activation, while overexpression of ACOXI increased CaMKII activation. Overall, our studies identify active CaMKII as a novel connection between organelle β-oxidation and acetyl-CoA transport with cell survival, migration, and PCa metastasis.
Keywords: prostate cancer, CaMKII, metastasis, ACOX, acetyl-CoA
Introduction
Calcium/calmodulin-dependent kinase II (CaMKII) is a family of multifunctional serine/threonine protein kinases that is important in diverse cellular processes (1,2). The canonical activation of CaMKII requires both Ca2+ and calmodulin. When calmodulin binds 4 molecules of Ca2+, it binds to CaMKII and induces autophosphorylation of CaMKII at the threonine 286 (T286) site. Phosphorylation of CaMKII at T286 has been established as a measure of CaMKII activation (3,4). Recent evidence suggests a potential role for CaMKII in cancer (5,6). However, its specific function and its regulation remain unclear.
Altered metabolism has been shown to drive cancer development (7,8). In addition to fueling rapid division, metabolites generated downstream of glucose breakdown, such as acetyl-CoA, can directly activate enzymes that promote cell survival. Previous studies by Nutt et al. (9) have shown that addition of glucose-6-phosphate (G6P) to X. laevis egg extract increases egg survival through activation of CaMKII. They further showed that glucose-6-phosphate (G6P) was metabolized to produce acetyl-CoA in X. laevis extracts. Free CoA, generated locally by a yet to be identified acetyl-CoA hydrolyzing reaction, then binds directly to the calmodulin-binding domain (CaMBD) of CaMKII to promote calmodulin (CaM) binding and activation of CaMKII at basal calcium concentrations (10). In X. laevis, activated CaMKII then phosphorylates and inhibits caspase-2 at serine 135 (S135) to promote oocyte survival (10). These studies demonstrate that acetyl-CoA, generated downstream of glucose breakdown, can directly regulate CaMKII activity to promote cell survival.
In a review by Nutt (11), comparing the metabolic pathways in X. laevis oocytes with those in cancer cells, it was found that X. laevis oocytes exhibit altered metabolism coupled to its apoptotic machinery in a similar fashion to cancer cells. If metabolic regulation in egg bears high similarity to tumor cells (11), it is possible that the metabolic activation of CaMKII, which plays a role in oocyte survival, may also be promoting proliferation and survival in prostate cancer (PCa). In this study, we demonstrate that CaMKII is activated in metastatic PCa cells and its activation enhances the metastasis of PCa. In addition, we show that one of the mechanisms of CaMKII activation is through organelle-derived acetyl-CoA production.
Materials and Methods
Cell lines, antibodies and reagents
Cell lines: Human PCa C4–2B4 (gift from Robert Sikes, University of Delaware, 2006), PC3-mm2 (gift from Isaiah J. Fidler, M.D. Anderson Cancer Center, 2005), PC3 (ATCC, 2011), and LNCaP (ATCC, 2015). C4–2B4-LT and PC3-mm2-LT cells, expressing luciferase and red fluorescence protein Tomato through a bi-cistronic retroviral vector pBMN-Luc-Tomato, were generated as described previously (12). STR was performed routinely on these cell lines to confirm their authenticity and mycoplasma was routinely tested. CaMKII antibody (611292) was purchased from BD Transduction Laboratories. Anti-pCaMKII (T286) antibody (sc-12886R) and ACOXIII antibody (sc-98756) were from Santa Cruz (Santa Cruz, CA). ACOXI antibody (H00000051-A01) was from Abnova (Taipei, Taiwan). CRAT antibody was from Proteintech (15170–1-AP).
Immunohistochemistry of human prostate cancer specimens.
Formalin-fixed, paraffin-embedded human PCa specimens from primary tumor (20 cases), lymph node metastasis (19 cases), and bone metastasis (19 cases) were obtained from MDACC Prostate Cancer Tissue Bank through an institutional approved IRB protocol. Immunohistochemistry using pCaMKII (T286) antibodies was performed using procedures described previously (13).
Knockout of CaMKII or Acox in PC3-mm2 cells using CRISPR-cas9 system
PC3-mm2 cells were transduced with a bicistronic retrovirus containing luciferase (Luc) and Tomato (PC3-mm2-LT). Knockout of CaMKII or AcoxI-III in PC3-mm2-LT were achieved by CRISPR/Cas9 system. PC3-mm2-LT cells were transfected with a mixture of plasmids. For CaMKII, h-CaMKII-gRNA-639 and h-CaMKII-gRNA-680 with nucleotide sequences listed in Supplementary Table S1 were used. These sequences are shared among CaMKII α, β, γ, δ isoforms and are expected to be able to knockout human CaMKII α, β, γ, and δ. The gRNAs were inserted into plasmids pSpCas9n(BB)-2A-puro(pX462) and the resulting plasmids, pSpCas9n(BB)-2A-puro(pX462)-h-CaMKII-gRNA-639 and pSpCas9n(BB)-2A-puro (pX462)-h-CaMKII-gRNA-680, were used to transfect PC3-mm2 cells and the transfected cells were selected with puromycin (2 μg/ml). For ACOX I-III, a total of six plasmids, including pSpCas9n(BB)-2A-puro(pX462)-hACOX(I-III)-gRNA and pSpCas9n(BB)-2A-HygroR(pX462)-hACOX(I-III)-gRNA were used. The gRNA sequences for AcoxI-III are listed in Supplementary Table S1. These plasmids were used to transfect PC3-mm2 cells and the transfected cells were selected with puromycin (2 μg/ml) and hygromycin (300 μg/ml). Genomic DNA was extracted from puromycin and hygromycin-resistant cells with FlexiGene DNA Kit (QIAGEN). Targeted cleavage of ACOXI-III genes was measured by PCR amplification using gene-specific primers ACOX1-gFW and ACOX1-gRev for Acox1, ACOX2-gFW and ACOX2-gRev for Acox2, ACOX3-gFW and ACOX3-gRev for Acox3 (Supplementary Table S1).
Generation of C4–2B4 cells overexpressing CaMKII or ACOXI.
cDNA encoding wild type, constitutively active (T286D), or inactive form (K42M) of CaMKII was inserted into bicistronic retroviral vector pBMN-I-NEO. cDNA encoding ACOXI was inserted into bicistronic retroviral vector pBMN-I-GFP. C4–2B4-LT cells were transduced with retrovirus generated from pBMN-CaMKII-NEO or pBMN-ACOXI-GFP and selected by resistance to G418 or FACS through GFP, respectively. C4–2B4-LT cells transduced with empty vector (C4–2B4-vector) were generated similarly.
Re-expression of constitutively active CaMKII in PC3-mm2 cells with knockout of CaMKII
PC3-mm2 clones #1, #6 and #10, with knockout of CaMKII, were transduced with retrovirus generated from pBMN-CaMKII-T286D-NEO, which contained cDNA for constitutively active form of CaMKII. Cells were selected by G418.
Western blotting analysis
Protein concentration was determined by Coomassie Plus assay. Proteins were separated in SDS-PAGE and immunoblotted as indicated.
Cell proliferation and soft agar colony assay
Cell proliferation was determined by viable cell counting. The soft agar colony assay was performed as described by Yu et al (14). In brief, cells (3 × 104 per well in a 6-well plate) were mixed with 0.35% agarose in growth medium with 5% FBS and plated on top of a solidified layer of 0.7% agarose in the same medium in a 6-well plate. The cells were fed every 3 days with growth medium for 14 days.
Cell migration and cell invasion assay
For cell migration assay, cells (3 × 105) in 300 μL of serum-free medium were seeded into FluoroBlock TM Cell Culture insert (BD Falcon). The lower chamber of a 24 well plate contained 500 μL of 5% FBS culture media. After incubation for 16 hours, the migrated cells were labeled with Calcein AM and were quantified in five randomly chosen visual fields. For cell invasion assay, cells (3 × 105) in 300 μL of serum-free medium were seeded into BioCoat Matrigel-coated invasion chamber (BD Bioscience). The lower chamber of a 24 well plate contained 500 μL of 5% FBS culture media. After incubation for 24 hours, the invaded cells were labeled with Calcein AM and were quantified in five randomly chosen visual fields.
Intraprostatic injection of tumor cells and bioluminescence imaging of mice
To determine the tumor growth in prostate and their metastasis to lymph nodes, PC3-mm2 cells (5 × 105 cells) with or without CaMKII or ACOX knockout were injected into prostates of SCID mice. Tumor growth in mice was monitored by bioluminescence imaging with an IVIS Imaging System (Xenogen, Alameda, CA). At the conclusion of the study, the tumors in the prostates and lymph nodes were removed and fixed in formaldehyde. Animal studies were performed in accordance with as well as approved by an Institutional Animal Care and Use Committee (IACUC).
Knockdown of CRAT by shRNA in PC3-mm2 cells
Lentivirus-expressing shRNAs were generated by cotransfecting CRAT shRNA plasmids with pCMV-dR8.2 dvpr and pCMV-VSVG packaging plasmids into 293FT cells. PC3-mm2-LT cells transduced with pGIPZ-shCRAT or pGIPZ vectors were selected by 5 μg/mL puromycin to generate PC3-shCRAT and PC3-vector (shNT), respectively.
Reverse transcription and quantitative PCR analysis
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen). Quantitative real-time RT-PCR (qRT-PCR) was performed using GAPDH as a control. The PCR primer sequences for AcoxI-III are listed in Supplementary Table S1.
Oil red O staining and lipidomics
For staining cells with Oil red O, cells were fixed with 4% paraformaldehyde for 15–30 min. The fixative was removed and cells were rinsed with phosphate buffered saline and allowed to air dry. Oil red O solution was then added to the cells. Lipidomics of PC3-mm2 cells with knockout of AcoxI-III, i.e., Acox#7, Acox#10, and Acox#21 cells, and vector control cells was performed by Lipotype GmbH (Germany). In brief, samples were spiked with lipid class-specific internal standards and lipids were extracted using chloroform and methanol (15). Lipid extracts were subjected to mass spectrometric analysis. Lipid quantification was based on the intensity of lipid class-specific internal standards. The levels of 10 classes of lipids, including 2 storage lipids, i.e., triacylglycerol (TAG) and cholesterol esters (CE), and 8 membrane lipids, i.e., diacylglycerol (DAG), phosphatidate (PA), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylserine (PS), and sphingomyelin (SM), were determined.
Results
Metastatic PCa express higher levels of active CaMKII
To elucidate the relationship between CaMKII activity and PCa progression, we surveyed several PCa cell lines with varying degrees of tumorigenic potential (16). We found that PCa cells with high tumorigenic potential, i.e., PC3 and PC3-mm2 cells, express high levels of CaMKII and active CaMKII, pCaMKII (T286) (3,4), compared with PCa cell lines with lower tumorigenic potential, i.e. LNCaP or C4–2B4 cells (Fig. 1A). Immunofluorescence staining of these cell lines with or without the peptide immunogen as competitive inhibitor further confirms the expression patterns observed in the Western blots (Fig. 1B, Supplementary Fig. S1). Next, we performed immunohistochemical analyses for the expression of pCaMKII (T286) in human PCa specimens from primary PCa tumors (20 cases), lymph node metastasis (19 cases), and bone metastasis (19 cases). The suitability of pCaMKII (T286) antibody for immunohistochemical analysis was first examined by using a xenograft PC3 tumor, which showed high levels of pCaMKII (T286) staining (Fig. 1C). The intense pCaMKII (T286) staining in the PC3 tumor was abolished in the presence of the competitive phospho-peptide that was used to generate the antibody (Fig.1C). Immunohistochemical staining showed that pCaMKII (T286) is not detected in prostate epithelial cells in normal prostate tissue, although it is expressed in the smooth muscle cells in the vascular wall and ganglion cells around the prostate (Supplementary Fig. S2). pCaMKII (T286) expression is detected in 5 of 20 (25%), 12 of 19 (63%), and 17 of 19 (89%) primary PCa, lymph node metastases, and bone metastases, respectively (Fig. 1D, Supplementary Fig. S2). The staining is mainly in the cytoplasm of tumor cells. The intensity of staining observed in primary prostate tumor cells is much weaker than those in lymph node and bone metastasis (Fig. 1D). Statistical analysis using chi-square test showed that the difference in pCaMKII (T286) expression between primary PCa and lymph node metastasis or bone metastasis is significant with p-value=0.016 and 0.00005, respectively. There is no significant difference in pCaMKII expression between lymph node metastasis and bone metastasis (p=0.056). Thus, the increase in the levels of pCaMKII correlates with PCa progression, suggesting that CaMKII activation may promote PCa progression.
Figure 1. pCaMKII expression in prostate cancer cell lines and human prostate cancer specimens.
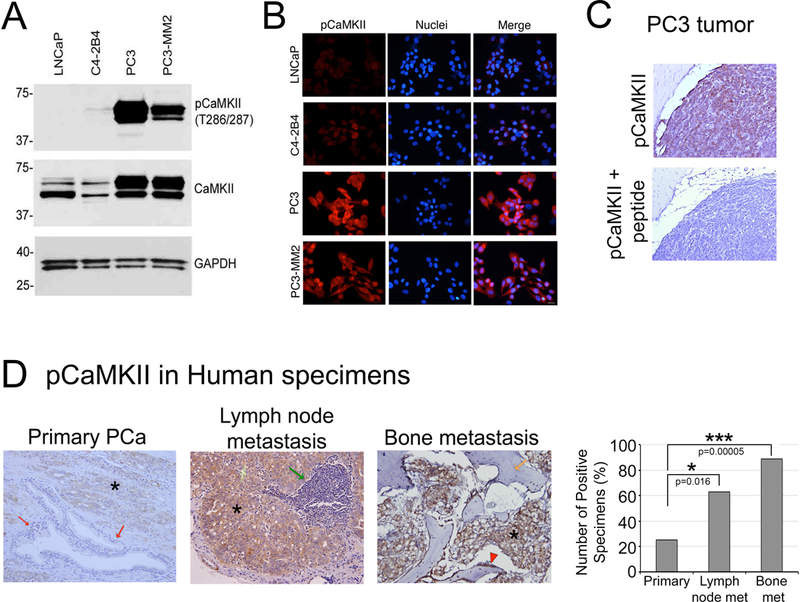
(A) Western blot of whole cell lysates from PCa cell lines. LNCaP, C4–2B4, PC3 and PC3-mm2 were immunoblotted for pCaMKII (T286) and total CaMKII. GAPDH was used as a loading control. (B) Immunofluorescence of LNCaP, C4–2B4, PC3 and PC3-mm2 cells. Cells were stained with antibody against pCaMKII (T286) (red) and DAPI (blue). Scale bar represents 25μm. (C) Immunohistochemistry of pCaMKII (T286), in the presence or absence of the peptide antigen, in a PC3 tumor generated subcutaneously. Magnification X200. (D) Immunohistochemistry for the expression of pCaMKII (T286) of specimens from primary prostate tumor, lymph node metastasis and bone metastasis. Left panels, Representative images. PCa cells in the primary tumor (asterisk) showed weak positivity while normal prostate epithelial cells are negative (red arrows). Metastatic PCa cells in lymph node metastasis (asterisk) and bone metastasis (asterisk) showed strong positivity. *, tumor; red arrows, non-neoplastic prostate glands; green arrow, lymphoid tissue; orange arrow, bone; red arrowhead, rimming cells (osteoblasts). Magnification x200. Right panel, graph of pCaMKII (T286) positive staining in specimens. Primary PCa, n=20; lymph node metastasis, n=19; bone metastasis, n=19. *, p <0.05, ***, p<0.001 by chi-square analysis.
CaMKII activation promotes survival, migration, and invasion of prostate cancer cells
To examine whether CaMKII plays a role in PCa progression, we deleted CaMKII genes using CRISPR/Cas9 that targets all four CaMKII isoforms to probe the role of CaMKII in PC3-mm2 cells. PC3-mm2 cells are highly tumorigenic and contain high levels of CaMKII and pCaMKII (Fig. 1A). PC3-mm2 clones #1, #6 and #10 contained significantly less total CaMKII protein and pCaMKII compared to vector control cells following transfection with CaMKII targeted CRISPR/Cas9 (Fig. 2A). Additionally, clones #1, #6 and #10 demonstrated significant decreases in proliferation in low serum (1%) (Fig. 2B), anchorage-independent growth (Fig. 2C), cell migration (Fig. 2D), and cell invasion (Fig. 2E). These results suggest that CaMKII activity promotes survival, anchorage-independent growth, migration, and invasion of PCa cells.
Figure 2. Knockout of CaMKII in PC3-mm2 cells.
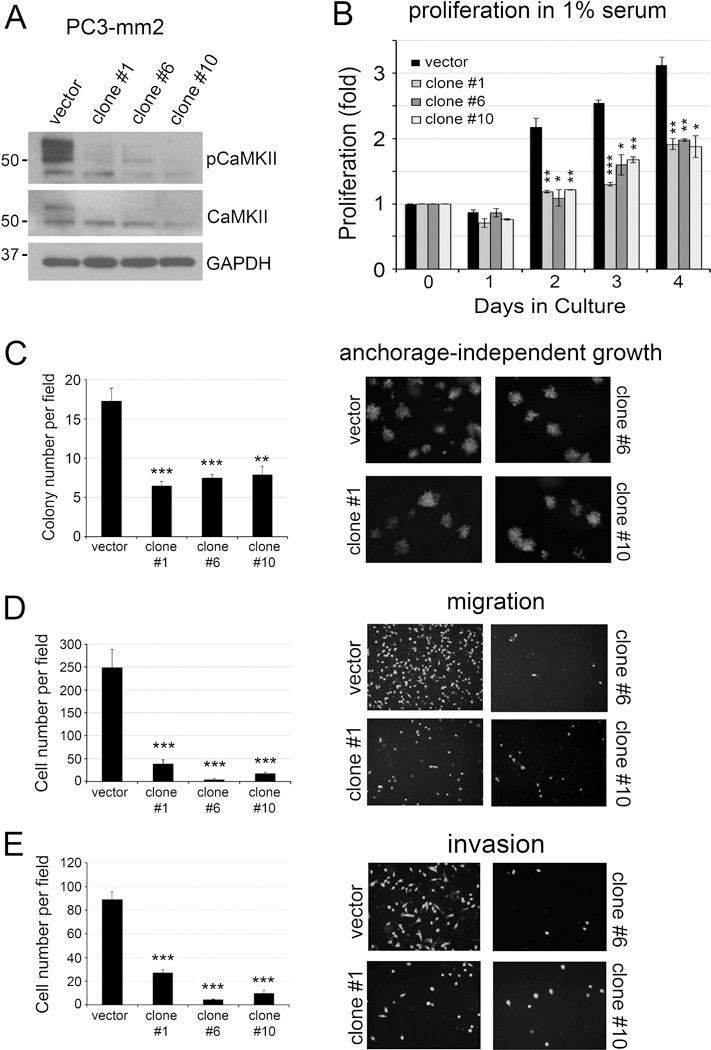
PC3-mm2 cells were transfected with empty vector (vector) or CRISPR/Cas9 targeting CaMKII genes (clones #1, #6, #10). (A) Total cell lysates were immunoblotted for pCaMKII and total CaMKII. GAPDH was used as a loading control. (B) Cells were cultured in 1% FBS and cell proliferation was measured by cell counting. The results were expressed as fold change of cell number counted on Day 0. n=3. The doubling time of the vector control PC3-mm2 cells was 18.2 ± 1.2 hours, and those for the CaMKII knockout clones #1, #6, #10 were 32.1 ± 1.0 hours, 43.5 ± 11.9 hours, and 35.6 ± 0.2 hours, respectively. The plating efficiency, determined at 24 hours after cell seeding, of the vector control cells was 87.5%, and for the CaMKII knockout clones #1, #6 and #10 were 70.7%, 86.5% and 76.0%, respectively. (C) Cells were grown in soft agar. Number of colonies per field was counted. n=3. (D) Cells were seeded onto a Boyden chamber. Cells migrated through the membrane were labeled with Calcein AM. n=2. (E) Cells were seeded into BioCoat Matrigel-coated invasion chamber. Cells invaded through the matrigel were labeled with Calcein AM. n=3. *, p<0.05, **, p<0.01, ***, p<0.001.
To further validate our genetic deletion studies, we overexpressed a constitutively active form of CaMKII (CaMKII-T286D), as well as a wild type and an inactive form of CaMKII (CaMKII-K42M) in C4–2B4 cells. Western blot showed an increase in the levels of total CaMKII in CaMKII-T286D, wild type, and CaMKII-K42M in CaMKII transfected cells compared to vector transfected cells (Fig. 3A). However, the level of CaMKII-T286D in C4–2B4 cells was consistently lower than those of wild type and CaMKII-K42M (Fig. 3A). Whether the lower level of CaMKII-T286D is due to the inability of C4–2B4 cells to tolerate high levels of CaMKII-T286D is unclear. Next, we examined the effect of CaMKII mutants on C4–2B4 cell proliferation. As shown in Fig. 3B, expression of CaMKII-T286D increased cell proliferation in low serum (1% FBS), but not in high serum (10% FBS), compared to vector control. Expression of wild type or CaMKII-K42M did not have significant effects on C4–2B4 cell proliferation in either 1% or 10% FBS (Fig. 3B). These results suggest that activated CaMKII, but not total CaMKII, plays a role in cell survival in low serum but not in cell proliferation. Consistently, expression of CaMKII-T286D, but not wild type or CaMKII-K42M, increased anchorage-independent growth of C4–2B4 cells as determined by soft agar colony assay (Fig. 3C). Although C4–2B4 cells are of low migratory potential, we found that expression of CaMKII-T286D, but not wild type or CaMKII-K42M, significantly increases C4–2B4 cell migration (Fig. 3D) and invasion (Fig. 3E). These results suggest that activation of CaMKII increases survival, anchorage-independent growth, migration, and invasion of C4–2B4 cells.
Figure 3. Overexpression of CaMKII in C4–2B4 cell line.
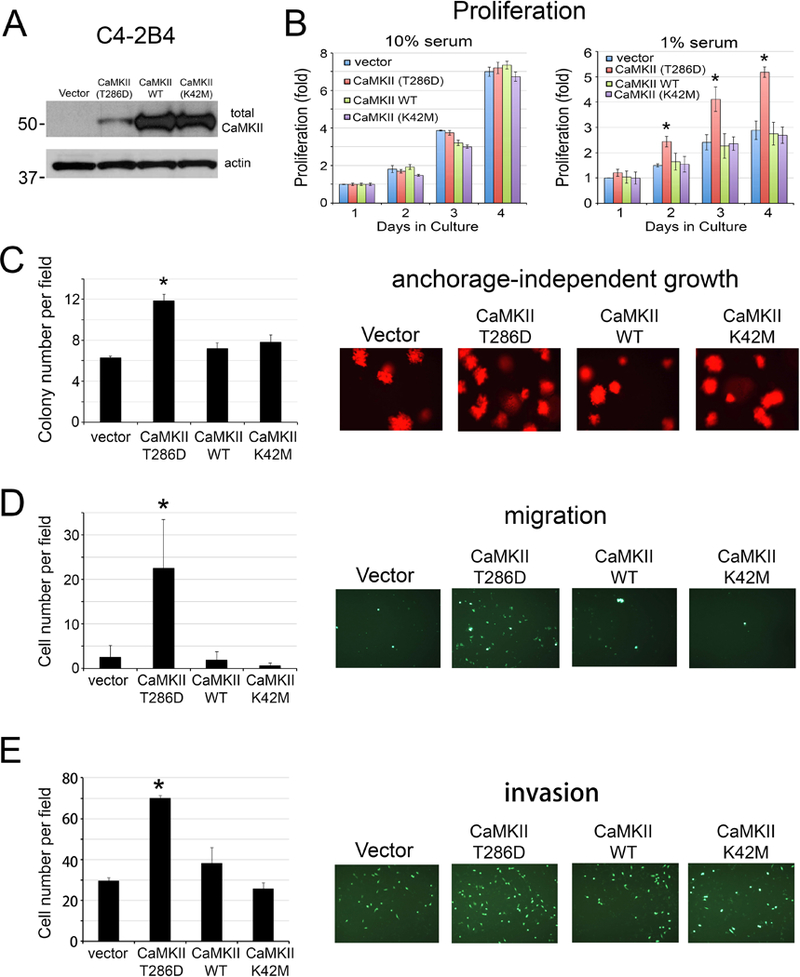
C4–2B4 cells were transfected with empty vector, plasmid containing constitutively active CaMKII-T286D, wild type CaMKII, or inactive mutant CaMKII-K42M. (A) Whole cell lysates were immunoblotted for total CaMKII. Actin was used as a loading control. (B) Cells were cultured in 10% (left panel) or 1% FBS (right panel). Cell proliferation is expressed as fold change of cell number compared to day 0. n=3. (C) Cells were grown in soft agar. Number of colonies per field was counted. n=3. (D) Cells were seeded onto a Boyden chamber. Cells migrated through membrane were labeled with Calcein AM. n=2. (E) Cells were seeded into BioCoat Matrigel-coated invasion chamber. Cells invaded through the matrigel were labeled with Calcein AM. n=3. *, p<0.05.
Ablation of CaMKII impairs prostate cancer metastasis
Our immunohistochemistry staining of human PCa specimens combined with our results demonstrating that CaMKII activation promotes PCa cell survival and migration suggested that CaMKII may play an important role in the metastasis of PCa. Thus, we examined whether tumorigenicity or metastasis are impaired when CaMKII was deleted. Luciferase-labeled PC3-mm2 cells with knockout of CaMKII, i.e., PC3-clone#1, PC3-clone#6, PC3-clone#10, or control PC3-vector cells were injected into the prostates of SCID mice. Tumor growth was assessed by bioluminescence in vivo. At 16 days post-injection, mice were euthanized and the tumors in the prostate were removed. Bioluminescence showed that PC3-vector cells were able to metastasize to lymph nodes, while PC3-clone#1, PC3-clone#6, and PC3-clone#10 cells had fewer metastases (Fig. 4A). Histological analyses of the dissected lymph nodes from PC3-vector injected mice showed the presence of tumor cells in the lymph nodes (Fig. 4B). The number of lymph node metastases, based on bioluminescence imaging and histological analyses, was significantly higher in mice injected with vector control cells compared to those injected with PC3-clone#1, #6, and #10 cells (Fig. 4B). Interestingly, there was no consistent difference in tumor weight between CaMKII knockout clones and the vector control (Fig. 4C), likely due to a lack of effect on cell proliferation by activated CaMKII. These observations suggest that deletion of CaMKII genes in PC3-mm2 cells reduced their metastasis to lymph nodes but had no effect on primary tumor growth.
Figure 4. Orthotopic injection of PC3-mm2 cells with knockout of CaMKII gene.
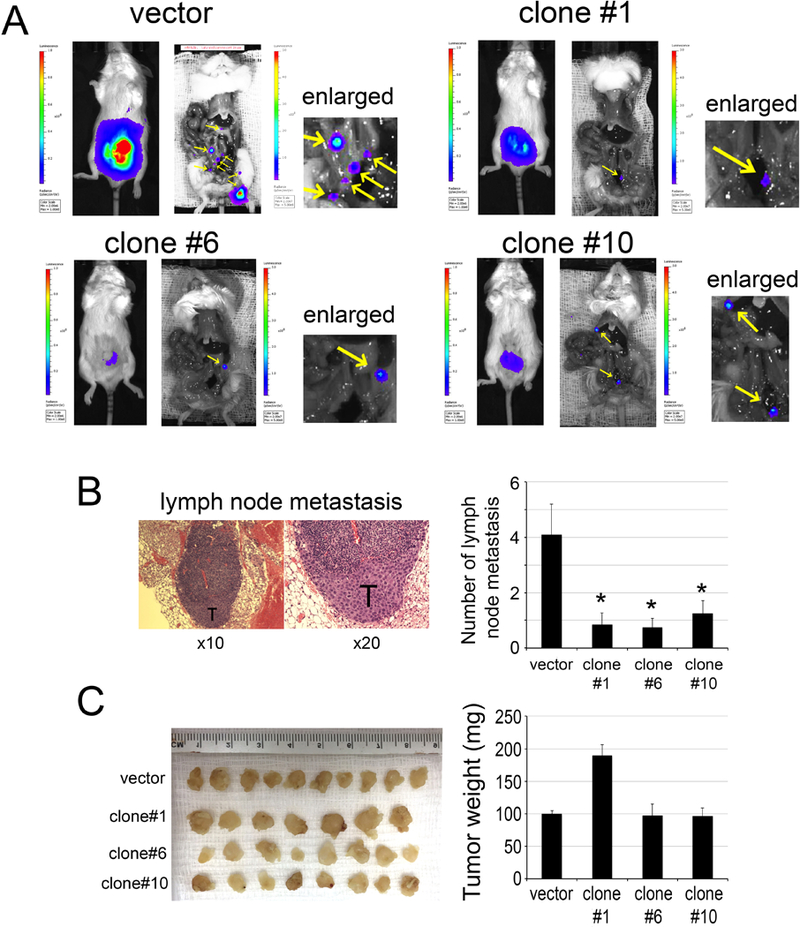
PC3-mm2 cells transfected with empty vector or CRIPSR/Cas9 that targets CaMKII genes (clones #1, #6, #10) were injected orthotopically into mouse prostate. (A) The growth and metastasis of tumor cells were monitored by bioluminescence. Yellow arrows, lymph nodes (see enlarged). (B) Left panel, histology for the presence of tumor cells in lymph nodes. T, tumor cells. Right panel, graph of average number of lymph node metastases in mice injected with PC3-mm2-vector or PC3-clones #1, #6, or #10. (C) Left panel, tumors removed from mouse prostates and fixed in formalin. Right panel, graph of average tumor weight from PC3-mm2-vector or PC3-clones #1, #6, or #10.
Re-expression of CaMKII-T286D in CaMKII knockout PC3-mm2 cells enhances metastasis to lymph nodes
To further examine whether the CaMKII knockout phenotypes could be rescued by re-expression, PC3-mm2 clones #1, #6 and #10, with knockout of CaMKII, were transduced with retrovirus generated from pBMN-CaMKII-T286D-NEO, which contained cDNA for the constitutively active form of CaMKII. We found that re-expression of CaMKII-T286D in CaMKII knockout cells reversed the biological changes including proliferation, anchorage-independent growth, and migration (Supplementary Fig. S3). In vivo metastasis studies by injecting PC3-mm2 clones #1, #6 and #10 with or without CaMKII-T286D re-expression intraprostatically showed that lymph node metastases were increased upon re-expression of CaMKII-T286D (Supplementary Fig. S4). These studies suggest that CaMKII plays a role in enhancing PCa metastasis to lymph nodes.
Acetyl CoA transport by carnitine acetyltransferase (CRAT) is required for CaMKII activation
Next, we examined the mechanisms by which CaMKII is activated during prostate tumorigenesis. Previous studies by McCoy et al. (10) have shown that coenzyme A, generated from acetyl-CoA, directly binds to the CaMKII regulatory domain and activates CaMKII. Thus, we examined the possibility that acetyl-CoA may activate CaMKII in PCa cells. CaMKII is a cytosolic protein. Cytosolic acetyl-CoA is produced in the cytoplasm from citrate or acetate or transported from intracellular organelles including mitochondria or peroxisomes to the cytoplasm. Acetyl-CoA generated in peroxisomes or mitochondria is generally thought to be compartmentalized. For acetyl-CoA to pass the peroxisomal or mitochondrial membrane into the cytosol, acetyl-CoA is converted to acetylcarnitine by the reversible enzyme, carnitine acetyltransferase (CRAT), which then releases acetyl-CoA to the cytosol (17). If acetyl-CoA derived from organelles is involved in CaMKII regulation, then CRAT would be required to transport acetyl-CoA from peroxisomes or mitochondria into the cytoplasm for CaMKII activation. To test this, we used shRNA to knockdown CRAT in PC3-mm2 cells. PC3-mm2 cells with CRAT knockdown were examined by immunoblot analysis. Of the four shRNAs tested, #1, #2, and #4 shRNA showed significant decrease of CRAT expression. Loss of CRAT resulted in a significant loss of CaMKII activation in all three shRNA knockdown cells (Fig. 5A). Correspondingly, a decrease in CRAT protein levels led to a reduction in the cell proliferation in low serum (Fig. 5B), colony formation (Fig. 5C), and cell migration (Fig. 5D). These data indicate that CRAT is necessary for CaMKII activation, and suggest that acetyl-CoA from organelles is one of the mechanisms that activate CaMKII (Fig. 5E).
Figure 5. Knockdown of CRAT in PC3-mm2 cells.
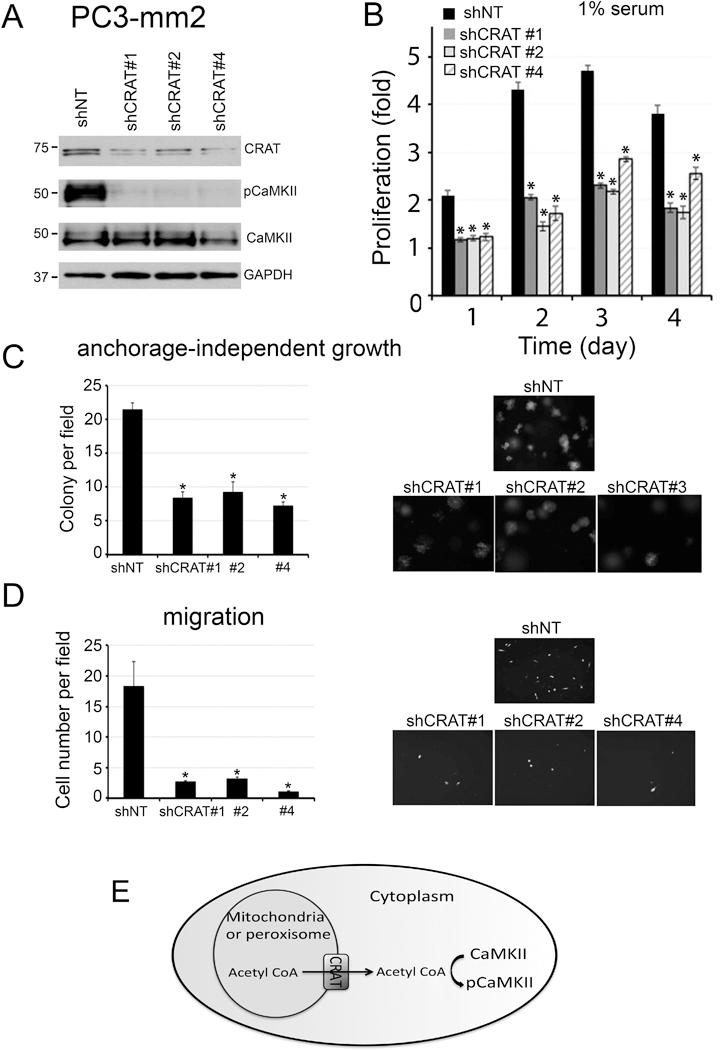
CRAT in PC3-mm2 cells was knocked down by using shRNAs #1, #2 or #4 in lentiviral vectors. (A) Total cell lysates were immunoblotted for CRAT, pCaMKII, and total CaMKII. GAPDH was used as a loading control. (B) Cells were cultured in 1% FBS. Cell proliferation is expressed as fold change of cell number compared to day 0. n=3. The doubling time of shNT was 23.0 ± 1.1 hours and the shCRAT #1, #2 and #4 were 29.5 ± 1.6 hours, 41.4 ± 2.3 hours and 32.9 ± 1.1 hours, respectively. The plating efficiency, determined at 24 hours after cell seeding, of shNT, shCRAT #1, #2 and #4 were 210.0%, 117.3%, 120.0% and 123.0% respectively. (C) Cells were grown in soft agar. Number of colonies per field was counted. n=3. (D) Cells were seeded onto a Boyden Chamber. Cells migrated through the membrane were labeled with Calcein AM. n=2. (E) Schematic illustration for a role of CRAT, expressed in peroxisome or mitochondria, in the transport of acetyl-CoA to the cytosol for CaMKII activation. *, p<0.05.
Organelle-generated acetyl-CoA activates CaMKII in prostate cancer cell lines
β-oxidation of long-chain acyl CoAs is one source of acetyl-CoA (18). We therefore examined whether organelle-generated acetyl-CoA could activate CaMKII. Acyl-CoA Oxidase 1, ACOXI, is the first and rate-limiting enzyme in organelle β-oxidation. We used genetic deletion of ACOXI to assess the requirement of β-oxidation for acetyl-CoA production. We found that knockdown of ACOXI by shRNA in PC3-mm2 was not sufficient to inhibit CaMKII activation (Supplementary Fig. S5). There are three separate ACOX gene products, which carry out oxidation of fatty acids in peroxisomes and mitochondria. Since decreasing ACOXI alone by shRNA is not sufficient to impair CaMKII activation, ACOXII and ACOXIII may be compensating in the absence of ACOXI enzyme activity. We therefore attempted to knockout all three ACOXI, ACOXII and ACOXIII genes together in PC3-mm2 cells through the CRISPR/Cas9 system using a mixture of 6 plasmids that target these three genes. Real-time RT-PCR for the messages of ACOX isoforms showed varied extent of knockdown in PC3-mm2 clones #7, #10 and #21 (Fig. 6A). Interestingly, no triple ACOXI-III knockout cells were generated. It is possible that Acox triple knockout cells were not viable. Western blot for ACOXI and ACOXIII antibodies showed a decrease in the protein levels of AcoxI and AcoxIII, respectively (Fig. 6B). Western blot for AcoxII was not shown due to the lack of working antibody. Importantly, all three clones also exhibited decreased pCaMKII (Fig. 6B). Consistently, all three clones showed decreased proliferation in low serum (Fig. 6C), colony formation in soft agar (Fig. 6D), and significant decreases in cell migration (Fig. 6E) and cell invasion (Fig. 6F). These observations suggest that β oxidation-regulated CaMKII activity is necessary for PCa cell survival, migration, and invasion.
Figure 6. Knockout of Acox in PC3-mm2 cells and overexpression of AcoxI in C4–2B4 cells.
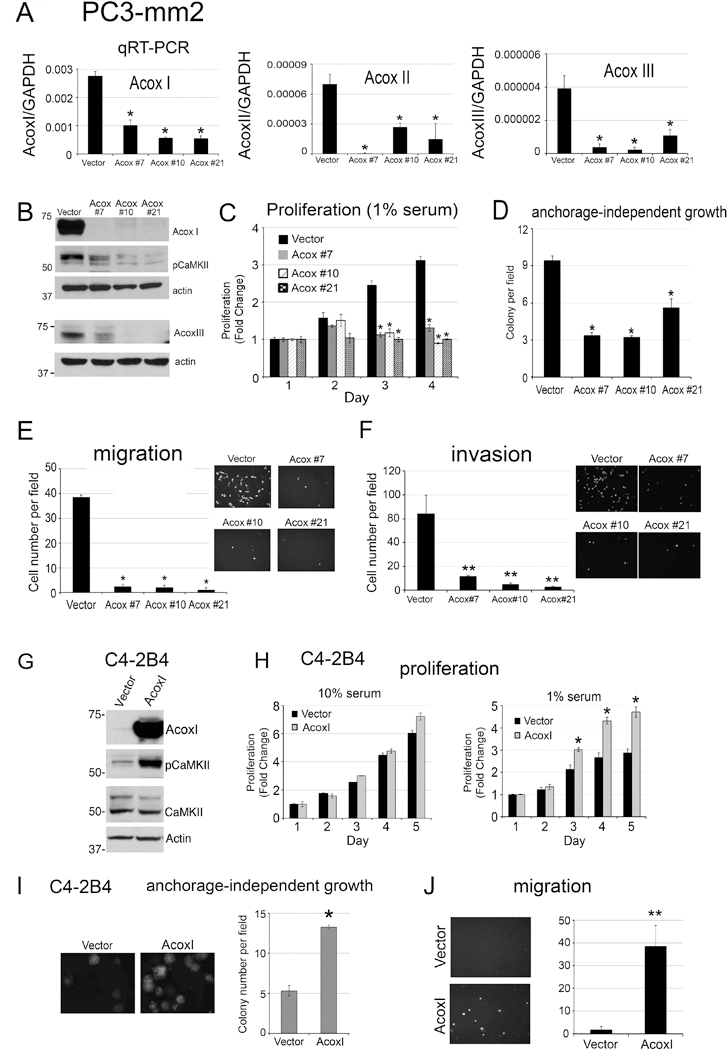
(A)-(F), PC3-mm2-LT cells were transfected with empty vector or CRISPR/Cas9 targeting all three AcoxI, AcoxII and AcoxIII genes (clones #7, #10, and #21). (A) Real-time RT-PCR was performed on total RNAs for the levels of AcoxI, AcoxII, and AcoxIII in the cells. (B) Whole cell lysates were immunoblotted for ACOXI, ACOXIII, and pCaMKII(T286). Actin was used as a loading control. (C) Proliferation of cells grown in 1% FBS expressed as fold change of cell number counted on day 0. n=3. (D) Number of colonies observed for cells grown in soft agar. n=3. (E) Number of cells migrated in a Boyden Chamber migration assay. Cells were labeled with Calcein AM. n=2. (F) Cells were seeded into BioCoat Matrigel-coated invasion chamber. Cells invaded through the matrigel were labeled with Calcein AM. n=3. (G)-(J), C4–2B4 cells were transfected with empty vector or plasmid containing ACOXI. (G) Total cell lysates were immunoblotted for AcoxI, pCaMKII and total CaMKII. Actin was used as a loading control. (H) Proliferation of empty vector or ACOXI overexpressing C4–2B4 cells grown in 10% or 1% FBS expressed as fold change of cell number counted in Day 0. n=3. (I) Number of colonies observed for vector or ACOXI overexpressing C4–2B4 cells grown in soft agar. n=3. (J) Number of cells migrated in a Boyden Chamber migration assay. n=2. *, p<0.05, **, p<0.01.
To complement our gene deletion studies, we also generated C4–2B4 cells overexpressing ACOXI (Fig. 6G). Increased expression of ACOXI was sufficient to activate CaMKII (Fig. 6G), increase cell proliferation in 1% serum but not in 10% serum (Fig. 6H), anchorage-independent growth (Fig. 6I), and cell migration (Fig. 6J). These properties are similar to those observed in C4–2B4 cells with activated CaMKII (Fig. 3B-D), suggesting that the β-oxidation generated acetyl-CoA-mediated increase in CaMKII activity is sufficient to increase cell survival, migration, and invasion activities in PCa cells.
Knockout of ACOX increases lipid content
Because Acox I-III play a role in β-oxidation of fatty acids, knockout of Acox I-III in PC3-mm2 cells may have an effect on their lipid content. We first used Oil Red O, which stains neutral triglycerides and lipids, to examine the lipid content of PC3-mm2 cells with knockout of ACOX, i.e., PC3-Acox #7, Acox #10, Acox #21, or control PC3-vector cells. We found that knockout of AcoxI-III increased the Oil Red O staining of the cells compared to control vector-transfected cells (Fig. 7A), consistent with a role of AcoxI-III in β-oxidation of fatty acids. We further determined the lipidomics/major lipid levels in these cell lines by using tandem mass spectrometry. The lipid composition of PC3-vector cells is shown in Supplementary Fig. S6. We found that the levels of one of the storage lipids, i.e., triacylglycerol, are significantly increased in all three AcoxI-III knockout cells compared to that in control vector-transfected cells (Fig. 7B), consistent with the results from Oil Red O stain. Among 87 triacylglycerol species, 15 of them showed significant increases in all of the three knockout cells compared to vector control (Fig. 7C). The effects of AcoxI-III knockout on other individual lipid components are shown in Supplementary Fig. S6. These data show that knockout of ACOX increases the lipid content in PCa cells, and support a role of β-oxidation of fatty acids in activating CaMKII.
Figure 7. Effect of knockout of Acox genes in PC3-mm2 cells on lipid content and prostate cancer metastasis.
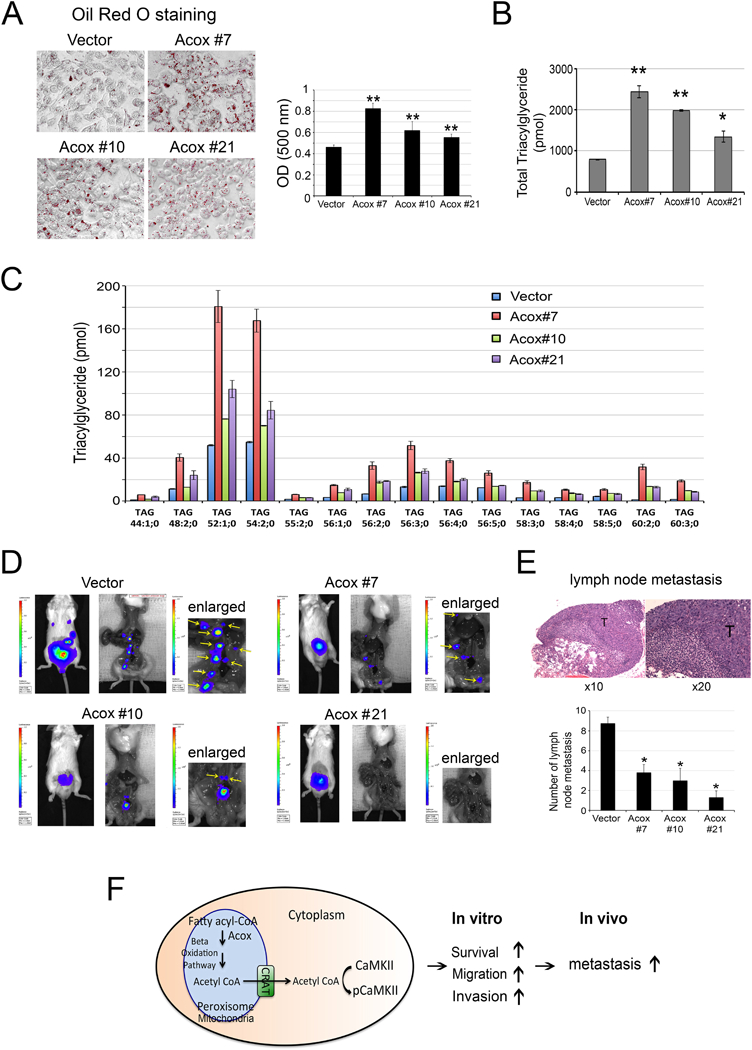
(A) Oil red O staining of PC3-mm2-vector and PC3 clones #7, #10, #21. Right panel, quantification of Oil red O staining. (B) Changes in the levels of triacylglycerol in PC3 clones #7, #10, #21 compared to PC3-mm2-vector cells. (C) The 15 species of triacylglycerol that showed significant increases in all three PC3 clones, i.e. Acox #7, #10, #21, compared to PC3-mm2-vector cells. Lipid species are annotated according to their molecular composition as [sum of the carbon atoms in the hydrocarbon moiety]:[sum of the double bonds in the hydrocarbon moiety]; [sum of hydroxyl groups].PC3-mm2 cells transfected with empty vector or CRIPSR/Cas9 that targets Acox genes (clones #7, #10, #21) were injected orthotopically into mouse prostate. (D) The growth and metastasis of tumor cells were monitored by bioluminescence. Yellow arrows, lymph nodes (see enlarged). (E) Upper panel, histology for the presence of tumor cells in lymph nodes. T, tumor cells. Lower panel, graph of average number of lymph node metastases in mice injected with PC3-mm2-vector or PC3 clones #7, #10, or #21. (F) Schematic illustration of a pathway that CaMKII activation increases metastasis of PCa cells. β-oxidation generated acetyl-CoA is transported out of peroxisomes or mitochondria through carnitine acetyltransferase (CRAT). The acetyl-CoA then activates CaMKII, which increases PCa cell survival, migration and invasion in vitro and metastasis in vivo. *, p<0.05, **, p<0.01.
Knockout of ACOX decreases prostate cancer metastasis
Next, we examined the effect of ACOX on tumorigenicity or lymph node metastasis. Luciferase-labeled PC3-mm2 cells with knockout of ACOX, i.e., PC3-Acox #7, Acox #10, Acox #21, or control PC3-vector cells were injected into the prostates of SCID mice. Tumor growth was assessed by bioluminescence in vivo. At 14 days post-injection, mice were euthanized and the tumors in the prostate were removed. Bioluminescence showed that the number of lymph node metastases, based on bioluminescence imaging and histological analyses, was significantly higher in mice injected with vector control cells compared to those injected with PC3-Acox #7, Acox #10 or Acox #21 cells (Fig. 7D). Histological analyses of the dissected lymph nodes from PC3-vector injected mice showed the presence of tumor cells in the lymph nodes (Fig. 7E). There was no consistent difference in tumor weight between ACOX knockout clones and the vector control (Supplementary Fig. S5). These observations suggest that deletion of ACOX genes in PC3-mm2 cells reduced their ability to metastasize to lymph nodes but had no effect on primary tumor growth. Collectively, these data show that ACOX increases β-oxidation of fatty acids to generate acetyl-CoA, which activates CaMKII and promotes PCa cell survival, migration, and invasion in vitro, and metastasis to lymph nodes in vivo (Fig. 7F).
Discussion
We have shown that CaMKII can be activated by an increase in the level of acetyl-CoA, with β-oxidation as one of the sources, and activated CaMKII promotes PCa cell survival, migration, and metastasis (Fig. 7F). Our studies provide a mechanism by which aberrant metabolism that increases β-oxidation promotes PCa survival and metastasis through the modulation of a kinase, and thus opening up the possibility of targeting this metabolic pathway for cancer treatment.
The finding that the organelle-derived acetyl-CoA metabolic pathway plays a role in regulating CaMKII activity uncovers a novel role for organelle-derived acetyl-CoA in cell signaling. It is possible that CaMKII may be associated with or in close proximity with organelle membranes where CaMKII is activated by acetyl-CoA transported out of the organelle by the transporter CRAT. Acetyl-CoA could influence cell signaling through protein binding or post-translational modification (10). CaMKII can be activated by Ca2+/CaM-dependent and -independent mechanisms. The Ca2+/CaM-independent mechanisms include threonine 287 autophosphorylation (19), methionine 281/282 oxidation (20), and serine 280 O-GlcNAclyation (21). S-nitrosylation of Cysteine 290 in the regulatory domain has also been shown to induce autonomous activation of CaMKII (22). Previous studies by McCoy et al. (10) showed that CoA bound to CaMKII through amino acids K292, R296, and K300 and CoA-induced CaMKII activation requires basal amounts of calcium. These observations suggest that methionine 28½82 oxidation and cysteine 290 may not be involved in the biological effects observed in this study. Thus, CoA-induced CaMKII activation represents an additional mechanism for CaMKII activation. Together, our findings uncover new roles of metabolism in generating cellular signals to control cell function.
The physiological or pathological conditions that regulate the acetyl-CoA-pCaMKII pathway are not clear. Previous studies by McCoy et al. (10), using X. laevis egg extracts, have shown that high glucose concentration in the cytosol stimulates acetyl-CoA production, which leads to activation of CaMKII and subsequent cell survival in oocytes. Their observations suggest that increased glucose metabolism may be one mechanism to activate the pathway. Because cellular uptake of glucose is augmented in cancer and increased levels of cytosolic acetyl-CoA have been detected in cancer (23,24), altered glucose metabolism in cancer cells may be one of the mechanisms that activate CaMKII.
Another relevant possibility that may regulate the pCaMKII pathway in PCa cells is androgen signaling. Rokhlin et al. (5) showed that androgen receptor activation decreases CaMKII activity while siRNA knockdown of AR led to an increase in CaMKII activity. These observations suggest that CaMKII activity can be controlled by AR activity. Consistently, we found that the levels of CaMKII and activated CaMKII in PCa cells are inversely correlated with the expression of AR, as LNCaP and C4–2B4 cells express AR while PC3 and PC3-mm2 cells do not. Studies by Rokhlin et al. (5,6) also showed that CaMKII activation plays a role in resistance to apoptosis following androgen deprivation therapy. However, it is not clear whether AR directly regulates CaMKII activity or AR regulates CaMKII activity through other intermediates including metabolites. These observations raise interesting questions concerning whether androgen depletion increases acetyl-CoA levels and pCaMKII(T286) in AR-dependent PCa cells. Interestingly, Vaz et al. (25,26) showed that androgen-responsive and nonresponsive PCa present distinct glycolytic metabolism profiles and that androgen enhances glycolytic metabolism. Tennakoon et al. (27) demonstrated that androgen increases β-oxidation. Further investigations into the role of androgen signaling on β oxidation-acetyl CoA-CaMKII pathway are warranted.
Our studies showed that CaMKII activation increases PCa cell survival in both low serum condition and in soft agar, raising an interesting question concerning the mechanism by which activated CaMKII increases cell survival. Our observations are consistent with previous studies that CaMKII activation leads to inhibition of apoptosis in oocytes (9). The mechanism by which activated CaMKII inhibits apoptosis in X. laevis oocyte extract is through phosphorylation of the prodomain of caspase-2, resulting in inactivation of caspase-2 and inhibition of oocyte apoptosis (9). Due to the lack of working antibody for human caspase-2, we did not test whether activated CaMKII phosphorylates caspase-2 in our PCa cell lines. On the other hand, Rokhlin et al. (5) showed that overexpression of CaMKII α and –β resulted in decreases in the protein levels of procaspase-7 and procaspase-8. Thus, it is likely that CaMKIIs exert their anti-apoptotic effects in part through regulation of caspases. Alternatively, Yang et al. (28) showed that CaMKII-mediated the expression and phosphorylation of c-FLIP (cellular Fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein) that inhibits apoptosis in glioma cells. Similarly, Xiao et al. (29) showed that CaMKII activity is upregulated in TRAIL-resistant melanoma cells, as inhibition of CaMKII activity downregulated c-FLIP protein expression and sensitized resistant cells to TRAIL-induced apoptosis. In human astrocytes, constitutively activated CaMKII in astrocytes protected cells from Fas-mediated apoptosis by regulating the expression of PEA-15/PED and c-FLIP (30). Further studies are needed to comprehensively determine how CaMKII activation promotes PCa cell survival.
There are several routes for the generation of intracellular acetyl-CoA. In addition to peroxisomes and mitochondria, acetyl-CoA can be generated from ATP-citrate lyase or acetyl-CoA synthetase in the cytosol. The acetyl-CoA transporter CRAT is expressed in both peroxisomes and mitochondria. Thus, knockdown of CRAT by shRNA may reduce the expression of CRAT in both organelles. While our data show that organelle-derived acetyl-CoA promotes CaMKII activation, we cannot exclude the involvement of cytosolic-generated acetyl-CoA in CaMKII activation. It would also be important to measure the levels of acetyl-CoA in the various PCa cell lines and in the ACOX knockout or overexpressing cell lines to assess whether the levels are correlated with CaMKII activation. However, due to the instability of acetyl-CoA and the presence of enzymes that can degrade acetyl-CoA in the cell lysates, attempts to measure acetyl-CoA levels in the PCa cell lysates were not successful.
We showed that, like CRAT deletion, knockout of ACOX enzymes reduced pCaMKII activity in PCa cells. We further addressed whether β-oxidation enzymes, e.g. ACOXI-III, correlate with CaMKII activation in human PCa cell lines or PCa specimens. We found that there is no correlation in the levels of ACOXI with pCaMKII in various PCa cell lines by real-time RT-PCR (Supplementary Fig. S5). Analyses for the levels of ACOXI-III in metastatic PCa compared to non-metastatic primary tissues or normal tissues in three human datasets, i.e., Taylor et al. (31), Lapointe et al. (32), and Tomlins et al. (33), also did not find a correlation between ACOX enzymes and PCa metastatic progression. It is likely that the regulation of acetyl-CoA levels lies in the levels of upstream substrate, e.g. glucose-6-phosphate, rather than in the ACOX enzymes. However, we cannot rule out at this time that the activity of ACOX enzymes might also be regulated via additional, unknown posttranslational mechanisms.
When PCa cells are dislodged from the primary tumor, the ability of tumor cells to survive in the circulation is critical for metastasis to occur (34–36). Our observations that knockout of CaMKII in PC3-mm2 cells led to a decrease in their metastasis to lymph nodes is consistent with a role of activated CaMKII in enhancing PCa cell survival in low serum culture conditions and in anchorage-independent growth as well as cell migration. This observation is also consistent with higher levels of pCaMKII in human PCa specimens from lymph nodes and bone metastasis than in primary tumors. Together, these observations suggest that aberrant metabolic activity enhances survival and metastatic potential of PCa cells by co-opting CaMKII signal transduction. The identification of a pathway by which metabolism targets specific proteins to support metastasis opens up the possibility of developing therapy strategies to target the aberrant metabolism for metastasis prevention. The data presented here suggest that the further development of inhibitors for CaMKII is warranted.
While the current study is focused on cytosolic CaMKII that was activated from CoA derived from possibly the mitochondria and/or peroxisomes, mitochondrial CaMKII has been shown to play a central role in the pathogenesis of cardiac disease (37,38). Recently, Sebag et al. (39) also showed that mitochondria CaMKII in airway epithelium plays a role in asthma. These studies raise an interesting question on the role of mitochondrial CaMKII in cancer. CaMKII inhibitor that specifically targets mitochondria was used to address the function of CaMKII in mitochondria. Such an approach can be applied in future studies to address whether mitochondrial CaMKII is also involved in PCa metastasis.
Supplementary Material
Significance:
This study identifies a cell metabolic pathway that promotes prostate cancer metastasis and suggests prostate cancer may be susceptible to beta-oxidation inhibitors.
Acknowledgements:
This work was supported by grants from National Cancer Institute of the National Institutes of Health under Award Number NIH P50 CA140388, CA174798 (S.-H.Lin), CA16672, Cancer Prevention Research Institute of Texas CPRIT RP150179 (S.-H.Lin) and CPRIT RP150282 (G.E.Gallick), and American Cancer Society RSG-14–226-01-CCG (L.K.Nutt).
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.Shonesy BC, Jalan-Sakrikar N, Cavener VS, Colbran RJ. CaMKII: a molecular substrate for synaptic plasticity and memory. Progress in molecular biology and translational science 2014;122:61–87. [DOI] [PubMed] [Google Scholar]
- 2.Skelding KA, Rostas JA, Verrills NM. Controlling the cell cycle: the role of calcium/calmodulin-stimulated protein kinases I and II. Cell cycle 2011;10(4):631–9. [DOI] [PubMed] [Google Scholar]
- 3.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J 2002;364(Pt 3):593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colbran RJ, Schworer CM, Hashimoto Y, Fong YL, Rich DP, Smith MK, et al. Calcium/calmodulin-dependent protein kinase II. Biochem J 1989;258(2):313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rokhlin OW, Taghiyev AF, Bayer KU, Bumcrot D, Koteliansk VE, Glover RA, et al. Calcium/calmodulin-dependent kinase II plays an important role in prostate cancer cell survival. Cancer biology & therapy 2007;6(5):732–42. [DOI] [PubMed] [Google Scholar]
- 6.Cohen MB, Rokhlin OW. Mechanisms of prostate cancer cell survival after inhibition of AR expression. J Cell Biochem 2009;106(3):363–71. [DOI] [PubMed] [Google Scholar]
- 7.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11(2):85–95. [DOI] [PubMed] [Google Scholar]
- 8.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 2008;13(6):472–82. [DOI] [PubMed] [Google Scholar]
- 9.Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 2005;123(1):89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCoy F, Darbandi R, Lee HC, Bharatham K, Moldoveanu T, Grace CR, et al. Metabolic activation of CaMKII by coenzyme A. Mol Cell 2013;52(3):325–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nutt LK. The Xenopus oocyte: a model for studying the metabolic regulation of cancer cell death. Seminars in cell & developmental biology 2012;23(4):412–8. [DOI] [PubMed] [Google Scholar]
- 12.Huang CF, Lira C, Chu K, Bilen MA, Lee YC, Ye X, et al. Cadherin-11 increases migration and invasion of prostate cancer cells and enhances their interaction with osteoblasts. Cancer Res 2010. 70:4580–9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye X, Lee YC, Choueiri M, Chu K, Huang CF, Tsai WW, et al. Aberrant expression of katanin p60 in prostate cancer bone metastasis. Prostate 2012;72(3):291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu G, Lee YC, Cheng CJ, Wu CF, Song JH, Gallick GE, et al. RSK promotes prostate cancer progression in bone through ING3, CKAP2, and PTK6-mediated cell survival. Molecular cancer research : MCR 2015;13(2):348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, et al. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci U S A 2011;108(5):1903–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell PJ, Kingsley EA. Human prostate cancer cell lines. Methods in molecular medicine 2003;81:21–39. [DOI] [PubMed] [Google Scholar]
- 17.Jogl G, Tong L. Crystal structure of carnitine acetyltransferase and implications for the catalytic mechanism and fatty acid transport. Cell 2003;112(1):113–22. [DOI] [PubMed] [Google Scholar]
- 18.Lazarow PB. Rat liver peroxisomes catalyze the beta oxidation of fatty acids. J Biol Chem 1978;253(5):1522–8. [PubMed] [Google Scholar]
- 19.Schworer CM, Colbran RJ, Soderling TR. Reversible generation of a Ca2+-independent form of Ca2+(calmodulin)-dependent protein kinase II by an autophosphorylation mechanism. J Biol Chem 1986;261(19):8581–4. [PubMed] [Google Scholar]
- 20.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008;133(3):462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013;502(7471):372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coultrap SJ, Bayer KU. Nitric oxide induces Ca2+-independent activity of the Ca2+/calmodulin-dependent protein kinase II (CaMKII). J Biol Chem 2014;289(28):19458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrer A, Wellen KE. Metabolism and epigenetics: a link cancer cells exploit. Curr Opin Biotechnol 2015;34:23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell 2014;159(7):1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaz CV, Alves MG, Marques R, Moreira PI, Oliveira PF, Maia CJ, et al. Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int J Biochem Cell Biol 2012;44(11):2077–84. [DOI] [PubMed] [Google Scholar]
- 26.Vaz CV, Marques R, Alves MG, Oliveira PF, Cavaco JE, Maia CJ, et al. Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. Journal of cancer research and clinical oncology 2016;142(1):5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 2014;33(45):5251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang BF, Xiao C, Roa WH, Krammer PH, Hao C. Calcium/calmodulin-dependent protein kinase II regulation of c-FLIP expression and phosphorylation in modulation of Fas-mediated signaling in malignant glioma cells. J Biol Chem 2003;278(9):7043–50. [DOI] [PubMed] [Google Scholar]
- 29.Xiao C, Yang BF, Song JH, Schulman H, Li L, Hao C. Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Experimental cell research 2005;304(1):244–55. [DOI] [PubMed] [Google Scholar]
- 30.Song JH, Bellail A, Tse MC, Yong VW, Hao C. Human astrocytes are resistant to Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. The Journal of neuroscience : the official journal of the Society for Neuroscience 2006;26(12):3299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A 2004;101(3):811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, et al. Integrative molecular concept modeling of prostate cancer progression. Nature genetics 2007;39(1):41–51. [DOI] [PubMed] [Google Scholar]
- 34.Fidler IJ. Critical factors in the biology of human cancer metastasis: twenty-eighth G.H.A. Clowes memorial award lecture. Cancer Res 1990;50(19):6130–8. [PubMed] [Google Scholar]
- 35.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009;9(4):274–84. [DOI] [PubMed] [Google Scholar]
- 36.Zetter BR. The cellular basis of site-specific tumor metastasis. N Engl J Med 1990. 322:605–12. [DOI] [PubMed] [Google Scholar]
- 37.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012;491(7423):269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng N, Anderson ME. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. Journal of molecular and cellular cardiology 2017;103:102–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sebag SC, Koval OM, Paschke JD, Winters CJ, Jaffer OA, Dworski R, et al. Mitochondrial CaMKII inhibition in airway epithelium protects against allergic asthma. JCI insight 2017;2(3):e88297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.