

Abstract
Background:
The incidence of eosinophilic esophagitis (EoE) is greater in male than female subjects, and the underlying molecular basis for this sex bias remains unclear.
Objective:
We sought to delineate the contribution of the sex hormone estrogen to the EoE phenotype and esophageal epithelial barrier function and remodeling.
Methods:
We performed demographic and incidence analyses of EoE in male and female subjects from a single-center pediatric cohort. Estrogen-responsive gene expression analyses and estrogen receptor (ESR) immunofluorescence staining of esophageal biopsy specimens from patients with EoE and control subjects were performed. The effect of 17β-estradiol (E2) on IL-13–induced signaling pathways, gene expression, and esophageal epithelial architecture and barrier function in a primary human esophageal keratinocyte cell (EPC2) culture system (EPC2–air-liquid interface) was examined.
Results:
We observed a male predominance in patients with EoE. Analyses of RNA sequencing data sets revealed a significant dysregulation of the estrogen-responsive gene network and expression of ESR1 and ESR2 in esophageal biopsy specimens from patients with EoE compared with control subjects. IL-13 stimulation of EPC2–air-liquid interface cells led to altered cellular architecture with induced dilation of intercellular spaces and barrier dysfunction. Pretreatment of EPC2s with E2 prior to IL-13 exposure abrogated IL-13– induced architectural changes and esophageal barrier dysfunction. Mechanistically, E2-protective effects were dependent on ESR2 and associated with diminishing of IL-13– induced tyrosine kinase 2 and signal transducer and activator of transcription 6 phosphorylation and EoE-dysregulated gene expression.
Conclusions:
Estrogen-responsive genes are modified in patients with EoE compared with control subjects. E2 attenuated IL-13– induced architectural changes and esophageal epithelial barrier dysfunction through inhibition of the IL-13/tyrosine kinase 2/ signal transducer and activator of transcription 6 pathway via ESR2-dependent process. Estrogen hormone signaling may protect against development of EoE in female subjects.
Keywords: Eosinophilic esophagitis, IL-13, estrogen, hormone, barrier dysfunction
GRAPHICAL ABSTRACT
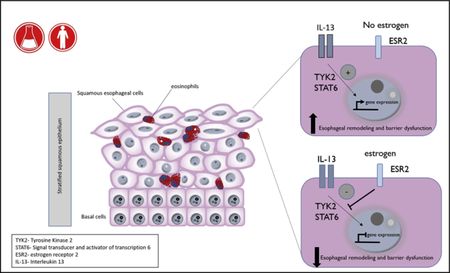
Eosinophilic esophagitis (EoE) is a chronic immune-mediated disease characterized by eosinophil infiltration of the esophageal epithelium, resulting in impairment of esophageal function and development of feeding intolerance, dysphagia, food impaction, or strictures.1,2 Epidemiologic studies in North America, Europe, and Asia have identified that EoE is 2 to 4 times more frequent in male than female subjects.3–6 The EoE male predominance appears to persist across demographic groups, regardless of age, geographic region, socioeconomic status, or race.4,7,8 Clinical studies indicate that although there is no difference in disease severity between male and female subjects, the clinical and histologic presentation of EoE can differ between the sexes.4 Moreover, adult male patients with EoE are more likely to have stricturing disease and a longer duration of symptoms before presentation.4 Furthermore, male pediatric patients with EoE are more likely to present with food impaction and feeding refusal, whereas female children with EoE report more abdominal pain.4 Intriguingly, male subjects are also at greater risk than female subjects of having other esophageal diseases, including gastroesophageal reflux disease, Barrett esophagus, and esophageal adenocarcinoma (1.7:1, 2:1, and 8–9:1, respectively).9,10 The underlying cause of increased risk of esophagus-related disorders, such as EoE, in male subjects and whether dissimilarities in EoE between male and female subjects represent differences in disease endotypes or reflective of differential underlying disease-driving mechanisms remains unknown.
Clinical and experimental evidence suggests that the manifestations of EoE are a consequence of chronic esophageal allergic inflammation and impaired epithelial barrier function, which drive esophageal remodeling and disease onset.11 Dysregulation of expression of a number of key epithelial barrier regulatory genes, including the desmosomal cadherin desmoglein 1 (DSG1),12 leucine-rich repeat-containing protein 31,13 kallikrein serine proteases,13 and calpain-14,14 have been linked to EoE and shown to alter esophageal epithelial barrier function. Furthermore, IL-13, a known proallergic cytokine, has been linked to dysregulation of expression of esophageal epithelial barrier genes in patients with EoE.15,16 Indeed, administration of IL-13 to esophageal epithelial cells is sufficient to dysregulate epithelial barrier regulatory gene expression and esophageal barrier function.12 IL-13 binds to either the type II IL-4 receptor (IL-4 receptor [IL-4R] α and IL-13 receptor [IL-13R] α1 subunits) or the type II IL-13 receptor (IL-13Rα2),17 which leads to phosphorylation of receptor-associated Janus kinases (JAKs), including tyrosine kinase 2 (TYK2) and JAK2.18,19 Activation of TYK2 or JAK2 promotes phosphorylation and activation of the transcription factor signal transducer and activator of transcription 6 (STAT6)20–22 and induction and suppression of STAT6-dependent gene expression, including inflammation-related genes, such as eotaxin-3 (CCL26), and genes related barrier function, such as the desmosome-related protein DSG1.12,23
There is emerging evidence that the underlying basis for sex-related susceptibility to various inflammatory conditions are sex hormones.24 Sex hormones influence the onset and severity of immune-mediated pathologic conditions by modulating innate and adaptive immunity during prenatal, prepubertal, and postpubertal stages of life.25–27 Endogenous estrogen hormones, of which 17β-estradiol (E2) is the predominant form, can affect physiologic processes beyond reproductive function, including modulation of inflammation and tissue differentiation, through genomic and nongenomic pathways.28–31 Indeed, many hematopoietic cells, including B and T lymphocytes, granulocytes, myeloid cells, and natural killer cells, express estrogen receptors (ESRs).32 Estrogens are bound in the blood to hormone-binding proteins28 and can dissociate from these binding proteins, cross plasma membranes, and stimulate gene expression through ESR-dependent and independent pathways.29,30 Estrogen canonical signaling occurs through binding of estrogens to the cytosolic ESR1 and ESR2, which can form homodimers or heterodimers.29 Ligand binding and dimerization of ESRs leads to translocation of the ESR to the nucleus and induction of gene expression through binding DNA estrogen response elements or indirectly through interactions with other transcription factors in a concentration- and tissue-specific manner.28,30 Estrogen can also bind to cytoplasmic endoplasmic reticulum or membrane-bound G protein-coupled receptors leading to activation of signal transduction pathways, such as mitogenactivated protein kinases, resulting in modulation of gene expression.29,30,33,34
Here we examine the male/female ratio of EoE and association with age of diagnosis. We explore the involvement of signaling by the sex hormone estrogen in the EoE transcriptome and define the effect of estrogen on IL-13–induced esophageal barrier function. We observed differences in estrogen-responsive gene expression in patients with EoE compared with control subjects and show altered ESR expression in esophageal tissue of patients with EoE compared with control subjects.
By using an in vitro EoE model system, we show that pretreatment of primary esophageal cells with the estrogen hormone E2 abrogated IL-13–induced barrier dysfunction and associated architectural changes. Suppression of IL-13–induced barrier function was dependent on ESR2 signaling and associated with decreased activation of IL-13–induced TYK2 and STAT6 signaling and downstream IL-13–mediated transcriptional events. Our data suggest that E2 can exert protective effects on the esophageal epithelium through downregulation of inflammation-induced esophageal barrier dysfunction. Collectively, these studies identify potential pathways that can explain the reduced incidence of EoE in female subjects.
METHODS
Cincinnati Center for Eosinophilic Disease cohort analysis
A retrospective review was performed of the electronic medical record database at the Cincinnati Center for Eosinophilic Disease (CCED). The CCED is part of a pediatric tertiary care referral center and is affiliated with an adult tertiary care academic medical center (https://www.cincinnatichildrens.org/cced). The medical record database includes demographic and histologic data of all patients with a biopsy-confirmed diagnosis of EoE treated at the CCED. Inclusion criteria for the query included patients of any age with a diagnosis of EoE and a clinical visit between January 2000 and January 2017. Patients were further divided by sex and age at initial diagnosis.
RNA sequencing and bioinformatics analysis
RNA sequencing (RNA-seq) results of human esophageal biopsy specimens were obtained from a previous analysis.35 Samples were from patients aged 1 to 34 years at the time of biopsy, with all but 3 patients being less than 18 years of age. For RNA-seq of primary human esophageal keratinocyte cell (EPC2)–air-liquid interface (ALI) cells, RNA was isolated from cells by using the RNEasy Kit (Qiagen, Germantown, Md), according to the manufacturer’s protocol. RNA quality was assessed by using the Agilent 2100 Expert bioanalyzer (Agilent Technologies, Santa Clara, Calif), and only the samples with an RNA integrity number of greater than 8 were processed for sequencing. RNA samples were subjected to RNA-seq at the Cincinnati Children’s Hospital Medical Center Gene Discovery and Genetic Variation Core, as previously described.36 The sequencing reads were aligned against the GRCh37 genome model by using TopHat2.04 with Bowtie 2.03,37,38 whereas the separated alignments were unified with Cuffmerge39 by using the University of California, Santa Cruz, model as reference. Raw data were then uploaded on Biowardrobe40 (http://biowardrobe.com) and analyzed by using a differential sequencing tool.
RNA-seq and gene expression quantification
Transcriptomic analyses were performed in Strand NGS (Strand Life Sciences, Hebbal, Bangalore). After removal of barcodes and primers, raw reads were aligned to the mm10 genome by using annotations provided by the University of California, Santa Cruz, and the following parameters: (1) minimum percentage identified, 90%; (2) maximum percentage gaps, 5%; (3) minimum aligned read length, 25; (4) number of matched to output per reads, 1; and (5) ignore reads with more than 5 matches. The aligner (COBWeb) was based on the Burrow-Wheeler transform method. Reads per kilobase per million (RPKM) were computed by using aligned reads and the expectation-maximization algorithm for the maximum likelihood estimation of expression. Furthermore, the RPKM minimal threshold was defined as 1 and normalized by using the DESeq algorithm, which computes a normalization factor (NF) for each sample. Within each sample, each transcript is divided by that transcript’s geometric mean across samples. The within-sample median of these values is that sample’s NF. To obtain normalized counts, a sample’s raw RPKM values are divided by that sample’s NF.
Finally, normalized per-transcript RPKM values were baselined to the median of all samples (n = 40,448 transcripts). Reasonably expressed transcripts (raw RPKM > 3 in 100% of samples in ≥ 1 condition) were included for differential analysis (n = 21,096 transcripts). Differential expression was determined through t test ANOVAs with a P value cutoff of .05 and a fold change (FC) requirement of greater than 1.5 (male vs female in control subjects, n = 63; male vs female in patients with EoE, n = 191; and patients with EoE vs control subjects, n = 6647). Data can be accessed through the National Center for Biotechnology Information’s Gene Expression Omnibus repository at GSE58640.
Pathway analysis was performed with toppgene.cchmc.org, which collects ontologic data from more than 30 individual repositories. The heat maps were generated by using R software’s heatmap.2 defaults, in which the Euclidean measure is used to obtain the distance matrix and complete agglomeration method for clustering.
Immunofluorescent staining
Formalin-fixed, paraffin-embedded, deidentified human esophageal biopsy samples of postpubertal (age, 15.2–17.9 years at time of biopsy) male and female healthy control subjects and patients with EoE were obtained from the CCED slide repository. Slides were deparaffinized and then underwent antigen retrieval with Tris-EDTA (pH 9). Samples were rinsed with PBS and then underwent heat-induced epitope retrieval by using a pressure cooker. Tissue was blocked with 4% donkey serum for 1 hour and then exposed to primary antibodies diluted in PBS. Primary antibodies included ESR1 (rabbit anti-human, 50 ng/mL; Invitrogen, Carlsbad, Calif) and ESR2 (rabbit anti-human, 500 ng/mL; Invitrogen). Biopsy specimens were treated with primary antibodies at 4°C overnight, rinsed with PBS, and then exposed to secondary antibody for 1 hour at room temperature (goat anti-rabbit Alexa Fluor 488 nm, 1:1,000 dilution; Invitrogen). The samples were exposed to chromosome counterstain 4ʹ,6-diamidino-2-phenylindole, and digital fluorescence images were recorded with Zeiss Apotome fluorescent microscope (AxioVision; Zeiss, Oberkochen, Germany) using NIKON elements software.
ALI cell-culture system
EPC2s, a human telomerase reverse transcriptase–immortalized esophageal epithelial cell line that has been characterized in multiple studies, was used for all experiments.41,42 Cells were seeded at a concentration of 1 × 104 cells per well on a 0.4-mm pore-size permeable membrane support system (Corning, Corning, NY) and then grown in low-calcium (0.09 mmol/L [Ca+2]) keratinocyte serum-free media. After cells reached confluence, typically day 3 of culture, the medium was changed to high-calcium keratinocyte serum-free medium (1.8 mmol/L [Ca+2]) for 3 additional days. On day 7, medium was removed from the inner chamber of the ALI system to expose cells to air and subsequently induce epithelial stratification and differentiation. Treatment was started on day 10 or 11 of culture, with 24 to 48 hours of exposure to IL-13 (100 ng/mL), E2 (1–100 nmol/L), or both.
Hematoxylin and eosin staining
Cells were fixed by adding 4% paraformaldehyde directly to the tops and bottoms of transwells. Cell plates were placed on ice, and support membranes were removed from the transwells. Cells on the support membranes were dehydrated with 70% ethanol and then paraffin embedded. Cells and support membranes were cut, placed on slides, deparaffinized, and stained with hematoxylin and eosin by the Cincinnati Children’s Hospital Medical Center Tissue Processing Core.
Quantification of dilated intercellular spaces
Dilated intercellular space (DIS) values represent automated space measurement quantification of a compilation of 340 images from hematoxylin and eosin–stained samples from each treatment group by using Image-Pro Plus software (Media Cybermetrics, Rockville, Md). DIS percentages represent a ratio of DIS area to total tissue area.
Western blotting
Cells were rinsed with cold PBS, and the culture plate was placed on ice. The cell-culture membrane support system was cut from the plastic transwells and placed in lysis buffer with protease inhibitor. Cells were scraped from the membranes, and the suspension was centrifuged at 4°C at 10,000g for 10 minutes. The supernatant was removed, and protein quantification was performed by using a serial bicinchoninic acid assay. The samples in SDS sample buffer were boiled for 10 minutes, separated by using SDS-PAGE gradient gels (Novex Life Technologies, Invitrogen), followed by Western blot analysis. The membranes were blocked with 5% milk and then incubated with primary antibody overnight at 4°C. Primary antibodies included STAT6 (rabbit anti-human, 1:1,000 dilution; Santa Cruz Biotechnology, Dallas, Tex), phosphorylated STAT6 (pSTAT6; rabbit anti-human, 1:1,000 dilution; Santa Cruz Biotechnology), TYK2 (rabbit anti-human, 1:1,000 dilution; Thermo Fisher Scientific, Waltham, Mass), and phosphorylated TYK2 (pTYK; rabbit anti-human, 1:500 dilution; Thermo Fisher Scientific). After serial rinsing with TBS with 20% Tween, the membranes were exposed to secondary antibody (LI-COR Biosciences, Lincoln, Neb) for 1 hour at room temperature. Quantitative expression analysis with 2-color infrared imaging was used to compare protein expression (Odyssey Imager; LI-COR Biosciences).
RNA-seq and quantitative PCR
After completion of the treatment course, cells were treated with TRIzol, scraped from the support membrane, and then frozen in TRIzol suspension at ‒80°C. RNA was extracted with the RNA isolation kit (Thermo Fischer Scientific) and then analyzed for purity and quantified with the Nanodrop 2000c (Thermo Scientific). mRNA was either submitted to the Cincinnati Children’s Hospital Medical Center Bioprocessing CORE for RNA-seq or amplified to cDNA. RNA-seq results are expressed in RPKM values.
Quantitative PCR was performed with SYBR Green. Suppressor of cytokine signaling 1 (SOCS1) forward primers (5ʹ-TCC CCT TCC AGA TTT GAC CG-3ʹ) and reverse primers (5ʹ-CCA CAT GGT TCC AGG CAA GT-3ʹ) were designed by Integrated DNA Technologies (Coralville, Iowa). Relative expression of SOCS1 was compared with baseline HPRT expression by using real-time PCR (CFX96R; Bio-Rad Laboratories, Hercules, Calif).
Transepithelial resistance analysis
Cells were grown on EPC2-ALI transwells, as previously described.43 After completion of the treatment course, the tops of the transwells were removed. The bottom portion of the transwells were placed in chamber sliders (P2320T slider, 6.5 mm diameter, sized for a 0.33-cm2 growth area) inserted into the dual Ussing chambers (P2300 dual Ussing chamber; Physiologic Instruments, San Diego, Calif) containing Ringer buffer (pH 7.4) and mounted on an 8-chamber Ussing system (EM-CSYS-8 EasyMount Ussing Chamber System; Physiologic Instruments, San Diego, Calif).
The transepithelial potential difference was measured by using 2 paired Ag and AgCl electrode tips made from 4% agar dissolved in 3 mol/L KCl. The electrodes were connected to a voltage clamp amplifier (VCC-MC8 multi-channel voltage clamp; Physiologic Instruments). The potential difference of the electrodes and the fluid resistance compensation were calibrated before insertion of the transwells into the chamber. The potential difference was monitored continuously with an open circuit for 15 to 30 minutes before establishing equilibrium. Rapid-pulse voltage pulses of 620-ms cycles were delivered every 1 second to yield a current response for calculation of resistance across the membrane by using Ohm’s law. Output was collected by using Acquire & Analyze Rev II software. In some experiments mature EPC2-ALI cells (>1500 Ω · cm2) were treated with either vehicle (0.1% dimethyl sulfoxide) or the highly selective ESR1 antagonist 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride (MPP; 100 pmol/L) or 4-[2-phenyl-5,7-bis(trifluoromethyl) pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP; 10 μmol/L) 30 minutes before exposure to E2 (100 nmol/L). Twenty-four hours later, EPC2-ALI cells were stimulated with IL-13 (100 ng) in the presence of vehicle, MPP, or PHTPP and E2 for 24 hours, and transepithelial resistance (TER) measurements were performed with an EVOM (World Precision Instruments, Sarasota, Fla).
Statistical analysis
Categorical data were expressed as percentages or ratios, and continuous data were expressed as means 6 SDs, unless otherwise stated. All analyses were performed with Prism 7.0 software (Prism Software, Irvine, Calif). Comparisons of categorical data, such as sex or age groupings, were performed by using x2 or Fisher exact tests. Statistical significance for continuous data was determined by using the Student t test or Mann-Whitney test for nonparametric data, such as age at diagnosis. In experiments comparing multiple experimental groups, analysis was performed by using 1-way ANOVA with the Bonferroni or Holm-Sidak posttest. Significance was considered for a P value of .05 or less.
RESULTS
Increased incidence of EoE in male subjects
To determine the prevalence of EoE in male and female subjects in our local cohort, we examined the number of patients with a diagnosis of EoE cared for at the CCED. A retrospective review of the electronic medical record database from January 2000 through January 2017 found that 2013 patients were given a diagnosis of EoE (n = 2013; male subject, n = 1485; female subjects, n = 528; see Fig E1, A, in this article’s Online Repository at www.jacionline.org). The male/female ratio for all patients of all ages was 2.85 (see Fig E1, B).
To compare the effect of age at diagnosis between sexes in pediatric patients, the cohort was stratified by age at initial diagnosis to groups of less than 5 years, 5 to 9 years, 10 to 14 years, and 15 to 19 years (see Fig E1, C and D). Examination of the male/female ratio revealed that male predominance was most pronounced in patients receiving a diagnosis at less than 5 years of age, with a male/female ratio of 3.8 (37% of cohort, n = 666; male subjects, n = 527; female subjects, n = 139; see Fig E1, C and D). The male sex predominance decreased as age of initial diagnosis increased, with a male/female ratio of 2.95 in patients aged 5 to 10 years (23.5% of the cohort, n = 423; male subjects, n = 316; female subjects, n = 107), 2.93 for patients aged 10 to 14 years (21.0% of cohort, n = 377; male subjects, n = 281; female subjects, n = 96), and 2.31 for patients aged 15 to 19 years (11.2% of the cohort, n = 167; male subjects, n = 90; female subjects, n = 77; see Fig E1, C and D). The male/female ratio of patients receiving a diagnosis at less than 5 years was statistically different than the male/female ratio of patients aged 15 to 19 years (P < .01). We observed no statistically significant differences between other age divisions. When stratified by sex, the average age of diagnosis for male subject was significantly lower than that of female subjects (8.7 ± 7.6 vs 11.9 ± 10.8 years, male and female subjects, respectively [mean ± SD]; P < .001). These analyses demonstrates the increased incidence of EoE in male patients in our cohort (male/female ratio, 3:1 in patients younger than 20 years) and that the male sex predominance was more pronounced in children less than 5 years of age.
Altered expression of estrogen-responsive genes in patients with EoE
To begin to determine the contribution of sex hormones to the increased male predominance in EoE, we examined expression of the sex hormone–responsive genes in esophageal biopsy samples from 4 female and 6 male patients with EoE and 3 of both healthy female and male control subjects by using RNA-seq data from a previously described cohort of patients (see Table E1 in this article’s Online Repository at www.jacionline.org).35 The estrogen-responsive genes were derived from the estrogen-responsive gene reference database (http://datam.i2r.a-star.edu.sg/ergdbV2/),44 which consists of 1069 validated estrogen-responsive human genes. Although this gene list does not contain all estrogen-responsive genes,45,46 it serves as an initial screen to identify the potential involvement of estrogen-responsive gene expression in the pathogenesis of EoE. Of the 1069 estrogen-responsive genes in the published reference database, 1057 (98.8%) were present in the University of California, Santa Cruz, gene model–based RNA-seq data set. Analysis showed that 29.9% (317/1057 genes, P < .05, FC > 1.5) of estrogen-responsive genes were significantly dysregulated (up or down) in male or female patients with EoE compared with control subjects (Fig 1). This is significantly different than the 5% of genes (n = 53 genes) anticipated to be dysregulated by chance (P <.01, 104 permutations of 30,000 genes). Of the 317 dysregulated genes, 58.7% (n = 186 genes) were upregulated in patients with EoE compared with control subjects, whereas 41.3% of genes (n = 131) were downregulated in patients with EoE (Fig 1, A). Fifty-eight of these genes were altered specifically in female subjects with EoE (n = 43 upregulated and n = 15 downregulated), and 44 were altered in male subjects with EoE (n = 13 upregulated and n = 31 downregulated, Table I). When examining the entire dysregulated transcriptome of patients with EoE compared with control subjects, 8% of all dysregulated genes were predicted to be estrogen responsive (172/2199 genes, P < .01, FC > 2; see Table E2 in this article’s Online Repository at www.jacionline.org).
FIG 1.

Estrogen-responsive genes in EoE. A, RNA-seq expression analysis of genes included in the estrogen-responsive gene database (n = 317/1057 genes, P < .05, FC > 1.5, 104 permutations of the 30,000-gene data set) from esophageal biopsy specimens of 10 patients with EoE (numbers 1–10; female patients, n = 4; male patients, n = 6) and 6 control subjects (numbers 11–16; female subjects, n = 3; male subjects, n = 3). Upregulated genes are represented in yellow, and downregulated genes are represented in blue. The magnitude of expression is represented by darkness of the color. F, Female; M, male. B, String protein-protein interaction network analysis of the 172 differentially expressed estrogen response genes in patients with EoE. Network nodes represent proteins encoded by the genes. Functional partner predictions are based on available experimental data, databases, text mining, and homology. Cluster nodes 1 to 3 are defined by using identified gene pathway analyses of the associated genes. C, Heat map analysis of expression of 10 estrogen-responsive genes (determined by the estrogen-responsive gene database) included in the EDP (94 genes) isolated from esophageal biopsy specimens of 6 control subjects (numbers 1–3 and 8–11; female subjects, n = 3; male subjects, n = 4) and 10 patients with EoE (patients 4–7 and 12–17; female patients, n = 4; male patients, n = 6). The EDP expression heat maps were generated by using R software’s heatmap.2 defaults, as described in the Methods section. The 7 genes are significantly expressed (log FC > 1.5, P < .05) in patients with EoE compared with control subjects. Differences in expression (log FC) of male patients with EoE and control subjects and female patients with EoE and control subjects are also shown. Upregulated genes are represented in yellow, and downregulated genes are representred in blue. The magnitude of expression is represented by darkness of the color.
TABLE I.
Gene ontology of estrogen-responsive genes in patients with EoE
| Group | Ontology | Activity | No. of genes | P value | Specific genes |
|---|---|---|---|---|---|
| Upregulated (male and female patients with EoE [n = 130]) | Biological process | Inflammatory response | 14/714 | 1.75E-07 | CYBB, SAA1, THBS1, SOCS3, CD44, ICAM1, CCL5, IFII6, PBK, PTGES, ITGAV, STAB1, CXCL1, BCL6 |
| Biological process | Regulation of cell cycle | 21/1012 | 2.26E-11 | PKMYT1, PMP22, FOXM1, NUSAP1, CKS2, PTTG1, THBS1, SFRP1 PTPRK, ANLN, BE2C, TPX2, BAK1, BIRC5, TACC3, CDC20, TOP2A, KIF11, KIFC1, CCNB1, AURKB | |
| Molecular function | Kinase binding | 12/693 | 5.29E-06 | TOP2A, ITGAV, PRC1, PKP2, CD44, TPX2, KIF20A, PTPRK, TRIB2, FOXM1 | |
| Downregulated (male and female patients with EoE [n = 85]) | Biological process | Response to hormone | 18/1035 | 9.15E-06 | CDKN1A, NCOA2, ANXA2, FOSL2, NUCB2, BCL2, TRIM24, ME1, RXRA, AREG, CITED2, GJA1, TIMP3, PIK3R1, DHCR24, PRKCE, BAIAP2L2, MARCKS |
| Biological process | Regulation of membrane permeability | 5/76 | 5.55E-05 | BCL2, BCL2L1, PPIF, IER3, PMAIP1 | |
| Biological process | Reponses to wounding | 16/973 | 6.00E-05 | KRT6B, ACHE, ANXA2, ACTG1, BCL2, IL1RL2, RXRA, ZFP36, S100A9, GJA1, TIMP3, IER3, PIK3R1, PRKCE, PARD3, MVK | |
| Upregulated (male patients with EoE [n = 13]) | Biological process | Steroid hormone receptor activity | 2/59 | 3.43E-04 | RARA, NR2C1 |
| Molecular function | Positive regulation of signal transduction | 7/1488 | 6.03E-07 | LGALS1, SLA, DUSP6, IGF1R, RARA, NR2C1, CD8A | |
| Downregulated (male patients with EoE [n = 31]) | Biological process | Gland development | 5/153 | 1.56E-06 | SOX9, ZNF703, CCND1, B4GALT1, TGFA |
| Biological process | Epithelial cell proliferation | 5/394 | 1.49E-04 | SOX9, ZNF703, CCND1, B4GALT1, TGFA | |
| Upregulated (female patients with EoE [n = 43]) | Biological process | DNA replication | 8/346 | 1.46E-07 | RAD51, CDC6, RFC3, BRCA1, POLA2, RFC5, RPA3, FEN1 |
| Biological process | Serine/threonine/tyrosine kinase activity | 2/49 | 3.53E-03 | MAP2K6, RPS6KA1 | |
| Downregulated (female patients with EoE [n = 15]) | Cellular component | Adherens junctions | 4/484 | 1.69E-04 | PTPN12, PGM5, EFNB2, NDRG1 |
| Pathways | NF-κB signaling pathway | 4/205 | 2.13E-06 | BLNK, IL1R1, CXCL2, NFKBIA |
Gene ontology of upregulated and downregulated gene expression from RNA-seq analysis of genes included in the estrogen-responsive gene database (total n = 317, FC > 1.5, P < .05) in patients with EoE (female patients, n = 4; male patients, n = 6) compared with control subjects (female subjects, n = 3; male subjects, n = 4) is shown.
NF-κB, Nuclear factor κB.
String protein-protein interaction network analysis47 of the 172 differentially expressed estrogen response genes in patients with EoE revealed 1 major cluster/node for interactions of genes associated with regulation of DNA synthesis (CDC45, RRM1, and RRM2), cell-cycle checkpoints (CCNB1 and CCNG2), and mitosis progression (TPX2, AURKA, MAD2L1, BUB1B, CDC20, and KIF2C; Fig 1, B). This major cell-cycle regulatory network was enjoined to a cluster of innate immune genes (cluster node 2) involved in chemoattraction (CXCL1, CCR1, ITGAE, and ICAM1), innate immune recognition (TLR2 and IRF9), and cell activation (CD44 and CD69; Fig 1, B). A confirmatory gene expression cohort of 10 patients with EoE and 10 control subjects showed that approximately 8% of dysregulated genes in patients with EoE were estrogen responsive (Gene Expression Omnibus deposition GSE113341).
Gene ontology analysis of the estrogen-responsive genes altered in male and female patients with EoE (Table I) demonstrated numerous altered pathways in several broad categories, including cell-cycle regulation (PKMYT1, PMP22, FOXM1, NUSAP1, CKS2, PTTG1, THBS1, SFRP1, PTPRK, ANLN, BE2C, TPX2, BAK1, BIRC5, TACC3, CDC20, TOP2A, KIF11, KIFC1, CCNB1, and AURKB), kinase binding (TOP2A, ITGAV, PRC1, PKP2, CD44, TPX2, KIF20A, PTPRK, TRIB2, and FOXM1), and inflammatory processes (CYBB, SAA1, THBS1, SOCS3, CD44, ICAM1, CCL5, IFII6, PBK, PTGES, ITGAV, STAB1, CXCL1, and BCL6). Both male and female patients with EoE compared with control subjects had decreased expression of estrogen-responsive genes related to regulation of membrane permeability (BCL2, BCL2L1, PPIF, IER3, and PMAIP1), response to hormones (CDKN1A, NCOA2, ANXA2, FOSL2, NUCB2, BCL2, TRIM24, ME1, RXRA, AREG, CITED2, GJA1, TIMP3, PIK3R1, DHCR24, PRKCE, BAIAP2L2, and MARCKS), and response to wounding (KRT6B, ACHE, ANXA2, ACTG1, BCL2, IL1RL2, RXRA, ZFP36, S100A9, GJA1, TIMP3, IER3, PIK3R1, PRKCE, PARD3, and MVK; P <.05; Table I). Female patients with EoE had increased expression of DNA-binding and repair genes (RAD51, CDC6, RFC3, BRCA1, POLA2, RFC5, RPA3, and FEN1) and decreased expression of genes related to cellular adherence and cell-cell junctions (PTPN12, PGM5, EFNB2, and NDRG1) and nuclear factor kB signaling (BLNK, IL1R1, CXCL2, and NFKBIA) compared with male patients (P < .05). Male patients with EoE had increased expression of genes related to steroid hormone receptor activity (RARA and NR2C1) and signal transduction (LGALS1, SLA, DUSP6, IGF1R, RARA, NR2C1, and CD8A) and decreased expression of a set of genes related to epithelial cell proliferation (SOX9, ZNF703, CCND1, B4GALT1, and TGFA) compared with female patients (P <.05, Table I). These findings suggest numerous genes altered in patients with active EoE are estrogen responsive, including gene groups related to membrane permeability and cell-cell junctions and that there is differential gene dysregulation between male and female subjects that could contribute to the observed male predominance of EoE.
Eosinophilic diagnostic panel and estrogen-responsive genes in patients with EoE
Recently, the CCED group developed an eosinophilic diagnostic panel (EDP) consisting of 94 dysregulated genes in the EoE transcriptome and 2 housekeeping genes (GAPDH and 18S).48 The EDP has shown utility as a diagnostic and disease activity surveillance test and could serve as an indicator of how changes in estrogen-responsive genes might correlate with disease activity.48,49 We found that 10 (10.6%) of the 94 EDP genes were part of the estrogen-responsive gene database (CA2, CFB, CITED2, CRISP3, CXCL1, EML1, FKBP5, KRT23, PHLDB2, and SLC16A6).
We reviewed the RNA-seq analysis of biopsy specimens from the male and female patients with EoE versus the control cohort described above to identify overlap with the EDP gene list and found that 7 of the 10 EDP genes (CA2, CFB, CITED2, CRISP3, CXCL1, EML1, and SLC16A6) were significantly dysregulated (either upregulated or downregulated) between the EoE and control populations (Fig 1, C, and see Table E3 in this article’s Online Repository at www.jacionline.org). Interestingly, 2 of these estrogen-responsive genes (CXCL1 and CRISP3) were also some of the most altered genes in the patients with EoE, with FCs of 56.9 (P < .001) and −621.5 (P < .001), respectively.
Esophageal gene expression in healthy female versus male subjects
Male and female patients with EoE had differences in esophageal biopsy gene expression profiles (Table I). To evaluate what differences might be sex specific, we compared esophageal biopsy gene expression between healthy female and male control subjects from the same cohort described above (female subjects, n = 3; male subjects n = 3; not age controlled). We found 128 genes with FCs of greater than 3 (upregulated, n = 66; downregulated, n = 62; 104 permutations) that differed between female and male control subjects, but none reached statistical significance. The most altered genes were sex chromosome specific (XIST: FC = +113, female vs male; RPS4Y1: FC = −162, female vs male). Gene ontology analysis identified molecular function genes related to glutathione transferase activity (GSTT2B, GSTM1, GSTM3, and GSTT2). Two of the differently expressed genes were part of the EDP (GLDC: FC = −5.84, female vs male; UPK1A: FC = +3.36, female vs male). GLDC is glycine decarboxylase, part of the mitochondrial glycine degradation system. UPK1A is a transmembrane protein that modulates membrane permeability and is also one of the most downregulated genes included in the EDP. Although the +3 FC difference in expression did not reach statistical significance (P = .1), the degree of dysregulation of UPK1A in patients with EoE merits further investigation.
Patients with EoE have increased expression of ESRs compared with healthy control subjects
Given these differences in expression of estrogen-responsive genes in patients with EoE, we next examined expression of the hormone receptors ESR1 and ESR2 in esophageal tissue from postpubertal (age, 15.2–17.9 years) male and female patients with EoE and control subjects (Fig 2). RNA expression analyses revealed that both ESR1 and ESR2 were expressed at low levels in both male and female control subjects and that levels were not significantly different between the sexes (Fig 2, A-D). Levels of expression of both ESR1 and ESR2 were increased in both male and female patients with EoE compared with control subjects; however, gene expression levels were low, with RPKM values of less than 1 for ESR1 and ESR2 in esophageal tissue (Fig 2, A-D).
FIG 2.

ESR1 and ESR2 gene expression. A and B, RNA-seq gene expression represented by RPKM values of ESR1 (Fig 2, A) and ESR2 (Fig 2, B) in esophageal biopsy specimens from healthy control subjects (female subjects, n = 3; male subjects, n = 3) and patients with EoE (female patients, n = 4; male patients, n = 6). ESR1 and ESR2 expression was increased in patients with EoE compared with that in control subjects. C and D, There was no difference in expression between male and female subjects in the EoE or control groups. The RPKM value was less than 1 for ESR1 and ESR2. E and F, ESR1 and ESR2 immunofluorescence staining of esophageal biopsy specimens from male and female patients with EoE and healthy control subjects: results of immunofluorescence analyses for ESR1 and ESR2 in control subjects (no eosinophils were present in the biopsy specimen) and a representative active EoE biopsy specimen (EoE; maximum of 62 [male] and 90 [female] eosinophils per high-power field) are shown. The bar is 20 μm for ×20 images (left panels) and 50 μm for ×40 images (right panels). A total of 2 control and 4 active biopsy specimens from patients with EoE were stained. The white line indicates a single-cell basal layer. The white arrow indicates a positive staining cell within the single-cell basal layer. The yellow arrow indicates positive staining within the basal proliferative zone. DAPI, 4ʹ,6-Diamidino-2-phenylindole; F, female; M, male.
Examination of the cellular distribution and localization of the ESRs in primary esophageal biopsy specimens from male and female control subjects and patients with EoE by using immunofluorescence revealed expression of ESR1 and ESR2 at baseline (Fig 2, E and F). Notably, expression of ESR1 and ESR2 appeared to be restricted to the basal layer of the esophageal epithelium (Fig 2, E and F, a and c, white arrows). We did not observe differences in levels of expression of ESR1 and ESR2 between male and female control subjects (Fig 2, E and F, upper and lower left panels). In patients with EoE, we observed an altered expression pattern of ESR1 and ESR2 in both male and female patients with EoE compared with control subjects (Fig 2, E and F). Notably, ESR1 and ESR2 expression was preserved within the basal layer of the esophageal epithelium; however, positive staining was also observed within the basal proliferative zone of the esophageal epithelium (Fig 2, E and F, yellow arrows). We did not observe differences in levels of expression of ESR1 and ESR2 between male and female patients with EoE within the basal layer and basal proliferative zone (Fig 2, E and F, upper and lower left panels and upper and lower right panels).
E2 pretreatment protects against IL-13–induced changes in esophageal barrier dysfunction and remodeling
Given the observed alteration in estrogen-responsive genes in patients with EoE coupled with differential expression of ESRs in esophageal tissues of patients with EoE, we examined the effects of E2 on IL-13–induced esophageal epithelial dysfunction in an in vitro model developed from esophageal epithelial cells (EPC2s) grown at the ALI. We have previously reported that IL-13 stimulation of EPC2-ALI cells induces a gene expression profile resembling that of EoE, with a 22% overlap of dysregulated genes.16 IL-13 also induces esophageal epithelial dysfunction (barrier impairment and proliferation) similar to that observed in patients with EoE.16 We have previously demonstrated that IL-13 stimulation of EPC2-ALI cells leads to changes in esophageal architecture, including DISs, epithelial proliferation, and barrier dysfunction.12 IL-13 stimulation of EPC2 cells significantly decreased TER compared with control values (Fig 3, A), which is indicative of barrier dysfunction (2021 ± 516 vs 1258 ± 334 Ω · cm2, vehicle vs IL-13 [mean ± SD]; n = 10; P < .001). Pretreatment of EPC2 cells with E2 (100 nmol/L) for 24 hours before IL-13 exposure (Fig 3, A) attenuated the IL-13–induced decrease in TER (1291 ± 190 vs 1597 ± 363 Ω · cm2, vehicle plus IL-13 vs E2 plus IL-13 [mean ± SD]; n = 10; P < .05). Treatment of EPC2 cells with E2 alone had no effect on esophageal epithelial barrier function (Fig 3, A). These findings indicate that estrogen mitigates IL-13–induced esophageal epithelial barrier dysfunction.
FIG 3.

TER and cellular architecture of esophageal epithelial cells exposed to IL-13 with or without E2. A, TER of the vehicle (Veh)– or E2-pretreated EPC2-ALI monolayer after IL-13 exposure. B and C, Quantification of DISs (Fig 3, B) and representative hematoxylin and eosin staining (Fig 3, C) of EPC2-ALI multicell layer cultures of the vehicle- or E2-pretreated EPC2-ALI monolayer after IL-13 exposure. D, RNA-seq expression analysis represented by RPKM values of esophageal epithelial cells (EPC2-ALI) exposed to vehicle or IL-13 with or without E2. Upregulated genes are represented in yellow, and downregulated genes are represented in blue. Magnitude of expression is represented by darkness of the color. EPC2-ALI monolayers were pretreated with vehicle (0.01% EtOH) or E2 (100 nmol/L) for 24 hours and subsequently stimulated with vehicle (PBS) or IL-13 (100 ng/mL) in the presence of E2 for 24 hours, and TER measurements were performed by using an 8-chamber EasyMount Ussing Chamber System, as described in the Methods section. C, DIS formation (percentage of total area) was quantitated by using morphometric analyses and expressed as means ± SEMs (n = 3 independent experiments). *P < .05. Fig 3, C, Magnification ×100.
The observed reduction in IL-13–induced esophageal epithelial barrier dysfunction by estrogen led us to examine the effect of estrogen on IL-13–induced esophageal architectural changes. We show that IL-13 exposure led to an overall disruption of cellular architecture, with increased dilation of intercellular spaces and irregularities of apical cellular layers in EPC2-ALI cells compared with vehicle-treated cells (Fig 3, B and C). Notably, these morphologic changes were ameliorated with pretreatment of cells with 100 nmol/L E2 for 24 hours before IL-13 exposure. In fact, the esophageal architecture of the EPC2-ALI cells stimulated with IL-13 in the presence of E2 resembled that of the group receiving vehicle treatment only (Fig 3, B and C). DIS quantification (Fig 3, B) showed that IL-13 induced an increased percentage of DISs compared with vehicle (8.2% ± 4.1% vs 3.9% ± 2.7%, vehicle plus IL-13 vs vehicle along [mean ± SD]; n = 10; P < .05), whereas E2 pretreatment prior to IL-13 exposure attenuated IL-13–induced DISs (8.2% ± 4.1% vs 2.4% ± 2.2%, vehicle plus IL-13 vs E2 plus IL-13 [mean ± SD]; n = 10; P < .05; Fig 3, B). We concluded that E2 promotes a protective effect against IL-13– induced esophageal barrier dysfunction and architectural changes in EPC2-ALI cells.
E2 pretreatment abrogates IL-13–induced activation of the TYK2/STAT6 signaling pathway
To begin to unravel the molecular basis of E2 suppression of IL-13–induced esophageal epithelial architectural remodeling and barrier dysfunction, we examined the gene expression profile of IL-13–stimulated EPC2-ALI cells in the presence and absence of E2 using RNA-seq analysis. Cells exposed to IL-13 for 24 hours had alteration of 246 genes (upregulated, n = 105; downregulated, n = 141; FC > 3), including genes related to epithelial barrier function (CA2), inflammatory response (TNFAIP6 and SOCS1), chemokines (CCL26), protease activity (CAPN14), and ion transport (ANO1; Fig 3, D, and Table II).14,50 Gene ontology demonstrated gene expression related to cytokine signaling, regulation of cell adhesion, and endothelial cell proliferation (Table III). Pretreatment of EPC2-ALI cells with E2 before IL-13 exposure led to an alteration in the IL-13–induced gene expression profile. Moreover, we observed abrogation of some IL-13–induced genes, including decreased expression of ANO1 and SOCS1. There were similar levels of expression of other IL-13–induced genes despite E2 pretreatment, including CCL26 (Table II). E2 pretreatment increased expression of CA2, TNFAIP6, and CAPN14 (Table II). This indicates that E2 treatment before IL-13 exposure diminishes the induction of some, but not all, IL-13–related genes. Interestingly, cells pretreated with E2 and then exposed to IL-13 had alteration in nearly 10 times more genes compared with cells after vehicle than IL-13 treatment alone compared with vehicle (total, n = 1077; upregulated, n = 519; downregulated, n = 588; FC > 3), with gene ontology being related to cell proliferation and keratinocyte differentiation (Table III). E2 treatment alone altered expression of 125 genes (upregulated, n = 42; downregulated, n = 83; FC > 3), as well as enhancement of expression of the desmosome-associated protein DSG1 (FC = 1.73). Gene ontology (Table III) showed E2 treatment alone induced genes related to cellular oxidant detoxification and gene silencing (P < .05). Pretreatment with E2 for 24 hours before IL-13 exposure resulted in differential expression of 100 genes compared with IL-13 treatment alone (upregulated, n = 45; downregulated, n = 55; FC > 3; P < .05), and gene ontology demonstrated genes related to responses to oxidative stress (Table III).
TABLE II.
Gene expression of EoE-related genes in esophageal epithelial cells exposed to IL-13 with or without E2
| Gene | Function | Vehicle + IL-13 at 24 h vs vehicle FC | E2 + IL-13 at 24 h vs E2 FC | E2 + IL-13 at 24 h vs vehicle + IL-13 at 24 h FC |
|---|---|---|---|---|
| ALOX15 | Inflammation process | 2.6 | 2.9 | 1.1 |
| ANO1 | Ion channel | 5.5 | 8.4 | −1.6 |
| ARG1 | Inflammation process | −6.7 | −6.7 | 1.9 |
| CA2† | Epithelium related | 3.1 | 3.1 | 1.6 |
| CCL26 | Chemokine | 3.8 | 4.8 | −1.2 |
| CDA | Pyrimidine salvage | −2.9 | −2.7 | −1.2 |
| CDH26 | Cell adhesion | 2.2 | 2.2 | 1.2 |
| CTSC | Tissue remodeling | 3.9 | 3.2 | 1.3 |
| FLG | Epithelium related | −3.3 | −2.9 | −1.2 |
| GCNT3 | Epithelium related | 5.3 | 6.0 | 1.0 |
| H19 | Steroid-responding element | −2.2 | −2.1 | −1.8 |
| IGFL1 | Proliferation/growth | −3.5 | −4.0 | 1.2 |
| PMCH | Inflammation process | 2.5 | 3.8 | 1.5 |
| PNLIPRP3 | Lipoprotein metabolism | −2.3 | −2.4 | 1.8 |
| SLC16A6† | Ion channel | −2.8 | −3.2 | 1.4 |
| SPINK7 | Epithelium related | 1.3 | 1.6 | 2.0 |
| TNFAIP6 | Inflammation process | 36.6 | 59.1 | 1.6 |
| CAPN14* | Endopeptidase activity | 120.0 | 128.1 | 1.1 |
| SLC9A3* | Ion channel | 1.1 | 1.2 | −2.2 |
| SOCS1* | Inflammation response | 6.9 | 9.9 | −2.0 |
Gene expression of EoE-related IL-13–induced genes (column 1) and gene function (column 2) from RNA-seq of EPC2-ALI cells treated with E2 (100 nmol/L) or vehicle (0.01% EtOH) for 48 hours, with exposure to IL-13 (100 ng/mL) at 24 hours is shown. Included are genes with FC expression of greater than 2 in either vehicle compared with vehicle plus IL-13 (column 3) or E2 pretreatment plus IL-13 at 24 hours compared with E2 alone (column 4) or vehicle plus IL-13 at 24 hours (column 5).
Known IL-13–induced genes not included in EDP.
EDP gene also included in the estrogen-responsive gene database.
TABLE III.
Gene ontology profile of esophageal epithelial cells exposed to IL-13 with or without E2
| Group | Ontology | Activity | No. of genes | P value | Specific genes |
|---|---|---|---|---|---|
| Vehicle vs vehicle plus IL-13 (n = 246) | Biological process | Regulation of cell adhesion | 17/709 | 9.38E-05 | CYP1B1, C1QTNF1, COL1A1, DPP4, SFRP1, TNFSF18, TNC, C13orf15, MAP3K14, ST6GAL1, APOD, TGM2, RASGRP1, THBS1, EGR3, CD59, B4GALNT2 |
| Biological process | Regulation of endothelial cell proliferation | 6/103 | 2.27E-04 | CCL26, CCL24, C13orf15, ARG1, THBS1, EGR3 | |
| Biological process | Response to cytokine | 18/825 | 1.89E-04 | CHI3L1, CCL26, IL22RA1, CISH, MME, CCL24, PID1, CARD14, COL1A1, SFRP1, TNFSF18, MAP3K14, BIRC3, IFI35, ARG1, ENTPD2, OASL, SOCS1 | |
| Vehicle vs E2 (n = 125) | Biological process | Cellular oxidant detoxification | 4/87 | 1.61E-04 | SZT2, HBA1, HBA2, HBB |
| Biological process | Gene silencing | 6/257 | 1.52E-04 | KMT2B, POLR2A, TERT, KMT2D, HIST1H4H, MSL3P1 | |
| Vehicle + IL-13 vs E2 + IL-13 (n = 100) | Biological process | Response to oxidative stress | 4/440 | 4.32E-04 | KLF2, HBA1, HBA2, HBB |
| Cellular component | Innate immune response in mucosa | 2/24 | 1.86E-04 | HIST1H2BJ, HIST1H2BC | |
| Vehicle vs E2 plus IL-13 (n = 1107) | Biological process | Mitotic cell cycle process | 96/932 | 3.39E-11 | NUSAP1, KIF18A, MIS18BP1, SPC25, MCM6, CDC7, CDC45, FZR1, ANAPC10, NDC80, ANXA1, RGCC, BIRC3, STIL, SMC2, NUP35, NDC1, HAUS1, MARK4, REEP4, PLK4, TJP3, PBK, NEDD1, BID, NCAPG, DBF4, BUB1, BUB1B, NPM1, CALM2, NUMA1, TERT, ZWINT, CCNB1, ITGB3BP, ORC4, ORC5, CCNE2, BRD4, MASTL, CDK1, CDC6, CDC34, TOP2A, PCNA, CDK7, CDKN3, PTTG1, CENPC, CENPE, ORC3, CETN2, CETN3, CHEK1, HMMR, TTK, CKS2, ANLN, PIM1, NUP37, ASPM, DSCC1, HSPA2, PLK3, HSP90AA1, TOM1L2, VRK1, KIF23, POLE2, NABP1, EPGN, KIF20B, PRIM1, TNKS1BP1, TCF7L1, KNTC1, ERCC6L, PSMA2, PSMA3, PSMA4, DLGAP5, PSMC6, NUF2, SPDL1, KNL1, ZWILCH, NUP107, RBBP8, RPS27L, CEP55, FANCI, CKAP2, SKA3, RPA3, MAD2L1 |
| Biological process | Keratinocyte differentiation | 19/140 | 9.55E-05 | EVPL, ALOX15B, FLG, FOXN1, ANXA1, TMEM79, ADAM9, CASP3, LCE1A, TGM1, C1orf68, CDSN, CASP14, OVOL2, PPL, CSTA, DNASE1L2, KRT10, LOR | |
| Biological process | Skin development | 33/280 | 7.14E-06 | EVPL, ALOX12B, ALOX15B, FLG, FOXN1, ANXA1, TMEM79, GRHL3, KRT27, ADAM9, SLC27A4, CASP3, LCE1A, TGM1, C1orf68, CDSN, CASP14, OVOL2, COL1A1, WNT7A, COL5A3, CLDN4, PPARD, PPL, IGFBP5, CSTA, TCF7L1, PRSS8, JUP, DNASE1L2, KRT10, LOR, ALOXE3 |
Column 1 is a comparison between the vehicle (0.01% EtOH) or E2 (100 nmol/L E2) treatment groups for 48 hours with or without addition of IL-13 (100 ng/mL) at 24 hours. Columns 2 and 3 represent gene ontology grouping and associated biological or cellular activity. Column 4 is the number of genes dysregulated per ontologic grouping. Column 5 is the P value of an ontologic grouping association using the hypergeometric probability mass function. Column 6 represents individual genes from the ontologic grouping.
The EDP-derived EoE transcriptome has been shown to correlate with clinical EoE disease activity,49 we therefore next examined for changes in EDP-related genes in our RNAseq analyses of IL-13-stimulated EPC2-ALI cells in the presence and absence of E2 to provide additional insight into the effects of E2 on genes related to the EoE transcriptome. We found 17 of the 94 EDP-related genes were altered, including ALOX15, ANO1, ARG1, CA2, CCL26, CDA, CDH26, CTSC, FLG, GCNT3, H19, IGFL1, MSRB3, PMCH, PNLIPRP3, SLC16A6, and TNFAIP6 (FC > 2; P < .05). Of the 17 genes, 2 were E2-responsive genes (CA2 and SLC16A6). The effect of E2 pretreatment before IL-13 exposure varied, with some genes having decreased expression, whereas no effect was observed in others (Table II).
The IL-13–induced EoE gene transcriptome is predominantly STAT6 dependent.51,52 Given the observed effect of E2 on IL-13– induced genes, we hypothesized that E2-protective effects occurred through alteration of IL-13–induced STAT6 signaling. To test this, we examined IL-13–induced STAT6 phosphorylation in EPC2-ALI cells using Western blot analyses. We show that IL-13 stimulation of EPC2-ALI cells induced STAT6 phosphorylation (Fig 4, A). Notably, IL-13 stimulation led to increased levels of pSTAT6 within 5 minutes, and maximal pSTAT6 was observed within 30 minutes (Fig 4, A). Pretreatment with E2 decreased IL-13–induced phosphorylation of STAT6 in EPC2 cells compared with IL-13 treatment alone at 30 minutes (FC = −0.495; n = 3; P < .05; Fig 4, A). Importantly, we observed no effect of E2 on total STAT6 levels, suggesting that E2 suppressed STAT6 phosphorylation and activation (Fig 4, A, and see Fig E2 in this article’s Online Repository at www.jacionline.org). Consistent with this observation, we observed a reduced expression of IL-132induced genes in EPC2-ALI cells after E2 exposure, including CCL26 (eotaxin-3; IL-13 induction of CCL26: FC = +4.0; E2 plus IL-13: FC = −1.0) and SOCS1 (IL-13 induction of SOCS1: FC = +7.0; E2 plus IL-13, FC = 22.0).53 The reduction in IL-13 signaling was not attributed to reduced receptor expression because our EPC2-ALI cell RNA-seq analyses did not reveal any significant effect of E2 on the mRNA expression of the IL-13 receptor subunits (IL-4Rα, IL-13Rα1, or IL-13Rα2; see Fig E3 in this article’s Online Repository at www.jacionline.org).
FIG 4.

TYK2 and STAT6 phosphorylation after exposure to IL-13 with or without E2 in esophageal epithelial cells. A, Representative Western blot of EPC2 lysate for phosphorylated STAT6 (P-STAT6) and STAT6 at 0, 5, 30, and 60 minutes after IL-13 (100 ng/mL) exposure with or without 24 hours of pretreatment with E2 (100 nmol/L) or vehicle (0.01% EtOH) analyzed by means of immunoblotting with anti-STAT6, anti–P-STAT6, and anti-GAPDH. Quantitative expression of P-STAT6 levels corrected to the housekeeping gene (GAPDH; n = 3). *P < .05, Student t test. B, TYK2 phosphorylation after exposure to IL-13 with or without E2 in esophageal epithelial cells. Representative Western blot of EPC2-ALI cell lysate for phosphorylated TYK2 (P-TYK2) and TYK2 at 0, 5, 30, and 60 minutes after IL-13 (100 ng/mL) exposure with or without 24 hours of pretreatment with E2 (100 nmol/L) or vehicle (0.01% EtOH). Analysis was done by means of immunoblotting with anti-TYK2, anti–P-TYK2, and anti-actin. Quantitative expression of P-TYK2 levels corrected to total TYK2 levels (n = 3). *P < .05, Student t test.
IL-13 induction of STAT6-dependent pathways requires activation of receptor-associated tyrosine kinases, including JAK2 and TYK2.19,23 TYK2 specifically has been implicated with IL-13–induced activation of STAT6.19 To determine whether E2 inhibits the IL-13–STAT6 pathway through TYK2, we examined the level of TYK2 phosphorylation in EPC2-ALI cells after E2 exposure. IL-13 treatment of EPC2-ALI cells induced a 7.55-fold increase in the actin-corrected ratio of pTYK2 to total TYK2 at 5 minutes after exposure to IL-13 (0.017 ± 0.015 vs 0.128 ± 0.05, vehicle vs vehicle plus IL-13 at 5 minutes [mean ± SD]; n = 3; P < .05; Fig 4, B). Pretreatment with E2 before IL-13 exposure resulted in only an FC of 1.4 in pTYK2/TYK2 at 5 minutes after IL-13 exposure (0.017 ± 0.015 vs 0.024 ± 0.02, vehicle vs E2 plus IL-13 at 5 minutes; n = 3; not significant). Levels of pTYK2 in the E2-pretreated group were decreased after 5 minutes of exposure to IL-13 compared with vehicle pretreatment (0.128 ± 0.05 vs 0.024 ± 0.02 [mean ± SD]; n = 3; P < .05). This decreased pTYK2 level compared with the total TYK2 level in E2-pretreated cells indicates E2-mediated suppression of TYK2 phosphorylation and activation.
E2-mediated abrogation of IL-13–induced changes in esophageal barrier dysfunction is ESR2 dependent
To determine the requirement of ESR1 or ESR2 in E2-mediated abrogation of IL-13–induced changes in esophageal barrier dysfunction, we used the highly selective ESR1 and ESR2 receptor antagonists MMP and PHTPP.54–58 Recent studies have demonstrated that antagonism of the ESR pathway inhibits esophageal epithelial proliferation54; therefore we generated mature EPC2-ALI monolayers and then pretreated the monolayers with MMP or PHTPP for 24 hours and subsequently examined the effect of E2 on IL-13–induced esophageal epithelial barrier dysfunction. As anticipated, IL-13 stimulation of EPC2s significantly decreased TER compared with the vehicle control (Fig 5), and pretreatment of EPC2-ALI cells with E2 inhibited the IL-13–mediated response (Fig 5). Pretreatment of EPC2 cells with the ESR1 antagonist MMP had no effect on E2-mediated inhibition; however, exposure of EPC2 cells to PHTPP released the E2-mediated inhibition of IL-13–induced esophageal epithelial barrier dysfunction (Fig 5). These findings indicate that estrogen mitigates IL-13–induced esophageal epithelial barrier dysfunction through ESR2.
FIG 5.
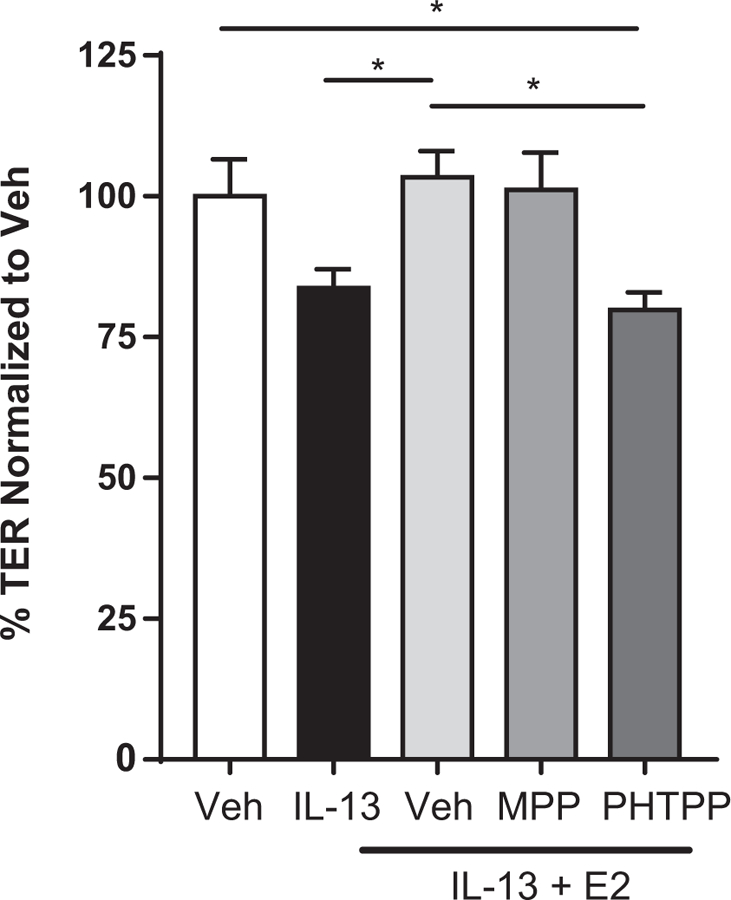
E2-mediated abrogation of IL-13–induced changes in esophageal barrier dysfunction is ESR2-dependent. Mature EPC2-ALI cells (>1500 Ω · cm2) were treated with either vehicle (0.1% dimethyl sulfoxide) or MPP (100 pmol/L) or PHTPP (10 μmol/L) 30 minutes before exposure to E2 (100 nmol/L). Twenty-four hours later, EPC2-ALI cells were stimulated with IL-13 (100 ng/mL) in the presence of vehicle, MPP, or PHTPP and E2 for 24 hours, and TER measurements were performed, as described in the Methods section. Data are represented as averages ± SEMs of 6 to 13 individual cultures per group. *P < .05 compared with vehicle plus IL-13 plus E2, multigroup ANOVA analyses and Holm-Sidak posttest.
DISCUSSION
EoE is more frequently diagnosed in male than female subjects, and the underlying cause of increased risk in male subjects has not been fully delineated. Here we confirmed the male predominance in EoE in a single-center pediatric cohort and also confirmed that the male predominance is most pronounced in patients given a diagnosis at less than 5 years of age. We reveal that that esophageal epithelium expresses both ESR1 and ESR2 and that 29.9% of the genes dysregulated in patients with EoE are estrogen-responsive genes involved in multiple pathways, including inflammation, membrane permeability, and cell-cell junctions. Mechanistic studies revealed that E2 attenuated IL-13–induced esophageal epithelial barrier dysfunction and architectural remodeling and that suppression of IL-13–induced esophageal dysfunction by E2 was associated with decreased IL-13–induced TYK2/STAT6 phosphorylation and activation and was ESR2-dependent. Based on our findings, we propose that estrogen hormone signaling confers a protective effect against the development of EoE.
The approximate 3:1 male predominance in patients with EoE observed in our CCED cohort is consistent with previous epidemiologic studies from around the world.7–9 Analyses of clinical registries from 5 tertiary care institutions in the United States have revealed a 72% male EoE predominance in the United States.4 Consistent with this, van Rhijn et al3 reported a 74% male predominance in an adult and pediatric European cohort, and a systematic review of EoE studies performed in Asian countries reported a 73% male predominance among patients with EoE of all ages.5 Interestingly, we show that the increased male/female ratio in patients with EoE was most pronounced in children given a diagnosis at less than 5 years of age, with 3.8 male subjects for every female subject. Consistent with this, previous studies have reported that male subjects are more likely than female subjects to receive a diagnosis as children of a patient with EoE (48% of male subjects vs 36% of female subjects received a diagnosis before age 18 years of age)7 and that male subjects received a diagnosis at an average younger age than female subjects (25 ± 19 vs 29 ± 20 years).
There is anecdotal clinical evidence supporting a role for sex hormones in the development of EoE or atopic diseases. For example, during infancy and childhood, asthma, atopic dermatitis (AD), and allergic rhinitis are more common in male than female subjects59; however, a female predominance develops in patients with asthma and AD around the age of puberty and continues throughout adulthood until menopause.60 Furthermore, male patients with AD had lower levels of circulating estradiol than healthy control subjects.61 We propose that female subjects can be protected from EoE or atopic disease through circulating estrogens. Estrogen levels are greater on average in female than male subjects during all Tanner stages of development, including infants and prepubescent children.62 However, a direct correlation between total circulating estrogen levels and risk of EoE seems unlikely to completely explain the sex bias in patients with EoE because our cohort showed a decrease in the male predominance for age of initial diagnosis during the teenage years, and clinical experience does not show a corresponding change in symptoms and disease activity in female patients with EoE as estrogen levels increase during puberty. The observations that female subjects have an alleviation of asthma symptoms during life phases associated with higher levels of circulating sex hormones, such as pregnancy, and others can have an exacerbation of symptoms63 further suggests a more complex role for sex hormones possibly related to dose and tissue specificity to confer protective effects in onset and development of allergic diseases.63
Our molecular studies revealed expression of ESR1 and ESR2 in the esophageal epithelium in healthy control subjects and that receptor expression is increased in patients with active EoE. Previous studies have demonstrated moderate positive staining for ESR1 and weak positive staining for ESR2 in normal esophageal tissue.64 We observed comparable levels of expression of both ESR1 and ESR2 in healthy subjects, with increased expression of both in male and female patients with active EoE. Our demonstration of induction of E2 genes in our EPC2-ALI model after E2 exposure indicates the presence of ESR receptors, and active estrogen-signaling pathways in esophageal cells supports the concept that estrogens exert physiologic effects in esophageal tissue through ESR-related signaling. Consistent with this argument, we show that pharmacologic antagonism of ESR2 released the E2-mediated protective response against IL-13–mediated barrier dysfunction. ESR2 has previously been reported to play a pivotal role in E2-mediated enhancement of intestinal epithelial barrier function. Although the molecular basis of ESR2-mediated protection of intestinal epithelial barrier properties is not fully elucidated, the E2-ESR2 axis is thought to stimulate upregulation of the tight and adherence junctional proteins including occludin and junctional adhesion molecule (JAM)-A in intestinal epithelial cells to reinforce the epithelial barrier.33 Interestingly, experimental analyses have identified the existence of ESR1 and ESR2 isoforms.65 The biological properties of the ESR1 and ESR2 isoforms are still unclear; however, they have been shown to have differential expression in various tissues and cell lines.65,66 RNA-seq analyses of esophageal biopsy samples revealed increased mRNA expression of ESR2 isoform 4 in patients with EoE compared with control subjects (see Table E4 in this article’s Online Repository at www.jacionline.org). Previous studies have reported that ESR2 isoform 1 is the full functional isoform and that ESR2 isoforms 2 to 5 do not possess intrinsic activity. However, these isoforms can interact with ESR2 isoform 1 and form functional heterodimers that possess altered tissue distribution and ligand binding affinity and functionality.67 Future analyses of ESR2 isoform 1 and 4 interactions in esophageal epithelial barrier function are currently under investigation.
The molecular basis of E2 suppression of IL-13–induced barrier dysfunction appears to relate to dampening of the IL-13–induced TYK2/STAT6 signaling axis. Moreover, we showed that E2 pretreatment suppressed IL-13–induced phosphorylation of TYK2 and STAT6 and subsequent activation of some IL-13–induced genes. RNA-seq analyses revealed that the effects of E2 occurred independently of any changes in IL-13 receptor subunit (IL4RA, IL13RA1, and IL13RA2) mRNA, which suggests the observed loss of IL-13–mediated effects are not a consequence of alterations in receptor expression. Furthermore, the observed comparable levels of total TYK2 between vehicle and E2 exposure suggests that suppression is not related to alteration in total TYK2 protein levels. TYK2, a non–receptor tyrosine-protein kinase, is known to phosphorylate downstream STATs, including STAT6, triggering STAT dimerization, nuclear translocation which leads to DNA binding, and activation of gene expression.68 TYK2 undergoes posttranslational modification to induce positive and negative regulatory effects, including phosphorylation, dephosphorylation, and ubiquitination.69 SOCS proteins are a family of molecules known to be induced by JAK-STAT signaling to act as a negative feedback loop.70 However, we observed no effect of E2 on expression of the SOCS inhibitors of TYK2, such as SOCS1 and SOCS3, in EPC2-ALI cells, suggesting that the E2-mediated suppression is not through upregulation of this counterregulatory mechanism.71,72 TYK2 can also be negatively regulated by phosphatases, such as Src homology 2 domain containing protein tyrosine phosphatase 1 and protein tyrosine phosphatase 1B, which can directly interact with TYK2 and modulate TYK2 signaling functions.73 Analyses of the EPC2-ALI plus E2 gene expression studies did not reveal any obvious alterations in phosphatase pathways, and currently, we are examining the molecular basis of E2 suppression of TYK2 phosphorylation. Given the experimental evidence indicating intricate cross-talk between JAKs and ESRs, the molecular basis of suppression might be through multiple different signaling mechanisms.
We have previously reported that IL-13 stimulation of primary esophageal and EPC-ALI cells induces esophageal barrier dysfunction,12 which is thought to be in part due to decreased expression of intercellular adhesion molecules, such as DSG1. Consistent with this, DSG1 has been shown to be downregulated in patients with EoE compared with control subjects.12 RNA-seq data from our EPC2-ALI model revealed that E2 alone was sufficient to induce esophageal DSG1 expression. Estrogens have been implicated in protection against esophageal epithelial barrier dysfunction through direct induction of major components of cellular tight junctions, including occludin.74–76 Bioinformatics analyses of the dysregulated estrogen-responsive genes in patients with EoE and E2-stimulated EPC-ALI cells identified other estrogen-responsive genes involved in preservation and maintenance of the esophageal barrier. One such gene was UPK1A, which encodes for a transmembrane protein that modulates membrane permeability. UPK1A is one of the most downregulated genes in patients with EoE, with a 38-fold reduction (P < .01) in esophageal expression in patients with EOE compared with control subjects.48 Although UPK1A was not included in the Estrogen Gene Database used in this study, UPK1A has been shown to be an estrogen-responsive gene in the genitourinary system, with a −71-fold decrease in expression in ESR1 knockout mice.77 We observed a +3.36 FC increased expression on UPKA1 in esophageal tissue of healthy female subjects compared with healthy male control subjects. UPK1A is active in esophageal tissue as a tumor suppressor of esophageal squamous cell carcinoma, which, interestingly, is 2:1 more common in male than female subjects78 and is an antagonist to matrix metalloproteinases.78,79 Increased expression of matrix metalloproteinases 2 and 14 have been implicated in patients with EoE.80 Although we did not observe an effect on UPK1A mRNA expression after E2 exposure in our EPC2-ALI model, the difference in expression of this gene between healthy male and female subjects, coupled with the marked dysregulation in patients with EoE compared with control subjects indicative of variable UPK1A expression, and the known connection to estrogen might contribute to male predominance in patients with EoE. Collectively, these data suggest that although E2 can dampen the IL-13−induced TYK2/ STAT6 signaling axis and reduce IL-13−induced barrier dysfunction, some of the protective effects of estrogen might extend beyond just suppression of IL-13/STAT6 signaling to include direct regulation of barrier enhancer gene expression.
In conclusion, we demonstrate the increased incidence of EoE in male subjects and that this increase was associated with dysregulation of estrogen-responsive gene expression, including genes related to inflammation, cell-cell junctions, and the EDP genes CA2, CFB, CITED2, CRISP3, CXCL1, EML1, and SLC16A6. Using an EPC2-ALI model, we showed that E2 exposure abrogated IL-132induced architectural changes of DISs and barrier dysfunction in esophageal epithelial cells. Mechanistically, we show that E2 suppression of IL-13’s effects were associated with decreased phosphorylation of TYK2 and STAT6 and dampened the IL-13 response. These studies suggest that estrogen hormone signaling can play a role in protecting against the development of EoE in female subjects.
Supplementary Material
{kind=link}
{kind=link}
Key messages.
EoE is 3:1 more common in male than female subjects, and the underlying molecular basis for the increased male incidence in patients with EoE is unclear.
Based on demographic, incidence, and RNA-seq analyses of patients with EoE in male and female subjects from single-center pediatric cohort, we revealed significant dysregulation of expression of the ESRs and estrogenresponsive gene network in patients with EoE.
Mechanistic analyses using a differentiated EPC2 culture system (EPC2-ALI) revealed that the sex hormone estrogen protected the esophageal epithelium from IL-13−induced esophageal architectural changes and barrier dysfunction.
Estrogen hormone signaling may help protect against development of EoE in female subjects, and targeting this pathway may be an approach for therapeutic intervention for suppression of esophageal inflammation and development of the EoE phenotype.
Acknowledgments
Supported by Food Allergy Research & Education, the National Institutes of Health (grant nos. AI112626 and DK090119 to S.P.H., T32 DK007727 to J.C.W., and P30 DK078392), the Campaign Urging Research for Eosinophilic Disease (CURED) Foundation; and the Sunshine Charitable Foundation and its supporters, Denise A. Bunning and David G. Bunning and the Mary H Weiser Food Allergy Center.
Abbreviations used
- AD
Atopic dermatitis
- ALI
Air-liquid interface
- CCED
Cincinnati Center for Eosinophilic Disease
- DIS
Dilated intercellular space
- DSG1
Desmoglein 1
- E2
17β-Estradiol
- EDP
Eosinophilic diagnostic panel
- EoE
Eosinophilic esophagitis
- EPC2
Primary human esophageal keratinocyte cell
- ESR
Estrogen receptor
- FC
Fold change
- IL-4R
IL-4 receptor
- IL-13Rα
IL-13 receptor α
- JAK
Janus kinase
- MPP
1,3-Bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride
- NF
Normalization factor
- PHTPP
4-[2-Phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol
- pSTAT6
Phosphorylated STAT6
- pTYK2
Phosphorylated tyrosine kinase 2
- RNA
seq RNA sequencing
- RPKM
Reads per kilobase per million
- SOCS
Suppressor of cytokine signaling
- STAT6
Signal transducer and activator of transcription 6
- TER
Transepithelial resistance
- TYK2
Tyrosine kinase 2
Footnotes
Disclosure of potential conflict of interest: V. Mukkada is a consultant and receives research funding from Shire. M. E. Rothenberg is a consultant for PulmOne, Spoon Guru, ClostraBio, Celgene, Shire, AstraZeneca, GlaxoSmithKline, Allakos, Adare, Regeneron, and Novartis and has an equity interest in the first 4 listed and Immune Pharmaceuticals and receives royalties from reslizumab (Teva Pharmaceuticals) and UpToDate; he is also an inventor of patents owned by Cincinnati Children’s Hospital Medical Center. S. P. Hogan is an inventor of patents owned by Cincinnati Children’s Hospital Medical Center. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Furuta GT, Liacouras CA, Collins MH, Gupta SK, Justinich C, Putnam PE, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007;133:1342–63. [DOI] [PubMed] [Google Scholar]
- 2.Dellon ES, Gonsalves N, Hirano I, Furuta GT, Liacouras CA, Katzka DA, et al. ACG clinical guideline: evidenced based approach to the diagnosis and management of esophageal eosinophilia and eosinophilic esophagitis (EoE). Am J Gastroenterol 2013;108:679–93. [DOI] [PubMed] [Google Scholar]
- 3.van Rhijn BD, Verheij J, Smout AJ, Bredenoord AJ. Rapidly increasing incidence of eosinophilic esophagitis in a large cohort. Neurogastroenterol Motil 2013;25: 47–52.e5. [DOI] [PubMed] [Google Scholar]
- 4.Moawad FJ, Dellon ES, Achem SR, Ljuldjuraj T, Green DJ, Maydonovitch CL, et al. Effects of race and sex on features of eosinophilic esophagitis. Clin Gastroenterol Hepatol 2016;14:23–30. [DOI] [PubMed] [Google Scholar]
- 5.Kinoshita Y, Ishimura N, Oshima N, Ishihara S. Systematic review: eosinophilic esophagitis in Asian countries. World J Gastroenterol 2015;21:8433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishimura N, Shimura S, Jiao D, Mikami H, Okimoto E, Uno G, et al. Clinical features of eosinophilic esophagitis: differences between Asian and Western populations. J Gastroenterol Hepatol 2015;30(suppl 1):71–7. [DOI] [PubMed] [Google Scholar]
- 7.Sperry SL, Woosley JT, Shaheen NJ, Dellon ES. Influence of race and gender on the presentation of eosinophilic esophagitis. Am J Gastroenterol 2012;107: 215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franciosi JP, Tam V, Liacouras CA, Spergel JM. A case-control study of socio-demographic and geographic characteristics of 335 children with eosinophilic esophagitis. Clin Gastroenterol Hepatol 2009;7:415–9. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, Sukocheva OA, Hussey DJ, Watson DI. Estrogen, male dominance and esophageal adenocarcinoma: is there a link? World J Gastroenterol 2012;18: 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu L, Chirala M, Younes M. Expression of estrogen receptor-beta isoforms in Barrett’s metaplasia, dysplasia and esophageal adenocarcinoma. Anticancer Res 2004;24:2919–24. [PubMed] [Google Scholar]
- 11.Abdulnour-Nakhoul SM, Al-Tawil Y, Gyftopoulos AA, Brown KL, Hansen M, Butcher KF, et al. Alterations in junctional proteins, inflammatory mediators and extracellular matrix molecules in eosinophilic esophagitis. Clin Immunol 2013;148:265–78. [DOI] [PubMed] [Google Scholar]
- 12.Sherrill JD, Kc K, Wu D, Djukic Z, Caldwell JM, Stucke EM, et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol 2014;7:718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Mello RJ, Caldwell JM, Azouz NP, Wen T, Sherrill JD, Hogan SP, et al. LRRC31 is induced by IL-13 and regulates kallikrein expression and barrier function in the esophageal epithelium. Mucosal Immunol 2016;9:744–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis BP, Stucke EM, Khorki ME, Litosh VA, Rymer JK, Rochman M, et al. Eosinophilic esophagitis-linked calpain 14 is an IL-13-induced protease that mediates esophageal epithelial barrier impairment. JCI Insight 2016;1:e86355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hebenstreit D, Luft P, Schmiedlechner A, Regl G, Frischauf AM, Aberger F, et al. IL-4 and IL-13 induce SOCS-1 gene expression in A549 cells by three functional STAT6-binding motifs located upstream of the transcription initiation site. J Immunol 2003;171:5901–7. [DOI] [PubMed] [Google Scholar]
- 16.Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, et al. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol 2007;120:1292–300. [DOI] [PubMed] [Google Scholar]
- 17.Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res 2011; 50:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murata T, Puri RK. Comparison of IL-13- and IL-4-induced signaling in EBV-immortalized human B cells. Cell Immunol 1997;175:33–40. [DOI] [PubMed] [Google Scholar]
- 19.Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic Biol Med 2013;54: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 1999;17: 701–38. [DOI] [PubMed] [Google Scholar]
- 21.Hershey GK. IL-13 receptors and signaling pathways: an evolving Web. J Allergy Clin Immunol 2003;111:677–90. [DOI] [PubMed] [Google Scholar]
- 22.Chatila TA. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol Med 2004;10:493–9. [DOI] [PubMed] [Google Scholar]
- 23.Cheng E, Zhang X, Wilson KS, Wang DH, Park JY, Huo X, et al. JAK-STAT6 pathway inhibitors block eotaxin-3 secretion by epithelial cells and fibroblasts from esophageal eosinophilia patients: promising agents to improve inflammation and prevent fibrosis in EoE. PLoS One 2016;11:e0157376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verthelyi D Sex hormones as immunomodulators in health and disease. Int Immunopharmacol 2001;1:983–93. [DOI] [PubMed] [Google Scholar]
- 25.Bereshchenko O, Bruscoli S, Riccardi C. Glucocorticoids, sex hormones, and immunity. Front Immunol 2018;9:1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaillon S, Berthenet K, Garlanda C. Sexual dimorphism in innate immunity. Clin Rev Allergy Immunol 2017[Epub ahead of print]. [DOI] [PubMed]
- 27.Ghosh S, Klein RS. Sex drives dimorphic immune responses to viral infections. J Immunol 2017;198:1782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vrtacnik P, Ostanek B, Mencej-Bedrac S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb) 2014;24:329–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 2006;7:497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev 2007;87: 905–31. [DOI] [PubMed] [Google Scholar]
- 31.Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. N Engl J Med 2002;346:340–52. [DOI] [PubMed] [Google Scholar]
- 32.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiol Rev 2001;81:1535–65. [DOI] [PubMed] [Google Scholar]
- 33.Braniste V, Leveque M, Buisson-Brenac C, Bueno L, Fioramonti J, Houdeau E. Oestradiol decreases colonic permeability through oestrogen receptor beta-mediated up-regulation of occludin and junctional adhesion molecule-A in epithelial cells. J Physiol 2009;587:3317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verdu EF, Deng Y, Bercik P, Collins SM. Modulatory effects of estrogen in two murine models of experimental colitis. Am J Physiol Gastrointest Liver Physiol 2002;283:G27–36. [DOI] [PubMed] [Google Scholar]
- 35.Sherrill JD, Kiran KC, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014;15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu TX, Sherrill JD, Wen T, Plassard AJ, Besse JA, Abonia JP, et al. MicroRNA signature in patients with eosinophilic esophagitis, reversibility with glucocorticoids, and assessment as disease biomarkers. J Allergy Clin Immunol 2012; 129:1064–75.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7:562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods 2011; 8:469–77. [DOI] [PubMed] [Google Scholar]
- 40.Kartashov AV, Barski A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol 2015;16:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res 2003;1:729–38. [PubMed] [Google Scholar]
- 42.Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem 2003;278:1824–30. [DOI] [PubMed] [Google Scholar]
- 43.Zeng C, Vanoni S, Wu D, Caldwell JM, Wheeler JC, Arora K, et al. Solute carrier family 9, subfamily A, member 3 (SLC9A3)/sodium-hydrogen exchanger member 3 (NHE3) dysregulation and dilated intercellular spaces in patients with eosinophilic esophagitis. J Allergy Clin Immunol 2018;142:1843–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang S, Han H, Bajic VB. ERGDB: estrogen responsive genes database. Nucleic Acids Res 2004;32:D533–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ikeda K, Horie-Inoue K, Inoue S. Identification of estrogen-responsive genes based on the DNA binding properties of estrogen receptors using high-throughput sequencing technology. Acta Pharmacol Sin 2015;36:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 2006;38:1289–97. [DOI] [PubMed] [Google Scholar]
- 47.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wen T, Stucke EM, Grotjan TM, Kemme KA, Abonia JP, Putnam PE, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology 2013;145:1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dellon ES, Veerappan R, Selitsky SR, Parker JS, Higgins LL, Beitia R, et al. A gene expression panel is accurate for diagnosis and monitoring treatment of eosinophilic esophagitis in adults. Clin Transl Gastroenterol 2017;8:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kc K, Rothenberg ME, Sherrill JD. In vitro model for studying esophageal epithelial differentiation and allergic inflammatory responses identifies keratin involvement in eosinophilic esophagitis. PLoS One 2015;10:e0127755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neilsen CV, Bryce PJ. Interleukin-13 directly promotes oesophagus production of CCL11 and CCL24 and the migration of eosinophils. Clin Exp Allergy 2010;40: 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003;125:1419–27. [DOI] [PubMed] [Google Scholar]
- 53.Hebenstreit D, Luft P, Schmiedlechner A, Duschl A, Horejs-Hoeck J. SOCS-1 and SOCS-3 inhibit IL-4 and IL-13 induced activation of Eotaxin-3/CCL26 gene expression in HEK293 cells. Mol Immunol 2005;42:295–303. [DOI] [PubMed] [Google Scholar]
- 54.Al-Khyatt W, Tufarelli C, Khan R, Iftikhar SY. Selective oestrogen receptor antagonists inhibit oesophageal cancer cell proliferation in vitro. BMC Cancer 2018;18:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sobrino A, Oviedo PJ, Novella S, Laguna-Fernandez A, Bueno C, Garcia-Perez MA, et al. Estradiol selectively stimulates endothelial prostacyclin production through estrogen receptor-a. J Mol Endocrinol 2010;44:237–46. [DOI] [PubMed] [Google Scholar]
- 56.Kim SC, Boese AC, Moore MH, Cleland RM, Chang L, Delafontaine P, et al. Rapid estrogen receptor-alpha signaling mediated by ERK activation regulates vascular tone in male and ovary-intact female mice. Am J Physiol Heart Circ Physiol 2018;314:H330–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo F, Huang WY, Guo Y, Ruan GY, Peng R, Li XP. 17beta-estradiol lowers triglycerides in adipocytes via estrogen receptor alpha and it may be attenuated by inflammation. Lipids Health Dis 2017;16:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fatima LA, Campello RS, Santos RS, Freitas HS, Frank AP, Machado UF, et al. Estrogen receptor 1 (ESR1) regulates VEGFA in adipose tissue. Sci Rep 2017;7: 16716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Osman M, Hansell AL, Simpson CR, Hollowell J, Helms PJ. Gender-specific presentations for asthma, allergic rhinitis and eczema in primary care. Prim Care Respir J 2007;16:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Postma DS. Gender differences in asthma development and progression. Gend Med 2007;4(suppl B):S133–46. [DOI] [PubMed] [Google Scholar]
- 61.Ebata T, Itamura R, Aizawa H, Niimura M. Serum sex hormone levels in adult patients with atopic dermatitis. J Dermatol 1996;23:603–5. [DOI] [PubMed] [Google Scholar]
- 62.Janfaza M, Sherman TI, Larmore KA, Brown-Dawson J, Klein KO. Estradiol levels and secretory dynamics in normal girls and boys as determined by an ultrasensitive bioassay: a 10 year experience. J Pediatr Endocrinol Metab 2006; 19:901–9. [DOI] [PubMed] [Google Scholar]
- 63.Keselman A, Heller N. Estrogen signaling modulates allergic inflammation and contributes to sex differences in asthma. Front Immunol 2015;6:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong J, Jiang SW, Niu Y, Chen L, Liu S, Ma T, et al. Expression of estrogen receptor alpha and beta in esophageal squamous cell carcinoma. Oncol Rep 2013; 30:2771–6. [DOI] [PubMed] [Google Scholar]
- 65.Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, et al. Cloning and characterization of human estrogen receptor beta isoforms. Biochem Biophys Res Commun 1998;247:75–8. [DOI] [PubMed] [Google Scholar]
- 66.Tong D, Schuster E, Seifert M, Czerwenka K, Leodolte S, Zeillinger R. Expression of estrogen receptor beta isoforms in human breast cancer tissues and cell lines. Breast Cancer Res Treat 2002;71:249–55. [DOI] [PubMed] [Google Scholar]
- 67.Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proc Natl Acad Sci U S A 2006;103: 13162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol 2000;105:1063–70. [DOI] [PubMed] [Google Scholar]
- 69.Leitner NR, Witalisz-Siepracka A, Strobl B, Muller M. Tyrosine kinase 2— Surveillant of tumours and bona fide oncogene. Cytokine 2017;89:209–18. [DOI] [PubMed] [Google Scholar]
- 70.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007;7:454–65. [DOI] [PubMed] [Google Scholar]
- 71.Piganis RA, De Weerd NA, Gould JA, Schindler CW, Mansell A, Nicholson SE, et al. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon alpha receptor (IFNAR1)-associated tyrosine kinase Tyk2. J Biol Chem 2011;286:33811–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 2012;36:239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol 2003;3:900–11. [DOI] [PubMed] [Google Scholar]
- 74.Grishina I, Fenton A, Sankaran-Walters S. Gender differences, aging and hormonal status in mucosal injury and repair. Aging Dis 2014;5:160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Masaka T, Iijima K, Endo H, Asanuma K, Ara N, Ishiyama F, et al. Gender differences in oesophageal mucosal injury in a reflux oesophagitis model of rats. Gut 2013;62:6–14. [DOI] [PubMed] [Google Scholar]
- 76.Honda J, Iijima K, Asanuma K, Ara N, Shiroki T, Kondo Y, et al. Estrogen enhances esophageal barrier function by potentiating occludin expression. Dig Dis Sci 2016;61:1028–38. [DOI] [PubMed] [Google Scholar]
- 77.Cerny KL, Ribeiro RA, Jeoung M, Ko C, Bridges PJ. Estrogen receptor alpha (ESR1)-dependent regulation of the mouse oviductal transcriptome. PLoS One 2016;11:e0147685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kong KL, Kwong DL, Fu L, Chan TH, Chen L, Liu H, et al. Characterization of a candidate tumor suppressor gene uroplakin 1A in esophageal squamous cell carcinoma. Cancer Res 2010;70:8832–41. [DOI] [PubMed] [Google Scholar]
- 79.Zheng Y, Wang DD, Wang W, Pan K, Huang CY, Li YF, et al. Reduced expression of uroplakin 1A is associated with the poor prognosis of gastric adenocarcinoma patients. PLoS One 2014;9:e93073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beppu L, Yang T, Luk M, Newbury RO, Palmquist J, Dohil R, et al. MMPs-2 and −14 are elevated in eosinophilic esophagitis and reduced following topical corticosteroid therapy. J Pediatr Gastroenterol Nutr 2015;61:194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.