

Abstract
Background:
Epigenetic mechanisms, including methylation, can contribute to childhood asthma. Identifying DNA methylation profiles in asthmatic patients can inform disease pathogenesis.
Objective:
We sought to identify differential DNA methylation in newborns and children related to childhood asthma.
Methods:
Within the Pregnancy And Childhood Epigenetics consortium, we performed epigenome-wide meta-analyses of school-age asthma in relation to CpG methylation (Illumina450K) in blood measured either in newborns, in prospective analyses, or cross-sectionally in school-aged children. We also identified differentially methylated regions.
Results:
In newborns (8 cohorts, 668 cases), 9 CpGs (and 35 regions) were differentially methylated (epigenome-wide significance, false discovery rate < 0.05) in relation to asthma development. In a cross-sectional meta-analysis of asthma and methylation in children (9 cohorts, 631 cases), we identified 179 CpGs (false discovery rate < 0.05) and 36 differentially methylated regions. In replication studies of methylation in other tissues, most of the 179 CpGs discovered in blood replicated, despite smaller sample sizes, in studies of nasal respiratory epithelium or eosinophils. Pathway analyses highlighted enrichment for asthma-relevant immune processes and overlap in pathways enriched both in newborns and children. Gene expression correlated with methylation at most loci. Functional annotation supports a regulatory effect on gene expression at many asthma-associated CpGs. Several implicated genes are targets for approved or experimental drugs, including IL5RA and KCNH2.
Conclusion:
Novel loci differentially methylated in newborns represent potential biomarkers of risk of asthma by school age. Cross-sectional associations in children can reflect both risk for and effects of disease. Asthma-related differential methylation in blood in children was substantially replicated in eosinophils and respiratory epithelium.
Keywords: Epigenetics, methylation, asthma, childhood, newborn, drug development
Asthma is the most common chronic disease of childhood,1 but the underlying mechanisms remain poorly understood. Genome-wide association study (GWAS) meta-analyses have identified many loci related to asthma,2 but these explain only a modest proportion of variation in asthma risk.3 Increasing evidence suggests that epigenetic variation can play a role in asthma pathogenesis.4 DNA methylation is the most studied epigenetic modification in human subjects. Prospective examination of methylation patterns in newborns in relation to asthma development might identify genes and mechanisms involved in the developmental origins of asthma.5
Epigenome-wide association studies (EWASs) of DNA methylation in blood in relation to asthma (numbers of cases range from 16-149)6–12 have identified differential methylation at some specific gene regions. The only meta-analysis of epigenome-wide methylation in childhood asthma included 392 cases but did not examine newborn methylation.13 A larger meta-analysis, including both methylation in newborns and at later ages, would increase the power for identification of novel loci.
Using the Illumina HumanMethylation450K BeadChip (Illumina450K; Illumina, San Diego, Calif), we performed a large-scale meta-analysis of childhood asthma in relation to whole-blood DNA methylation in newborns to evaluate whether methylation patterns at birth relate to disease development. We separately examined cross-sectional associations between whole-blood DNA methylation and the presence of asthma in children of at least school age. We investigated the association of DNA methylation in blood and asthma at both individual sites and over genomic regions and evaluated the potential functional effect of findings by integrating gene expression, pathway analyses, detailed functional annotation, and the search for druggable targets of differentially methylated loci. We also followed up our findings using methylation data in eosinophils and from nasal respiratory epithelium.
METHODS
The Methods section in this article’s Online Repository at www.jacionline.org provides additional details on the methods used in this study.
Study population
The Pregnancy and Childhood Epigenetics (PACE) consortium is an international consortium of cohorts with Illumina450K DNA methylation data at birth (newborns) or in childhood.14 In prospective analyses we evaluated childhood asthma at school age in relation to blood DNA methylation data from newborns (8 cohorts: Avon Longitudinal Study of Parents and Children [ALSPAC], Children’s Health Study [CHS], Etude des Déterminants pré et post natals du développement et de la santé de l’Enfant [EDEN] birth cohort, Generation R, Genetics of Overweight Young Adults [GOYA], Norwegian Mother and Child [MoBa] cohort 1, MoBa2, and Newborn Epigenetics STudy [NEST]). We also conducted cross-sectional analyses of methylation measured in children in relation to asthma status at that same time point (9 cohorts: Children, Allergy, Milieu, Stockholm, Epidemiology [BAMSE] EpiGene; BAMSE MeDALL; European Childhood Obesity Project [CHOP]; Genes-environments & Admixture in Latino Americans [GALA II]; Inner City Asthma Consortium [ICAC]; Northern Finland Birth Cohort [NFBC] 1986; Prevention and Incidence of Asthma and Mite Allergy [PIAMA]; the Raine study; and Swedish Twin study On Prediction and Prevention of Asthma [STOPPA]). To avoid problems from small numbers, we set a minimum of 15 cases for participating cohorts to perform analyses.
Harmonization of childhood asthma variables
We developed a harmonized definition of asthma based on the questionnaire data available in each cohort. Asthma was assessed at school age, which was defined as 5 years or older, and varied by cohort. Asthma was defined by a doctor’s diagnosis of asthma and the report of at least 1 of the following: (1) current asthma, (2) asthma in the past year, or (3) asthma medication use in the last year. Noncases were children who had never had asthma.
Methylation data measurement and quality control
DNA methylation was measured with the Illumina450K platform. Cohorts performed their own quality control, normalization, and analysis of untransformed β values. Previously, we found that the use of different preprocessing or normalization methods did not influence meta-analysis results.15,16 Probes on the X and Y chromosomes were removed, as were those in which a single nucleotide polymorphism (SNP) was present in the last 5 bp of the probe, which could interfere with binding. Rather than remove probes a priori that have appeared on various published lists of potentially cross-reactive probes or probes near SNPs, we examined post hoc those that appear in statistically significant results.17,18
Annotation of CpGs
This article’s tables include the University of California, Santa Cruz (UCSC) RefGene name from Illumina’s annotation file and enhanced annotation to the UCSC Known Gene. UCSC Known Gene annotations include the nearest gene within 10 Mb of each CpG and fill in many missing gene names. All annotations use the human February 2009 (GRCh37/hg19) assembly.
Cohort-specific statistical analyses
The association of methylation and asthma was assessed by using logistic regression. Covariates included in adjusted models were maternal age, sustained maternal smoking during pregnancy,15 maternal asthma, socioeconomic status, and child’s sex. Cohorts adjusted for batch effects by using ComBat19 or SVA20 or by including a batch covariate in their models. We also adjusted for potential cell-type confounding by including estimated proportions calculated by using the Houseman method,21 with a cord blood reference panel22 for newborn cohorts or an adult blood reference panel23 for child cohorts. The primary models presented include adjustment for covariates and cell type; reduced models are presented for comparison.
Meta-analyses
As in other consortium genomic analyses,24,25 we meta-analyzed the study-specific results using inverse variance weighting, which is also referred to as fixed-effects meta-analysis, with METAL.26 We accounted for multiple testing by controlling for the false discovery rate (FDR) at 0.05.27 To enable readers to assess whether the results across studies are consistent, we provide forest plots of the study-specific effect estimates and 95%CIs. As another way to visualize meaningful heterogeneity or influential results, we also provide plots for all significant CpGs of regression coefficients and 95% CIs where we leave out 1 cohort at a time. Although inverse variance-weighted meta-analysis does not require the assumption of homogeneity,25 where there is even nominal evidence for heterogeneity (Pheterogeneity < .05 without correction for multiple testing) for any CpG we report as genome-wide significant, we also provide meta-analysis P values from standard random-effects meta-analysis by using METASOFT.28
Analyses of differentially methylated regions
Differentially methylated regions (DMRs) were identified by using 2 methods: comb-p29 and DMRcate.30 To correct for multiple comparisons, comb-p uses a 1-step Sidak correction,29 and DMRcate uses an FDR correction.30 Each method requires the input of parameters to be used in selecting the regions. DMRcate30 has default values for the minimum number of CpGs in a region (ie, 2) and a minimum length of 1000 nucleotides; we used these values in comb-p to maximize comparability. To be conservative, we set the significance threshold at .01 rather than .05 and only considered a DMR to be statistically significant if it met this threshold in both packages (Šideák-corrected P < .01 from comb-p and FDR < 0.01 from DMRcate). DMRcate annotates DMRs to UCSC RefGene from the Illumina annotation file.
Functional follow-up of significant DNA methylation findings
Correlation of differentially methylated sites with expression of nearby genes.
To examine whether differentially methylated sites affect gene expression, we analyzed paired methylation and gene expression data, both of which were measured in blood, from several data sets (see this article’s Online Repository at www.jacionline.org)31–37: 2 with methylation and gene expression in newborns (Gene Expression Omnibus [GSE62924 and GSE48354], n 5 38; and Isle of Wight 3rd Generation Study [IoW], n = 157),32–34 1 with newborn methylation and gene expression at age 4 years (Infancia y Medio Ambiente [INMA], n = 113), 5 another with gene expression and methylation both measured at age 4 years (INMA, n = 112),35 1 with both measured at age 16 years (BAMSE; n = 248),38 and the largest with both measured in adults (BIOS consortium, n = 3096).36,37 For each of our significant CpGs, we examined the association with expression of transcripts within a 500-kb window (±250 kb from the CpG). For DMRs, we used a window 250 kb upstream and downstream of the end and start site of each region. A given CpG or region might have more than 1 gene transcript in this window. In the smaller datasets of paired gene expression and methylation in newborns or children, we report nominal evidence for significance (P < .05); for the much larger adult data set, we report associations based on FDRs of less than 0.05.
Functional annotation.
To identify tissue- or cell type-specific signals in significant EWAS results, we used eFORGE.39 Pathway and network analyses were conducted by using Ingenuity Pathway Analysis (Qiagen, Venlo, The Netherlands; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis).40 Because of possible uncertainty regarding genome annotation of probes flagged in the literature as potentially cross-reactive,41 we excluded those from pathway analyses. We also compared our methylation findings with those from published studies of methylation in relation to asthma and evaluated whether the implicated genes overlap with loci identified in GWASs.42,43 Additionally, we matched the genes to which our asthma-associated CpGs and DMRs annotated against the ChEMBL database (version 22.1) to identify whether any are targets of approved drugs or drugs in development.44
Look-up replication of significant DNA methylation findings in nasal respiratory epithelium and eosinophils
We examined the cell-type specificity of significant findings in whole blood in childhood by doing a look-up in 2 data sets, with methylation measured with the Illumina450K in respiratory epithelium collected by means of nasal brushing (455 sixteen-year-old Dutch children [37 with asthma] from the PIAMA study13 and 72 African-American children [36 asthmatic patients and 38 nonasthmatic subjects],45 as well as a study with methylation measured with the Illumina450K in eosinophils isolated from blood [16 asthmatic patients and 8 nonasthmatic subjects aged 2-56 years from the Saguenay-Lac-Saint-Jean [SLSJ] region in Canada).13,46,47
RESULTS
Prospective analysis of newborn methylation in relation to asthma development included 8 cohorts; the cross-sectional analysis of methylation in children in relation to asthma included 9 cohorts, with mean ages at assessment of both asthma status and methylation ranging from 7 to 17 years (Table I contains counts by cohort and Table E1 in this article’s Online Repository at www.jacionline.org contains descriptive statistics). Because newborn DNA methylation is measured at birth, age at asthma assessment is the time between assessment of methylation and asthma status in prospective analyses. All models included covariates and cell type, unless otherwise noted. Some studies over-sampled asthmatic patients within their population-based cohorts using a nested case-control or case-cohort design for methylation measurement, and therefore the case/control ratio varies across studies.
TABLE I.
Sample sizes by cohort for epigenome-wide association analyses of asthma in relation to DNA methylation in newborns or children
| Age group | Cohort | No. | No. of cases |
|---|---|---|---|
| Newborns | ALSPAC | 688 | 88 |
| CHS | 229 | 39 | |
| EDEN | 150 | 34 | |
| Generation R | 661 | 37 | |
| GOYA | 507 | 37 | |
| MoBa1 | 666 | 149 | |
| MoBa2 | 458 | 239 | |
| NEST | 213 | 45 | |
| Meta-analysis | 3572 | 668 | |
| Children | BAMSE EpiGene | 307 | 93 |
| BAMSE MeDALL | 214 | 47 | |
| CHOP | 382 | 19 | |
| GALA II | 193 | 106 | |
| ICAC | 187 | 92 | |
| NFBC 1986 | 413 | 17 | |
| PIAMA | 197 | 15 | |
| Raine study | 509 | 105 | |
| STOPPA | 460 | 137 | |
| Meta-analysis | 2862 | 631 |
Cohort-specific information on covariates is provided in Table E1.
Asthma in relation to newborn DNA methylation
Meta-analysis of asthma and newborn methylation (668 cases and 2904 noncases; 8 cohorts: ALSPAC, CHS, EDEN, Generation R, GOYA, MoBA1, MoBa2, and NEST) identified 9 statistically significant (FDR < 0.05) individual CpGs (Manhattan and volcano plots in Fig 1). The 9 CpGs include 2 that have appeared on a list of poorly hybridizing probes41 and thus must be regarded with caution (ch.11.109687686R and ch.6.1218502R). The other 7 CpGs annotated to the following genes: CLNS1A, MAML2/Mir_548, GPATCH2/SPATA17, SCOC/LOC100129858, AK091866, SUB1, and WDR20 (Table II). We identified 35 significant DMRs (Table III and see Table E2 in this article’s Online Repository at www.jacionline.org for individual CpGs within DMRs); DMRs did not overlap the significant CpGs. Seven of the 9 significant CpGs showed greater methylation in children with asthma than in noncases. All 9 CpGs had P values of 3.55 × 10−3 or less in a crude model and P values of 4.16 × 10−4 or less in the covariate-adjusted models that did not include cell type (see Table E3 in this article’s Online Repository at www.jacionline.org). None of the 9 CpGs had been previously reported in the literature (see Table E4 in this article’s Online Repository at www.jacionline.org).
FIG 1.
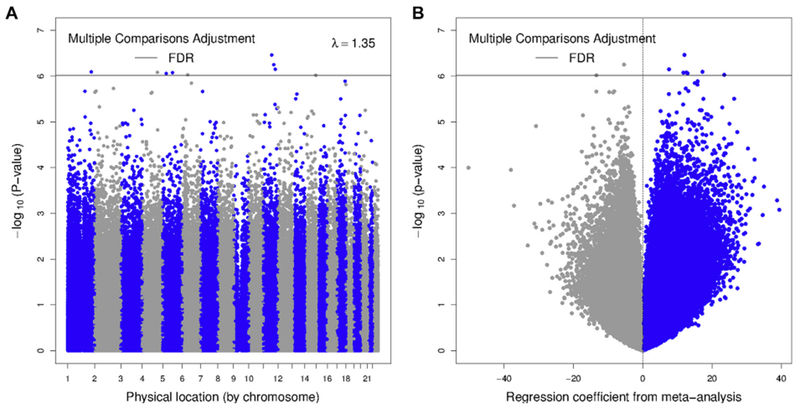
Meta-analysis of asthma in relation to newborn methylation: A, Manhattan plot; B, volcano plot. The model is adjusted for covariates and cell types.
TABLE II.
Nine significant CpGs (FDR < 0.05) from the meta-analysis of asthma in relation to newborn methylation
| CpG* | chromosome:position | UCSC RefGene name | UCSC Known Gene† | Average methylation | OR‡ (CI) | P value | Direction§ |
|---|---|---|---|---|---|---|---|
| cg21486411 | Chr 11:77348243 | CLNS1A | CLNS1A | 0.089 | 1.13 (1.08-1.18) | 3.43E-07 | +?++++++ |
| cg16792002 | Chr 11:95788886 | MAML2 | Mir_548 | 0.840 | 0.95 (0.93-0.97) | 5.59E-07 | −−−−−−−+ |
| ch.11.109687686R | Chr 11:110182476 | 0.085 | 1.08 (1.05-1.11) | 7.06E-07 | +??+++++ | ||
| cg13427149 | Chr 1:217804379 | GPATCH2; SPATA17 | GPATCH2 | 0.063 | 1.19 (1.11-1.27) | 8.04E-07 | ++++++++ |
| cg17333211 | Chr 4:141294016 | SCOC | LOC100129858 | 0.074 | 1.13 (1.08-1.19) | 8.25E-07 | −+−+++++ |
| cg02331902 | Chr 5:90610303 | AK091866 | 0.089 | 1.12 (1.07-1.18) | 8.37E-07 | −−++++++ | |
| cg13289553 | Chr 5:32585524 | SUB1 | SUB1 | 0.085 | 1.14 (1.08-1.20) | 8.68E-07 | +++++++− |
| ch.6.1218502R | Chr 6:51250028 | 0.054 | 1.27 (1.15-1.39) | 9.32E-07 | +??+++++ | ||
| cg07156990 | Chr 14:102685678 | WDR20 | WDR20 | 0.930 | 0.87 (0.83-0.92) | 9.54E-07 | −++−−−−− |
OR, Odds ratio.
ch probes (ch.11.109687686R and ch.6.1218502R) have been reported to be cross-hybridizing, and thus UCSC Known Gene is intentionally left blank.
Annotation based on UCSC Known Gene also fills in the nearest gene within 10 MB.
Odds ratio of having asthma for a 1% absolute increase in methylation. Adjusted for covariates and cell type.
For each cohort participating in the analysis, + indicates a positive direction of effect, − indicates a negative direction of effect, and ? indicates missing information for that CpG in a given cohort. Cohort order is as follows: ALSPAC, CHS, EDEN, Generation R, GOYA, MoBa1, MoBa2, and NEST.
TABLE III.
DMRs (n = 35) for asthma in relation to newborn methylation identified by using both comb-p (P < .01) and DMRcate (FDR < 0.01) methods
| chromosome:position | Gene name* | No. of CpGs in region | P value from comb-p† | FDR from DMRcate‡ |
|---|---|---|---|---|
| Chr 1: 59280290-59280842 | LINC01135 | 5 | 1.23E-03 | 1.01E-03 |
| Chr 1: 220263017-220263699 | BPNT1; RNU5F-1 | 11 | 4.49E-04 | 7.74E-05 |
| Chr 1: 1296093-1296489 | MXRA8 | 2 | 9.83E-03 | 3.86E-04 |
| Chr 2: 202097062-202097608 | CASP8 | 5 | 1.14E-03 | 1.64E-05 |
| Chr 2: 235004843-235005012 | SPP2 | 2 | 6.22E-03 | 1.15E-03 |
| Chr 3: 194188646-194189444 | ATP13A3 | 3 | 1.06E-03 | 7.14E-04 |
| Chr 4: 113218385-113218525 | ALPK1 | 3 | 2.00E-03 | 3.69E-04 |
| Chr 5: 158526108-158526694 | EBF1 | 6 | 9.56E-04 | 2.16E-05 |
| Chr 5: 81573780-81574461 | RPS23 | 11 | 3.75E-03 | 1.47E-04 |
| Chr 5: 64777678-64778186 | ADAMTS6 | 10 | 7.09E-03 | 9.97E-05 |
| Chr 6: 291687-292824 | DUSP22 | 9 | 6.69E-06 | 1.18E-05 |
| Chr 6: 32799997-32801050 | TAP2 | 13 | 1.27E-03 | 6.66E-05 |
| Chr 6: 26234819-26235610 | HIST1H1D | 9 | 6.12E-03 | 7.67E-05 |
| Chr 6: 29648161-29649085 | ZFP57 | 22 | 1.82E-08 | 3.13E-11 |
| Chr 6: 31055396-31055503 | C6orf15 | 5 | 3.61E-04 | 7.05E-05 |
| Chr 7: 106694832-106695007 | PRKAR2B | 2 | 6.86E-03 | 7.92E-04 |
| Chr 7: 87974722-87975316 | STEAP4 | 4 | 2.32E-03 | 7.44E-05 |
| Chr 7: 158045980-158046359 | PTPRN2 | 6 | 1.98E-03 | 5.94E-04 |
| Chr 8: 127889010-127889296 | PCAT1 | 4 | 2.68E-05 | 1.44E-05 |
| Chr 8: 33370172-33371226 | TTI2 | 9 | 1.08E-04 | 6.40E-06 |
| Chr 10: 71871364-71871634 | H2AFY2 | 4 | 8.06E-03 | 6.19E-04 |
| Chr 10: 65028929-65029169 | JMJD1C | 5 | 8.56E-03 | 6.12E-04 |
| Chr 11: 268923-269469 | NLRP6 | 5 | 3.71E-03 | 1.42E-03 |
| Chr 11: 107328442-107328915 | CWF19L2 | 10 | 5.10E-03 | 2.13E-05 |
| Chr 12: 74931289-74932008 | ATXN7L3B | 10 | 1.03E-03 | 2.81E-06 |
| Chr 12: 58329764-58330116 | LOC100506844 | 5 | 1.58E-03 | 5.22E-04 |
| Chr 13: 108953659-108954055 | TNFSF13B | 2 | 5.19E-03 | 2.37E-03 |
| Chr 13: 31618695-31618744 | TEX26 | 2 | 4.63E-03 | 2.09E-04 |
| Chr 14: 69341139-69341739 | ACTN1 | 4 | 1.36E-03 | 9.96E-04 |
| Chr 16: 20774873-20775353 | ACSM3 | 5 | 3.47E-03 | 1.58E-03 |
| Chr 17: 74667833-74668253 | LOC105274304 | 6 | 2.13E-03 | 8.34E-07 |
| Chr 17: 21029189-21029296 | DHRS7B | 2 | 7.18E-03 | 5.11E-05 |
| Chr 18: 47813745-47815431 | CXXC1 | 10 | 2.58E-05 | 1.68E-03 |
| Chr 21: 36421467-36421956 | RUNX1 | 6 | 2.23E-03 | 1.67E-04 |
| Chr 22: 24372913-24374013 | LOC391322 | 12 | 3.21E-04 | 1.35E-07 |
DMRcate annotates to UCSC RefGene from the Illumina annotation file.
Comb-p uses a 1-step Sidak multiple-testing correction on the regional P value assigned by using the Stouffer-Liptak method.
DMRcate takes the minimum Benjamini-Hochberg FDR-corrected P value in the region as representative after recalculating P values by using Gaussian kernel smoothing.
Forest plots showing cohort-specific odds ratios and 95% CIs for the 9 CpGs are shown in Fig E1 in this article’s Online Repository at www.jacionline.org. Two cohorts in the newborn analysis include subjects of non-European ancestry (NEST and CHS), and therefore we evaluated whether these were influential. The forest plots (Fig E1) suggest that for just 1 of the 9 CpGs (cg07156990), the size of the effect estimate was larger in NEST than in other studies, but the P value for heterogeneity was not close to statistically significant (Pheterogeneity = .26), and after removing NEST, the meta-analysis P value was attenuated only slightly to 2.8 × 10−6 from 9.5 × 10−7. When we repeated the meta-analysis removing both NEST and CHS, results were very consistent with those from all cohorts (correlation of regression coefficients = 0.996). With respect to tests of heterogeneity, only 1 of the 9 CpGs, cg13289553, produced a P value for heterogeneity that was even nominally significant (Pheterogeneity = .04, Table E3 includes Pheterogeneity values for all 9 CpGs and the random-effects meta-analysis results for this CpG); GOYA had the largest magnitude of association, but effect estimates were in the same positive direction across studies (see Fig E1). Analyses leaving out 1 cohort at a time do not suggest that any of the results are driven by a single cohort (plots of untransformed effect estimates and 95% CIs are shown in Fig E2 in this article’s Online Repository at www.jacionline.org).
Asthma in relation to childhood DNA methylation
In a meta-analysis of asthma in relation to DNA methylation measured in childhood (631 cases and 2231 noncases; 9 cohorts: BAMSE EpiGene, BAMSE MeDALL, CHOP, GALA II, ICAC, NFBC, PIAMA, Raine study, and STOPPA), we identified 179 CpGs at genome-wide significance (FDR < 0.05, Manhattan and volcano plots in Fig 2; results for all 179 CpGs are shown in Table E5 in this article’s Online Repository at www.jacionline.org). Nearly all (173/179) showed decreased methylation in asthma cases versus noncases; similar predominant directionality was seen in a recent study.13
FIG 2.

Meta-analysis of asthma in relation to childhood methylation: A, Manhattan plot; B, volcano plot. The model is adjusted for covariates and cell types. CpGs corresponding to more than 1 gene with significant CpGs (FDR < 0.05) are highlighted in red.
As in the newborn analysis, results were consistent across studies for the 179 significant CpGs (forest plots are shown in Fig E3 in this article’s Online Repository at www.jacionline.org, and plots of regression coefficients and 95% CIs from analyses leaving one cohort out at a time are shown in Fig E4 in this article’s Online Repository at www.jacionline.org). Two of the cohorts were adolescents (NFBC: mean age, 16.0 years; SD, 0.4 years; Raine study: mean age, 17.0 years; SD, 0.2 years); repeating the meta-analysis without these 2 cohorts provided high correlations with values for our FDR-significant findings from all cohorts (correlation of coefficients = 0.96). Because 2 studies (ICAC and GALA) included subjects who were not of European ancestry, we compared significant results with and without including these 2 studies and found them to be very similar (correlation of coefficients = 0.99). Table E5 provides P values for heterogeneity and, where those are even nominally significant (Pheterogeneity < .05), random-effects meta-analysis results.
Of the 179 FDR-significant CpGs, 34 CpGs were not singletons (ie, >1 significant CpG annotated to a given gene). These 34 nonsingleton CpGs correspond to 13 genes: ACOT7, LOC100189589, IL5RA, SLC25A26/LRIG1, RPS6KA2, KCNH2, ZNF862/BC045757, AK096249, PRG2, EVL/AX747103, KIAA0182, ZFPM1, and EPX (Table IV). We identified 36 significant DMRs by using both calling methods (Table V). Of the 179 FDR-significant CpGs, 31 fell within one of these 36 DMRs, and 21 of the 36 DMRs contained at least 1 FDR-significant CpG.
TABLE IV.
Thirty-four CpGs annotated to 13 genes with more than 1 significant CpG (FDR < 0.05) from the meta-analysis of asthma in relation to childhood methylation
| CpG | chromosome:position | UCSC RefGene name | UCSC Known gene* | P value | Average methylation | OR† (CI) | Direction‡ |
|---|---|---|---|---|---|---|---|
| cg13066938 | Chr 1: 6341140 | ACOT7 | ACOT7 | 1.67E-05 | 0.682 | 0.91 (0.88-0.95) | −−+?−−+−− |
| cg21220721 | Chr 1: 6341230 | ACOT7 | ACOT7 | 1.02E-08 | 0.763 | 0.94 (0.92-0.96) | −−+−−−−−− |
| cg09249800 | Chr 1: 6341287 | ACOT7 | ACOT7 | 1.19E-08 | 0.916 | 0.88 (0.84-0.92) | ???−−−?−− |
| cg11699125 | Chr 1: 6341327 | ACOT7 | ACOT8 | 7.54E-10 | 0.799 | 0.90 (0.87-0.93) | −−+−−−−−− |
| cg00043800 | Chr 2: 74612144 | LOC100189589 | LOC100189589 | 1.32E-05 | 0.585 | 0.91 (0.87-0.95) | −−−−−++−− |
| cg17988187 | Chr 2: 74612222 | LOC100189589 | LOC100189590 | 1.21E-06 | 0.699 | 0.90 (0.86-0.94) | −−+?−−+−− |
| cg01310029 | Chr 3: 3152374 | IL5RA | IL5RA | 4.18E-06 | 0.744 | 0.89 (0.85-0.94) | −−−?−−+−− |
| cg10159529 | Chr 3: 3152530 | IL5RA | IL5RA | 4.48E-06 | 0.736 | 0.90 (0.86-0.94) | −−−?−−−− |
| cg07410597 | Chr 3: 66404129 | SLC25A26 | LRIG1 | 2.70E-07 | 0.773 | 0.88 (0.84-0.93) | −−+−−−+−− |
| cg04217850 | Chr 3: 66428294 | SLC25A26 | LR1G2 | 2.35E-06 | 0.747 | 0.88 (0.83-0.93) | −−+−−−−−− |
| cg15304012 | Chr 6: 166876490 | RPS6KA2 | RPS6KA2 | 1.86E-05 | 0.697 | 1.08 (1.04-1.13) | +++++++++ |
| cg19851574 | Chr 6: 167178233 | RPS6KA2 | RPS6KA2 | 3.42E-06 | 0.671 | 0.95 (0.94-0.97) | −−+−−−−−− |
| cg03329755 | Chr 6: 167189272 | RPS6KA2 | RPS6KA2 | 6.14E-06 | 0.818 | 0.91 (0.88-0.95) | −++−−−−−− |
| cg05184016 | Chr 7: 149543136 | ZNF862 | BC045757 | 2.74E-08 | 0.817 | 0.85 (0.80-0.90) | −−+−−−−−− |
| cg07970948 | Chr 7: 149543165 | ZNF862 | BC045757 | 6.39E-08 | 0.771 | 0.91 (0.88-0.94) | −−−+−−+−− |
| cg06558622 | Chr 7: 149543177 | ZNF862 | BC045757 | 3.39E-09 | 0.818 | 0.88 (0.85-0.92) | −−−−−−−−− |
| cg24576940 | Chr 7: 150648283 | KCNH2 | KCNH2 | 1.83E-05 | 0.848 | 0.87 (0.81-0.93) | −−−−−−−−− |
| cg23147443 | Chr 7: 150649655 | KCNH2 | KCNH2 | 1.83E-06 | 0.842 | 0.89 (0.85-0.93) | ???−−−?−− |
| cg18666454 | Chr 7: 150651937 | KCNH2 | KCNH2 | 1.46E-07 | 0.761 | 0.89 (0.86-0.93) | −−−−−−−−− |
| cg13850063 | Chr 9: 138362321 | AK096249 | 5.49E-08 | 0.819 | 0.78 (0.71-0.85) | −−+?−−−−− | |
| cg14011077 | Chr 9: 138362327 | AK096249 | 7.02E-09 | 0.797 | 0.86 (0.82-0.90) | −−−?−−−−− | |
| cg15700636 | Chr 11: 57156050 | PRG2 | PRG2 | 2.35E-07 | 0.746 | 0.89 (0.85-0.93) | −−+−−−−−− |
| cg08773180 | Chr 11: 57157607 | PRG2 | PRG2 | 1.77E-07 | 0.741 | 0.89 (0.85-0.93) | −−+−−−+−− |
| cg12819873 | Chr 11: 57157632 | PRG2 | PRG2 | 9.55E-06 | 0.760 | 0.90 (0.86-0.94) | −−−−−−+−− |
| cg16409452 | Chr 14: 100610186 | EVL | AX747103 | 4.89E-07 | 0.770 | 0.91 (0.87-0.94) | −−+−−−−−− |
| cg14084609 | Chr 14: 100610407 | EVL | AX747103 | 2.96E-09 | 0.780 | 0.89 (0.85-0.92) | −−−−−−−−− |
| cg18550847 | Chr 14: 100610570 | EVL | AX747103 | 7.10E-07 | 0.730 | 0.91 (0.88-0.94) | −−+?−−−−− |
| cg08640475 | Chr 16: 85551478 | KIAA0182 | 2.36E-06 | 0.815 | 0.92 (0.89-0.95) | −−+−−−−−− | |
| cg10099827 | Chr 16: 85551514 | KIAA0182 | 1.32E-06 | 0.808 | 0.92 (0.89-0.95) | −−−−−−−−− | |
| cg08940169 | Chr 16: 88540241 | ZFPM1 | ZFPM1 | 2.93E-07 | 0.778 | 0.91 (0.87-0.94) | −−−−−−+−− |
| cg04983687 | Chr 16: 88558223 | ZFPM1 | ZFPM1 | 1.33E-10 | 0.744 | 0.93 (0.90-0.95) | −−−−−−−−− |
| cg25173129 | Chr 17: 56269410 | EPX | EPX | 8.09E-07 | 0.753 | 0.88 (0.84-0.93) | −−+−−−+−− |
| cg02970679 | Chr 17: 56269818 | EPX | EPX | 9.99E-07 | 0.776 | 0.88 (0.83-0.92) | −−−−−−+−− |
| cg17374802 | Chr 17: 56270828 | EPX | EPX | 2.06E-06 | 0.713 | 0.90 (0.86-0.94) | −−−?−−+−− |
OR, Odds ratio.
Annotation based on UCSC Known Gene also fills in nearest gene within 10 MB.
Odds ratio of having asthma for a 1% absolute increase in methylation. Adjusted for covariates and cell type.
For each cohort, + indicates a positive direction of effect, − indicates a negative direction of effect, and ? indicates missing information for that CpG. Cohort order is as follows: BAMSE EpiGene, BAMSE MeDALL, CHOP, GALAII, ICAC, NFBC1986, PIAMA, RAINE, and STOPPA.
TABLE V.
DMRs for asthma in relation to childhood methylation with adjustment for covariates and cell type identified by using both comb-p (P < .01) and DMRcate (FDR < 0.01) methods
| chromosome:position | Gene name* | No. of CpGs in region | P value from comb-p† | FDR from DMRcate‡ |
|---|---|---|---|---|
| Chr 1: 161575716-161576323 | HSPA7 | 4 | 8.61E-03 | 1.24E-03 |
| Chr 1: 209979111-209979780 | IRF6 | 13 | 4.62E-04 | 1.90E-04 |
| Chr 1: 2036283-2036644 | PRKCZ | 4 | 2.00E-04 | 3.14E-05 |
| Chr 1: 87596820-87596935 | LINC01140 | 3 | 1.58E-03 | 2.79E-05 |
| Chr 2: 149639612-149640260 | KIF5C | 4 | 3.50E-03 | 1.14E-05 |
| Chr 2: 11917490-11917788 | LPIN1 | 3 | 4.81E-03 | 6.25E-04 |
| Chr 3: 195974258-195974330 | PCYT1A | 3 | 5.07E-05 | 2.00E-05 |
| Chr 3: 3151795-3152917 | IL5RA | 6 | 1.35E-08 | 2.12E-09 |
| Chr 5: 38445220-38446193 | EGFLAM | 9 | 5.11E-06 | 1.28E-05 |
| Chr 5: 132008525-132009631 | IL4 | 4 | 5.36E-07 | 3.11E-05 |
| Chr 6: 112688010-112688931 | RFPL4B | 4 | 4.89E-05 | 5.19E-04 |
| Chr 6: 166876490-166877039 | RPS6KA2;RPS6KA2-IT1 | 8 | 3.08E-05 | 1.74E-06 |
| Chr 7: 156735383-156735657 | NOM1 | 3 | 7.11E-03 | 2.82E-03 |
| Chr 7: 149543136-149543178 | ZNF862 | 3 | 3.85E-16 | 1.43E-16 |
| Chr 7: 65419185-65419289 | VKORC1L1 | 7 | 2.82E-03 | 1.04E-03 |
| Chr 8: 832917-833049 | ERICH1-AS1;DLGAP2 | 3 | 6.15E-03 | 6.44E-03 |
| Chr 8: 141046436-141046853 | TRAPPC9 | 5 | 8.93E-07 | 3.45E-09 |
| Chr 9: 138362321-138362505 | PPP1R26-AS1 | 3 | 2.72E-05 | 1.44E-09 |
| Chr 9: 130859454-130859607 | SLC25A25 | 2 | 2.69E-08 | 5.84E-08 |
| Chr 11: 65545808-65547173 | AP5B1 | 8 | 1.31E-10 | 9.73E-12 |
| Chr 11: 69291998-69292065 | LINC01488 | 3 | 4.55E-04 | 1.65E-04 |
| Chr 11: 59856225-59856359 | MS4A2 | 2 | 1.50E-03 | 3.25E-04 |
| Chr 12: 15125458-15126021 | PDE6H | 4 | 6.93E-03 | 7.65E-06 |
| Chr 14: 100610071-100610668 | EVL | 6 | 7.79E-16 | 1.24E-19 |
| Chr 15: 64275810-64275854 | DAPK2 | 2 | 4.91E-04 | 2.04E-04 |
| Chr 15: 99443213-99443667 | IGF1R | 4 | 6.57E-05 | 2.41E-04 |
| Chr 16: 875257-875627 | PRR25 | 4 | 3.34E-03 | 3.21E-03 |
| Chr 16: 88539861-88540397 | ZFPM1 | 5 | 1.09E-04 | 1.13E-05 |
| Chr 16: 615709-616221 | PRR35 | 5 | 1.62E-04 | 2.70E-07 |
| Chr 16: 85551478-85551749 | GSE1 | 3 | 5.27E-07 | 2.37E-07 |
| Chr 17: 56269410-56270829 | EPX | 5 | 6.20E-11 | 1.41E-08 |
| Chr 17: 78682785-78683458 | RPTOR | 5 | 1.18E-04 | 4.03E-04 |
| Chr 19: 51961666-51961938 | SIGLEC8 | 3 | 2.37E-04 | 5.07E-04 |
| Chr 19: 50553682-50554511 | LOC400710 | 10 | 1.78E-07 | 3.81E-06 |
| Chr 20: 35503832-35504554 | TLDC2 | 8 | 1.23E-03 | 5.90E-08 |
| Chr 21: 42520365-42520903 | LINC00323 | 3 | 1.41E-04 | 2.64E-05 |
DMRcate annotates to UCSC RefGene from Illumina annotation file. The first listed gene is shown.
Comb-p uses a 1-step Sidak multiple-testing correction on the regional P value assigned by using the Stouffer-Liptak method.
DMRcate takes the minimum Benjamini-Hochberg FDR-corrected P value in the region as representative after recalculating P values by using Gaussian kernel smoothing.
Three studies in our meta-analysis of asthma in relation to childhood methylation (PIAMA, BAMSE MeDALL, and BAMSE Epigene) also contributed to a recent meta-analysis of both preschool and school-aged asthma outcomes13; these studies contributed only a quarter (n = 155) of the 636 cases in our meta-analysis. That EWAS meta-analysis of asthma at preschool and school age13 identified 14 CpGs at genome-wide significance; 7 were among our 179 genome-wide significant findings for childhood methylation (cg13835688, cg14011077, cg03131767, cg13628444, cg10142874, cg01901579, and cg01445399), and 6 others represented in our data set (cg15344640, cg11456013, cg01770400, cg19764973, cg08085199, and cg16592897) were nominally statistically significant (P < .05) and direction matched for all 13. When repeating the meta-analysis excluding those 3 studies, 13 of the 14 CpGs had P values of less than .05 and directions of association matched; only cg06483820 produced no evidence for association (P = .74). In additional comparison with the literature, differential methylation in ACOT7 and ZFPM1 was previously identified in an EWAS of blood in relation to IgE48 and in 2 of our contributing studies, ICAC and ALSPAC, to asthma,10,12 as well as in an EWAS of nasal epithelium to asthma.45
Comparing newborn and childhood methylation models, none of the 9 FDR-significant CpGs for newborn methylation were nominally significant (P < .05) in the childhood methylation analysis. Only 6 of the 179 CpGs significant for asthma in relation to childhood methylation were at least nominally significant for newborn methylation; 2 of these had consistent directions of effect (cg16409452 [EVL] and cg09423651 [NCK1]).
Replication of findings for asthma in relation to childhood methylation in nasal epithelium
We assessed whether the 179 CpGs differentially methylated in blood in relation to asthma in childhood were also differentially methylated in relation to current asthma in nasal epithelium from 2 studies (see Table E6 in this article’s Online Repository at www.jacionline.org). Among 455 Dutch children (37 with asthma) studied at age 16 years,13 we found evidence for replication for 20 CpGs, matching direction-of-effect estimates and nominal significance (P < .05). Among African American children aged 10 to 12 years with persistent asthma plus atopy (36 cases) compared with 36 nonasthmatic nonatopic children, 128 of the 179 CpGs produced effect estimates for asthma in the same direction and also had P values of less than .05 for association.
Replication of findings for asthma in relation to childhood methylation in eosinophils
We looked up the 179 CpGs differentially methylated in childhood in relation to asthma in EWASs of 16 asthma cases and 8 noncases in whom methylation had been measured in purified eosinophils. Of the 177 CpGs included in this data set, all directions of association with asthma were the same as in the PACE consortium and 148 produced P values of less than .05 (see Table E7 in this article’s Online Repository at www.jacionline.org).
Functional annotation
For the newborn analysis, among the 7 significant CpGs (after removing the 2 “ch”-probes), all 7 were near a transcription factor binding site, and 6 were in a DNase hypersensitivity site identified in at least 1 ENCODE cell line, supporting a potential functional relevance to transcriptional activity (see Fig E5 in this article’s Online Repository at www.jacionline.org).
Among the 179 CpGs significantly differentially methylated in childhood in relation to asthma, there was significant depletion of localization to CpG islands (17 CpGs, 9.5%, P = 1.09 × 10−11) and promoters (34 CpGs, 19.0%, P = 1.10 × 10−4). Functional annotation plots are shown in Fig E6 in this article’s Online Repository at www.jacionline.org for the 13 gene regions to which the 34 nonsingleton CpGs annotate. Among the 179 CpGs, 113 were in DNAse hypersensitivity sites. Using eFORGE39 to examine enrichment of all 179 significant CpGs for histone marks (H3K27me3, H3K36me3, H3K4me3, H3K9me3, and H3K4me1), we found significant enrichment for H3K4me1 in blood and lung tissue and H3K36me3 in blood (see Fig E7 in this article’s Online Repository at www.jacionline.org).
Association of methylation and gene expression
For the CpGs and regions we identified as differentially methylated in either newborns or children in relation to asthma, we assessed association between paired levels of blood DNA methylation and whole-blood gene expression for nearby transcripts defined as within a 500-kb window of the significant CpG or DMR in newborns (Gene Expression Omnibus, n = 38; INMA, n = 113; IoW, n = 157), children (4-year-olds in INMA, n = 112; 16-year-olds in BAMSE, n = 248), and adults (BIOS consortium, n = 3096).
Among 9 CpGs differentially methylated in newborns in relation to asthma, 3 were associated with expression of a nearby transcript in 3 data sets (cg17333211 in newborns, 4-year-olds, and adults and cg02331902 and cg07156990 in 2 newborn data sets and 4-year-olds), and an additional 3 CpGs were associated with expression in 2 data sets (cg13427149 in 16-year-olds and adults and cg13289553 and cg21486411 in newborns and 4-year-olds; see Table E8, A, in this article’s Online Repository at www.jacionline.org.). All regions differentially methylated in newborns in relation to asthma were related to expression in at least 1 data set (see Table E8, B).
For methylation in childhood, nearly all (176/179) CpGs related to asthma also associated with expression in at least 1 data set (Table E8, C). CpGs annotated to IL5RA were significantly associated with expression in 4 cohorts (BIOS consortium, INMA, IoW, and BAMSE). All 36 regions differentially methylated in childhood were associated with expression of a nearby transcript in at least 1 data set (see Table E8, D).
Pathway analysis
Using Ingenuity Pathway Analysis, we identified pathways, as well as disease processes and biological functions, significantly enriched (P < .05) for the genes to which significant individual CpGs or DMRs annotated in the meta-analysis of asthma in relation to newborn or childhood methylation (see Tables E9 and E10 in this article’s Online Repository at www.jacionline.org). Genes to which the 7 significant CpGs (after removing “ch”-probes) and 35 significant DMRs in newborn methylation analysis were annotated were significantly enriched (P < .05) for canonical pathways relevant to immune function in asthmatic patients, including endothelial nitric oxide synthase (eNOS) signaling, the inflammasome, and nuclear factor κB (NF-kB) signaling (see Table E9). Enriched disease processes and biologic functions included several involving immune function and others involving immune and organ development (see Table E9). Given the larger number of implicated genes for childhood methylation, many more pathways, disease processes, and biological functions were enriched (see Table E10). There was substantial overlap in newborns and children in the significantly enriched pathways and diseases and biological function relevant to immune function, immunologic disease, and development (see Fig E8 in this article’s Online Repository at www.jacionline.org). As an example, Fig 3 shows the network of 4 overlapping disease and biological processes between newborns and children: tissue morphology, immunological disease, inflammatory disease, and cell-mediated immune response.
FIG 3.
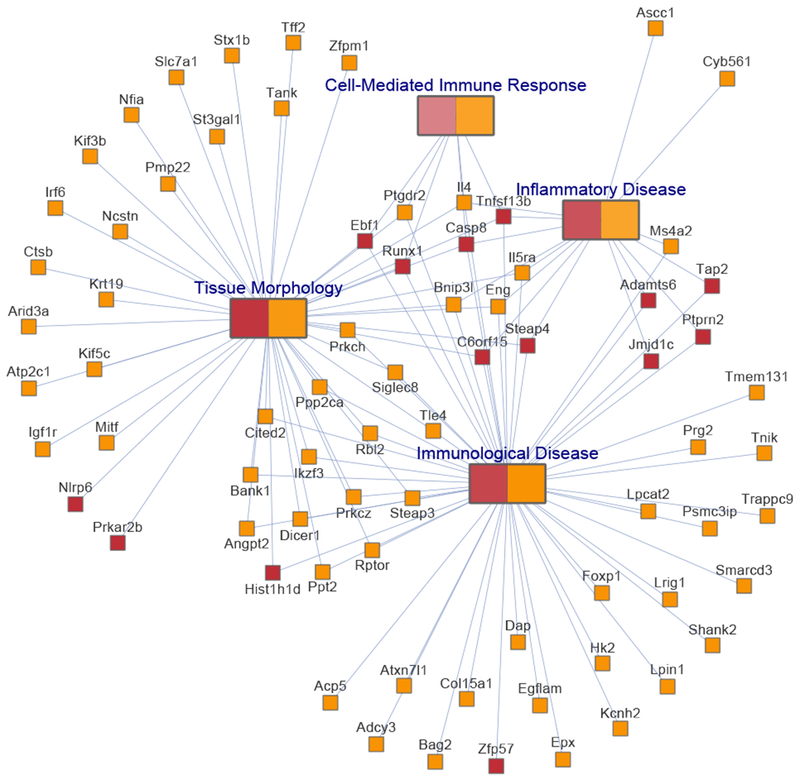
A network is shown for 4 categories of disease and biological functions overlapping between analyses of asthma in relation to either newborn or childhood methylation: immunological disease, cell-mediated immune response, inflammatory disease, and tissue morphology. A gene is connected to a disease or function if it has been previously shown to be involved in it. All genes marked in red are implicated from newborn methylation analyses, and those marked in in orange are implicated from childhood methylation analyses.
Druggable targets
Among regions differentially methylated in newborns in relation to later asthma, RUNX1 is a target of the agent CHEMBL2093862 and CASP8 is the target of CHEMBL2105721 (Nivocasan), an inhibitor of this caspase and 2 others (1 and 9). Among genes with individual CpGs significantly differentially methylated in childhood in relation to asthma, KCNH2 (3 significant CpGs) is a target of several approved drugs with mechanism of action of blocking HERG (human Ether-à-go-go-related gene), including the antiarrhythmic agents amiodarone hydrochloride, dofetilide, and sotalol. Notably, sotalol is also a β-adrenergic receptor antagonist. IL5RA (2 significant CpGs) is the target for a drug approved for use in patients with severe asthma, benralizumab, the mechanism of action of which is antagonism of this gene.49 Several other genes implicated by either an individual CpG (16 genes) or DMR analysis (5 genes, including IGF1R) are targets for approved or potential drugs (see Tables E11 and E12 in this article’s Online Repository at www.jacionline.org).
DISCUSSION
This epigenome-wide meta-analysis of the association between childhood asthma and DNA methylation measured at birth or childhood identified numerous novel CpGs and regions differentially methylated in relation to this common health outcome. The 9 CpGs and 35 regions significantly differentially methylated in relation to asthma in newborn blood DNA are potential markers of risk for disease development. There were many more statistically significant associations of asthma in relation to childhood DNA methylation, with 179 CpGs and 36 regions; these might reflect both the risk for and effects of this disease.50
Among the significant CpGs in newborns, 6 were in DNAse hypersensitivity sites, supporting a potential regulatory effect on gene function. Additionally, genes to which cg13427149 (GPATCH2/SPATA17) and cg16792002 (MAML2) annotate have previously been associated with obesity phenotypes,51,52 conditions related to childhood asthma. This supports the potential functional importance and asthma relevance of our newborn findings.
Some CpGs on the 450K array have been reported as potentially polymorphic by virtue of location near SNPs.41 Given that many of the nearby SNPs are low frequency and most will not interfere with probe binding, which would generate a truly spurious result rather than filter these in advance; in the PACE consortium we examined statistically significant CpGs post hoc for occurrence on lists of potentially problematic CpGs in the literature, as recently recommended by others.17,18 Lists of potentially problematic probes change over time, as do underlying gene annotations.53 We note that 2 of the 9 significant CpGs in newborn methylation (ch.11.109687686R and ch.6.1218502R) were flagged as potentially nonspecific (“ch”) probes by Chen et al.41 We provide association results for these because they might be useful to others but, acknowledging this caveat, do not include them in downstream analyses that assume certainty regarding gene localization. With respect to the issue of CpGs previously reported as near SNPs, we visually assessed plots of all significant CpGs in 3 of our largest cohorts (MoBa1 and Generation R for newborn methylation [see Fig E9 in this article’s Online Repository at www.jacionline.org] and STOPPA for childhood methylation [see Fig E10 in this article’s Online Repository at www.jacionline.org]) to verify unimodal distributions.
We identified many more CpGs and DMRs associated with later asthma, likely because these also capture disease effects. Our findings might also reflect different pathophysiologic mechanisms related to newborn versus childhood methylation and asthma. A comprehensive search for methylation signals at birth that predict later development of asthma likely requires much larger sample sizes given the intervening effects of exposures and developmental processes that may outweigh effects of small methylation differences present at birth.54 However, although overlap at the level of specific CpGs or DMRs was low, there was substantial overlap at the pathway and network levels (Fig 3 and see Fig E8).
To follow-up our differentially methylated signals for potential functional effect, we examined correlations with gene expression. Because of the relatively small sizes of the paired gene expression data sets in newborns or children, we also examined a much larger data set of adults to increase power. Although the number of subjects in data sets of newborns or children with both gene expression and methylation data were modest (range, 38-248), limiting power to find correlations, we found that a high proportion of CpGs and DMRs related to asthma were also correlated with gene expression in at least 1 data set in this age range. This further supports the functional effect of our methylation findings.
Our search for druggable targets identified 2 genes from the newborn DMR analysis that are targets for either approved or potential drugs. The childhood analysis identified more drug targets. One of these genes, IL5RA, already has an approved asthma drug that inhibits its product. This analysis further supports the relevance to asthma pathogenesis and the potential clinical usefulness of these findings. Investigating the potential to repurpose approved drugs for new indications has been recently highlighted as a cost-effective way to develop new therapeutic modalities.55
We meta-analyzed results across studies by using fixed-effects meta-analysis with inverse variance weighting. Recently, Rice et al25 have summarized issues regarding the choice of meta-analytic models for combining study-specific results in genomic analyses and show that the inverse variance-weighted average estimates a reasonable and interpretable parameter, even under the assumption that effect sizes differ. Furthermore, they point out that a fixed-effects meta-analysis does not require the assumption of homogeneity. Rice et al25 also emphasize the importance of evaluating meta-analysis effect estimates and significance tests along with visualization of study-specific estimates rather than relying on a single statistical estimate of heterogeneity. Accordingly, we provide forest plots to show the consistency of study-specific findings for all significant meta-analysis results (see Fig E1 for newborn methylation and Fig E3 for childhood methylation). Furthermore, we performed a systematic leave-one-out meta-analysis for all significant CpGs, in which we leave each cohort out one by one (see Fig E2 for newborn methylation and Fig E4 for childhood methylation). In addition, where there is even nominal evidence for heterogeneity (Pheterogeneity < .05), we provide random-effects results in Tables E3 (newborn methylation) and E5 (childhood methylation).
We recognize various limitations. As in most EWASs,13 as well as GWAS meta-analyses,56 asthma was defined by questionnaire. As in Xu et al,13 we used a reported doctor’s diagnosis combined with symptoms and medication use. Although use of self-reported outcomes can lead to misclassification, this should be nondifferential with respect to methylation and thus should lead to bias toward the null rather than create false-positive findings. We did not stratify the analyses by allergic status because most cohorts do not have objective measures of atopy, and in many cohorts sample size would have been inadequate for stratification.
We also note that the diverse cohorts included in the analysis could have introduced heterogeneity based on ancestry or, in the analysis of methylation in older children, 2 studies in older adolescents. However, in the studies of older children, non-European ancestry of older children did not appear to be influential in sensitivity analyses. Although magnitudes of the associations are modest, this is consistent with other genome-wide analyses of methylation in newborns and children in relation to various exposures.15,57,58 These effect sizes are not surprising given that highly reproducible genetic signals discovered in asthma GWASs, such as ORMDL3,59 are also modest.
We used logistic regression in the prospective analyses of newborn methylation in relation to asthma rather than Cox regression, which is not commonly used in high-dimensional genomic studies. If time to asthma were available or could be estimated reliably, a Cox model would be more efficient. However, for asthma, the exact time to disease development is poorly estimated. Thus epidemiologic studies generally use age at diagnosis, but there can be a very long lag between disease onset and diagnosis. In our scenario, where the exact time to asthma is unknown, using error-prone outcomes can actually result in larger bias. Thus, considering the tradeoff between bias and efficiency, logistic regression is the better option. We also note that where the condition under study has less than 10% prevalence, as is the case for our outcome of asthma diagnosed at school age, the odds ratio is a good approximation of the hazard ratio.60 To address the important aspect of age at diagnosis of asthma, we used age at diagnosis for the harmonized definition of asthma. With the exception of a couple of studies, in which sensitivity analyses removing them did not suggest undue influence, the range of mean ages was not large.
Unmeasured confounding is a concern in all analyses of observational data. With high-dimensional genomic data, variability caused by batch effects is an additional potential source of unmeasured confounding.61 In this meta-analysis each cohort corrected for batch effects by using methods most suitable for their own data. In most studies methylation analyses were completed over a short period of time, which greatly reduces batch effects.61 When using methods such as adjustment for batch variables or ComBat, one must specify the putative batch variables. To the extent that there are unknown factors contributing to laboratory variability, there might be residual confounding. Various methods have been proposed to attempt to address unmeasured confounding in high-dimensional data. However, in meta-analyses findings tend to be significant because they are consistent across studies. Thus the chance that unmeasured confounding is operating in the same manner across studies done in different countries with methylation measured in different laboratories and at different times, resulting in false-positive significant associations in the meta-analysis, is greatly reduced. Furthermore, in the childhood methylation analysis we have substantial replication of findings from a recently published meta-analysis,13 even after overlapping subjects are removed. In addition, the consistency of our findings from blood DNA with results for DNA isolated from 2 tissues highly relevant for asthma, eosinophils and nasal respiratory epithelium, provides compelling evidence that our findings are not driven by unmeasured confounding.
Identification of DMRs provides a way to reduce the dimensionality of the epigenome-wide methylation data and can identify associations at the regional level, where there are not individually significant CpGs. The 2 methods that we used for DMR identification, DMRcate and comb-p, are the only 2 published methods available for use with results of meta-analyses.29,30 A recent review noted that the various methods published for identifying DMRs use different assumptions and statistical approaches and thus rarely identify exactly the same regions.62 Accordingly, to reduce false-positive results, we reported only DMRs identified as statistically significant by both methods.
We measured DNA methylation in whole blood, a mix of cell types. Cell counts were not measured, but we adjusted our models for estimated cell counts using established reference-based methods to address confounding by cell-type differences.21 For childhood, as opposed to newborn, methylation, we used an adult reference panel because a suitable one is not available for children. Notably, the considerable overlap between our findings in whole blood and smaller studies of 2 highly asthma-relevant tissues, nasal epithelium, an excellent proxy for airway epithelium in studies of asthma,63 and purified eosinophils, greatly reduces the concern that our findings are false-positive results because of failure to fully account for the influence of asthma on white blood cell proportions.
In addition to confirmation of findings in studies of eosinophils and nasal respiratory epithelium and the high power resulting from meta-analyses, other strengths of the study include our efforts to standardize the definition of asthma across studies, the large sample size provided by meta-analyses, and evaluation of potential biological implications of our findings through detailed examination of functional annotation, pathway analysis, correlation of differentially methylated sites with gene expression, and consideration of potential druggable targets.
In summary, we identified numerous novel CpGs and regions associated with childhood asthma in relation to DNA methylation measured either at birth in prospective analyses or in childhood in cross-sectional analyses. Many of the genes annotated to these CpGs and regions are significantly enriched for pathways related to immune responses crucial in asthmatic patients; several genes are targets for either approved or investigational drugs. Most differentially methylated CpGs or regions correlated with expression at a nearby gene. Many more individual CpGs were differentially methylated in childhood in relation to their current asthma status. There was appreciable overlap with findings in nasal respiratory epithelium and purified eosinophils. The CpGs and regions identified in newborns might be potential biomarkers of later asthma risk; those identified in childhood likely reflect both processes that affect disease risk and effects of having the disease. The novel genes implicated by this study might shed new light on asthma pathogenesis.
Supplementary Material
Key message.
This large-scale genome-wide meta-analysis of DNA methylation and childhood asthma identified novel epigenetic variations related to asthma in newborns and children.
Acknowledgments
We thank Dr Frank Day (National Institute of Environmental Health Sciences [NIEHS]) and Jianping Jin of Westat (Durham, NC) for expert computational assistance and Erin Knight (NIEHS) for assistance with the literature review. See the supplementary materials in this article’s Online Repository at www.jacionline.org for complete acknowledgements.
Supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. See supplemental materials in this article’s Online Repository at www.jacionline.org for complete funding information for individual studies.
Abbreviations used
- ALSPAC
Avon Longitudinal Study of Parents and Children
- BAMSE
Children, Allergy, Milieu, Stockholm, Epidemiology
- BIOS
Biobank-based Integrative Omics Studies
- CHOP
European Childhood Obesity Project
- CHS
Children’s Health Study
- DMR
Differentially methylated region
- EDEN
Etude des Déterminants pré et post natals du développement et de la santé de l’Enfant
- EWAS
Epigenome-wide association study
- FDR
False discovery rate
- GALA II
Genes-environments & Admixture in Latino Americans
- GOYA
Genetics of Overweight Young Adults
- GWAS
Genome-wide association study
- ICAC
Inner City Asthma Consortium
- IoW
Isle of Wight 3rd Generation Study
- INMA
Infancia y Medio Ambiente
- MoBa
Norwegian Mother and Child
- NEST
Newborn Epigenetics STudy
- NFBC
Northern Finland Birth Cohort
- PACE
Pregnancy And Childhood Epigenetics
- PIAMA
Prevention and Incidence of Asthma and Mite Allergy
- SNP
Single nucleotide polymorphism
- STOPPA
Swedish Twin study On Prediction and Prevention of Asthma
- UCSC
University of California, Santa Cruz
Footnotes
Disclosure of potential conflict of interest: C. Ruiz-Arenas receives grant support from Agència de Gestió d’Ajuts Universitaris i de Recerca. S. S. Oh, C. Eng, and E. G. Burchard receive grant support from the NIH and the Tobacco-Related Disease Research Program. I. V. Yang and C. V. Breton receive grant support from the National Institutes of Health (NIH). C. Söderhäll receives grant support from several competitive grants from public and private sources and receives royalties from book chapters in study material. R. Arathimos and G. C. Sharp receive support from the Medical Research Council. E. Kajantie receives grant support from the European Commission, Academy of Finland, Foundation for Pediatric Research, Sigrid Jus×lius Foundation, Signe and Ane Gyllenberg Foundation, and Juho Vainio Foundation. G. Pershagen receives grant support from the Swedish Research Council. C. L. Relton receives grant support from Wellcome Trust. C. Almqvist receives grant support from the Swedish Research Council through the Swedish Initiative for Research on Microdata in the Social And Medical Sciences (SIMSAM) framework, Stockholm County Council (ALF-projects), Swedish Heart-Lung Foundation, and Swedish Asthma and Allergy Association’s Research Foundation. A. J. Henderson receives grant support from the Medical Research Council and Wellcome Trust. E. Melén received grant support from the European Research Council during conduct of the study and lecture fees from Thermo Fisher Scientific and Meda outside the submitted work. G. H. Koppelman receives grant support from the Lung Foundation of the Netherlands, MEDALL EU FP7, the UBBO EMMIUS Foundation, TEVA The Netherlands, Vertex, GlaxoSmithKline, and the TETRI Foundation. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Bisgaard H, Szefler S. Prevalence of asthma-like symptoms in young children. Pediatr Pulmonol 2007;42:723–8. [DOI] [PubMed] [Google Scholar]
- 2.Wjst M, Sargurupremraj M, Arnold M. Genome-wide association studies in asthma: what they really told us about pathogenesis. Curr Opin Allergy Clin Immunol 2013;13:112–8. [DOI] [PubMed] [Google Scholar]
- 3.Weiss ST, Silverman EK. Pro: genome-wide association studies (GWAS) in asthma. Am J Respir Crit Care Med 2011;184:631–3. [DOI] [PubMed] [Google Scholar]
- 4.DeVries A, Vercelli D. Epigenetic mechanisms in asthma. Ann Am Thorac Soc 2016;13(suppl 1):S48–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma S, Chhabra D, Kho AT, Hayden LP, Tantisira KG, Weiss ST. The genomic origins of asthma. Thorax 2014;69:481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunawardhana LP, Gibson PG, Simpson JL, Benton MC, Lea RA, Baines KJ. Characteristic DNA methylation profiles in peripheral blood monocytes are associated with inflammatory phenotypes of asthma. Epigenetics 2014;9:1302–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy TM, Wong CC, Arseneault L, Burrage J, Macdonald R, Hannon E, et al. Methylomic markers of persistent childhood asthma: a longitudinal study of asthma-discordant monozygotic twins. Clin Epigenetics 2015;7:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicodemus-Johnson J, Naughton KA, Sudi J, Hogarth K, Naurekas ET, Nicolae DL, et al. Genome-wide methylation study identifies an IL-13-induced epigenetic signature in asthmatic airways. Am J Respir Crit Care Med 2016;193:376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rastogi D, Suzuki M, Greally JM. Differential epigenome-wide DNA methylation patterns in childhood obesity-associated asthma. Sci Rep 2013;3:2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang IV, Pedersen BS, Liu A, O’Connor GT, Teach SJ, Kattan M, et al. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol 2015;136:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeVries A, Wlasiuk G, Miller SJ, Bosco A, Stern DA, Lohman IC, et al. Epigenome-wide analysis links SMAD3 methylation at birth to asthma in children of asthmatic mothers. J Allergy Clin Immunol 2017;140:534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arathimos R, Suderman M, Sharp GC, Burrows K, Granell R, Tilling K, et al. Epigenome-wide association study of asthma and wheeze in childhood and adolescence. Clin Epigenetics 2017;9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu CJ, Soderhall C, Bustamante M, Baiz N, Gruzieva O, Gehring U, et al. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med 2018;6:379–88. [DOI] [PubMed] [Google Scholar]
- 14.Felix JF, Joubert BR, Baccarelli AA, Sharp GC, Almqvist C, Annesi-Maesano I, et al. Cohort profile: Pregnancy And Childhood Epigenetics (PACE) consortium. Int J Epidemiol 2018;47:22–23u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet 2016;98:680–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet 2016; 9:436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews SV, Ladd-Acosta C, Feinberg AP, Hansen KD, Fallin MD. “Gap hunting” to characterize clustered probe signals in Illumina methylation array data. Epigenetics Chromatin 2016;9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu MC, Kuan PF. A guide to Illumina BeadChip data analysis. Methods Mol Biol 2018;1708:303–30. [DOI] [PubMed] [Google Scholar]
- 19.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8:118–27. [DOI] [PubMed] [Google Scholar]
- 20.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet 2007;3:1724–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, LMcKenney S, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics 2016;11:354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012;7:e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Psaty BM, Sitlani C. The Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium as a model of collaborative science. Epidemiology 2013;24:346–8. [DOI] [PubMed] [Google Scholar]
- 25.Rice K, Higgins J, Lumley T. A re-evaluation of fixed effect(s) meta-analysis. J R Stat Soc A 2018;181:205–27. [Google Scholar]
- 26.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc B 1995;57:289–300. [Google Scholar]
- 28.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet 2011;88: 586–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 2012;28:2986–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, VLord R, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gref A, Merid SK, Gruzieva O, Ballereau S, Becker A, Bellander T, et al. Genome-wide interaction analysis of air pollution exposure and childhood asthma with functional follow-up. Am J Respir Crit Care Med 2017;195:1373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arshad SH, Karmaus W, Zhang H, Holloway JW. Multigenerational cohorts in patients with asthma and allergy. J Allergy Clin Immunol 2017;139:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rager JE, Bailey KA, Smeester L, Miller SK, Parker JS, Laine JE, et al. Prenatal arsenic exposure and the epigenome: altered microRNAs associated with innate and adaptive immune signaling in newborn cord blood. Environ Mol Mutagen 2014;55:196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rojas D, Rager JE, Smeester L, Bailey KA, Drobna Z, Rubio-Andrade M, et al. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol Sci 2015;143:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guxens M, Ballester F, Espada M, Fernandez MF, Grimalt JO, Ibarluzea J, et al. Cohort profile: the INMA—Infancia y Medio Ambiente—(Environment and Childhood) project. Int J Epidemiol 2012;41:930–40. [DOI] [PubMed] [Google Scholar]
- 36.Bonder MJ, Luijk R, Zhernakova DV, Moed M, Deelen P, Vermaat M, et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat Genet 2017;49:131–8. [DOI] [PubMed] [Google Scholar]
- 37.Zhernakova DV, Deelen P, Vermaat M, van Iterson M, van Galen M, Arindrarto W, et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat Genet 2017;49:139–45. [DOI] [PubMed] [Google Scholar]
- 38.Wickman M, Kull I, Pershagen G, Nordvall SL. The BAMSE project: presentation of a prospective longitudinal birth cohort study. Pediatr Allergy Immunol 2002;13(suppl 15):11–3. [DOI] [PubMed] [Google Scholar]
- 39.Breeze CE, Paul DS, van Dongen J, Butcher LM, Ambrose JC, Barrett JE, et al. eFORGE: a tool for identifying cell type-specific signal in epigenomic data. Cell Rep 2016;17:2137–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014;30:523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013;8:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Research 2017;Vol. 45(Database issue):D896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leslie R, O’Donnell CJ, Johnson AD. GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 2014;30:i185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res 2014;42:D1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang IV, Pedersen BS, Liu AH, O’Connor GT, Pillai D, Kattan M, et al. The nasal methylome and childhood atopic asthma. JAllergy Clin Immunol 2017;139:1478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferland C, Guilbert M, Davoine F, Flamand N, Chakir J, Laviolette M. Eotaxin promotes eosinophil transmigration via the activation of the plasminogen-plasmin system. J Leukoc Biol 2001;69:772–8. [PubMed] [Google Scholar]
- 47.Laprise C The Saguenay-Lac-Saint-Jean asthma familial collection: the genetics of asthma in a young founder population. Genes Immun 2014;15:247–55.24646526 [Google Scholar]
- 48. Chen W, Wang T, Pino-Yanes M, Forno E, Liang L, Yan Q, et al. An epigenome-wide association study of total serum IgE in Hispanic children. J Allergy Clin Immunol 2017;140:571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nair P, Wenzel S, Rabe KF, Bourdin A, Lugogo NL, Kuna P, et al. Oral glucocorticoid-sparing effect of benralizumab in severe asthma. N Engl J Med 2017;376:2448–58. [DOI] [PubMed] [Google Scholar]
- 50.Tost J A translational perspective on epigenetics in allergic diseases. J Allergy Clin Immunol 2018;142:715–26. [DOI] [PubMed] [Google Scholar]
- 51.Velez Edwards DR, Naj AC, Monda K, North KE, Neuhouser M, Magvanjav O, et al. Gene-environment interactions and obesity traits among postmenopausal African-American and Hispanic women in the Women’s Health Initiative SHARe Study. Hum Genet 2013;132:323–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fox CS, Liu Y, White CC, Feitosa M, Smith AV, Heard-Costa N, et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet 2012;8:e1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yandell M, Ence D. A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet 2012;13:329–42. [DOI] [PubMed] [Google Scholar]
- 54.Xu CJ, Bonder MJ, Soderhall C, Bustamante M, Baiz N, Gehring U, et al. The emerging landscape of dynamic DNA methylation in early childhood. BMC Genomics 2017;18:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sachs RE, Ginsburg PB, Goldman DP. Encouraging new uses for old drugs. JAMA 2017;318:2421–2. [DOI] [PubMed] [Google Scholar]
- 56.Demenais F, Margaritte-Jeannin P, Barnes KC, Cookson WOC, Altmuller J, Ang W, et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet 2018;50:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joubert BR, den Dekker HT, Felix JF, Bohlin J, Ligthart S, Beckett E, et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun 2016;7:10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharp GC, Salas LA, Monnereau C, Allard C, Yousefi P, Everson TM, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet 2017;26:4067–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007;448:470–3. [DOI] [PubMed] [Google Scholar]
- 60.Zhang J, Yu KF. What’s the relative risk? A method of correcting the odds ratio in cohort studies of common outcomes. JAMA 1998;280:1690–1. [DOI] [PubMed] [Google Scholar]
- 61.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 2010;11:733–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teschendorff AE, Relton CL. Statistical and integrative system-level analysis of DNA methylation data. Nat Rev Genet 2018;19:129–47. [DOI] [PubMed] [Google Scholar]
- 63.Yang IV, Richards A, Davidson EJ, Stevens AD, Kolakowski CA, Martin RJ, et al. The nasal methylome: a key to understanding allergic asthma. Am J Respir Crit Care Med 2017;195:829–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.