

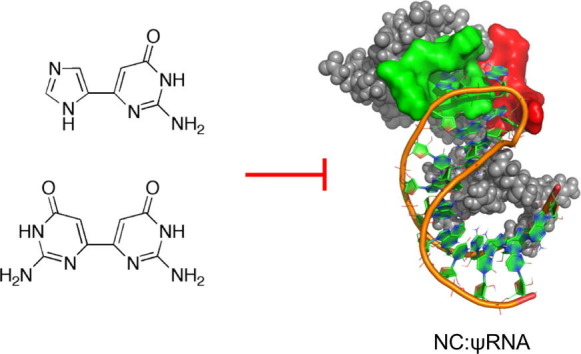
Graphical abstract
Keywords: Fleximers, Pyrimidine, HIV-1, NC, Synthesis
Abstract
Anti-HIV-1 drug design has been notably challenging due to the virus’ ability to mutate and develop immunity against commercially available drugs. The aims of this project were to develop a series of fleximer base analogues that not only possess inherent flexibility that can remain active when faced with binding site mutations, but also target a non-canonical, highly conserved target: the nucleocapsid protein of HIV (NC). The compounds were predicted by computational studies not to function via zinc ejection, which would endow them with significant advantages over non-specific and thus toxic zinc-ejectors. The target fleximer bases were synthesized using palladium-catalyzed cross-coupling techniques and subsequently tested against NC and HIV-1. The results of those studies are described herein.
1. Introduction
For some time, the Seley-Radtke group has designed and synthesized various classes of flexible purine nucleos(t)ides, or “fleximers”.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 These novel nucleosides were designed to investigate how flexibility in the nucleobase could potentially affect receptor-ligand recognition and function. In addition, their flexible design allows them to overcome issues with binding site mutations thus retaining their activity.
To date, fleximers have shown key advantages over the corresponding purine-base nucleosides.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 For example, the distal guanosine fleximer (Flex-G, Fig. 1 ) proved to be an inhibitor of S-adenosyl-l-homocysteine hydrolase by adapting a syn conformation, thereby placing the exocyclic amino group such that it mimicked the amino group from an adenosine nucleobase.3, 4 Moreover, the guanosine fleximer triphosphate (Flex-GTP) was shown to be a superior substrate of human GDP-l-fucose pyrophosphorylase compared to the natural substrate GTP,5 likely due to the fleximer’s ability to interact with amino acids in the active site not accessible by GTP.6 This also allowed Flex-GTP to retain all activity when essential catalytic residues needed for GTP binding were mutated.5, 6 Recently, a series of fleximers possessing acyclic sugars exhibited broad spectrum antiviral activities against coronaviruses, flaviviruses and filoviruses, further supporting their significance.10, 11
Fig. 1.

Guanosine distal and proximal Fleximers.
In an effort to explore the fleximer approach to other viruses such as HIV-1, a virus with the ability to readily mutate and develop antiviral resistance, a series of fleximers were designed and studied in silico for their ability to inhibit the virus’ nucleocapsid protein, NC.14, 15, 16, 17
NC plays several key roles in HIV-1 replication. Through non-specific binding, it acts as a chaperone protein, partially protecting the viral nucleic acids (NAs).18 During reverse transcription, NC directs the annealing of cellular tRNA(Lys,3) primer to the HIV-1 primer binding site, thus initiating the synthesis of the (−)-strong stop DNA.19, 20 NC then facilitates the two strand transfers required for (−) and (+) strand synthesis. It is also implicated to be a vital element in vDNA integration.21, 22 Prior to encapsidation, NC discriminates viral from host NA by selectively binding to the HIV-1 Ψ-encapsidation signal sequence.23, 24, 25 In addition, in vitro studies have shown that NC may chaperone the dimerization of the two copies of HIV-1 viral genomic RNA by rearranging the kissing complex into an extended duplex through a series of stabilizing and destabilizing events, an important step prior to encapsidation.26, 27
Because of NC’s interaction with multiple highly conserved sequences of the HIV-1 genome, and being essential in all HIV-1 subtypes, NC represents a powerful drug target for developing novel antivirals.28, 29, 30, 31 More importantly, it is thought to be highly resistant to mutation due to its multifunctional role, thus providing a significant advantage over other protein targets.26, 32 Thus, inhibitors of the interaction between NC and the viral nucleic acids could provide a new approach to antiretroviral therapy.14, 33 For this purpose, a series of fleximers were computationally designed with that goal in mind. Herein we report the synthesis and biological results for a series of compounds that were predicted to interact with NC.
2. Computational studies
In many structural studies done to date on NC,16, 17, 34, 35, 36 a guanosine residue was shown to consistently stack with the W37 residue, whether bound to DNA (PBS-DNA) or RNA (Ψ-RNA). As such, the inherent flexibility of the fleximer guanine analogue was predicted to affect binding and potentially result in inhibition.
Based on this hypothesis, a number of fleximer nucleosides were initially designed, however the early results showed that the sugar moiety on the fleximer nucleoside provided no benefit over the fleximer base itself. As a result, the corresponding fleximer base analogues were therefore pursued, since this would signfiicantly shorten the synthetic route. Thus, the fleximer bases were then tested computationally against the NMR structure of the NC in complex with a small molecule inhibitor (Figs. 2 and 3 ).37 To this end, a computational protocol was established and refined in the group of Botta.14, 33, 38 Several NC binding small molecules have already been discovered through this protocol, supporting its validity.14, 15, 33, 38
Fig. 2.

Target guanine fleximer bases and bipyrimidines.
Fig. 3.

Docking-based predicted binding conformation of 1–4 and guanine within the hydrophobic pocket of the NC.14, 33, 38 A) distal fleximer guanine base (1), B) proximal fleximer guanine base (2), C) distal guanine bipyrimidine (3), D) proximal guanine bipyrimidine (4) and E) guanine bound to NC. The protein is shown as green cartoon and lines (residues within 4 Å from each ligand are shown and labelled). H-bonds are highlighted by black dashed lines. Zn ions are shown as grey spheres.
The docking results of the fleximer bases on NC revealed several key advantages for the proposed target compounds. The docking conformation of fleximer bases 1–4 (Fig. 3) within the hydrophobic pocket that is located in correspondence of W37 showed an excellent structural overlay with respect to the guanine base. Moreover, all of the fleximer bases were able to establish a network of H-bond interactions with the backbone atoms of key residues in the hydrophobic pocket (i.e. K33, G35, W37, and M46, Fig. 3A–D) that is highly comparable to that established by the guanine base (Fig. 3E). Additionally, 1–4 adopted a similar stacking conformation to W37 as is observed with the natural guanine. However the additional rotatable bond allowed for the pyrimidine moiety to extend and interact with the neighboring residues (i.e. K47 in the binding mode of 4, Fig. 3D) into the solvent area.
The distal guanine fleximer base (1) was predicted to bind stronger than the proximal base (2) and, notably, slightly stronger than the guanine base (Table 1 ), although scoring values calculated by the FRED docking program with the Chemgauss4 function were highly comparable to each other.39, 40 Thus, the observed differences between guanine, 1 and 2 may not be significant.
Table 1.
FRED scores.
| Compound | FRED score (Chemgauss4 function)a |
|---|---|
| Guanine | −6.36 |
| 1 | −6.77 |
| 2 | −6.26 |
| 3 | −6.08 |
| 4 | −6.14 |
Adimensional, the lower the score, the stronger affinity.
In addition, the docking study surprisingly showed that the bipyrimidine scaffold (3 and 4, Fig. 2), would also be highly advantageous, although to a slightly lesser extent than the parent imidazole-pyrimidine scaffold (Table 1), thus those targets were pursued as well.
As a result of this molecular modeling analysis, compounds 1–4 (Fig. 2) were selected as the best starting candidates as proof of concept.
3. Chemistry
The intended route to achieve both sets of flexible purine and bipyrimidine bases utilized palladium-catalyzed cross-coupling, with the pyrimidine as the organometallic coupling partner and the imidazole as the halogenated coupling partner. The goal was to achieve two products from one reaction as the organometallic moiety has been shown to undergo homocoupling during cross-coupling reactions.41, 42, 43, 44, 45
As the distal compounds were predicted to be better binders to NC, the first goal was to install the organometal on the C-6 of 5 to avoid protecting the exocyclic amine. To obtain the iodinated intermediate, commercially available 2-amino-6-chloro-4-methoxypyrimidine was iodinated using hydroiodic acid (Scheme 1 ). The reaction was then neutralized and filtered, and the precipitate recrystallized in ethanol to obtain 5.
Scheme 1.
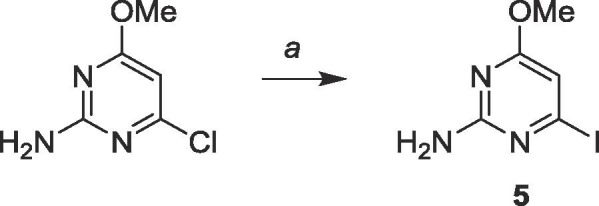
Reagents and conditions: a. HI (57%), 0 °C to rt, 72 h.
Since the proximal intermediate 2-amino-4-methoxy-5-tributylstannylpyrimidine was easily achievable starting with 2-amino-5-iodo-4-methoxypyrimidine,9, 10, 11 similar methodologies were applied to obtain the 2-amino-4-methoxy-6-tributylstannylpyrimidine intermediate 6, using various palladium catalysts as well as a range of temperatures, but none of the conditions yielded the desired organostannane 6 (Scheme 2 ).
Scheme 2.

Attempted synthesis of 6.
The organoborane 7 was then considered for subsequent Suzuki coupling (Scheme 3 ), however, the boronic ester could not be generated under any conditions.
Scheme 3.

Attempted synthesis of 7.
Because the cross-coupling intermediates could not be obtained through palladium catalysis, harsher conditions were used to achieve the desired coupling partners. The exocyclic amine was protected (Scheme 4 ) with TMS as it is immune to Grignard and lithium reagents but can be cleaved easily in mildly acidic conditions.46 Each TMS group was added sequentially as opposed to simultaneously to form 8 in situ. Lithium halogen exchange using n-BuLi, followed by metalation with tributyltin chloride finally produced deprotected organostannane 6. Characterization through 1H and 13C NMR in addition to MS confirmed the presence of the product however 6 proved highly unstable and decomposed rapidly.
Scheme 4.

Reagents and conditions: a. EtMgBr, TMSCl, THF, −78 °C; b. (i) n-BuLi, THF, −78 °C, 10 min, (ii) SnBu3Cl, −78 °C-rt, 18 h.
As installation of the metal on the pyrimidine proved to be difficult, and literature showed that an organozinc intermediate could be generated in situ with the imidazole moiety for subsequent Negishi coupling, the previous methodology was abandoned.47
A benzyl (Bn) protecting group was initially chosen as Bn’s are robust against many conditions.46 The organozinc 10 was generated in situ and subsequent Negishi coupling with 5 (Scheme 5 ) yielded the protected distal fleximer 11, albeit in poor yield (24%).
Scheme 5.

Reagents and conditions: a. NaH (95%), BnBr, TBAI, THF, reflux, 18 h; b. (i) EtMgBr, THF, −78 °C, (ii) ZnCl2, 2 h, rt; c. 5, Pd(PPh3)4, CuI, THF, reflux, 18 h.
Moreover, the Bn and methyl groups were extremely difficult to deprotect (Scheme 6 ). Hydrogenation using Pd/C in the presence of H2 at room temperature was thought to be sufficient to remove the Bn group, however no reaction occurred. This was ultimately achieved by heating the reaction mixture of 11 with ammonium formate and Pd/C in EtOH to 120 °C. Boron tribromide (BBr3) was employed to deprotected the methyl, however, a solubility problem arose when attempting to demethylate 13 in DCM, and therefore deprotection of the methyl was more efficient when performed prior to deprotection of the Bn (Scheme 6).
Scheme 6.
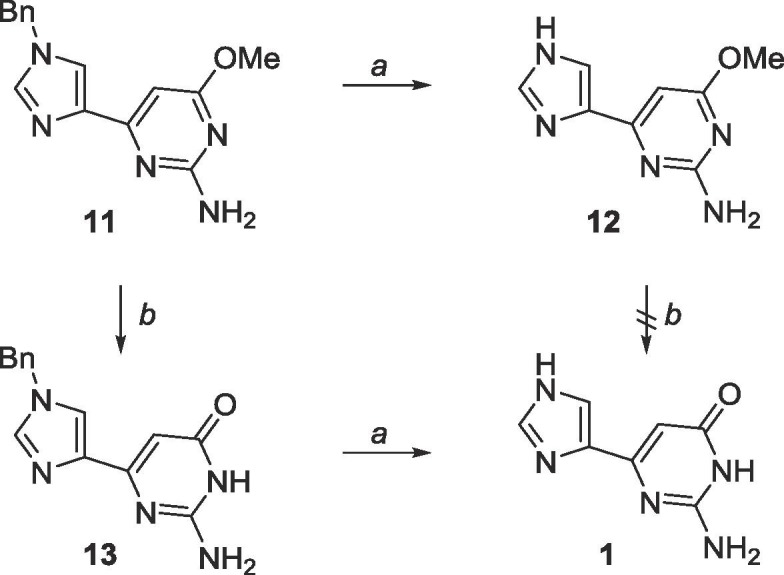
Reagents and conditions: a. ammonium formate, Pd/C, EtOH, 120 °C, 48 h; b. BBr3, CH2Cl2, rt, 72 h.
Unfortunately, these molecules proved difficult to purify via column chromatography, likely due to either their polar nature or their ability to stack efficiently from the presence of two heteroaromatic moieties and multiple hydrogen bonding elements. To bypass these challenges, a trityl protecting group was employed to protect the imidazole, and the exocyclic amine of the pyrimidine was protected with tert-butyloxycarbonyl (Boc, Schemes 7 and 8 ).
Scheme 7.

Reagents and conditions: a. Di-tert-butyl dicarbonate, DMAP, CH2Cl2, rt, 18 h.
Scheme 8.

Reagents and conditions: a. (i) EtMgBr, THF, −78 °C, (ii) ZnCl2, 2 h, rt; b. 14, Pd(PPh3)4, CuI, THF, rt, 18 h; c. acetic acid, rt, 18 h; d. TFA, rt, 18 h; e. BBr3, EtOAc, rt, 72 h.
Negishi coupling using these two heterocycles proved more facile and the cross-coupling reaction proceeded at room temperature (Scheme 8).47 Trityl deprotection was accomplished using acetic acid while Boc deprotection required trifluoroacetic acid. As previously mentioned, the methyl protected fleximer guanine 12 was insoluble in DCM, however, it was soluble in EtOAc, and final deprotection using BBr3 in EtOAc was successful.
Since 1 was ultimately obtained through Negishi coupling, a similar strategy was employed to obtain the analogous bipyrimidine (Scheme 9 ). Unexpectedly, the amine-linked compound 23 was produced instead. The compound was then subjected to aminolysis to convert the chloro group to an exocyclic amine, however, no starting material or product was recovered (Scheme 9).
Scheme 9.

Reagents and conditions: a. (i) EtMgBr, THF, −78 °C, 10 min (ii) ZnCl2, 2 h, rt; b. 2-amino-4-chloro-6-methoxypyrimidine, PdCl2(PPh3)2, CuI, THF, reflux, 18 h; c. NH3, CH3OH, 120 °C, Parr bomb, 48 h.
In hindsight, the presence of a palladium catalyst likely induced a Buchwald-Hartwig amination,48 leading to the synthesis of 22 and the absence of compound 21.
Because the correct cross-couplings procedures could not be easily performed on the C-6 of 5, 6-bromo-2,4-dimethoxypyrimidine (25, Scheme 10 ) was pursued instead, which which was envisioned by first brominating barbituric acid with POBr3 followed by nucleophilic substitution using sodium methoxide to afford compound 25.
Scheme 10.

Reagents and conditions: a. POBr3, N,N-dimethylaniline, toluene, 110 °C, 3 h; b. sodium methoxide, methanol, 0 °C to rt, 18 h.
To achieve the targeted bipyrimidine 3 through Stille coupling, 25 was first converted to stannane 26. Interestingly, 26 was never synthesized, however the bipyrimidine 27 was recovered at good yields (80%, Scheme 11 ). From there, two strategies were attempted to attain the desired bipyrimidine 3. First, removal of the methyl protecting groups followed by chlorination via POCl3 was tried, and intermediate 28 was used crude as it was insoluble in various purification solvents. The tetrachlorinated intermediate 29 could not be isolated as the crude reaction mixture was difficult to purify. The next approach was to directly convert the methoxy groups to amines (30) and enzymatically convert the C4-NH2 group to an —OH using adenosine deaminase. Neither the starting material nor product were recovered, likely due to the harsh conditions used.
Scheme 11.

Reagents and conditions: a. bis(tributyltin), Pd(PPh3)2Cl2, 1,4-dioxane, 120 °C, 18 h; b. BBr3, CH2Cl2, rt, 48 h; c. NH3, CH3OH, 120 °C, 72 h; d. POCl3, reflux, 18 h; e. ADA.
Since homocoupling of the pyrimidines had occurred with the dimethoxypyrimidines, it was speculated that the same conditions would likely produce a homocoupled product for the 2-amino-4-methoxypyrimidines as well, which fortuitously proved true (Scheme 12 ). Unfortunately, the product 21 proved to be highly insoluble, and impossible to purify, and was only observed via MS.
Scheme 12.

Reagents and conditions: a. bis(tributyltin), Pd(PPh3)2Cl2, 1,4-dioxane, 130 °C, 48 h; b. BBr3, CH2Cl2, rt, 48 h.
As achieving the distal analogues proved challenging, the focus then turned to synthesizing the proximal analogues. Considering the synthesis of compound 31 was facile, a straightforward Stille with the halogenated pyrimidine produced the bipyrimidine 33 (Scheme 13 ), however, similar purification and solubility issues as 21 occurred.
Scheme 13.

Reagents and conditions: a. Pd(PPh3)4, DMF, 90 °C, 18 h; b. BBr3, −78 °C-rt, CH2Cl2, 48 h.
To complete this series, the proximal fleximer guanine 2 was also pursued. Instead of using Stille cross-coupling techniques, the Negishi method used for achieving the distal fleximer guanine was employed (Scheme 14 ).47 Surprisingly, no product was observed when the reaction was allowed to stir at room temperature, and poor yields were found even after reflux.
Scheme 14.
Reagents and conditions: a. (i) EtMgBr, THF, −78 °C, (ii) ZnCl2, 2 h, rt, (iii) 32, Pd(PPh3)4, CuI, THF, rt and reflux, 18 h.
The failure of the reaction was speculated to be due to the placement of the halogen on the electron-rich carbon of the C-5 position of the pyrimidine. As palladium prefers to react with electron-deficient carbons, the oxidative addition reaction between the electron-rich halogenated coupling partner and palladium likely did not occur, which led to the absence of the desired coupled product.49 Hence, the organozinc was placed on the pyrimidine in subsequent reactions.
As predicted, the organozinc on the pyrimidine was successfully synthesized in situ, and Negishi cross-coupling followed by deprotection of the trityl group was accomplished to yield 37 (Scheme 15 ).47 Regrettably, methyl deprotection to produce the proximal fleximer guanine 2 was unsuccessful using BBr3 in either CH2Cl2 or EtOAc as the intermediate was insoluble in both. Deprotection using catalytic sulfuric acid and TMSCl in acetic anhydride also did not produce 2 (confirmed through MS). The hypothesis to explain this phenomenon ties into the in silico results by Bardon et al. that showed that the most thermodynamically stable conformation of the proximal fleximer bases is in a planar form.50 This conformation could be promoting the stacking of the bases that consequently does not allow 37 to solubilize in usual solvents.
Scheme 15.

Reagents and conditions: a. EtMgBr, TMSCl, THF, −78 °C; b. (i) EtMgBr, (ii) ZnCl2, 2 h, rt; c. 16, Pd(PPh3)4, CuI, THF, 40 °C, 18 h; d. AcOH, rt, 48 h; e. BBr3, CH2Cl2/EtOAc, or TMSCl, cat. H2SO4, Ac2O, rt, 48 h.
The dimethoxypyrimidine series was also pursued and proved more facile to obtain as no additional protection steps were required. Following the approach to realize the 2-amino-4-methoxy series, the dimethoxy proximal compounds were synthesized using the pyrimidine as the organometallic moiety (Scheme 16 ). Deprotection of the methyl groups were attempted, however, the resulting crude mixture was insoluble and thus purification was not possible.
Scheme 16.

Reagents and conditions: a. (i) EtMgBr, THF, −78 °C, (ii) ZnCl2, 2 h, rt; b. 4(5)-iodoimidazole, Pd(PPh3)4, CuI, THF, 60 °C, 18 h; c. bis(pinacolato)diboron, KOAc, PdCl2(dppf)2·CH2Cl2, 100 °C, 1 h; d. 5-bromo-2,4-dimethoxypyrimidine, PdCl2(dppf)2·CH2Cl2, Cs2CO3, 105 °C, 1 h.
Similar conditions were used to synthesize the dimethyl protected distal fleximer xanthosine as Scheme 8 (Scheme 17 ). The yield of 42 (68%) was higher than that of 18 (43%), with the major structural difference being the C-2 substitution (OMe versus NHBoc).
Scheme 17.

Reagents and conditions: a. 25, Pd(PPh3)4, CuI, THF, 6 h; b. AcOH, rt, 48 h.
4. Results
With compound 1 in hand, a 1H NMR experiment was performed with NC. If the fleximer did bind to the NA binding site of NC where W37 resides, a shift should be observed in the aromatic region of the protein, similar to that shown in Goudreau et al.37 After recording the 1H NMR spectra of NC in D2O without 1, a one equivalent molar ratio of 1 was added to the sample (dissolved in DMSO-d6). Both the aliphatic region and aromatic region (Fig. 4 ) were recorded but no significant changes in the NC signals were detected. This study does not necessarily exclude 1 as an NC binder, but does prove that the interaction is at most very weak, thus undetectable via NMR experiments.
Fig. 4.

1H NMR of NC (red) and NC with 1 (blue), aromatic region.
Unfortunately, none of the other products that were successfully purified would dissolve in the NMR solvent or in the presence of the protein, therefore the NMR studies were abandoned.
Finally, all of the target compounds were sent to the National Cancer Institute (NCI) to be tested against HIV-1 by Dr. Eric Freed. Disappointingly, none of the analogues exhibited any meaningful antiviral activity.
5. Conclusion
A number of fleximer bases were successfully synthesized via palladium-catalysed cross-couplings in this study, albeit with more difficulty than anticipated, especially during purification. The synthetic routes developed for the distal fleximer base bypassed the classic tricyclic route that has been used in the Seley-Radtke group for over a decade. If yields could be improved, this would potentially be useful for synthesis of distal fleximer nucleosides in the future. Biologically however, these molecules were not recognized by NC based on the NMR experiment performed, and were inactive against HIV-1.
While the biological results were disappointing, the project did provide a better understanding of palladium-catalysed cross-coupling strategies for fleximer bases in terms of choosing the optimum cross-coupling partners. This which has proven advantageous for other projects ongoing in our group and others, the results of which will be published as they become available.
6. Experimental section
All chemicals were obtained from commercial sources and used without further purification unless otherwise noted. Anhydrous DMF, CH3OH, DMSO and EtOH were purchased from Fisher Scientific. Anhydrous THF, acetone, CH2Cl2, CH3CN, and ether were obtained using a solvent purification system (mBraun Labmaster 130). NMR solvents were purchased from Cambridge Isotope Laboratories (Andover, MA). All 1H and 13C NMR spectra were obtained either on a JEOL ECX 400 MHz NMR, operated at 400 and 100 MHz, respectively, or a Bruker AVANCE III HD 500 MHz NMR, operated at 500 and 125 MHz, respectively, and referenced to internal tetramethylsilane (TMS) at 0.0 ppm. The spin multiplicities are indicated by the symbols s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). Reactions were monitored by thin-layer chromatography (TLC) using 0.25 mm Whatman Diamond silica gel 60-F254 pre-coated plates. Purification was performed on a Teledyne Isco CombiFlash Rf 200, and eluted with the indicated solvent system. Yields refer to chromatographically and spectroscopically (1H and 13C NMR) homogeneous materials. Mass Spectra were recorded at the UMBC MCAC for nominal using Bruker APOLLO™ II ESI/APCI - MALDI Dual Source for apex(R)-Qe FTMS or Johns Hopkins Mass Spectrometry Facility for high resolution using VG Analytical VG-70SE Magnetic Sector Mass Spectrometer.
6.1. Synthesis of 2-amino-4-iodo-6-methoxypyrimidine (5)
Commercially available 2-amino-4-chloro-6-methoxypyrimidine (5.0 g, 31.3 mmol) was suspended in 20 mL of 57 wt% HI in H2O at 0 °C. The mixture was stirred at room temperature for 72 h. The resulting sludge was diluted in 20 mL H2O and neutralized to pH 7–8 using sat. Na2CO3. The precipitate was filtered and recrystallized in EtOH to yield a white solid (4.1 g, 16.3 mmol, 52%). 1H NMR (400 MHz, DMSO-d6) δ 3.78 (s, 3H), 6.07 (s, 1H), 7.15 (br, 2H). 13C NMR (100 MHz, DMSO-d6) δ 40.7, 94.7, 160.3, 163.4, 171.5. MS (ESI, pos, CH3OH) calculated for C5H6IN3O [M+H]+ 251.96, found 251.96.
6.2. Synthesis of 2-amino-4-methoxy-6-tributylstannylpyrimidine (6)
5 (139 mg, 0.55 mmol) was dissolved under N2 in 20 mL of anhydrous THF and cooled to −78 °C. EtMgBr (3.0 M, 0.20 mL, 0.61 mmol) was added dropwise and allowed to stir for 2 min. TMSCl (0.08 mL, 0.61 mmol) was added and allowed to stir for 5 min. Again, EtMgBr (3. 0 M, 0.20 mL, 0.61 mmol) was added dropwise and allowed to stir for 2 min, then TMSCl (0.08 mL, 0.61 mmol) was added and allowed to stir for 5 min. n-Butyllithium (1.6 M, 0.4 mL, 0.61 mmol) was added dropwise and allowed warm to room temperature and stirred for 3.5 h. Tributyltin chloride (0.3 mL, 1.11 mmol) was added and the mixture was stirred for 18 min. The reaction was quenched using 10 mL NH4Cl and the solvent was removed in vacuo. The crude material was extracted into CH2Cl2 (20 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 9:1–3:1) to yield a yellow oil (126 mg, 0.30 mmol, 55 % yield). Rf = 0.80, (hexanes/EtOAc = 4:1). Compound decomposed rapidly. 1H NMR (400 MHz, CDCl3) δ 0.85–0.93 (m, 9H), 1.04–1.08 (m, 6H), 1.25–1.39 (m, 6H), 1.50–1.67 (m, 6H), 3.84 (s, 3H), 4.93 (br, 2H), 6.26 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 9.8, 13.7, 27.4, 29.0, 52.9, 107.6, 161.9, 168.4, 183.8. MS (APCI, pos, CH3CN) calculated for C17H34N3OSn [M+H]+ 416.17, found 416.17.
6.3. Synthesis of 4-(1-benzyl-1H-imidazol-4-yl)-6-methoxy-2-pyrimidinylamine (11)
1-Benzyl-4-iodo-1H-imidazole 9 (118 mg, 0.42 mmol)51 was dissolved under N2 in 25 mL of anhydrous THF and cooled to −78 °C. EtMgBr (3.0 M, 0.15 mL, 0.44 mmol) was added dropwise and allowed to stir for 10 min. ZnCl2 (0.7 M in THF, 1.2 mL, 0.84 mmol) was subsequently added dropwise, stirred at −78 °C for 10 min, warmed to room temperature and stirred for 2 h. The organozinc was added dropwise to a mixture of 5 (105 mg, 0.42 mmol), Pd(PPh3)4 (48 mg, 0.04 mmol), CuI (19 mg, 0.1 mmol) in 40 mL of anhydrous THF and allowed to stir at room temperature for 24 h. The reaction was quenched using 10 mL sat. EDTA solution and THF was removed in vacuo. The crude material was extracted into CH2Cl2 (50 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 1:1–0:1) to yield a white solid (25 mg, 0.10 mmol, 24% yield). Rf = 0.35, (EtOAc). 1H NMR (400 MHz, DMSO-d6) δ 3.78 (s, 3H), 5.23 (s, 2H), 6.41 (s, 1H), 6.46 (br, 2H), 7.29–7.37 (m, 5H), 7.66 (s, 1H), 7.89 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 51.4, 53.8, 92.2, 120.3, 127.6, 128.6, 129.2, 135.5, 138.0, 140.6, 160.6, 162.6, 171.9. HRMS (FAB) calculated for C15H15N5O [M+H]+ 282.1355, found 282.1351.
6.4. Synthesis of 4-(3H-imidazol-4-yl)-6-methoxy-2-pyrimidinylamine (12)
Synthetic procedure related to of Scheme 8 reported. 19 (100 mg, 0.34 mmol) was dissolved in 20 mL trifluoroacetic acid and stirred for 48 h. The solvent was removed and the crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 19:1–4:1) to yield a white solid (59 mg, 0.31 mmol, 91% yield). Rf = 0.50, (CH2Cl2/CH3OH = 4:1). 1H NMR (400 MHz, CF3COOD) δ 3.81 (s, 3H), 6.59 (s, 1H), 8.18 (s, 1H), 8.72 (s, 1H). 13C NMR (100 MHz, CF3COOD) δ 55.7, 97.4, 121.6, 123.8, 136.8, 141.5, 156.4, 172.8. 1H NMR (400 MHz, DMSO-d6) δ 3.77 (s, 3H), 6.44 (s, 1H), 7.56 (s, 1H), 7.70 (s, 1H), 10.62 (br, 1H). 13C NMR (100 MHz, DMSO-d6) δ 53.3, 90.3, 119.6 (m), 136.9, 137.7 (m), 160.9, 163.7, 171.2043. MS (APCI, pos, CH3OH) calculated for C8H9N5O [M+H]+ 192.09, found 192.1.
6.5. 2-Amino-6-(1-benzyl-1H-imidazol-4-yl)-3H-pyrimidin-4-one (13)
11 (25 mg, 0.10 mmol) was dissolved in 15 mL anhydrous CH2Cl2 under N2 and cooled to −78 °C. BBr3 (3 M, 0.4 mL, 1.2 mmol) was added dropwise and the reaction was allowed to warm to room temperature and stirred for 72 h. The mixture was dripped slowly into 20 mL iced water and stirred for 30 min. The solvent was removed in vacuo and the crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 9:1–4:1) to yield a white solid (16 mg, 0.06 mmol, 60% yield). Rf = 0.70, (CH2Cl2/CH3OH = 9:1). 1H NMR (400 MHz, CD3OD) δ 5.29 (s, 2H), 6.23 (s, 1H), 7.32–7.38 (m, 5H), 7.74 (s, 1H), 7.96 (s, 1H). MS (APCI, pos, CH3OH) calculated for C14H14N5O [M+H]+ 268.12, found 268.11.
6.6. Synthesis of 2-(di-tert-butoxycarbonylamino)-4-iodo-6-methoxypyrimidine (14) & 4-iodo-6-methoxy-2-(tert-butoxycarbonylamino)pyrimidine (15)
2-amino-4-iodo-6-methoxypyrimidine 5 (4.1 g, 16.3 mmol) was suspended in 50 mL CH2Cl2 under N2. Di-tert-butyl decarbonate (8.9 g, 40.8 mmol) and 4-dimethylaminopyridine (5.0 g, 40.9 mmol) were added. The reaction was allowed to stir at room temperature for 18 h. TLC showed absence of starting material and two products. The solvent was removed under pressure and crude material was purified using silica gel column chromatography (hexanes/EtOAc = 4:1–3:2) to yield 14 as a colorless oil (2.9 g, 6.5 mmol, 40% yield). Rf = 0.85, (hexanes/EtOAc = 4:1). 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 18H), 3.85 (s, 3H), 7.01 (s, 1H). 13C NMR (100 MHz, CDCl3) 27.9, 54.5, 83.6, 116.4, 126.8, 150.2, 156.4, 169.8. MS (APCI, pos) calculated for C15H23IN3O5 [M+H]+ 452.07, found 452.05, and 15 as a white solid (2.8 g, 8.0 mmol, 49%). Rf = 0.70, (hexanes/EtOAc = 4:1). 1H NMR (400 MHz, CDCl3) δ 1.47 (s, 9H), 3.93 (s, 3H), 6.35 (s, 1H), 7.68 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 28.2, 54.6, 81.8, 101.5, 150.0, 157.0, 161.0, 171.4. MS (APCI, pos, CH3CN) calculated for C10H15IN3O3 [M+H]+ 352.02, found 351.98.
6.7. Synthesis of 6-methoxy-2-(tert-butoxycarbonylamino)-4-(1-trityl-1H-imidazol-4-yl)pyrimidine (18)
Commercially available 4-iodo-1-trityl-1H-imidazole (500 mg, 1.15 mmol) was dissolved under N2 in 25 mL of anhydrous THF and cooled to −78 °C. EtMgBr (3.0 M, 0.4 mL, 1.20 mmol) was added dropwise and allowed to stir for 10 min. ZnCl2 (0.7 M in THF, 3.3 mL, 2.3 mmol) was subsequently added dropwise, stirred at −78 °C for 10 min, warmed to room temperature, and stirred for 2 h. The organozinc was added dropwise to a mixture of 15 (451 mg, 1.0 mmol), Pd(PPh3)4 (115 mg, 0.1 mmol) and CuI (10 mg, 0.05 mmol) in 40 mL of anhydrous THF and allowed to stir at room temperature for 24 h. The reaction was quenched using 10 mL sat. EDTA solution and THF was removed in vacuo. The crude material was extracted into CH2Cl2 (50 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 1:1–0:1) to yield a yellow oil (267 mg, 0.5 mmol, 43% yield). Rf = 0.60, (hexanes/EtOAc = 1:2). 1H NMR (500 MHz, CDCl3) δ 1.45 (s, 9H), 3.98 (s, 3H), 6.99 (s, 1H), 7.14–7.16 (m, 6H), 7.32–7.33 (m, 9H), 7.47 (br, 1H), 7.60 (s, 1H), 7.61 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 28.2, 53.9, 75.8, 80.8, 95.9, 122.2, 128.2, 128.2, 129.8, 139.0, 139.9, 142.0, 150.7, 157.3, 161.4, 171.5. MS (APCI, pos, CH3CN) calculated for C32H32N5O3 [M+H]+ 534.25, found 534.24.
6.8. Synthesis of 4-(3H-imidazol-4-yl)-6-methoxy-2-(tert-butoxycarbonylamino)-pyrimidine (19)
18 (267 mg, 0.50 mmol) was dissolved in 20 mL acetic acid and stirred for 48 h. The solvent was removed and the crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 19:1–4:1) to yield an off-white solid (140 mg, 0.48 mmol, 96% yield). Rf = 0.85, (CH2Cl2/CH3OH = 9:1). 1H NMR (400 MHz, DMSO-d6) δ 1.43 (s, 9H), 3.87 (s, 3H), 6.80 (s, 1H), 7.27 (br, 1H), 7.64 (s, 1H), 7.76 (s, 1H), 9.67 (br, 1H). 13C NMR (100 MHz, DMSO-d6) δ 28.52, 54.02, 79.82, 95.05, 119.68, 128.94, 133.60, 137.51, 151.56, 158.09, 171.01. MS (APCI, pos, CH3OH) calculated for C13H18N5O3 [M+H]+ 292.14, found 292.12.
6.9. Synthesis of 2-amino-6-(3H-imidazol-4-yl)-3H-pyrimidin-4-one (1)
12 (41 mg, 0.21 mmol) was dissolved in 20 mL anhydrous EtOAc under N2 and cooled to −78 °C. Boron tribromide (1.0 M, 0.6 mL, 0.6 mmol) was added dropwise. The reaction was allowed to warm to room temperature and stirred for 36 h. The mixture was dripped slowly into 20 mL iced water and stirred for 30 min. The solvent was removed in vacuo and the crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 9:1–2:1) to yield a white solid (32 mg, 0.18 mmol, 86% yield). Rf = 0.25, (CH2Cl2/CH3OH = 2:1). 1H NMR (400 MHz, CD3COOD) δ 6.42 (s, 1H), 8.12 (s, 1H), 8.61 (s, 1H). 13C NMR (100 MHz, CD3COOD) δ 97.6, 120.1, 131.0, 137.0, 150.7, 155.1, 164.4. HRMS (FAB) calculated for C7H7N5O [M+H]+ 178.0729, found 178.0727.
6.10. Synthesis of 6,6′-dimethoxy-4,4′-bipyrimidine-2,2′-diamine (21)
5 (101.3 mg, 0.40 mmol) was dissolved in 30 mL degassed 1,4-dioxane in a glass tube. Bis(tributyltin) (0.2 mL, 0.4 mmol) was added, followed by Pd(PPh3)2Cl2 (28.3 mg, 0.04 mmol). The glass tube was sealed and heated to 130 °C for 48 h. The tube was cooled to 0 °C, opened and warmed to room temperature. The crude content was filtered over a pad of Celite and the solvent was removed. The crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 9:1–2:1) to yield an impure mixture. Another attempt at purification with silica gel column chromatography using the Pharmasset conditions (EtOAc/CH3OH/acetone/H2O = 6:1:1:0.5) still yielded impure mixture. Rf = 0.15, (EtOAc/CH3OH/acetone/H2O = 6:1:1:0.5). MS (APCI, pos, DMSO/CH3OH = 1:1) calculated for C12H14N4O4 249.11 (M+H+), found: 249.1.
6.11. Synthesis of 4-(4-chloro-6-methoxy-2-pyrimidinylamino)-6-methoxy-2-pyrimidinamine (22)
5 (212 mg, 0.84 mmol) was dissolved under N2 in 20 mL of anhydrous THF and cooled to −78 °C. EtMgBr (3.0 M, 0.3 mL, 0.93 mmol) was added dropwise and allowed to stir for 2 min. TMSCl (0.1 mL, 0.93 mmol) was added and allowed to stir for 5 min. Again, EtMgBr (3.0 M, 0.3 mL, 0.93 mmol) was added dropwise and allowed to stir for 2 min, then TMSCl (0.1 mL, 0.93 mmol) was added and allowed to stir for 5 min. EtMgBr (3.0 M, 0.3 mL, 0.93 mmol) was added dropwise and allowed to stir for 10 min followed by addition of ZnCl2 (1 M in THF, 1.7 mL, 1.7 mmol) dropwise, stirred at −78 °C for 10 min, warmed to room temperature and stirred for 2 h. The organozinc was added dropwise to a mixture of 2-amino-6-chloro-4-methoxypyrimidine (80 mg, 0.50 mmol), PdCl2(PPh3)2 (59 mg, 0.08 mmol) and CuI (16 mg, 0.08 mmol) in 30 mL of anhydrous THF and allowed to stir at reflux for 24 h. The reaction was quenched using 20 mL sat. EDTA solution and THF was removed in vacuo. The crude material was extracted into CH2Cl2 (30 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 1:3) to yield a white solid (107 mg, 0.38 mmol, 76% yield). Rf = 0.50, (hexanes/EtOAc = 1:3). 1H NMR (400 MHz, DMSO-d6) δ 3.76 (s, 3H), 3.91 (s, 3H), 6.30 (br, 2H), 6.56 (s, 1H), 6.85 (s, 1H), 9.80 (br, 1H). 13C NMR (100 MHz, DMSO-d6) δ 53.7, 54.8, 84.7, 99.7, 158.2, 159.5, 160.6, 162.4, 171.2, 172.2. HRMS (FAB) calculated for C10H11ClN6O2 [M+H]+ 283.0710, found 283.0788.
6.12. Synthesis of 2,4,6-tribromopyrimidine (24)
Commercially available barbituric acid (2.0 g, 15.6 mmol) was suspended in 30 mL of anhydrous toluene under N2 and cooled to 0 °C. Phosphorus (V) oxybromide (17.9 g, 62.4 mmol) was added and N,N-dimethylaniline (3.6 mL, 28.4 mmol) was added dropwise. The mixture was heated to 110 °C and stirred vigorously for 3 h. The reaction was cooled to room temperature and quenched with 30 mL iced water. The mixture was transferred to a separatory funnel and the remaining insoluble gum was washed with EtOAc (10 mL × 3). All organic layers were combined and washed with sat. NaHCO3 (10 mL × 3), then brine (10 mL × 2), and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 99:1–9:1) to yield 25 as a while solid (3.6 g, 11.4 mmol, 73% yield). Rf = 0.80, (hexanes/Et2O = 4:1). 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 128.2, 150.8, 153.2. Agrees with literature values.9
6.13. Synthesis of 4-bromo-2,6-dimethoxypyrimidine (25)
2,4,6-Tribromopyrimidine 24 (1.04 g, 3.28 mmol) was dissolved in 50 mL methanol and cooled to 0 °C. A sodium methoxide solution (0.5 M, 13.2 mL, 6.60 mmol) was added dropwise and the mixture was warmed to room temperature and stirred for 18 h. The reaction was quenched using 20 mL NH4Cl and the solvent was removed. The crude material was extracted into CH2Cl2 (50 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 19:1–4:1) to yield a white solid (575 mg, 2.62 mmol, 80% yield). Rf = 0.75, (hexanes/Et2O = 4:1). 1H NMR (400 MHz, CDCl3) δ 3.8900 (s, 3H), 3.9244 (s, 3H), 6.51 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 54.4, 55.4, 105.0, 152.0, 164.5, 171.7. MS (APCI, pos, CH3CN) calculated for C6H7BrN2O2 [M+H]+ 218.98 and 220.97, found 218.94 and 220.93.
6.14. Synthesis of 2,2′,6,6′-tetramethoxy-4,4′-bipyrimidine (27)
25 (157 mg, 0.72 mmol) was dissolved in 20 mL degassed 1,4-dioxane in a glass tube. Bis(tributyltin) (0.4 mL, 0.72 mmol) was added, followed by Pd(PPh3)4 (83 mg, 0.07 mmol). The glass tube was sealed and heated to 120 °C for 18 h. The tube was cooled to 0 °C, opened and warmed to room temperature. The crude content was filtered over a pad of Celite and the solvent was removed. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 1:0–9:1) to yield a white fluffy solid (81 mg, 0.29 mmol, 80% yield). Rf = 0.80, (hexanes/EtOAc = 9:1). 1H NMR (400 MHz, CDCl3) δ 4.01 (s, 6H), 4.06 (s, 6H), 7.41 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 54.3, 55.0, 99.4, 162.8, 165.5, 173.1. MS (ESI, pos, CH3CN) calculated for C12H14N4O4 [M+H]+ 279.11, found 279.1.
6.15. Synthesis of 4,4′-dimethoxy-5,5′-bipyrimidine-2,2′-diamine (33)
31 (206 mg, 0.5 mmol)9 and 32 (124 mg, 0.5 mmol) were dissolved in 50 mL degassed DMF. Pd(PPh3)4 (57 mg, 0.05 mmol), CuI (19 mg, 0.1 mmol) and CsF (150 mg, 0.99 mmol) were added. The reaction was allowed to stir at 90 °C for 18 h. The contents were cooled and filtered over Celite. The solvent was removed, and the crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 4:1–2:1) to yield contaminated 33. The contaminated sample was recrystallized in EtOAc, followed by ethanol, followed by methanol and finally DMSO to obtain a white solid (15 mg, 0.06 mmol, 12 % yield). 1H NMR (400 MHz, DMSO-d6) δ 3.73 (s, 6H), 6.54 (br, 4H), 7.78 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 53.5, 103.9, 158.9, 163.3, 167.4. MS (APCI, pos, DMSO/CH3OH = 1:1) calculated for C10H12N6O2 [M+H]+ 249.11, found 249.0.
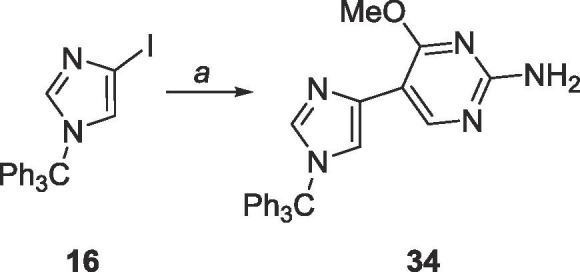
6.16. Synthesis of 4-methoxy-5-(1-trityl-1H-imidazol-4-yl)-2-pyrimidinylamine (34)
32 (500 mg, 1.99 mmol) was dissolved under N2 in 50 mL of THF and cooled to −78 °C. EtMgBr (3.0 M, 0.7 mL, 2.10 mmol) was added dropwise and allowed to stir for 2 min. TMSCl (0.3 mL, 2.19 mmol) was added and allowed to stir for 5 min. Again, EtMgBr (3. 0 M, 0.7 mL, 2.10 mmol) was added dropwise and allowed to stir for 2 min, then TMSCl (0.3 mL, 2.19 mmol) was added and allowed to stir for 5 min. EtMgBr (3.0 M, 0.7 mL, 2.10 mmol) was added dropwise and allowed to stir for 10 min followed by addition of ZnCl2 (0.7 M in THF, 5.7 mL, 3.98 mmol) dropwise, stirred at −78 °C for 10 min, warmed to room temperature and stirred for 2 h. The organozinc was added dropwise to a mixture of 16 (850 mg, 1.95 mmol), Pd(PPh3)4 (230 mg, 0.2 mmol), CuI (20 mg, 0.1 mmol) in 80 mL of THF and allowed to stir at room temperature for 24 h. The reaction was quenched using 10 mL sat. EDTA solution and THF was removed in vacuo. The crude material was extracted into CH2Cl2 (50 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 1:1–0:1) to yield a yellow solid (252 mg, 0.58 mmol, 29% yield). Rf = 0.45, (EtOAc). 1H NMR (500 MHz, CDCl3) δ 3.86 (s, 3H), 4.92 (br, 2H), 7.28 (s, 1H), 7.33–7.37 (m, 15H), 7.46 (s, 1H), 8.86 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 53.4, 75.4, 106.5, 119.9, 128.0, 129.9, 135.0, 138.5, 142.5, 155.3, 161.0, 166.1.
6.17. Synthesis of 5-(1H-imidazol-4-yl)-4-methoxy-2-pyrimidinylamine (37)
34 (252 mg, 0.58 mmol) was dissolved in 20 mL acetic acid and stirred for 48 h. The solvent was removed and the crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 19:1–4:1) to yield an off-white solid (108 mg, 0.56 mmol, 97% yield). Rf = 0.60, (CH2Cl2/CH3OH = 4:1). 1H NMR (500 MHz, CD3OD) δ 4.04 (s, 3H), 7.36 (d, 1H, J = 1.10 Hz), 7.70 (d, 1H, J = 1.15 Hz), 8.50 (s, 1H). 13C NMR (125 MHz, CD3OD) δ 52.7, 104.4, 116.8, 134.9, 147.1, 153.6, 161.7, 166.3. MS (APCI, pos, DMSO/CH3OH = 1:5) calculated for C8H9N5O [M+H]+ 192.09, found 192.04.
6.18. Synthesis of 5-(1H-imidazol-4-yl)-2,4-dimethoxypyrimidine (39)
Commercially available 5-bromo-2,4-dimethoxypyrimidine (200 mg, 0.91 mmol) was dissolved under N2 in 20 mL of anhydrous THF and cooled to −78 °C. EtMgBr (3.0 M, 0.3 mL, 0.96 mmol) was added dropwise and allowed to stir for 10 min. ZnCl2 (0.7 M in THF, 2.6 mL, 1.83 mmol) was subsequently added dropwise, stirred at −78 °C for 10 min, warmed to room temperature and stirred for 2 h. The organozinc was added dropwise to a mixture of 4(5)-iodo-1H-imidazole (155 mg, 0.8 mmol), Pd(PPh3)4 (92 mg, 0.08 mmol) and CuI (8 mg, 0.04 mmol) in 30 mL of THF and allowed to stir at reflux for 18 h. The reaction was quenched using 10 mL sat. EDTA solution and THF was removed in vacuo. The crude material was extracted into CH2Cl2 (50 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 1:0–9:1) to yield a white solid (72 mg, 0.35 mmol, 44% yield). Rf = 0.75, (CH2Cl2/CH3OH = 9:1). 1H NMR (400 MHz, DMSO-d6) δ 3.87 (s, 3H), 4.00 (s, 3H), 7.41 (s, 1H), 7.72 (s, 1H), 8.81 (s, 1H), 12.25 (br, 1H). 13C NMR (100 MHz, DMSO-d6) δ 54.5, 54.9, 110.1, 117.0, 131.3, 136.2, 154.9, 163.3, 166.7. MS (APCI, pos, CH3OH) calculated for C9H10N4O2 [M + H]+ 207.09, found 207.03.
6.19. Synthesis of 2-(2,4-dimethoxy-5-pyrimidinyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (40)
Commercially available 5-bromo-2,4-dimethoxypyrimidine (578 mg, 2.64 mmol) was suspended in 30 mL degassed DMF under N2, followed by addition of bis(pinacolato)diboron (804 mg, 3.17 mmol), potassium acetate (777 mg, 7.92 mmol) and PdCl2(dppf)2·CHCl3 (108 mg, 0.13 mmol). The mixture was heated to 100 °C and stirred for 1 h. The reaction was cooled and transferred to 50 mL dH2O. The mixture was extracted in EtOAc/toluene (1:1, 20 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The solvent was removed in vacuo and the crude product was used without further purification.
6.20. Synthesis of 2,2′,4,4′-tetramethoxy-5,5′-bipyrimidine (41)
Commercially available 5-bromo-2,4-dimethoxypyrimidine (250 mg, 1.14 mmol) and crude 40 (2.64 mmol) were suspended in 25 mL degassed 1,4-dioxane/dH2O (4:1) under N2 in a sealed glass flask. Cs2CO3 (1.12 g, 3.44 mmol) and PdCl2(dppf)2·CHCl3 (46 mg, 0.06 mmol). The glass flask was sealed, heated to 105 °C and stirred for 1 h. The flask was chilled to 0 °C, opened, and warmed to room temperature. The crude content was filtered over a pad of Celite and the solvent was evaporated in vacuo. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 2:1–3:7) to yield a white solid (212 mg, 0.76 mmol, 67% yield). Rf = 0.40, (hexanes/EtOAc = 2:1). 1H NMR (400 MHz, CDCl3) δ 3.96 (s, 6H), 4.02 (s, 6H), 8.18 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 54.2, 55.0, 108.2, 158.8, 165.1, 168.6. MS (ESI, pos, CH3CN) calculated for C12H14N4O4 [M+H]+ 279.11, found 279.19.
6.21. Synthesis of 2,4-dimethoxy-6-(1-trityl-1H-imidazol-4-yl)pyrimidine (42)
4-iodo-1-trityl-1H-imidazole (500 mg, 1.15 mmol) was dissolved under N2 in 25 mL of THF and cooled to −78 °C. EtMgBr (3.0 M, 0.4 mL, 1.20 mmol) was added dropwise and allowed to stir for 10 min. ZnCl2 (0.7 M in THF, 3.3 mL, 2.3 mmol) was subsequently added dropwise, stirred at −78 °C for 10 min, warmed to room temperature and stirred for 2 h. The organozinc was added dropwise to a mixture of 26 (219 mg, 1.0 mmol), Pd(PPh3)4 (115 mg, 0.1 mmol), CuI (10 mg, 0.05 mmol) in 40 mL of THF and allowed to stir at room temperature for 6 h. The reaction was quenched using 10 mL sat. EDTA solution and THF was removed in vacuo. The crude material was extracted into CH2Cl2 (50 mL × 3), washed with brine (10 mL × 2) and the organic layer was dried over MgSO4. The crude material was purified using silica gel column chromatography (hexanes/EtOAc = 2:1–1:4) to yield a white solid (307 mg, 0.68 mmol, 68% yield). Rf = 0.50, (hexanes/EtOAc = 1:1). 1H NMR (400 MHz, CDCl3) δ 3.89 (s, 3H), 3.91 (s, 3H), 6.98 (s, 1H), 7.12–7.13 (m, 6H), 7.27–7.28 (m, 9H), 7.46 (d, 1H, J = 1.36 Hz), 7.6518 (d, 1H, J = 1.84 Hz). 13C NMR (100 MHz, CDCl3) δ 53.9, 54.6, 75.9, 95.0, 122.6, 128.3, 129.8, 139.3, 139.8, 142.1, 162.4, 165.4, 171.1, 172.5.
6.22. Synthesis of 4-(1H-imidazol-4-yl)-2,6-dimethoxypyrimidine (43)
42 (307 mg, 0.68 mmol) was dissolved in 30 mL acetic acid and stirred for 48 h. The solvent was removed and the crude material was purified using silica gel column chromatography (CH2Cl2/CH3OH = 1:0–9:1) to yield a white solid (133 mg, 0.64 mmol, 95% yield). Rf = 0.80, (CH2Cl2/CH3OH = 9:1). 1H NMR (400 MHz, CD3COOD) δ 4.00 (s, 3H), 4.03 (s, 3H), 6.95 (s, 1H), 8.22 (s, 1H), 9.03 (s, 1H). 13C NMR (100 MHz, CD3COOD) δ 54.0, 54.7, 97.0, 119.6 (m), 130.7, 136.0, 154.2, 165.8, 172.9. MS (APCI, pos, CH3OH) calculated for C9H10N4O2 [M+H]+ 207.09, found 207.04.
7. Computational and modelling studies
Molecular modeling study was performed as described in previous studies.14, 38 Briefly, the NMR structure of the NC in complex with a small molecule inhibitor was used as rigid receptor in molecular docking simulations,37 which were carried out by the FRED docking program from OpenEye, version 3.0.1.39, 40 Ligand conformational analysis was performed with OMEGA from OpenEye.52, 53
8. Nucleocapsid NMR studies
8.1. Expression and purification of recombinant HIV-1 NC protein54
The HIV-1 NC coding region in pNL4-355 was PCR amplified using the 5′-primer CCAGCTACCATACATATGCAGAAAGGC (NdeI site underlined) and the 3′-primer GGCCGGATCCTCCCTAACTAATTAGCCTGTC-TCTC (BamHI and stop codon underlined). The expression vector pET-3a (Novagen, Madison, WI) was doubly digested with NdeI and BamHI and treated with calf intestinal alkaline phosphate. The PCR product was purified by phenol-extraction and ethanol-precipitation and doubly digested with NdeI and BamHI. The insert and vector were ligated using phage T4 DNA ligase at 16 °C for five hours and transformed into competent HMS174. DNA from transformants were sequenced and found to be identical with the HIV-1 NC coding sequence in pNL4-3. A clone, designated as pRD2, overexpressed the 55-residue NC protein with the sequence M Q K G N F R N Q R K T V K C F N C G K E G H I A K N C R A P R K K G C W K C G K E G H Q M K D C T E R Q A N (the two zinc knuckles are underlined). Ion-spray mass spectrometry confirmed the mass of the apoprotein to be 6369(±2) Da (calculated 6369 Da) and 6501(±2) Da (calculated 6500 Da) for the Zn-bound protein.
For protein expression of HIV-1 NC in Escherichia coli, pRD2 was transformed into BL21(DE3) pLysE. The purification scheme for the recombinant HIV-1 NC was adapted from Ji et al. 56 and You & McHenry.57 Culture media were supplemented with 100 μg/l ampicillin and 34 μg/L chloramphenicol. A starter culture of 20 ml of ZB58 inoculated from a single colony was grown at 37 °C overnight. The starter culture was added to 2 l of M9ZB58 supplemented with 0.1 mM ZnCl2 and grown at 37 °C to an absorbance at 600 nm of 0.5 to 0.6 before induction with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After three hours, the cells were harvested by centrifugation, resuspended in 30 ml of lysis buffer (50 mM Tris-HCl (pH 8.0), 10% (v/v) glycerol, 0.1 M NaCl, 0.1 mM ZnCl2, 5 mM dithiothreitol, 2 mM EDTA), and stored at 70 °C. To lyse the cells, the cells were thawed in ice-water, and 172 ml of 10 mM PMSF (phenylmethylsulfonyl fluoride), 30 ml of 1 mg/ml pepstatin A, and 2.1 ml of 1% (w/v) sodium deoxycholate were added. The cells were sonicated by five bursts of 20 seconds to reduce the viscosity. The nucleic acids were precipitated by adding 4% (w/v) polyethyleneimine (pH 7.9) dropwise to a final concentration of 0.4% and stirred for 15 minutes before centrifugation at 23,000 g for 30 minutes at 4 °C. The supernatant was collected (42 ml), filtered (0.45 μm pore size), and loaded at 1 ml/minute onto a 20 mL Q-Sepharose and a 20 ml SP-Sepharose column (Pharmacia) connected in series and previously equilibrated with 200 ml of buffer A (50 mM Tris-HCl (pH 8.0), 10% glycerol, 0.1 M NaCl, 0.1 mM ZnCl2, 10 mM BME (β-mercaptoethanol)). After washing with 60 ml of buffer A, the Q-Sepharose column was detached, and the SP-Sepharose column was washed with 1.5 column volumes of buffer A. A ten column volume linear gradient from 40% to 50% buffer B (50 mM Tris-HCl (pH 8.0), 10% glycerol, 1.0 M NaCl, 0.1 mM ZnCl2, 10 mM BME) was applied to elute the HIV-1 NC protein. The protein fractions were pooled (15 ml) and loaded at 0.5 ml/minute onto a 320 ml Sephadex G-50 column (Pharmacia) pre-equilibrated with two volumes of buffer C (50 mM Tris-HCl (pH 7.0), 10% glycerol, 0.1 M NaCl, 0.1 mM ZnCl2, 10 mM BME). The NC protein eluted at 175 ml and fractions were pooled (35 ml) for concentration and dialysis into NMR buffer (see below).
8.2. Sample preparation54
NMR buffer (10 mM Tris-HCl, pH 7.0, 140 mM KCl, 10 mM NaCl, 1 mM MgCl2) was deoxygenated by sparging with argon for 15 minutes and filter-sterilized (0.2 μm pore size). The protein sample was dialyzed using Centricon-3 by adding NMR buffer five or six times (total volume 40–50 ml). The buffered protein sample was lyophilized for ease of storage.
8.3. NMR data collection and analysis
25 μM protein samples were made in 500 μl of D2O and loaded into a 5 mm NMR tube. After taking the blank 1H spectrum, the test compound was titrated into the protein sample such that 1:1 ratio of compound/protein could be established. Data for 1H NMR signal assignments were collected at a sample temperature of 10 °C with a Bruker DMX 600 MHz (1H) NMR and Bruker AVANCE III HD 500 MHz NMR spectrometers.
Declaration of Competing Interest
The authors have no conflicts to declare.
Acknowledgements
This work was supported by the National Institutes of Health T32 GM066706, R21 AI118470 (KSR), and R01 GM42561, R25 GM055036 (MFS). The authors also wish to thank the OpenEye Free Academic Licensing Program for providing a free academic license for molecular modeling and chemoinformatics software.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2019.05.019.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Seley K.L., Zhang L., Hagos A. “Fleximers” Design and synthesis of two novel split nucleosides. Org Lett. 2001;3:3209–3210. doi: 10.1021/ol0165443. [DOI] [PubMed] [Google Scholar]
- 2.Seley K.L., Zhang L., Hagos A., Quirk S. “Fleximers” Design and synthesis of a new class of novel shape-modified nucleosides. J Org Chem. 2002;67:3365–3373. doi: 10.1021/jo0255476. [DOI] [PubMed] [Google Scholar]
- 3.Seley K.L., Quirk S., Salim S., Zhang L., Hagos A. Unexpected inhibition of S-adenosyl-L-homocysteine hydrolase by a guanosine nucleoside. Bioorg Med Chem Lett. 2003;13:1985–1988. doi: 10.1016/s0960-894x(03)00331-7. [DOI] [PubMed] [Google Scholar]
- 4.Polak M., Seley K.L., Plavec J. Conformational properties of shape modified nucleosides – Fleximers. J Am Chem Soc. 2004;126:8159–8166. doi: 10.1021/ja0498078. [DOI] [PubMed] [Google Scholar]
- 5.Quirk S., Seley K.L. Substrate discrimination by the human GTP fucose pyrophosphorylase. Biochemistry-Us. 2005;44:10854–10863. doi: 10.1021/bi0503605. [DOI] [PubMed] [Google Scholar]
- 6.Quirk S., Seley K.L. Identification of catalytic amino acids in the human GTP fucose pyrophosphorylase active site. Biochemistry-Us. 2005;44:13172–13178. doi: 10.1021/bi051288d. [DOI] [PubMed] [Google Scholar]
- 7.Seley K.L., Salim S., Zhang L. “Molecular chameleons”. Design and synthesis of C-4-substituted imidazole fleximers. Org Lett. 2005;7:63–66. doi: 10.1021/ol047895v. [DOI] [PubMed] [Google Scholar]
- 8.Seley K.L., Salim S., Zhang L., O'Daniel P.I. “Molecular chameleons”. Design and synthesis of a second series of flexible nucleosides. J Org Chem. 2005;70:1612–1619. doi: 10.1021/jo048218h. [DOI] [PubMed] [Google Scholar]
- 9.Wauchope O.R., Velasquez M., Seley-Radtke K. Synthetic routes to a series of proximal and distal 2 '-Deoxy fleximers. Synthesis-Stuttgart. 2012;44:3496–3504. doi: 10.1055/s-0032-1316791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters H.L., Jochmans D., de Wilde A.H. Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg Med Chem Lett. 2015;25:2923–2926. doi: 10.1016/j.bmcl.2015.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yates M.K., Raje M.R., Chatterjee P. Flex-nucleoside analogues – novel therapeutics against filoviruses. Bioorg Med Chem Lett. 2017;27:2800–2802. doi: 10.1016/j.bmcl.2017.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann S.C., Sadler J.M., Andrei G., Snoeck R., Balzarini J., Seley-Radtke K.L. Carbocyclic 5 '-nor “reverse” fleximers. Design, synthesis, and preliminary biological activity. Med Chem Comm. 2011;2:650–654. doi: 10.1039/C1MD00094B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmermann S.C., Sadler J.M., O'Daniel P.I., Kim N.T., Seley-Radtke K.L. “Reverse” carbocyclic fleximers: synthesis of a new class of adenosine deaminase inhibitors. Nucleos Nucleot Nucl. 2013;32:137–154. doi: 10.1080/15257770.2013.771187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori M., Kovalenko L., Malancona S. Structure-based identification of HIV-1 nucleocapsid protein inhibitors active against wild-type and drug-resistant HIV-1 strains. ACS Chem Biol. 2018;13:253–266. doi: 10.1021/acschembio.7b00907. [DOI] [PubMed] [Google Scholar]
- 15.Mori M., Schult-Dietrich P., Szafarowicz B. Use of virtual screening for discovering antiretroviral compounds interacting with the HIV-1 nucleocapsid protein. Virus Res. 2012;169:377–387. doi: 10.1016/j.virusres.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Mori M., Dietrich U., Manetti F., Botta M. Molecular dynamics and DFT study on HIV-1 nucleocapsid protein-7 in complex with viral genome. J Chem Inf Model. 2010;50:638–650. doi: 10.1021/ci100070m. [DOI] [PubMed] [Google Scholar]
- 17.Mori M., Manetti F., Botta M. Predicting the binding mode of known NCp7 inhibitors to facilitate the design of novel modulators. J Chem Inf Model. 2011;51:446–454. doi: 10.1021/ci100393m. [DOI] [PubMed] [Google Scholar]
- 18.Krishnamoorthy G., Roques B., Darlix J.L., Mély Y. DNA condensation by the nucleocapsid protein of HIV-1: a mechanism ensuring DNA protection. Nucleic Acids Res. 2003;31:5425–5432. doi: 10.1093/nar/gkg738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hargittai M.R., Mangla A.T., Gorelick R.J., Musier-Forsyth K. HIV-1 nucleocapsid protein zinc finger structures induce tRNA(Lys,3) structural changes but are not critical for primer/template annealing. J Mol Biol. 2001;312:985–997. doi: 10.1006/jmbi.2001.5021. [DOI] [PubMed] [Google Scholar]
- 20.Cen S., Khorchid A., Gabor J., Rong L., Wainberg M.A., Kleiman L. Genomic placement and the initiation step of reverse transcription in human immunodeficiency virus type 1. J Virol. 2000;74:10796–10800. doi: 10.1128/jvi.74.22.10796-10800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas J., Gorelick R. Nucleocapsid protein function in early infection processes. Virus Res. 2008;134:39–63. doi: 10.1016/j.virusres.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poljak L., Batson S.M., Ficheux D., Roques B.P., Darlix J.-L., Käs E. Analysis of NCp7-dependent activation of HIV-1 cDNA integration and its conservation among retroviral nucleocapsid proteins. J Mol Biol. 2003;329:411–421. doi: 10.1016/s0022-2836(03)00472-8. [DOI] [PubMed] [Google Scholar]
- 23.De Guzman R.N., Wu Z.R., Stalling C.C., Pappalardo L., Borer P.N., Summers M.F. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science. 1998;279:384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 24.Shubsda M.F., Paoletti A.C., Hudson B.S., Borer P.N. Affinities of packaging domain loops in HIV-1 RNA for the nucleocapsid protein. Biochemistry-Us. 2002;41:5276–5282. doi: 10.1021/bi016045+. [DOI] [PubMed] [Google Scholar]
- 25.Athavale S.S., Ouyang W., McPike M.P., Hudson B.S., Borer P.N. Effects of the nature and concentration of salt on the interaction of the HIV-1 nucleocapsid protein with SL3 RNA. Biochemistry-Us. 2010;49:3525–3533. doi: 10.1021/bi901279e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darlix J.-L., Godet J., Ivanyi-Nagy R., Fossé P., Mauffret O., Mély Y. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J Mol Biol. 2011;410:565–581. doi: 10.1016/j.jmb.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 27.Johnson S.F., Telesnitsky A. Retroviral RNA dimerization and packaging: the what, how, when, where, and why. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darlix J.L., Garrido J.L., Morellet N., Mely Y., de Rocquigny H. Properties, functions, and drug targeting of the multifunctional nucleocapsid protein of the human immunodeficiency virus. Adv Pharmacol. 2007;55:299–346. doi: 10.1016/S1054-3589(07)55009-X. [DOI] [PubMed] [Google Scholar]
- 29.Breuer S., Chang M.W., Yuan J., Torbett B.E. Identification of HIV-1 inhibitors targeting the nucleocapsid protein. J Med Chem. 2012;55:4968–4977. doi: 10.1021/jm201442t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Rocquigny H., Shvadchak V., Avilov S. Targeting the viral nucleocapsid protein in anti-HIV-1 therapy. Mini Rev Med Chem. 2008;8:24–35. doi: 10.2174/138955708783331603. [DOI] [PubMed] [Google Scholar]
- 31.Mori M., Kovalenko L., Lyonnais S. Nucleocapsid protein: a desirable target for future therapies against HIV-1. Curr Top Microbiol Immunol. 2015;389:53–92. doi: 10.1007/82_2015_433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldschmidt V., Miller Jenkins L.M., de Rocquigny H., Darlix J.-L., Mély Y. The nucleocapsid protein of HIV-1 as a promising therapeutic target for antiviral drugs. HIV Therapy. 2010;4:179–198. [Google Scholar]
- 33.Mori M., Dasso Lang M.C., Saladini F. Synthesis and evaluation of bifunctional aminothiazoles as antiretrovirals targeting the HIV-1 nucleocapsid protein. ACS Med Chem Lett. 2018 doi: 10.1021/acsmedchemlett.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bombarda E., Ababou A., Vuilleumier C. Time-resolved fluorescence investigation of the human immunodeficiency virus type 1 nucleocapsid protein: influence of the binding of nucleic acids. Biophys J. 1999;76:1561–1570. doi: 10.1016/S0006-3495(99)77315-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Paiva R.E.F., Du Z., Peterson E.J., Corbi P.P., Farrell N.P. Probing the HIV-1 NCp7 nucleocapsid protein with site-specific gold(I)-phosphine complexes. Inorg Chem. 2017;56:12308–12318. doi: 10.1021/acs.inorgchem.7b01762. [DOI] [PubMed] [Google Scholar]
- 36.Morellet N., Demene H., Teilleux V. Structure of the complex between the HIV-1 nucleocapsid protein NCp7 and the single-stranded pentanucleotide d(ACGCC) J Mol Biol. 1998;283:419–434. doi: 10.1006/jmbi.1998.2098. [DOI] [PubMed] [Google Scholar]
- 37.Goudreau N., Hucke O., Faucher A.M. Discovery and structural characterization of a new inhibitor series of HIV-1 nucleocapsid function: NMR solution structure determination of a ternary complex involving a 2:1 inhibitor/NC stoichiometry. J Mol Biol. 2013;425:1982–1998. doi: 10.1016/j.jmb.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 38.Gamba E., Mori M., Kovalenko L. Identification of novel 2-benzoxazolinone derivatives with specific inhibitory activity against the HIV-1 nucleocapsid protein. Eur J Med Chem. 2018;145:154–164. doi: 10.1016/j.ejmech.2017.12.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGann M. FRED pose prediction and virtual screening accuracy. J Chem Inf Model. 2011;51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 40.FRED 3.0.1 OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com.
- 41.Miller W.D., Fray A.H., Quatroche J.T., Sturgill C.D. Suppression of a palladium-mediated homocoupling in a Suzuki cross-coupling reaction. Development of an impurity control strategy supporting synthesis of LY451395. Org Process Res Dev. 2007;11:359–364. [Google Scholar]
- 42.Morenomanas M., Pajuelo F., Pleixats R. Preparation of 1,3-diarylpropenes by phosphine-free palladium(O)-catalyzed suzuki-type coupling of allyl bromides with arylboronic acids. J Org Chem. 1995;60:2396–2397. [Google Scholar]
- 43.Wallow T.I., Novak B.M. Highly efficient and accelerated suzuki aryl couplings mediated by phosphine-free palladium sources. J Org Chem. 1994;59:5034–5037. [Google Scholar]
- 44.Marck G., Villiger A., Buchecker R. Aryl couplings with heterogeneous palladium catalysts. Tetrahedron Lett. 1994;35:3277–3280. [Google Scholar]
- 45.Campi E.M., Jackson W.R., Marcuccio S.M., Naeslund C.G.M. High yields of unsymmetrical biaryls via cross-coupling of arylboronic acids with haloarenes using a modified suzuki-beletskaya procedure. J Chem Soc Chem Comm. 1994 2395 2395. [Google Scholar]
- 46.Wuts P.G.M., Greene T.W., Greene T.W. 4th ed. Wiley-Interscience; Hoboken, N.J.: 2007. Greene's protective groups in organic synthesis. p xxviii, 1082 p. [Google Scholar]
- 47.Bourderioux A., Naus P., Perlikova P. Synthesis and significant cytostatic activity of 7-hetaryl-7-deazaadenosines. J Med Chem. 2011;54:5498–5507. doi: 10.1021/jm2005173. [DOI] [PubMed] [Google Scholar]
- 48.Ruiz-Castillo P., Buchwald S.L. Applications of palladium-catalyzed C-N cross-coupling reactions. Chem Rev. 2016;116:12564–12649. doi: 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meijere A.D., Diederich F.O. Wiley-VCH:; Weinheim: 2004. Metal-catalyzed cross-coupling reactions. 2nd, completely rev. and enl ed. [Google Scholar]
- 50.Bardon A.B., Wetmore S.D. How flexible are fleximer nucleobases? A computational study. J Phys Chem A. 2005;109:262–272. doi: 10.1021/jp046957w. [DOI] [PubMed] [Google Scholar]
- 51.He Y., Chen Y.Z., Du H.W., Schmid L.A., Lovely C.J. A convenient synthesis of 1,4-disubstituted imidazoles. Tetrahedron Lett. 2004;45:5529–5532. [Google Scholar]
- 52.Hawkins P.C.D., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and cambridge structural database. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.OpenEye OMEGA 3.0.0.1: OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com.
- 54.Lee B.M., De Guzman R.N., Turner B.G., Tjandra N., Summers M.F. Dynamical behavior of the HIV-1 nucleocapsid protein. J Mol Biol. 1998;279:633–649. doi: 10.1006/jmbi.1998.1766. [DOI] [PubMed] [Google Scholar]
- 55.Adachi A., Gendelman H.E., Koenig S. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ji X., Klarmann G.J., Preston B.D. Effect of human immunodeficiency virus type 1 (HIV-1) nucleocapsid protein on HIV-1 reverse transcriptase activity in vitro. Biochemistry-Us. 1996;35:132–143. doi: 10.1021/bi951707e. [DOI] [PubMed] [Google Scholar]
- 57.You J.C., McHenry C.S. HIV nucleocapsid protein. Expression in Escherichia coli, purification, and characterization. J Biol Chem. 1993;268:16519–16527. [PubMed] [Google Scholar]
- 58.Studier F.W., Rosenberg A.H., Dunn J.J., Dubendorff J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.