

Abstract
The ability of cells to perceive and translate versatile cues into differential chromatin and transcriptional states is critical for many biological processes1–5. In plants, timely transition to a flowering state is crucial for successful reproduction6–9. EARLY BOLTING IN SHORT DAY (EBS) is a negative transcriptional regulator that prevents premature flowering in Arabidopsis thaliana10,11. We found that EBS contains bivalent bromo-adjacent homology (BAH)-plant homeodomain (PHD) reader modules that bind H3K27me3 and H3K4me3, respectively. We observed co-enrichment of a subset of EBS-associated genes with H3K4me3, H3K27me3, and Polycomb repressor complex 2 (PRC2). Notably, EBS adopted an autoinhibition mode to mediate its switch in binding preference between H3K27me3 and H3K4me3. This binding balance was critical because disruption of either EBS-H3K27me3 or EBS-H3K4me3 interaction induced early floral transition. Our results identify a bivalent chromatin reader capable of recognizing two antagonistic histone marks, and we propose a distinct mechanism of interaction between active and repressive chromatin states.
EBS possesses a putative N-terminal BAH domain, a PHD finger, and a C-terminal short loop (Fig. 1a). Although the PHD finger of EBS has been shown to bind H3K4me2 and H3K4me3 in vitro10, its BAH domain has not been characterized, despite such domains having been reported to function as histone mark readers12–15. To determine whether the EBS BAH domain can bind histone marks, we screened EBS on a histone peptide array16. EBS recognized the activation marks H3K4me2 and H3K4me3 (Supplementary Fig. 1a,b and Supplementary Table 1). To our surprise, EBS also bound to the repressive marks H3K27me2 and H3K27me3 (Supplementary Fig. 1c–e and Supplementary Table 1). Our isothermal titration calorimetry (ITC) data revealed that EBS had a stronger affinity for H3K27me3 (49.8 μM) than for H3K4me3 (86.8 μM) (Fig. 1b,c).
Fig. 1 |. Structural basis for recognition of H3K27me3 by BAH.
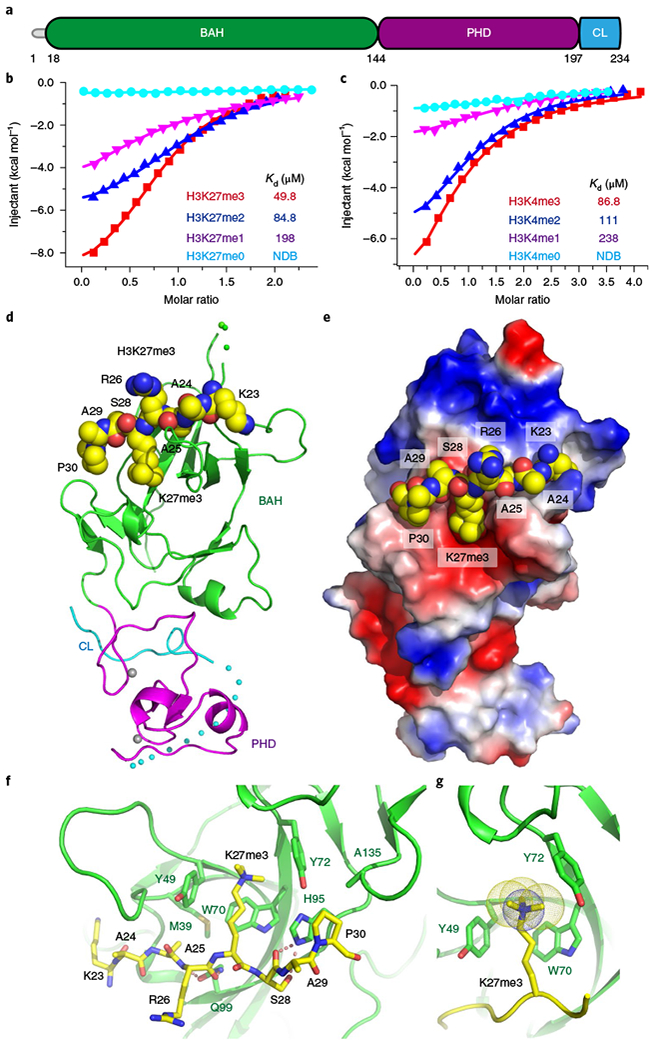
a, Domain architecture of Arabidopsis EBS. CL, C-terminal loop. b,c, ITC binding curves show that EBS recognizes H3K27me3 (b) and H3K4me3 (c) with preferences for higher methylation status. NDB, no detectable binding. The ITC experiments were repeated twice independently with similar results. d, Overall structure of EBS in complex with a H3K27me3 peptide. The peptide is shown in a space-filling representation. e, An electrostatic surface view showing the H3K27me3 peptide bound in a negatively charged surface cleft. f, Details of specific recognition of H3K27me3 by an aromatic cage of BAH. The hydrogen bonding interactions are highlighted with dashed red lines. g, An enlarged view is shown of the BAH aromatic cage accommodating H3K27me3.
Next, we determined the crystal structure of EBS in complex with a histone H3(20–35)K27me3 peptide at 2.0-Å resolution (Fig. 1d and Supplementary Table 2). A small hydrophobic loop of the PHD finger protruded into a hydrophobic pocket of the BAH domain to enable interactions between the two domains (Supplementary Fig. 2a). The H3K27me3 peptide bound along a negatively charged surface cleft on the BAH domain without contacting the PHD finger (Fig. 1d,e and Supplementary Fig. 2b). The side chain of H3K27me3 was inserted into a classic aromatic cage formed by Tyr49, Trp70, and Tyr72 involving both cation-π and hydrophobic interactions (Fig. 1f,g), similar to other methyl-lysine-binding modules17–19. The EBS BAH aromatic cage is conserved in other methyl-lysinebinding BAH domains, such as mouse ORC1 BAH recognizing H4K20me2 and maize ZMET2 BAH binding to H3K9me212,13. The main chain carbonyl and side chain hydroxyl groups of H3S28 formed two hydrogen bonds with the side chain imidazole group of His95, further fixing the conformation of this residue (Fig. 1f). The prolyl ring of H3P30 had a particular directionality whereby it stacked with the imidazole ring of His9520 (Fig. 1f). Notably, mutations of the key interacting residues resulted in impairment of binding between EBS and H3K27me3 (Supplementary Fig. 2c).
In the EBS–H3K27me3 structure, the PHD adopted a classic PHD finger fold resembling other H3K4me3-binding PHDs17,21,22. However, we failed to obtain crystals of full-length EBS in complex with H3K4me2 or H3K4me3 peptides. Notably, an EBS C-terminal loop folded back and interacted with the PHD finger (Fig. 2a). The canonical aromatic cage (formed by Tyr148, Tyr155, and Trp170) of PHD was occupied by Pro211, resulting in an autoinhibition mode that made the PHD finger inaccessible to methyl-lysine. The specific interaction between the proline ring and the aromatic cage has been observed in structures of peptide-L3MBTL1 complexes23–25. Consistent with this, we found that EBS without the C-terminal autoinhibition loop (EBSΔC, 1-199) bound to the H3K4me3 peptide with a higher binding affinity of 30.7μM (Fig. 2b) than that of full-length EBS (86.8μM; Fig. 1c), which supports the theory that the C-terminal loop inhibits EBS-H3K4me3 binding.
Fig. 2 |. Structural basis for an autoinhibition loop formed by the EBS C terminus.
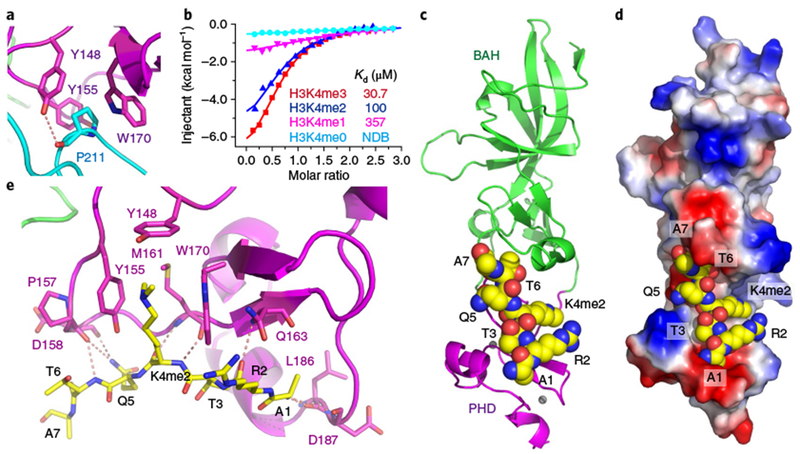
a, The C terminus of EBS folds back, with Pro211 inserted into the aromatic cage of the PHD finger. b, ITC binding curves show the binding affinity of EBSΔC for H3K4 peptides. The ITC experiments were repeated twice independently with similar results. c, The overall structure of EBSΔC in complex with the H3K4me2 peptide. The peptide is shown in a space-filling representation. d, An electrostatic surface view of EBSΔC showing the H3K4me2 peptide bound in a negatively charged surface cleft. e, Details of the interaction between EBS and the H3K4me2 peptide. The methyl-lysine is accommodated in a classic aromatic cage formed by three aromatic residues (Tyr148, Tyr155, and Trp170).
We next solved the crystal structure of EBSΔC in complex with an H3(1–15)K4me2 peptide at 3.1-Å resolution (Fig. 2c and Supplementary Table 2). The overall EBSΔC structure resembled that of full-length EBS with a root-mean-squared (r.m.s.) deviation of 0.6 Å on superposition (Supplementary Fig. 3a). The histone peptide bound along the negatively charged surface of the PHD finger, overlapping with the autoinhibition binding sites (Fig. 2d and Supplementary Fig. 3b,c). Recognition of H3K4me2 resulted in the N-terminal Ala1 residue anchoring in a negatively charged pocket and insertion of H3K4me2 into the canonical aromatic cage, leading to cation-π and hydrophobic interactions, similar to other methyl-lysine-binding modules (Fig. 2d,e and Supplementary Fig. 3d)17. Mutations of the key aromatic cage residues caused a substantial decrease in binding affinity (Supplementary Fig. 3e). We next assessed EBS binding toward an H3(1–35)K4me3K27me3 doubly methylated peptide and found that full-length EBS bound to H3K4me3K27me3 with a binding affinity of 44.3 μM (Supplementary Fig. 3f), similar to the affinity of EBS for H3K27me3 (49.8 μM; Fig. 1b). In contrast, EBSΔC bound to H3K4me3K27me3 with a binding affinity of 29.7 μM (Supplementary Fig. 3g), similar to the affinity of EBSΔC for H3K4me3 (30.7 μM; Fig. 2b). The analysis yielded an approximate 1:1 stoichiometry between peptides and proteins (Supplementary Fig. 3f,g). In addition, the distance between the two peptide binding sites of EBS and the conformation of the two histone peptides made it difficult for a single EBS protein to capture the H3K4me3 and H3K27me3 marks simultaneously (Supplementary Fig. 3h), suggesting that the PHD–H3K4me3 and BAH–H3K27me3 interactions are two independent events and mutually exclusive.
H3K4me3 and H3K27me3 are two histone marks that are well known for their antagonistic roles in balancing chromatin states and regulating gene expression1,26. The ability of a single EBS protein to recognize two antagonistic histone marks via two distinct domains suggests that there is crosstalk between active and repressive chromatin states. Previously reported H3K4me3–H3K27me3 interactions often involve multiple proteins or multi-protein complexes1,2,26. To investigate whether EBS colocalizes with H3K4me3 and H3K27me3 in vivo, we determined EBS genome-wide occupancy via chromatin immunoprecipitation (ChlP)-seq and identified 2,637 binding peaks (P < 1 × 10−3) corresponding to 2,432 genes (Supplementary Table 3). We also determined the genome-wide distribution of CURLY LEAF (CLF), an Arabidopsis H3K27me3 methyltransferase of PRC227,28. The majority of EBS binding peaks were enriched in the gene body proximal to the transcription start site (TSS), similar to CLF (Fig. 3a,b and Supplementary Fig. 4). A significant number of EBS-associated genes overlapped with those of CLF (P= 2.8× 10−85), as well as Polycomb protein LIKE HETEROCHROMATIN PROTEIN 1 (LHP1)29,30 and EMBRYONIC FLOWER 1 (EMF1)31,32 (Fig. 3b,c and Supplementary Fig. 5a,b). Target genes shared by EBS and CLF showed slightly higher H3K27me3 levels than CLF-only targets, but had higher levels of H3K27me3 than EBS-only targets (Fig. 3d,e). EBS target genes had higher H3K4me3 levels than CLF targets (Fig. 3e). Consistent with this, a large subset of EBS-bound genes (~90%) were co-marked with H3K27me3 or H3K4me3 (Fig. 3f,g and Supplementary Fig. 5c). Furthermore, EBS occupancy on several of its targets (FT, EMF1, and SOC1) was significantly reduced when H3K27me3 levels were diminished in the clf mutant (Fig. 3h,i).
Fig. 3 |. EBS colocalizes with H3K4me3 and H3K27me3.
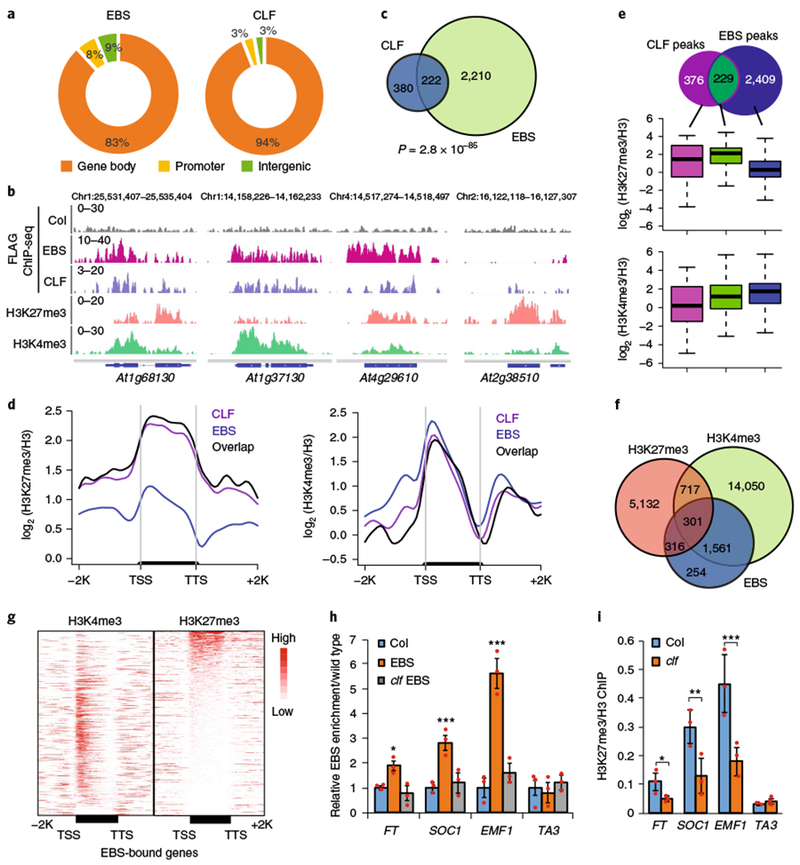
a, Genomic distribution of EBS and CLF ChIP-seq peaks. b, Browser views of normalized ChIP-seq peaks at representative loci. c, Venn diagram showing the overlap of EBS- and CLF-associated genes. The P value is based on the hypergeometric test. d, Metaplots showing average H3K27me3 and H3K4me3 levels along the transcription units of EBS, CLF, and their common target genes. e, Box plots showing average H3K27me3 and H3K4me3 levels in unique EBS-occupied genes and target genes shared with CLF. Box edges show the interquartile range (IQR), horizontal center lines denote medians, and whiskers denote±1.5 IQR. f, Venn diagram showing the overlap of EBS-associated genes with H3K27me3- and H3K4me3-marked genes. g, Heat maps of H3K27me3 and H3K4me3 occupancy along the transcription units of EBS-bound genes. TTS, transcription termination site; −2 K, 2 kb upstream of the TSS; + 2 K, 2 kb downstream of the TTS. h, Relative EBS enrichment at FT, SOC1, and EMF1 in Col, EBS-FLAG (EBS), and EBS-FLAG in clf (clf EBS). i, Relative H3K27me3 enrichment in clf at FT, SOC1, EMF1, and TA3 (a negative-control locus). Data are shown as means± s.d. from three independent experiments (red dots). Black asterisks indicate a significant difference from Col. Two-tailed Student’s t tests were used to calculated P values: *P< 0.05, **P<0.01, ***P< 0.001.
Our structural and biochemical data identify a unique dual BAH–PHD binding module of EBS with H3K4me3 and H3K27me3. To investigate the functional importance of this dual recognition, we generated point mutations in the aromatic cage that abolished BAH–H3K27me3 and PHD–H3K4me3 binding and tested their effect on flowering, as the loss-of-function EBS mutation induces early floral transition11. In comparison with wild-type EBS transgenes that rescued ebs, EBS mutants defective in H3K27me3 or H3K4me3 binding were unable to rescue the early-flowering phenotype in ebs (n = 6 independent transgenic lines; Fig. 4a,b and Supplementary Fig. 6a–c). Consistent with the role of EBS in controlling flowering by repressing FLOWERING TIME (FT) expression11, both BAH and PHD mutants showed a strong induction of FT expression that was similar to that in the ebs null mutant (Fig. 4c), demonstrating a critical role for BAH–H3K27me3 and PHD–H3K4me3 binding in EBS function in vivo.
Fig. 4 |. Functional analysis of EBS mutants defective in H3K4me3 or H3K27me3 binding and a C-terminus deletion mutant.
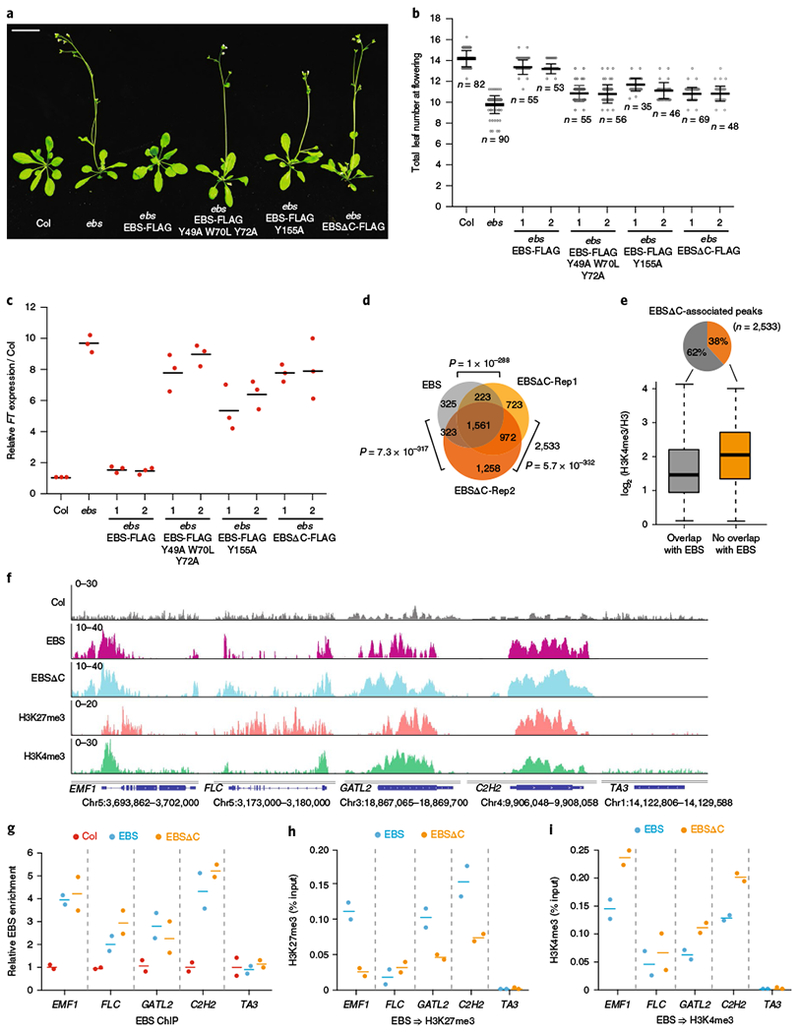
a, Plant phenotype of Col, ebs, ebs transformed with wild-type EBS (EBS-FLAG), H3K4 (EBS-FLAG Y155A) or H3K27 (EBS-FLAG Y49A W70L Y72A) binding-defective mutants, or EBSΔC (EBSΔC-FLAG). Scale bar, 1 cm. Images from three individual plants show similar results. b, Counting leaf number at flowering of long-day-grown plants described in a. The numbers 1 and 2 indicate two independent T2 lines. Black horizontal lines represent the mean, and the error bars represent ±s.d. from the number of plants counted for each line. c, Relative FT mRNA levels after normalization to ACTIN7 in plants described in a. Data points are from three independent experiments (bars denote mean). d, Overlap of EBS- and EBSΔC-associated genes. Two biological replicates for ChIP-seq are shown as EBSΔC-Rep1 and EBSΔC-Rep2. The P value is based on the hypergeometric test. e, Box plot showing the average H3K4me3 levels at unique EBSΔC-associated peaks compared with peaks overlapping with EBS. Box edges show the IQR, horizontal center lines denote medians, and whiskers denote±1.5 IQR. f, Browser view of normalized ChIP-seq peaks at representative loci. g, Relative enrichment of EBS and EBSΔC at the indicated loci in f. h,i, Sequential ChIP–qPCR of H3K4me3 (h) or H3K27me3 (i) enrichment relative to EBS or EBSΔC input after initial EBS/EBSΔC ChIP (TA3 is a negative-control locus). Bars denote the mean of two independent experiments.
To investigate the in vivo relevance of the C-terminal autoinhibition loop, we generated a C-terminal deletion mutant and found that it failed to complement the early flowering of ebs (n = 6 independent transgenic lines; Fig. 4a,b and Supplementary Fig. 6d), despite having a much higher protein level than wild-type EBS (Supplementary Fig. 6e). A similar early-flowering phenotype was observed when EBS was overexpressed in a wild-type background11. Notably, deleting the C-terminal loop also caused significant FT upregulation (Fig. 4c), suggesting that proper EBS levels are critical for EBS function. Despite having similar binding patterns at the gene body (Supplementary Fig. 7a), we found that a significant subset of EBSΔC-associated genes overlapped with those of EBS (Fig. 4d and Supplementary Table 4). A slightly higher H3K4me3 level was associated with unique EBSΔC-bound genes than with genes overlapping with EBS (Fig. 4e). We also noted a slightly higher H3K4me3 level for EBSΔC than for EBS at their common target genes (Supplementary Fig. 7b). One possible explanation is that deletion of the C-terminal loop exposes the PHD binding pocket and mediates a transition of the EBS binding preference from H3K27me3 to H3K4me3. To test this hypothesis, we performed sequential ChIP–qPCR in which chromatin was first enriched with EBS or EBSΔC and then with either H3K27me3 or H3K4me3 (Supplementary Fig. 7c). Despite having similar occupancy and similar H3K27me3 levels (Fig. 4f,g and Supplementary Fig. 7d,e), EBSΔC showed higher co-enrichment of H3K4me3 and lower levels of H3K27me3 overall than EBS at the EMF1, GATL2, and C2H2 loci (Fig. 4h,i). This indicates a binding preference switch of EBS from H3K27me3- to H3K4me3-marked chromatin at these loci when it lacks the C-terminal loop. Next, we performed immunoprecipitation coupled with mass spectrometry (IP–MS) and found that MULTICOPY SUPPRESSOR OF IRA 4 (MSI4), which is known to interact with CLF33, co-purified with EBS (Fig. 5a,b and Supplementary Table 5). The EBS–MSI4 interaction was independent of the C-terminal loop (Fig. 5c).
Fig. 5 |. EBS interacts with MSI4.
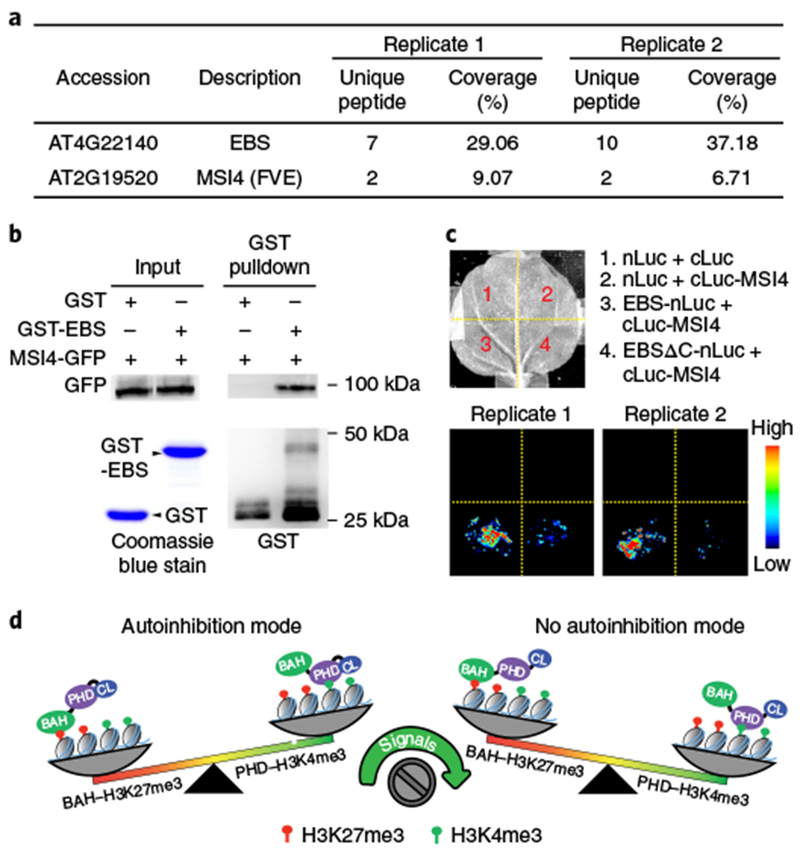
a, Summary of partial proteins associated with EBS identified by affinity purification and mass spectrometry analysis from two independent experiments. b, In vitro pulldown assay using recombinant purified GST-EBS and MSI4 proteins purified from transgenic plants expressing 35S::MSI4-GFP. Representative blots from two independent experiments with similar results are shown (uncropped images are shown in Supplementary Fig. 8). c, A split-luciferase complementation assay confirms EBS-MSI4 interaction. Nicotiana benthamiana leaves co-infiltrated with Agrobacteria containing the indicated constructs were used for imaging. Two independent experiments are shown. nLuc- and cLuc-only vectors served as negative controls. d, A working model depicting the two binding states of EBS. In autoinhibition mode, the C-terminal loop folds back to interact with the PHD finger and inhibits its accessibility to H3K4me3, which enables BAH binding to H3K27me3. Following release of the inhibitory loop by certain signals, EBS switches its binding preference toward H3K4me3.
In conclusion, we characterized EBS as a bivalent reader capable of switching its binding preference between H3K27me3- and H3K4me3-marked chromatin in a manner mediated by its C-terminal inhibition loop (Fig. 5d). We speculate that certain developmental (for example, small ligands and hormones) or molecular (for example, post-translational modifications and protein-protein interactions) signals might trigger the release of this inhibitory loop. In the future, it will be interesting to identify these signals and elucidate how they modulate the chromatin binding preference of EBS.
URLs
SERp server, http://services.mbi.ucla.edu/SER/; Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/; ProteomeXchange, http://proteomecentral.proteomexchange.org/; Sequence Read Archive, http://www.ncbi.nlm.nih.gov/sra; R packages, http://www.cran.r-project.org/; NGS plot, https://github.com/shenlab-sinai/ngsplot; BEDTools, http://bedtools.readthedocs.io/en/latest/.
Methods
Plant materials and growth conditions
All Arabidopsis thaliana lines were derived from ecotype Columbia-0 (Col-0). The loss-of-function ebs point-mutation line11 was a gift from M. Piñeiro (Centro de Biotecnología y Genómica de Plantas, UPM-INIA). The clf28 line (SALK_139371) was a gift from R. Amasino (University of Wisconsin–Madison). Seeds were sown in soil and kept at 4 °C for 2 d before transferring to constant (24 h light), long-day (16 h light/8 h dark), or short-day (8 h light/16 h dark) conditions at 22 °C. N. benthamiana plants were grown at 24 °C under constant light.
Construction of plasmid vectors and generation of transgenic plants
Genomic DNA sequences corresponding to EBS, EBSΔC, and CLF with their 1-kb promoters were amplified and cloned into the pENTR/D-TOPO vector (Thermo Fisher, K240020). These constructs were recombined into pEarleyGate302 binary vectors to create epitope-tagged 3 × FLAG fusions34,35 and were transformed into their respective mutant background: pEBS::EBS-3 × FLAG/ebs (abridged as EBS-FLAG), pEBS::EBSΔC-3 × FLAG/ebs (abridged as EBSΔC-FLAG), and pCLF::CLF-3 × FLAG/clf28 (abridged as CLF-FLAG). The EBS BAH and PHD binding-disruptive point mutations and C-terminal deletion constructs were generated in the pEBS::EBS-3 × FLAG backbone using a Q5 Site-Directed Mutagenesis kit (New England BioLabs, E0554S) and were transformed into the ebs mutant via Agrobacterium-mediated floral dip method. Western blotting and RT–qPCR experiments were conducted using tissue from the same individual plant lines in the T2 generation using anti-FLAG antibody (Sigma, A8592) and the primers described in Supplementary Table 6. Sequential ChIP experiments in the C-terminal deletion mutant lines were conducted in T2 plants. The pEBS::EBS-3 × FLAG construct was crossed into the clf28 mutant background, and F2 plants with loss-of-function CLF were used for ChIP–qPCR experiments.
GST protein purification and peptide microarray.
The full-length cDNA of EBS was amplified and cloned into GST-tagged protein expression vector pGOOD, modified from pGEX-6P (GE Healthcare) by adding a 6 × His tag at the C terminus. GST vector only and GST-EBS protein were induced with 200 mM IPTG (isopropyl β-d-1-thiogalac-topyranoside) for 3 h at 16 °C. Peptide microarray was performed as previously described36. Briefly, peptide binding assays were performed with a modified two-chamber simplex gasket (Intuitive Bioscience). After tightening the gasket onto the slide surface, the peptide array was blocked with blocking solution (1 × PBS, 0.05% Tween-20 pH 7.4, 1% BSA) at 4 °C overnight to reduce nonspecific binding. After washing three times with 1 ml of TTBS buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% Tween-20), 500 nM GST-tagged recombinant EBS protein in 1 ml of binding buffer (50 mM HEPES pH 7.5, 50 mM NaCl, 5% glycerol, 0.4% BSA, 2 mM DTT) was incubated with the peptide array at room temperature for 2 h with gentle rotation. The slide was then washed with TTBS three times and incubated with an anti-GST antibody (Thermo Fisher, CAB4169) in TBS with 1% BSA for 1 h at room temperature. After washing with TTBS three times, the slide was incubated with an anti-rabbit IRDye 650 secondary antibody (LI-COR, 926-65020) in 1 ml of TBS buffer with 1% BSA for 1 h. The slide was washed with TTBS three times, dried by centrifugation, and imaged at dual wavelengths of 532 and 635 nm on an Axon GenePix 4000B (Molecular Devices). The laser power was set to 100% with automatic gain adjustment (0.05% saturation tolerance) for dual photomultipliers. Features in each block were defined by manual adjustment of a 13 × 13 grid (feature diameter, 280 μm; column spacing and row spacing, 320 μm) to cover every spot. Signal intensities were quantified by GenePix Pro 6.1 software (Molecular Devices). For each spot, the mean intensities at the 635-nm wavelength were used for subsequent analysis. For each peptide species, an average was calculated from three replicate spots. The signal at the 532-nm wavelength was used as a control to identify misprinting events. Detailed information for peptides on the array can be found in Supplementary Table 1.
Immunoprecipitation and mass spectrometry
IP–MS was performed as previously described with slight modifications37. Nuclei were isolated from 15 g of Arabidopsis plants expressing EBS-FLAG with 150 ml of nuclear isolation buffer (10 mM HEPES pH 8, 1 M sucrose, 5 mM KCl, 5 mM MgCl2, 5 mM EDTA, 0.6% Triton X-100, 0.4 mM PMSF) with complete protease inhibitor cocktail (Roche) and then lysed with 15 ml of IP buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 5 mM MgCl2, 5% glycerol, 0.1% NP-40, 1 mM DTT, 1 mM PMSF, and protease inhibitor cocktail) by Dounce 20 times with a tight pestle. EBS protein complex was purified with 200 μl of FLAG beads (Sigma, M8823) and eluted with 400 μl of elution buffer (IP buffer without NP-40) containing 150 ng/μl 3 × FLAG peptide (Sigma, F4799) three times. Proteins were precipitated with 20% trichloroacetic acid and digested with trypsin for mass spectrometry analysis.
In vitro pulldown assays
GFP-tagged MSI4-transgenic Arabidopsis were generated by introducing GFP-tagged full-length MSI4 (MSI4-GFP) with expression driven by the 35 S promoter into the wild-type Col background. One gram of 3-week-old plants expressing MSI4-GFP was ground into fine powder with liquid nitrogen and resuspended in 5 ml of pulldown buffer (50 mM Tris-HCl pH 7, 150 mM NaCl, 1 mM EDTA, 5% glycerol, 0.5% Triton X-100, protease inhibitor). After centrifugation for 15 min at 12,000 r.p.m., the supernatant was evenly divided and transferred into two new tubes and incubated with 10 μg of GST-EBS or GST vector only for 3 h with constant rotation at 4 °C. Prewashed glutathione agarose bead slurry (GE Healthcare, 17-0756-01) was added to the MSI4-GFP extracts and incubated for 1 h. Beads containing protein complex were washed with pulldown buffer four times before boiling with 40 μl of SDS loading buffer for SDS–PAGE separation and western blots. The GFP antibody was from Roche (11814460001), and the anti-GST antibody was from Thermo Fisher (CAB4169).
Split-luciferase assays
The coding sequences of EBS and EBSΔC were cloned into the pCAMBIA-nLUC vector38 containing the N-terminal part of the luciferase gene, resulting in EBS-nLuc and EBSΔC-nLuc plasmid constructs. Full-length cDNA of MSI4 was cloned into the pCAMBIA-cLUC expression vector38 containing the C-terminal part of the luciferase gene, resulting in the cLuc-MSI4 construct. N. benthamiana leaves were co-infiltrated with Agrobacterium harboring the EBS-nLuc and cLuc-MSI4 constructs or the EBSΔC-nLuc and cLuc-MSI4 constructs. Primer details for the constructs are described in Supplementary Table 6. Infiltrated plants were kept in the dark for 24 h before being moved to a normal condition (16 h light/8 h dark) for an additional 24 h. Infiltrated leaves were detached and sprayed with 2 mM luciferin. Images were taken with ImageQuant LAS 4000 (GE Healthcare) and transferred to color images with Image Studio (LI-COR).
Protein expression and purification for protein crystallization
The Arabidopsis EBS sequence was cloned into a self-modified pET-Sumo vector to fuse a His6-SUMO tag to the N terminus of the target protein. The plasmid was transformed into Escherichia coli strain BL21(DE3) RIL (Stratagene). Cultured cells were grown in LB medium at 37 °C until the OD600 reached 0.8. Cells were then cooled to 18 °C, and expression was induced by adding IPTG to a final concentration of 0.2 mM. Recombinant expressed protein was purified using a nickel affinity column (GE Healthcare). The His6-SUMO tag was cleaved by a self-purified ULP1 protease and further removed by running an additional nickel affinity column (GE Healthcare). The protein was further purified using a heparin column (GE Healthcare) and a Superdex G200 gel-filtration column (GE Healthcare). All EBS point-mutant and deletion constructs were generated using a PCR-based mutagenesis method and were expressed and purified using the same protocol as for the wild-type protein. Primer details for the constructs are described in Supplementary Table 6. All peptides were ordered from GL Biochem and EpiCypher.
Crystallization, data collection, and structure determination
Before crystallization, purified protein was mixed with histone peptides at a molar ratio of 1:4 and further incubated at 4 °C for 1 h. Crystallization was carried out using the hanging drop vapor diffusion method at 20 °C. Despite efforts on many different EBS constructs and various lengths of the H3K27me3 peptide, we were unable to obtain crystals with diffraction quality. Alternatively, we introduced localized surface-entropy-reduction mutations to EBS39 and found that a K201A K202A double mutant of EBS in complex with the H3(20–35)K27me3 peptide successfully yielded crystals in a condition of 0.2 M sodium acetate and 20% PEG-3350. EBSΔC (1–199) in complex with H3K4me2 peptides was crystallized in a condition with 0.1 M MES, pH 6.5, and 40% PEG-200. All crystals were soaked in reservoir solution supplemented with 15% glycerol and flash cooled in liquid nitrogen. The Zn-SAD data for the ESB–H3K27me3 complex were collected at the peak wavelength for zinc (1.2827 Å) at beamline BL19U1 of the National Center for Protein Sciences Shanghai (NCPSS) at the Shanghai Synchrotron Radiation Facility (SSRF). All other data were collected at beamline BL17U1 at SSRF. The data were further processed using the HKL2000/3000 packages40. Statistics from data collection are listed in Supplementary Table 2.
The structure of EBS in complex with H3K27me3 peptide was solved using the SAD method as implemented in Phenix41. Structure refinement and model building were conducted using Phenix41 and Coot42, respectively. The structure of the EBSΔC–H3K4me2 complex was solved using the molecular replacement method with Phenix41 and with the structure of the EBS–H3K27me3 complex as a search model. Structure refinement and model building were carried out by Phenix41 and Coot42, respectively. Throughout refinement, 5% randomly chosen reflections were set aside for the free R validation. The geometry of the models was analyzed using Procheck43. The structural refinement statistics are summarized in Supplementary Table 2. The X-ray structure validation reports for the two structures can be found in Supplementary Data 1 and 2. All molecular graphics were generated using PyMOL (DeLano Scientific).
Isothermal titration calorimetry
ITC binding curves were measured using a Microcal PEAQ-ITC instrument (Malvern). Purified proteins were dialyzed against buffer containing 100 mM NaCl, 20 mM HEPES pH 7, and 2 mM β-mercaptoethanol. Peptide was dissolved in the same buffer. Titrations were performed at 20 °C. Data were analyzed using Origin 7.0. The peptides used in the ITC experiments were as follows: H3(1–15)K4me3, H3(1–15)K4me2, H3(1–15) K4me1, H3(1–15)K4me0, H3(20–35)K27me3, H3(20–35)K27me2, H3(20–35) K27me1, H3(20–35)K27me0, and H3(1–35)K4me3K27me3.
RNA extraction and qRT–PCR
Total RNA was extracted from 22-leaf-stage plants grown under long-day conditions harvested right before dusk using TRIzol reagent (Thermo Fisher, 15596026). One microgram of RNA was reverse-transcribed into cDNA with SuperScript III (Thermo Fisher, 18080093) followed by qPCR with SYBR Green Master Mix (Bio-Rad, 1725271) using CFX96 RealTime System 690 (Bio-Rad). Relative FT transcript level to ACTIN was calculated with the ΔCt method44. Detailed information for primers can be found in Supplementary Table 6.
Immunoblot analysis
FLAG-epitope-tagged proteins were detected with horseradish peroxidase (HRP)-conjugated anti-FLAG antibody (Sigma, A8592). The following histone antibodies were used: H3 (Abcam, ab1791), H3K4me3 (Millipore, 04-745), and H3K27me3 (Millipore, 07-449). All western blots were developed using the ECL Plus Western Blotting Detection System (GE Healthcare, RPN2132) and chemiluminescent imaging using an Imagequant LAS 4000 (GE Healthcare).
Chromatin immunoprecipitation
EBS, EBSΔC, and CLF ChIP assays were performed as previously described37,45. Briefly, 2 g of 3-week-old plants were ground into powder in liquid nitrogen and cross-linked in nuclear isolation buffer (10 mM HEPES pH 8, 1 M sucrose, 5 mM KCl, 5 mM MgCl2, 5 mM EDTA, 0.6% Triton X-100, 0.4 mM PMSF, and protease inhibitor cocktail tablet (Roche, 14696200)) with 1% formaldehyde for 20 min at room temperature. Cross-linking was quenched by adding glycine to a final concentration of 120 mM, and the homogenate was filtered through miracloth (Millipore, 475855). Samples were pelleted and resuspended with 0.3 ml of nuclear lysis buffer (50 mM Tris-HCl pH 8, 10 mM EDTA, 1% SDS, 0.4 mM PMSF, protease inhibitor cocktail tablet) and 0.7 ml of ChIP dilution buffer (1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8, 167 mM NaCl, 0.4 mM PMSF, and protease inhibitor cocktail tablet) before shearing the chromatin by sonication.
After centrifugation at 5,000 r.p.m. for 10 min, the supernatant was incubated with 40 μl of magnetic FLAG beads (Sigma) overnight with rotation at 4 °C.
After sequential washes with low-salt buffer (150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8), high-salt buffer (500 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8), LiCl buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8), and TE buffer (10 mM Tris-HCl pH 8, 1 mM EDTA), the DNA-protein complex was eluted with elution buffer (1% SDS, 0.1 M NaHCO3) and cross-linking was reversed at 65 °C overnight. After proteinase K and RNase treatment, DNA was purified by the standard phenol-chloroform method and used for qPCR or high-throughput sequencing.
Library construction, high-throughput sequencing, and data analysis
ChIP-seq libraries were constructed using the Ovation Ultralow DR Multiplex System (NuGEN, 0330) and sequenced using a HiSeq 2000 (Illumina) in the University of Wisconsin–Madison Biotechnology Center. Sequencing reads were aligned to the Arabidopsis TAIR10 genome using Bowtie2 (v2.1.0) with default parameters46. Reads mapping to identical positions in the genome were collapsed into one read. EBS and CLF ChIP-seq data were normalized to Col-0, and MACS47 was used for peak calling with P = 1 × 10−3. H3K4me3 ChIP-seq data were normalized to H3, and SICER48 was used for peak calling. BEDTools (2.17.0) and custom Perl scripts were used for further analysis. ChIP-seq peaks of H3K4me3 and H3K27me3 were obtained from a prior study49. 6,466 H3K27me3-marked genes were obtained by overlapping the two published H3K27me3 ChIP-seq datasets31,49. In the EBS and CLF genome-wide distribution pattern, the log2-transformed value of the normalized ChIP read density relative to wild-type Col-0 reads was calculated and binned at 100-kb intervals. For plots of H3K4me3 and H3K27me3 levels over genes, each gene was divided into 20 intervals (5% for each interval) separately for the gene body, the 2 kb upstream of the TSS, and the 2 kb downstream of the TTS. All statistical analysis and figures were done using R (3.2.3). ChIP-seq read numbers can be found in Supplementary Table 7.
Sequential chromatin immunoprecipitation
Four grams of the above ground tissues of Col, EBS-FLAG, and EBSΔC-FLAG from 3-week-old plants grown in long-day conditions was harvested. The first ChIP with FLAG beads was performed as described above. Instead of eluting protein–DNA complex with ChIP elution buffer, the FLAG immunoprecipitated DNA–protein complex was eluted twice with 250 μl of 150 ng/μl 3 × FLAG peptide for 15 min at room temperature each time. Of the 500 μl of total eluted EBS–DNA complex, 100 μl was used for DNA purification as EBS enrichment. The remaining 400 μl was divided equally into two tubes and incubated with 5 μl of either anti-H3K4me3 (Millipore, 04-745) or anti-H3K27me3 (Millipore, 07-449) antibody for 1 h at 4 °C with rotation. Samples were further incubated with 40 μl of prewashed magnetic protein A beads (Thermo Fisher, 10001D) overnight at 4 °C with rotation. The beads were washed twice with low-salt buffer, one time with high-salt buffer, one time with LiCl buffer, and one time with TE. DNA–protein complexes were then eluted with ChIP elution buffer, and cross-linking was reversed at 65 °C overnight. After proteinase K and RNase treatment, DNA was purified by standard phenol-chloroform method and used for qPCR.
Statistical analysis
Statistical test methods, sample sizes, and P values are indicated in the corresponding figure legends. The investigators were not blinded to group allocations during experiments and data analysis. The experiments were not randomized unless stated, and no statistical method was used to predetermine sample sizes. Two-tailed Student’s t tests were conducted using Excel. P values for the Venn diagram overlap analysis are based on hypergeometric tests. Box plots were generated using R (version 3.2.3) with the horizontal center lines denoting the median, box edges denoting the IQR, and whiskers denoting ± 1.5 IQR.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Code availability
Analyses were performed using publicly available software and software available upon request.
Data availability
The X-ray structures have been deposited in the Protein Data Bank with accession codes 5Z8L and 5Z8N. ChIP-seq data have been deposited into GEO with accession code GSE101428. The mass spectrometry datasets have been deposited into ProteomeXchange with accession code PXD009794.
Supplementary Material
Acknowledgements
We thank the staff at the Shanghai Synchrotron Radiation Facility for data collection, the staff at the UW-Madison Biotechnology Center for high-throughput sequencing, J. Jiang and J. Chen (UW-Madison) for MSI4 constructs, Z. Shen for help with the split-LUC assay, M. Piñeiro (Centro de Biotecnología y Genómica de Plantas, Spain) for ebs seeds, Y. He (Shanghai Center for Plant Stress Biology) for discussion, and R. Amasino and D. Patel for manuscript comments. This study was supported by the National Key R&D Program (2016YFA0503200), the National Science Foundation of China (31622032 and 31770782), and the Chinese Academy of Sciences to J.D.; the Alexander von Humboldt Foundation and NSF CAREER (MCB-1552455) and NIH-MIRA (R35GM124806) to X.Z.; the NIH (GM059785-15/P250VA) to J.M.D.; and the NIH-NCI (R01CA193481) to L.M.S.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41588-018-0187-8.
References
- 1.Piunti A & Shilatifard A Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352, aad9780 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Geisler SJ & Paro R Trithorax and Polycomb group-dependent regulation: a tale of opposing activities. Development 142, 2876–2887 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Moris N, Pina C & Arias AM Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet 17, 693–703 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Hepworth J & Dean C Flowering locus C’s lessons: conserved chromatin switches underpinning developmental timing and adaptation. Plant Physiol. 168, 1237–1245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao J, Jin R & Wagner D Developmental transitions: integrating environmental cues with hormonal signaling in the chromatin landscape in plants. Genome Biol. 18, 88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ietswaart R, Wu Z & Dean C Flowering time control: another window to the connection between antisense RNA and chromatin. Trends Genet. 28, 445–453 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Amasino R Seasonal and developmental timing of flowering. Plant J. 61, 1001–1013 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Hyun Y, Richter R & Coupland G Competence to flower: age-controlled sensitivity to environmental cues. Plant Physiol. 173, 36–46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang G & Kohler C Epigenetic processes in flowering plant reproduction. J. Exp. Bot 68, 797–807 (2017). [DOI] [PubMed] [Google Scholar]
- 10.López-González L et al. Chromatin-dependent repression of the Arabidopsis floral integrator genes involves plant specific PHD-containing proteins. Plant Cell 26, 3922–3938 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piñeiro M, Gómez-Mena C, Schaffer R, Martínez-Zapater JM & Coupland G Early Bolting In Short Days is related to chromatin remodeling factors and regulates flowering in Arabidopsis by repressing. Ft. Plant Cell 15, 1552–1562 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du J et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151, 167–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuo AJ et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier–Gorlin syndrome. Nature 484, 115–119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang N & Xu RM Structure and function of the BAH domain in chromatin biology. Crit. Rev. Biochem. Mol. Biol 48, 211–221 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Zhao D et al. The BAH domain of BAHD1 is a histone H3K27me3 reader. Protein Cell 7, 222–226 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su Z & Denu JM MARCC (matrix-assisted reader chromatin capture): an antibody-free method to enrich and analyze combinatorial nucleosome modifications. Curr. Protoc. Mol. Biol 111, 21.32.1–21.32.21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel DJ A structural perspective on readout of epigenetic histone and DNA methylation marks. Cold Spring Harb. Perspect. Biol 8, a018754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musselman CA, Khorasanizadeh S & Kutateladze TG Towards under standing methyllysine readout. Biochim. Biophys. Acta 1839, 686–693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews FH, Strahl BD & Kutateladze TG Insights into newly discovered marks and readers of epigenetic information. Nat. Chem. Biol 12, 662–668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zondlo NJ Aromatic-proline interactions: electronically tunable CH/π interactions. Acc. Chem. Res 46, 1039–1049 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez R & Zhou MM The PHD finger: a versatile epigenome reader. Trends Biochem. Sci 36, 364–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musselman CA & Kutateladze TG Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 39, 9061–9071 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H et al. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol. Cell 28, 677–691 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang WK et al. Malignant brain tumor repeats: a three-leaved propeller architecture with ligand/peptide binding pockets. Structure 11, 775–789 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Min J et al. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol 14, 1229–1230 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Voigt P, Tee WW & Reinberg D A double take on bivalent promoters. Genes Dev. 27, 1318–1338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodrich J et al. A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 386, 44–51 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Kohler C & Grossniklaus U Epigenetic inheritance of expression states in plant development: the role of Polycomb group proteins. Curr. Opin. Cell Biol. 14, 773–779 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Zhang X et al. The Arabidopsis LHP1 protein colocalizes with histone H3 Lys27 trimethylation. Nat. Struct. Mol. Biol 14, 869–871 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Berry S, Rosa S, Howard M, Bühler M & Dean C Disruption of an RNA-binding hinge region abolishes LHP1-mediated epigenetic repression. Genes Dev. 31, 2115–2120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SY, Lee J, Eshed-Williams L, Zilberman D & Sung ZR EMF1 and PRC2 cooperate to repress key regulators of Arabidopsis development. PLoS Genet. 8, e1002512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu F et al. Trithorax group proteins act together with a Polycomb group protein to maintain chromatin integrity for epigenetic silencing during seed germination in Arabidopsis. Mol. Plant 11, 659–677 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Pazhouhandeh M, Molinier J, Berr A & Genschik P MSI4/FVE interacts with CUL4-DDB1 and a PRC2-like complex to control epigenetic regulation of flowering time in Arabidopsis.Proc. Natl Acad. Sci. USA 108, 3430–3435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X et al. Canonical and noncanonical actions of Arabidopsis histone deacetylases in ribosomal RNA processing. Plant Cell 30, 134–152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu L, Chen X, Qian S & Zhong X The plant-specific histone residue Phe41 is important for genome-wide H3.1 distribution. Nat. Commun 9, 630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su Z et al. Reader domain specificity and lysine demethylase-4 family function. Nat. Commun 7, 13387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X et al. Powerdress interacts with histone deacetylase 9 to promote aging in Arabidopsis. eLife 5, e17214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen H et al. Firefly luciferase complementation imaging assay for protein-protein interactions in plants. Plant Physiol. 146, 368–376 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldschmidt L, Cooper DR, Derewenda ZS & Eisenberg D Toward rational protein crystallization: a Web server for the design of crystallizable protein variants. Protein Sci. 16, 1569–1576 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot.Acta Crystallogr.D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laskowski RA, Macarthur MW, Moss DS & Thornton JM Procheck: a program to check the stereochemical quality of protein structures. J. Appl. Cryst 26, 283–291 (1993). [Google Scholar]
- 44.Livak KJ & Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Lu L, Chen X, Sanders D, Qian S & Zhong X High-resolution mapping of H4K16 and H3K23 acetylation reveals conserved and unique distribution patterns in Arabidopsis and rice. Epigenetics 10, 1044–1053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zang C et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo C et al. Integrative analysis of chromatin states in Arabidopsis identified potential regulatory mechanisms for natural antisense transcript production. Plant J. 73, 77–90 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The X-ray structures have been deposited in the Protein Data Bank with accession codes 5Z8L and 5Z8N. ChIP-seq data have been deposited into GEO with accession code GSE101428. The mass spectrometry datasets have been deposited into ProteomeXchange with accession code PXD009794.