

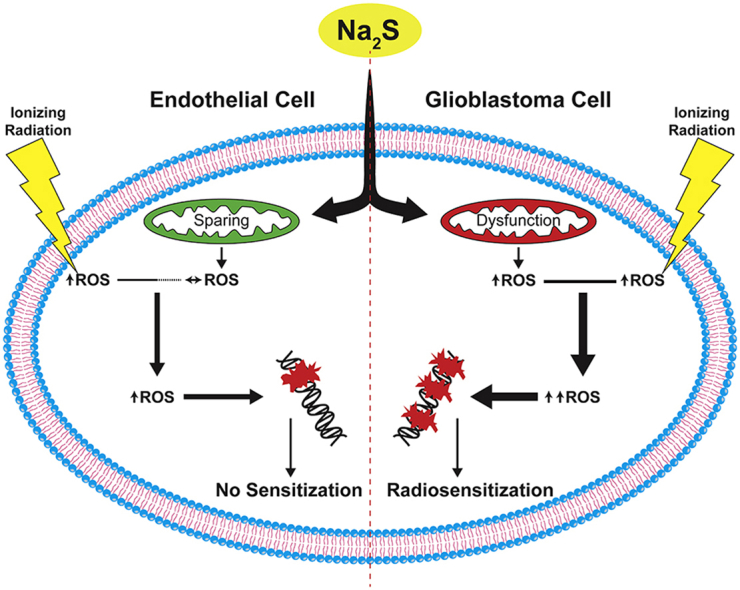
Abstract
Glioblastoma (GBM) has a poor prognosis despite intensive treatment with surgery and chemoradiotherapy. Previous studies using dose-escalated radiotherapy have demonstrated improved survival; however, increased rates of radionecrosis have limited its use. Development of radiosensitizers could improve patient outcome. In the present study, we report the use of sodium sulfide (Na2S), a hydrogen sulfide (H2S) donor, to selectively kill GBM cells (T98G and U87) while sparing normal human cerebral microvascular endothelial cells (hCMEC/D3). Na2S also decreased mitochondrial respiration, increased oxidative stress and induced γH2AX foci and oxidative base damage in GBM cells. Since Na2S did not significantly alter T98G capacity to perform non-homologous end-joining or base excision repair, it is possible that GBM cell killing could be attributed to increased damage induction due to enhanced reactive oxygen species production. Interestingly, Na2S enhanced mitochondrial respiration, produced a more reducing environment and did not induce high levels of DNA damage in hCMEC/D3. Taken together, this data suggests involvement of mitochondrial respiration in Na2S toxicity in GBM cells. The fact that survival of LN-18 GBM cells lacking mitochondrial DNA (ρ0) was not altered by Na2S whereas the survival of LN-18 ρ+ cells was compromised supports this conclusion. When cells were treated with Na2S and photon or proton radiation, GBM cell killing was enhanced, which opens the possibility of H2S being a radiosensitizer. Therefore, this study provides the first evidence that H2S donors could be used in GBM therapy to potentiate radiation-induced killing.
Keywords: Hydrogen sulfide, Glioblastoma, Ionizing radiation, DNA damage, DNA repair, Mitochondria, Reactive oxygen species
Graphical Abstract
Highlights
-
•
Sodium sulfide selectively kills GBM cells by inducing DNA damage.
-
•
Sodium sulfide induces mitochondrial dysfunction and oxidative stress in GBM cells.
-
•
Toxicity to sodium sulfide is dependent on mitochondrial respiration.
-
•
Sodium sulfide radiosensitizes GBM cells to photon and proton radiation.
1. Introduction
Glioblastoma multiforme (GBM) is the most common malignant central nervous system (CNS) cancer in adults accounting for 55% of all gliomas [1]. It is a rapidly progressing, life-threatening disease with a median survival of 14.6 months following maximal safe surgical resection and photon radiotherapy (60 Gy in 30 fractions) with concomitant and adjuvant temozolomide (TMZ), an alkylating agent [2]. The poor survival despite intensive treatment highlights a need for novel therapy. Several clinical trials in GBM have found that dose escalation with either intensity modulated radiation therapy (IMRT) or protons increases median survival up to 21 months; however, there is an increased risk of radiation necrosis that makes the survival benefit unclear [[3], [4], [5], [6]]. The brain vasculature is a critical mediator of radiation necrosis. Endothelial cell apoptosis after ionizing radiation (IR) can cause blood-brain barrier disruption and leukocyte extravasation leading to tissue necrosis [7]. Radiosensitization agents may overcome this problem by allowing for the use of lower doses to achieve comparable cytotoxic effects; however, no radiosensitizers are currently approved for the treatment of GBM [8].
Hydrogen sulfide (H2S) is the second leading cause of inhalational deaths and exposure to 500 ppm can be fatal [9]. However, it is now understood that H2S is an endogenous gasotransmitter [10]. It is synthesized in most cells of the body by three different enzymes: cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (3-MST). In aqueous solution, H2S dissociates at 37 °C, pH 7.4 and exists as H2S, HS−, or S2−. H2S, HS− and S2− collectively are known as hydrogen sulfide in biological systems. H2S in vivo primarily exists as HS− and can alter enzyme activity and cell signaling predominantly by the addition of sulfhydryl groups to proteins [11]. H2S is therefore involved in various physiologic processes and at low levels, protects the cardiovascular system against damage [12]. In the brain, H2S can act as an antioxidant. It increases cytoplasmic and mitochondrial glutathione in neurons and protects against glutamate toxicity [13,14]. It also attenuates methionine-induced oxidative stress in brain endothelial cells [15].
The role of H2S in cancer biology is less clear and has been a subject of continued debate with studies citing either pro-cancer or anti-cancer effects depending on the cancer type as well as H2S concentration [16]. Upregulation of CBS in colon cancer promotes proliferation and angiogenesis [17]. In contrast, 3-MST is downregulated in astrocytoma and knockdown of CBS promotes GBM tumorigenesis suggesting a tumor-suppressing role of H2S in the brain [18,19]. Use of H2S donors has also demonstrated anti-cancer effects. The slow-releasing GYY4137 selectively acidifies breast cancer and hepatocellular carcinoma cells but not breast epithelial cells or lung fibroblasts to promote cell death [20]. Several studies have also suggested H2S acts as a nuclear DNA damaging agent in lung fibroblasts and intestinal epithelial cells; however, this effect has not been studied in cancer [21,22].
To date, no studies have examined the effect of exogenous H2S on GBM. In the present study, we show that sodium sulfide (Na2S), a fast-releasing H2S donor, selectively kills GBM cells while sparing normal brain endothelial cells by increasing DNA damage through a ROS-dependent mechanism. Furthermore, this is the first work demonstrating that Na2S, and hence H2S, can selectively radiosensitize GBM cells in culture to photon or proton radiation. This therefore supports future studies into the development of H2S-releasing compounds as clinical radiosensitizers to selectively kill GBM tumor cells.
2. Materials and methods
2.1. Cell culture
Human T98G and U87 cells (ATCC) were cultured in EMEM medium supplemented with 10% fetal bovine serum (FBS). Human cerebral microvascular endothelial cells (hCMEC/D3) were acquired from Dr. Steven Alexander (LSU-Health Shreveport) and cultured in EndoGRO-MV (MilliporeSigma) between passages 32–37 [23]. LN18 and U87 human glioblastoma cell lines from ATCC were used to derive rho-zero (ρ0) sublines by An Tan (Malaghan Institute of Medical Research, New Zealand). The ρ+ and ρ0 LN18 and U87 cell lines were obtained from Michael Berridge (Malaghan Institute of Medical Research, New Zealand) and cultured in DMEM containing 1 mM pyruvate supplemented with 10% FBS and 50 μg/mL uridine. All cells were grown at 5% CO2 and routinely tested for mycoplasma.
2.2. PCR to detect mitochondrial DNA
Total DNA was isolated from cells using the QiaAmp DNA mini kit (Qiagen). PCR primers were obtained from Eurofins to amplify regions of DNA corresponding to β actin [Forward: d(ATCATGTTTGAGACCTTCAACA), Reverse: d(CATCTCTTGCTCGAAGTCCA)] in the nuclear genome or cytochrome b [Forward: d(CTAGCAACACTCCACCTCCTAT), Reverse: d(GTAAGCCGAGGGCGTCTTTGCTTG)] in the mitochondrial genome. PCR reactions were performed according to manufacturer's instructions with 1–2 μg total DNA, Taq DNA polymerase (Promega) and primers to amplify DNA corresponding to β actin (318 bp) or cytochrome b (123 bp) using annealing temperatures of 53 °C or 57 °C, respectively. PCR products were visualized following gel electrophoresis.
2.3. Hydrogen sulfide treatment
Sodium sulfide (Alfa Aesar), a fast-releasing H2S donor, was freshly prepared in degassed, deionized water before each treatment. Na2S from Alfa Aesar has high purity with minimal polysulfide contamination [24]. Cells were treated with either 476 μM Na2S or degassed water for 4 h at 37 °C (Fig. 1A). Cells were media changed and new Na2S added after 2 h due to its short half-life of 5–30 min in vitro. [25] Irradiation experiments were performed during the second 2 h incubation.
Fig. 1.

Sodium sulfide is cytotoxic to GBM but not human cerebral microvascular endothelial cells. Cells were treated with Na2S for 4 h with media change and replacement at 2 h (A). The dose response of T98G and U87 cells to Na2S was determined using clonogenic survival. The dashed green line indicates the position of Surviving Fraction = 1 on the graph. Data was analyzed using a one-way ANOVA with Tukey's post-hoc analysis (B). Free sulfide levels were measured after the 4 h treatment with 476 μM Na2S using HPLC. Data was analyzed using a Student's t-test comparing 0 and 476 μM for each cell line (C). The effect of 476 μM Na2S on hCMEC/D3 was also determined using clonogenic survival. Data was analyzed using a Student's t-test comparing 0 and 476 μM (D). All data are from 3 independent experiments. Error bars represent SD and * represents P < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.4. Hydrogen sulfide measurements
Free sulfide concentration was measured using HPLC [26]. Cells were treated with either degassed water or 476 μM Na2S for a total of 4 h (Fig. 1A) and collected in degassed stabilization buffer (SB; 100 mM Tris-HCl, 0.1 mM DTPA, 0.1% Triton X-100; pH 9.5). The protein concentration was measured using DC™ Protein Assay (Bio-Rad). Subsequent steps were performed in hypoxic conditions. The sample was incubated with 10 mM monobromobimane (MBB) for 30 min. The reaction was stopped with 200 mM 5-sulfosalicylic acid and the sample analyzed by RP-HPLC. Data is expressed as nmol of free sulfide/mg of protein.
2.5. Photon and proton irradiation
Cell cultures were individually irradiated with either proton or photon modalities to physical doses of 2 Gy, 4 Gy, 6 Gy or 10 Gy. For each modality and container geometry, a computed tomography (CT) radiotherapy treatment simulation was performed using a “blank” container. In each case, the simulation was analogous to that of an actual human patient, i.e. the CT imaging parameters were appropriate for dose calculation and the container was aligned using optical lasers to a setup position that was easily reproduced on the radiotherapy machines. For each simulation and subsequent radiation delivery, containers were sandwiched between slabs of radiotherapy-quality water-equivalent plastic to provide appropriate levels of radiation buildup and backscatter. These conditions were necessary for accurate dose calculation and to generate dose distributions that were robust to minor setup errors during radiation delivery. The CT simulation images of the containers were then imported into the RayStation™ treatment planning system (RaySearch Laboratories, Inc.; Stockholm, Sweden) and contoured as necessary (outer boundary, targets, etc.). Treatment plans were then generated for each modality and dose level. In all cases, a single, static beam (oriented above and perpendicular to the cell container) was utilized to deliver the desired dose. Targets were contoured in such a manner that minor setup errors would not affect the dose delivery. Photon irradiations were performed using a 6 MV beam energy on an Elekta VersaHD™ (Elekta, Inc.; Stockholm, Sweden) conventional radiotherapy linear accelerator. Proton irradiations were performed using an energy range of 72 MeV–131 MeV on an Ion Beam Applications (IBA) ProteusONE™ (IBA SA, Louvain-La-Neuve, Belgium) proton therapy gantry. The range of proton energies was necessary due to the pencil beam scanning delivery technique of the machine, which deposits dose at different depths by modulating proton energy.
2.6. Clonogenic assay and DEF calculation
Cells were seeded in cell culture flasks for 4 h before treatment with degassed water or Na2S. For experiments where cells were also treated with ionizing radiation (IR), cells were exposed to 0–10 Gy during the second Na2S treatment (Fig. 1A). Cells were then incubated for a defined period of time to allow formation of colonies with >50 cells (T98G: 10 days, U87: 11 days, hCMEC/D3: 12 days, LN18 WT: 6 days, LN18 ρ0: 21 days). Plating efficiency (PE) was calculated as . The surviving fraction (SF) was defined as . Survival curves following ionizing radiation were fit to the linear quadratic equation where D is the dose. SF for IR with Na2S was normalized to Na2S treatment alone to adjust for cytotoxicity from Na2S. The dose enhancement fraction at 10% survival (DEF10) for Na2S was defined as . The relative biological effectiveness at 10% survival (RBE10) for protons was defined as , where D10 is the treatment dose that produces 10% surviving fraction.
2.7. γH2AX staining
γH2AX foci were stained using immunohistochemistry. Briefly, cells were cultured on glass coverslips and grown to confluency. Following treatment with Na2S ± IR (Fig. 1A), cells were either immediately fixed in 4% paraformaldehyde or were incubated 2–24 h prior to fixing. Cells were then lysed, and blocked with 5% normal goat serum. Cells were incubated overnight at 4 °C in anti-γH2AX antibody (MilliporeSigma, 1:2000), stained with anti-mouse IgG Alexa Fluor 488 secondary antibody (Invitrogen, 1:500) for 1 h, and DAPI (Biotium, 1 μg/mL). Images were taken using a Nikon Eclipse Ti or Olympus BX43 microscope. The number of foci in 50 cells was counted in each of 3 independent experiments using JCountPro and the data pooled to determine the average number of foci per cell [27].
2.8. Alkaline comet assay
The FPG-modified alkaline comet assay was performed as previously described, with modification [28]. Following treatment with Na2S ± IR (Fig. 1A), samples of cells were divided in two and embedded in 6% low-melting point agarose, solidified on agarose coated slides, and incubated in lysis buffer (100 mM EDTA, 2.5 M NaCl, 10 mM Tris-HCl, 1% Triton-X; pH 10) overnight at 4 °C. Slides were equilibrated in enzyme reaction buffer (40 mM HEPES, 0.1 M KCl, 0,6 mM EDTA, 0.2 mg/mL BSA; pH 8). To detect oxidative base damage, one slide of each sample remained in only enzyme reaction buffer, while the second was treated with 8 U/mL of formamidopyrimidine [fapy]-DNA glycosylase (FPG, New England Biolabs). Both slides were incubated for 30 min at 37 °C. Slides were equilibrated and subjected to electrophoresis in alkaline buffer (0.3 M NaOH, 1 mM EDTA) for 20 min at 30 V and 300 mA. DNA was recondensed in neutralization buffer (0.4 M Tris-HCl, pH 7.5) for 20 min, dried overnight, and stained with propidium iodide (1 μg/mL). Comets were imaged using a Nikon Eclipse Ti and 15X objective. The tail moment was calculated from the tail length and staining intensity of the head and tail using OpenComet (ImageJ) [29]. Approximately 50 cells were analyzed for each of three independent experiments and the average tail moment calculated.
2.9. Quantification of glutathione
Cellular glutathione concentration was measured using high performance liquid chromatography (HPLC) as previously described, with modification [30]. Cells were treated with either degassed water or 476 μM Na2S (Fig. 1A) and collected immediately after treatment in 5% TCA. Samples were subjected to centrifugation and the cell pellet was dissolved in 1 M NaOH prior to the total amount of protein being measured using DC™ Protein Assay (Bio-Rad). The supernatant was derivatized with 80 mM iodoacetic acid (pH adjusted to 7–8) and 6% Sanger's dinitrofluorobenzene (pH adjusted to 7). Samples were filtered and applied to a 250 × 4.6-mm Alltech Lichrosorb NH2 10 μm anion-exchange column to separate reduced (GSH) and oxidized (GSSG) glutathione. The GSH and GSSG per mg protein was calculated.
2.10. ROS assay
CM-H2DCFDA (Molecular Probes) was used to measure general ROS with or without treatment with 2.5 mM TEMPOL (Enzo Life Sciences). Equal number of cells were seeded into 96 well plates one day prior to treatment. Cells were pre-treated with TEMPOL for 1 h and then 476 μM Na2S or degassed water was also added to the cells. After 2 h, the media was replaced and cells were treated again with only 476 μM Na2S or degassed water for 2 h. Cells were then incubated with 2 μM CM-H2DCFDA for 20 min at 37 °C and washed with PBS. Fluorescent intensity was measured at 485 nm excitation and 520 nm emission and normalized to the degassed water control to calculate relative fluorescent intensity.
2.11. Mitochondrial oxygen consumption
Determination of oxygen consumption rate (OCR) was performed as previously described, with modification [31]. 80,000 cells were seeded onto Seahorse Bioscience XF24 microplates and treated the following morning with either degassed water or 476 μM Na2S for a total of 4 h (Fig. 1A). OCR was measured using a Seahorse Bioscience XF24 Extracellular Flux Analyzer. Cells were cultured in DMEM containing 4 mM glutamine (Gibco) and supplemented with 50 mM glucose and 2 mM sodium pyruvate for basal measurements then treated with oligomycin (1 μM), FCCP (1 μM), and rotenone/antimycin A (0.5 μM) to measure OCR. The spare capacity was calculated as the FCCP OCR minus the basal OCR. After OCR measurements, attached cells were dissolved in 0.1 N NaOH for protein analysis with Pierce BCA Protein Assay (ThermoScientific). The OCR was calculated and is expressed as pmol of oxygen consumed per minute per mg protein.
2.12. Mitochondrial complex I and III activity
T98G and hCMEC/D3 cells were treated with either degassed water or 476 μM Na2S for a total of 4 h (Fig. 1A). Cell lysates were prepared in 20 mM hypotonic potassium phosphate buffer (pH 7.5) by freezing in liquid nitrogen and thawing at 37 °C three times. The protein concentration was measured using DC™ Protein Assay (Bio-Rad). Complex I and III activity was measured in the cell lysates according to Spinazzi, M et al. [32] Complex I activity was measured as the rotenone-sensitive oxidation of NADH at 340 nm in 50 μg of protein. Complex III activity was measured as the antimycin A-sensitive oxidation of cytochrome c at 550 nm in 20 μg of protein. Activities are expressed as nmol conversion of NADH to NAD/min/mg protein for complex I and nmol reduction of oxidized cytochrome c/min/mg for complex III.
2.13. DNA repair assays
Non-homologous end-joining (NHEJ) and base excision repair (BER) capacity were assessed using plasmid reporter assays. For NHEJ, the NHEJ-I vector [33] was obtained from Vera Gorbunova (University of Rochester, NY). The NHEJ-I vector was linearized with I-SceI and subjected to electrophoresis prior to purification using the Qiagen Gel Extraction Kit (Qiagen).
For BER, 8-oxoguanine (8-oxoG) was incorporated into the transcribed strand of the DNA sequence encoding the mOrange reporter [34]. By-pass of the 8-oxoG during transcription results in insertion of a C or an A. Only incorporation of an A results in transcript that encodes active mOrange reporter. Repair of 8-oxoG produces transcript with a C that produces inactive mOrange, resulting in decreased mOrange expression.
T98G cells were pre-treated with water or 476 μM Na2S for 2 h. Cells were removed from the dish and transfected using solution V, program O16 and the AMAXA Nucleofector (LONZA). At least two transfections for each type of vector and treatment were carried out in each experiment, and triplicate experiments were performed. Each transfection contained 1.2 × 106 cells and 1 μg linear NHEJ-I vector and 300 ng pDS2Rednuc (Clontech) for the NHEJ assay or 100 ng pMAX:mOrange 8oxoG, 1.8 μg carrier DNA and 100 ng pMAXBFP for the BER assay. Following transfection, cells were treated for 2 h with water or 476 μM Na2S. Flow cytometry was used to determine the GFP and RFP-expressing cells for NHEJ after 24 and 48 h or mOrange and BFP-expressing cells for BER after 17 h.
2.14. Statistical analysis
All experiments were repeated three independent times and data are presented as mean ± standard deviation (SD) unless otherwise indicated. Student's t-test was used for comparison between two groups. One way analysis of variance (ANOVA) with Tukey's post-hoc analysis was used for comparison of >2 groups. All statistical and regression analyses were performed in GraphPad Prism (GraphPad Software Inc.). Regression coefficients were compared using one-way ANOVA. A p-value less than 0.05 was considered statistically significant.
3. Results
3.1. Sodium sulfide selectively kills GBM cells
Studies on the role of H2S in cancer have yielded conflicting results with disparate studies claiming pro-cancer and anti-cancer effects. Published studies indicate that H2S may be detrimental for GBM. Knockdown of cystathionine beta synthase (CBS), the major H2S synthesis enzyme in the brain, increases tumor growth in a mouse xenograft model of GBM [19]. To determine whether H2S is able to kill GBM cells, T98G and U87 cells were treated with Na2S, a fast-releasing H2S donor. A dose dependent reduction in colony formation was measured for T98G and U87 cells treated with Na2S (Fig. 1B). 476 μM Na2S resulted in approximately 40% cell killing and was the dose chosen for subsequent experiments. Free sulfide levels in cells were elevated after the 4 h 476 μM Na2S treatment (Fig. 1C) despite the 5–30 min in vitro half-life of Na2S [25]. Since endothelial cells are important players in GBM treatment-associated toxicity [7] and are expected to be exposed to any adjuvant treatment, the cytotoxicity of 476 μM Na2S was assessed for cerebral microvascular endothelial cells (hCMEC/D3). Interestingly, treatment with 476 μM Na2S did not decrease hCMEC/D3 survival (Fig. 1D) even though free sulfide levels were elevated in the cells (Fig. 1C).
3.2. Sodium sulfide induces DNA damage in GBM
A limited number of studies have suggested H2S can act as a genotoxic agent [21,35]. Increased DNA damage can promote cell death and may contribute to Na2S GBM cell toxicity. To determine if Na2S is a genotoxic agent in GBM, DNA damage was quantified using γH2AX immuno-staining. While γH2AX foci are generally suggestive of double strand breaks (DSBs), γH2AX can also increase following other forms of DNA damage [36]. Therefore an increase in γH2AX is an indicator of an increase in DNA damage and possibly DSBs. Treatment with 476 μM Na2S increased the number of γH2AX foci per cell in T98G and U87 cells with no significant effect in hCMEC/D3 (Fig. 2A and B).
Fig. 2.

Sodium sulfide induces DNA double-strand break formation in GBM. Representative images of γH2AX foci following treatment with 476 μM Na2S or degassed water for 4 h are shown (A). The number of foci per cell was quantified with JCount Pro using the same parameters for all cell types and replicates (B). Analysis was performed on 150 cells pooled from 3 independent experiments and error bars represent standard error of the mean (SEM). * represents P < 0.05 using a Student's t-test.
Oxidative base lesions are the most prevalent type of DNA damage in cells with 8-oxoguanine (8-oxoG) being the most common [37]. Previous studies suggest H2S can induce radical-associated DNA damage in naked nuclei [38]; however, no published study has examined this in cells with functional antioxidant pathways. The FPG-modified alkaline comet assay was used to assess oxidative base damage, specifically 8-oxoG and formamidopyrimidines. Na2S increased oxidative base damage in T98G and U87 cells, and to a lesser degree in hCMEC/D3 cells (Fig. 3). This assay detects single strand breaks (SSB) and alkali-labile sites when the cells are not treated with FPG. T98G cells also had a significant increase in SSB following treatment with Na2S (Fig. 3B).
Fig. 3.

Sodium sulfide increases oxidative DNA base damage. The alkaline comet assay with FPG treatment was used to detect oxidative base damage following 0 or 476 μM Na2S treatment for 4 h (A) and the tail moment was measured using OpenComet (B). Analysis was performed on 3 independent experiments with at least 50 cells in each experiment. Error bars represent SD and * represents P < 0.05 using a Student's t-test.
3.3. Sodium sulfide does not inhibit NHEJ or BER repair
An increase in DNA damage can result from either an induction of damage or a decrease in repair. To determine whether Na2S inhibits DNA repair, we used DNA repair reporter plasmids to assess NHEJ, the major pathway for DSB repair, and 8-oxoguanine glycosylase (OGG1)-mediated BER in T98G cells. For NHEJ, the NHEJ-I vector [33] was linearized with I-SceI, which removes an adenoviral exon that disrupts the GFP coding region. Active GFP is only produced if the I-SceI DSB is repaired in the cells by NHEJ. GFP-expression was measured at 24 and 48 h and no significant difference was found between treated and untreated cells (Fig. 4A).
Fig. 4.

Sodium sulfide does not affect DSB repair and OGG1-directed BER. T98G cells were pretreated with 476 μM Na2S or degassed water for 2 h, transfected with reporter plasmid, and treated for an additional 2 h. For non-homologous end joining, repaired plasmids encode functional GFP and fluorescent expression was detected 24 and 48 h post-transfection using flow cytometry. Data is expressed as GFP+/RFP+ to normalize for transfection efficiency (A). To assess BER, mOrange plasmid containing an 8-oxoguanine was co-transfected with pMaxBFP into T98G cells. Transcriptional mutagenesis of 8-oxoguanine results in functional mOrange that was detected 17 h after transfection via flow cytometry. Data is expressed as mOrange+ x MFI normalized to BFP+ x MFI as a transfection control (B). Error bars represent SD. Data was analyzed using a Student's t-test to compare treated and untreated cells at a particular time point.
For BER, 8-oxoguanine (8-oxoG) was incorporated into the transcribed strand of the DNA sequence encoding the mOrange reporter [34]. Only incorporation of an A in the transcript during by-pass of the 8-oxoG produces active mOrange reporter. A decrease in BER prevents removal of 8-oxoG and results in an increase in mOrange expression. Initial experiments examined expression at ∼6, 17 and 40 h post-transfection of the T98G cells. The optimal mOrange expression was after 17 h. Triplicate experiments were performed measuring mOrange expression at 17 h post-transfection but no significant difference was found between treated and untreated cells (Fig. 4B).
3.4. Sodium sulfide increases oxidative stress in GBM
Excess ROS can increase oxidative DNA damage. Glutathione is an important cellular antioxidant that maintains redox balance and a lower GSH:GSSG ratio is an indicator of oxidative burden. T98G and U87 cells treated with Na2S had higher GSSG levels and lower GSH:GSSG ratio compared to untreated cells (Fig. 5A). Strikingly, Na2S had the reverse effect in hCMEC/D3: an increase in the GSH level was measured in treated cells that resulted in a higher GSH:GSSG ratio. An increase in total ROS in GBM cells was also measured using the redox-sensitive fluorescent probe, CM-H2DCFDA. This increase was attenuated by treatment with 2.5 μM TEMPOL (Fig. 5B).
Fig. 5.

Sodium sulfide increases oxidative stress and ROS formation in GBM. GSH and GSSG levels were measured using HPLC to calculate the GSH: GSSG ratio after 4 h of treatment with 0 or 476 μM Na2S (A). ROS was measured using CM-H2DCFDA. Cells were pretreated with TEMPOL for 1 h and then 476 μM Na2S or degassed water was also added to the cells. New media with only 476 μM Na2S or degassed water was added to the cells after 2 h. Data are expressed as a fold change relative to respective controls. N = 3 for U87, N = 4 for T98G and hCMEC/D3 cells (B). Error bars represent SD and * represents P < 0.05 using a paired Student's t-test.
3.5. Sodium sulfide has differential effects on mitochondrial function
The mitochondria are a major source of cellular ROS and complex I and III of the electron transport chain (ETC) are implicated in ROS production [39]. Electron leak at these complexes can result in one electron reductions of oxygen into superoxide (O2•-) and the probability of electron leak increases with decreased mitochondrial respiration [40]. T98G and U87 cells treated with Na2S had significantly lower basal OCR than untreated cells, while Na2S increased basal OCR in hCMEC/D3 (Fig. 6A and B). Na2S had no effect on spare capacity, which is defined as the FCCP OCR minus the basal OCR (Fig. 6C). To determine whether Na2S impairs complex I or III in Na2S-treated cells, complex I and III activities were measured in cell lysates from T98G and hCMEC/D3 cells treated with degassed water or Na2S. Na2S significantly decreased complex III but not complex I activity in T98G cells and had no effect on these activities in hCMEC/D3 (Fig. 6D and E).
Fig. 6.

Sodium sulfide alters mitochondrial function and inhibits complex III activity. Oxygen consumption rate (OCR) was measured in cells treated with 476 μM Na2S or degassed water for 4 h using a Seahorse Bioscience XF24 Extracellular Flux Analyzer (A). Basal respiration is the OCR prior to addition of oligomycin (B). The spare capacity was calculated as the FCCP OCR minus the basal OCR (C). Complex I activity was measured as the rotenone-sensitive oxidation of NADH at 340 nm in cell lysate (D). Complex III activity was measured as the antimycin A-sensitive oxidation of cytochrome c at 550 nm in cell lysate (E). All data are from 3 independent experiments. Error bars represent SD and * represents P < 0.05 using a Student's t-test. Refer to the web version for easier identification of cell lines in Figure 6A.
3.6. Sodium sulfide sensitivity in GBM is dependent on a functional electron transport chain
Thirteen components of the ETC are encoded by mitochondrial DNA (mtDNA) [41]. Rho zero (ρ0) cells lack mitochondrial DNA and a functional ETC. Attempts to produce ρ0 T98G cells were unsuccessful, but PCR to detect cytochrome b confirmed the ρ0 status of the U87 ρ0 and LN18 ρ0 cells obtained from Dr. Berridge (Fig. 7A). U87 ρ0 cells did not form colonies and could not be used for the clonogenic assay. However, in agreement with data from the T98G and U87 cells, LN18 ρ+ cells were sensitive to Na2S and 476 μM Na2S reduced survival by 30% (Fig. 7B). Na2S had no significant effect on LN18 ρ0 cell survival even at the higher dose of 909 μM Na2S, which reduced survival of the LN18 ρ+ cells by over 40%.
Fig. 7.

GBM sensitivity to sodium sulfide is dependent on the electron transport chain. U87 and LN-18 ρ0 were confirmed by PCR using mitochondria specific cytochrome b primers and β-actin as a control (A). Survival of LN18 ρ+ and ρ0 following treatment with Na2S or degassed water for 4 h was determined using clonogenic assay (B). U87 ρ0 cells did not form colonies. All data are from 3 independent experiments. Error bars represent SD and * represents P < 0.05 using a one-way ANOVA with Tukey's post-hoc analysis.
3.7. Sodium sulfide sensitizes GBM to ionizing radiation
Photon and proton ionizing radiation (IR) damages DNA to kill cells. Since Na2S induces DNA damage in T98G and U87 cells, 476 μM Na2S was investigated for its efficacy as a radiosensitizer. T98G cells were irradiated at 0–10 Gy using photons and protons in the presence or absence of Na2S and the data fitted to a linear quadratic model to calculate the RBE10 and DEF10. T98G cells had a proton RBE10 of 1.2, which is in close agreement with clinically accepted values [42]. Na2S had a DEF10 of 1.34 for both photons and protons at 10% survival (Fig. 8A and Table 1). Na2S did not sensitize hCMEC/D3 to photons or protons at a clinically relevant dose of 2 Gy (Fig. 8B). The α or β parameters for the LQ were compared for the IR survival curves with and without Na2S using one-way ANOVA and the P values for the α (Pα) or β (Pβ) parameters were <0.05, respectively, for both photons and protons (Table 1).
Fig. 8.

Sodium sulfide selectively radiosensitizes GBM to ionizing radiation. T98G cells were treated with 476 μM Na2S or degassed water for a total of 4 h and irradiated during the 4th hr of Na2S treatment. Surviving fraction at each radiation dose was normalized to their respective controls to account for Na2S induced killing (A). Data were curve fit to the linear quadratic (LQ) equation to calculate RBE10 and DEF10 at 10% surviving fraction (SF10). Protons had an RBE10 = 1.2. Photons and protons had a DEF10 = 1.34. hCMEC/D3 cells were similarly irradiated using a clinically relevant dose of 2 Gy (B) and data analyzed by two-way ANOVA with Tukey's post-hoc for multiple comparison.
Table 1.
| Type of IR | LQ (0 μM Na2S) | LQ (476 μM Na2S) | Pα | Pβ | SF10 (0 μM Na2S) | SF10 (476 μM Na2S) | DEF10 |
|---|---|---|---|---|---|---|---|
| Photon |
R2 = 0.9385 |
R2 = 0.9645 |
<0.05 | <0.05 | 8.30 Gy | 6.21 Gy | 1.34 |
| Proton |
R2 = 0.9734 |
R2 = 0.9183 |
<0.05 | <0.05 | 6.92 Gy | 5.17 Gy | 1.34 |
LQ is the equation for the surviving fraction obtained by fitting the survival data to the linear quadratic equation.
The α or β parameters for the LQ were compared for the IR survival curves with and without Na2S using one-way ANOVA to obtain the p values for the α (Pα) or β (Pβ) parameters, respectively.
SF10 is the IR dose that reduced survival by 90%.
DEF10 is the Dose Enhancement Factor at 10% survival and is calculated as follows.
3.8. DNA damage induction by ionizing radiation and sodium sulfide
To examine how the combination treatment altered the induction of DNA damage, the comet assay was performed and γH2AX foci/cell were measured (Fig. 9). Na2S tended to increase oxidative base damage following IR as assessed by the comet assay; however, it was not statistically significant (Fig. 9A).
Fig. 9.

DNA Damage Induction by Ionizing Radiation and Sodium Sulfide. The synergistic effect of 476 μM Na2S on IR-induced DNA damage was assessed using the alkaline comet assay with FPG treatment (A) or γH2AX foci/cell (B, C, D). T98G cells were treated with 0 or 476 μM Na2S for a total of 4 h and irradiated with either 1.9 Gy photon or 2 Gy proton during the 4th hour of Na2S treatment. The comet assay was performed immediately after this 4 h treatment schedule. Analysis was performed on 3 independent experiments with at least 50 cells in each experiment using a one-way ANOVA with Tukey's post-hoc analysis. Error bars represent SD (A). γH2AX foci was quantified immediately after the 4 h treatment schedule (0 h repair time, B) and at 2 (C) and 24 h (D) repair time to determine DSB repair kinetics. Analysis was performed on 150 cells pooled from 3 independent experiments and error bars represent SEM (B, C, D). For all experiments, * represents P < 0.05. Each time point was compared using a one-way ANOVA with Tukey's post-hoc analysis but only differences between 0 and 476 μM Na2S at each IR dose are indicated for simplicity. See text for explanation of the whole analysis.
Data from the measurement of γH2AX foci/cell at each time point were compared using a one-way ANOVA. As expected, both photons and protons induced γH2AX foci in T98G cells and levels were elevated after IR at 0 and 2 h compared to 0 Gy (P < 0.05 Fig. 9B and C). Treatment with 476 μM Na2S further increased γH2AX foci/cell immediately following proton but not photon radiation (P < 0.05 Fig. 9B). After 2 h of repair (Fig. 9C), a statistical difference was found between the Na2S-treated and untreated cells at all IR doses, but by 24 h the only difference was between the Na2S-treated and untreated cells irradiated with 2 Gy protons. This suggests that less repair occurred of the DNA damage induced by Na2S and protons compared to protons or photons alone, or Na2S and photons.
A two-way ANOVA with Tukey's post-hoc test was also used to analyze γH2AX foci/cell data for each treatment group at 0, 2 and 24 h to assess DNA repair over time. Without Na2S treatment, the γH2AX foci/cell induced by photons were significantly different between all the time points, indicating that repair was occurring between 0-24 h. However, for protons alone there was no significant difference for the number of γH2AX foci/cell between 0 and 2 h, but a significant difference was found for 0 and 24 h, and 2 and 24 h (P < 0.05). This indicates that repair occurred between 2 and 24 h, which is slower than photon damage. Addition of Na2S to either photons or protons resulted in no significant difference for the number of γH2AX foci/cell between 0 and 2 h, but a significant difference was found for 0 and 24 h, and 2 and 24 h (P < 0.05).
In summary for the γH2AX foci/cell, repair was detected for photon-induced damage from 0 to 24 h, and the analyses suggest that the addition of the Na2S inhibited/slowed repair in the first 2 h. Repair of proton or proton and Na2S-induced damage occurred at 2–24 h, but at 24 h there was still a higher level of γH2AX foci/cell in Na2S and proton-treated cells versus proton-treated cells (Fig. 9D). This suggests that repair of the damage induced by the combination treatment was slower and hence this damage may be more difficult to repair. An increase in oxidative base damage and DSBs increases the probability of formation of complex clustered lesions, which can be difficult to repair [43]. It is possible that addition of Na2S to IR increases the complexity of the damage induced.
4. Discussion
H2S is the third identified endogenous gasotransmitter. It is involved in physiologic processes such as vasodilation, inflammation, and neuromodulation [[44], [45], [46]]. Altered H2S metabolism has been identified in cancers including colon, breast, and GBM [16]. Since endogenously synthesized H2S has a tumor suppressing role in GBM and other brain cancers [18,19], the effect of exogenous H2S on GBM cells was tested. For the first time, the fast-releasing H2S donor, Na2S, was shown to selectively kill and radiosensitize GBM cells to IR by a mechanism requiring mitochondria and involving ROS and DNA damage.
In this study, Na2S exhibited a concentration dependent cytotoxic effect on GBM cells at doses below industry established lethal levels (476 μM Na2S; 37 ppm H2S), but at supraphysiologic levels since free sulfide concentrations vary between 20 nM to several micromolar in tissue [47]. Sulfide can exist as H2S, HS−, or S2− in solution. At physiologic conditions (pH 7.4, 37 °C), ∼20% of sulfide exists as H2S and 80% as HS− with minimal amounts of S2− [48]. Given this, 476 μM Na2S should increase free bioavailable sulfide levels by approximately 95 μM H2S and 380 μM HS−. This dose had no effect on cerebral microvascular cell survival, which is in agreement with published work demonstrating doses up to 1 mM Na2S are not cytotoxic to human umbilical vein endothelial cells (HUVEC) [49]. Although the effect of Na2S on neurons or astrocytes was not examined here, others have shown H2S as an antioxidant in the brain [14,50,51].
In contrast to H2S role as a cytoprotectant, several studies have shown H2S to be a genotoxic agent. H2S increases micronuclei formation in lung fibroblasts [21], generates FPG-sensitive lesions in naked nuclei [38], and increases SSBs in supercoiled plasmid under physiologically relevant conditions by a hydrogen peroxide dependent mechanism [52]. This latter study suggests H2S can produce ROS by chemical reactions independent of cellular mechanisms. Our work demonstrating the induction of γH2AX foci and oxidative base damage by Na2S in GBM cells with intact antioxidant protection mechanisms is in line with published work.
An increase in DNA damage can be due to induction of damage or inhibition of repair. H2S has been shown to alter DNA damage and repair signaling via ATR [53] and can also modulate DNA repair protein activity via post-translational sulfhydration [54,55]. However, we did not observe any differences in NHEJ or OGG1-mediated BER suggesting Na2S is inducing DNA damage. ROS is implicated in this DNA damage induction in T98G and U87 cells as Na2S decreased the GSH:GSSG ratio and increased CM-H2DCFDA fluorescence. Interestingly, Na2S induced a lower level of DNA damage in hCMEC/D3 cells and did not induce CM-H2DCFDA detectable ROS. This could be explained by the Na2S treatment increasing GSH levels in hCMEC/D3 cells, which protects the cells from ROS. Other studies have shown H2S to be protective against oxidative stress in endothelial cells [15,52]. Because the increased ROS in GBM cells was attenuated by TEMPOL, a superoxide dismutase mimetic, it is possible that Na2S promotes superoxide production in GBM cells.
Mitochondria are a major source of superoxide and ROS [40]. Superoxide is generated in the mitochondria by electrons that escape the ETC and reduce molecular oxygen. The probability of electron leak increases with electron occupancy time or impaired mitochondrial respiration [39,40]. Conversely, increased mitochondrial respiration may decrease superoxide formation. H2S is a known modulator of mitochondrial activity. At low concentrations, it donates electrons to the ETC while higher concentrations inhibit complex IV [56]. Therefore, mitochondrial stimulation of basal respiration by Na2S in hCMEC/D3 may be due to electron donation.
Complex I and III are the main generators of mitochondrial ROS [40], but complex I activity was not altered by Na2S in GBM cells; only complex III activity was diminished. H2S has a modest reducing potential with a two-electron redox potential of 0.17 V at pH 7 [57]. Inhibition of complex IV by H2S involves reduction of ferric (Fe3+) heme a3 to ferrous (Fe2+) heme a3 [58]. Interestingly, complex III has a mobile Rieske protein with a unique FeS cluster where one Fe is stabilized by histidines instead of cysteines thus giving it a higher midpoint potential (Em = ∼300 mV) that is comparable to cytochrome a3 in complex IV (Em = ∼220 mV) [59,60]. Complex I also contains Fe–S clusters; however, their negative midpoint potentials make them more resistant to reduction by H2S [61]. Upon reduction by ubiquinol, the Rieske protein in complex III transiently moves from heme bL to the “c1-position” to facilitate electron transfer to cytochrome c1. Impaired oxidation of the Rieske protein as well as maintenance of the Rieske protein away from heme bL promotes ROS production in Rhodobacter capsulatus [62]. It is possible that H2S reduces complex III activity in GBM cells since it has critical iron containing subunits with appropriate midpoint potentials. Therefore, ROS generation in GBM cells is likely due to complex III inhibition. To our knowledge, this is the first report of H2S-mediated complex III inhibition. Inhibition of complex III by Na2S could increase production of superoxide that is subsequently reduced to H2O2 and hydroxyl radicals that damage DNA and promote GBM cell death. To support this, GBM cells without functional ETC (LN-18 ρ0 cells) are protected from Na2S cytotoxicity.
The contrasting effects of H2S on ROS production, DNA damage, and cell death in GBM and normal brain endothelial cells highlight fundamental differences between cancer and normal cells. Cancer cells preferentially use aerobic glycolysis over oxidative phosphorylation for ATP production [63]. In the present study, complex I and III activity were lower in hCMEC/D3 compared to T98G cells. While this appears contradictory to OCR results where GBM cells had a lower OCR, this difference may be accounted for by inefficient transport of electrons to complex IV where oxygen is reduced and ETC dysfunction in GBM cells consistent with the Warburg effect. The Warburg effect, confers a competitive growth advantage to cancer cells by not only providing energy but also maintaining redox balance and conserving carbons for anabolic metabolism. Specifically, cancer cells have elevated NADPH from the pentose phosphate pathway. NADPH is a critical cofactor in the reduction of GSSG into GSH and oxidized thioredoxin (TRX) to reduced TRX [64]. In fact, we observed higher GSH levels in GBM cells compared to hCMEC/D3. These antioxidants reduce excess reactive oxygen species (ROS) produced by cancer cells and maintain an intermediate level of ROS that allow for normal proliferative signaling as well as tumorigenesis [65]. ROS-directed therapies selectively disrupt redox homeostasis in cancer cells by pushing already-elevated, tumor promoting oxidative stress to even higher, tumor suppressing levels with minimal effect on normal cells with comparatively lower oxidative burden [66]. H2S may be acting similarly to selectively promote oxidative stress and cell death in GBM.
Several GBM clinical trials have shown increased survival following dose-escalated radiation therapy; however, increased risk of radiation necrosis makes the survival benefit unclear [[3], [4], [5], [6]]. Radiosensitizers could increase the gap in radiation response between cancer and normal tissue to reduce radiation necrosis. In this study, Na2S radiosensitized T98G cells (DEF10 = 1.34) to both photons and protons likely by enhancing DSBs and oxidative base damage. A dose enhancing factor of 1.34 could significantly affect tumor treatment. An example of an approved and much used radiosensitizer is cisplatin, which is a chemotherapy used in combination with radiation as the standard of care for the treatment of a number of cancers. Cisplatin has a DEF = 1.14 in breast cancer cells [67] and a DEF = 1.4 in squamous cell carcinoma [68].
IR is a DNA damaging agent capable of generating multiple DNA lesions in close proximity (1–2 helical turns) [69]. These clustered lesions are difficult to repair [70,71] and more prevalent following charged particle radiation such as protons [69]. γH2AX foci formed by protons in conjunction with Na2S were more persistent than foci formed by radiation alone. There was also a significant increase in γH2AX foci/cell between Na2S and photon compared to photon alone at the 2 h timepoint. This suggests Na2S may be increasing DNA lesion complexity. Oxidative lesions can occur near DSBs, increasing lesion complexity, and delaying repair that ultimately promotes cell death. Also, repair of closely opposed oxidative base lesions can form DSBs during BER initiation, which may increase γH2AX foci formation at later timepoints [72,73]. This increase in lesion complexity cannot be detected using NHEJ or BER reporter plasmids which only examine repair of the single lesion present in the plasmid. Finally, Na2S did not synergize with 2 Gy IR in hCMEC/D3. This could be explained by the finding that Na2S alone did not induce high levels of DNA damage in these cells. This is a critical consideration in radiation therapy since brain endothelial cells are a key player in white matter necrosis [7].
In summary, we show that Na2S selectively kills GBM cells in culture by inhibiting mitochondrial function, promoting ROS formation, and radiosensitizing GBM cells to both photon and proton therapy. As a CNS targeted drug, H2S has several pharmacologically favorable properties. As a gas, it is freely diffusible across the blood brain barrier. Furthermore, inhalation of H2S gas immediately prior to radiation could rapidly increase its concentration in the brain and promote its use as a radiosensitizer. There are also other sulfide donors with different kinetics (GYY4137), subcellular targeting (AP39), and release mechanisms (DATS) that may potentiate anti-cancer effects [25]. Ideal anti-cancer compounds should also preferentially kill tumor cells and spare normal cells. While the mechanism behind the differential effect of Na2S in cancer versus normal cells remains unclear, H2S may be leveraging a fundamental difference in cancer mitochondria resulting from the Warburg effect to increase ROS beyond tolerable levels. It is important to mention that H2S may have tumorigenic effects at lower concentrations [16]. While 5 μM and 10 μM of Na2S had no effect on GBM cell survival, we did not examine the effect of lower concentrations on DNA damage and mitochondrial function and this is a limitation of the present study. Future studies are needed to better understand the divergent effects of H2S in GBM versus normal tissue as well as determine the optimal donor, concentration, and its efficacy in vivo.
Disclosures
MRM reports grants from Ion Beam Applications (IBA), outside the submitted work; LRR receives honorarium and is a general stockholder of IBA.
Acknowledgements
This work was supported by the Carroll Feist Predoctoral Fellowship awarded to AYX, an Institutional Development Award (IDeA) from the National Institutes of General Medical Sciences of the NIH under grant number P20GM121307 (CGK), and National Institute of Health grant number CA092584 (ZDN). The authors would also like to thank Creighton France for performing the PCR confirming the rho status of cells and Sophia Nicolosi for preparation of linear DNA for the NHEJ-I plasmid.
References
- 1.Thakkar J.P., Dolecek T.A., Horbinski C., Ostrom Q.T., Lightner D.D., Barnholtz-Sloan J.S. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014;23(10):1985–1996. doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Fitzek M.M., Thornton A.F., Rabinov J.D., Lev M.H., Pardo F.S., Munzenrider J.E. Accelerated fractionated proton/photon irradiation to 90 cobalt gray equivalent for glioblastoma multiforme: results of a phase II prospective trial. J. Neurosurg. 1999;91(2):251–260. doi: 10.3171/jns.1999.91.2.0251. [DOI] [PubMed] [Google Scholar]
- 4.Iuchi T., Hatano K., Kodama T., Sakaida T., Yokoi S., Kawasaki K. Phase 2 trial of hypofractionated high-dose intensity modulated radiation therapy with concurrent and adjuvant temozolomide for newly diagnosed glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2014;88(4):793–800. doi: 10.1016/j.ijrobp.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka M., Ino Y., Nakagawa K., Tago M., Todo T. High-dose conformal radiotherapy for supratentorial malignant glioma: a historical comparison. Lancet Oncol. 2005;6(12):953–960. doi: 10.1016/S1470-2045(05)70395-8. [DOI] [PubMed] [Google Scholar]
- 6.Mizumoto M., Yamamoto T., Ishikawa E., Matsuda M., Takano S., Ishikawa H. Proton beam therapy with concurrent chemotherapy for glioblastoma multiforme: comparison of nimustine hydrochloride and temozolomide. J. Neuro Oncol. 2016;130(1):165–170. doi: 10.1007/s11060-016-2228-4. [DOI] [PubMed] [Google Scholar]
- 7.Yang I., Aghi M.K. New advances that enable identification of glioblastoma recurrence. Nat. Rev. Clin. Oncol. 2009;6(11):648–657. doi: 10.1038/nrclinonc.2009.150. [DOI] [PubMed] [Google Scholar]
- 8.Masashi Mizumoto Y.O., Tsuboi Koji. Proton beam therapy for intracranial and skull base tumors. Transl. Cancer Res. 2013;2(2):87–96. [Google Scholar]
- 9.Jiang J., Chan A., Ali S., Saha A., Haushalter K.J., Lam W.-L.M.L. Hydrogen sulfide--mechanisms of toxicity and development of an antidote. Sci. Rep. 2016;6:20831. doi: 10.1038/srep20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16(13):1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 11.Mustafa A.K., Gadalla M.M., Sen N., Kim S., Mu W., Gazi S.K. H2S signals through protein S-sulfhydration. Sci. Signal. 2009;2(96) doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabil O., Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxidants Redox Signal. 2014;20(5):770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura Y., Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18(10):1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 14.Kimura Y., Goto Y.-I., Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxidants Redox Signal. 2010;12(1):1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi N., Moshal K.S., Sen U., Vacek T.P., Kumar M., Hughes W.M. H2S protects against methionine–induced oxidative stress in brain endothelial cells. Antioxidants Redox Signal. 2009;11(1):25–33. doi: 10.1089/ars.2008.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu D., Si W., Wang M., Lv S., Ji A., Li Y. Hydrogen sulfide in cancer: friend or foe? Nitric Oxide. 2015;50:38–45. doi: 10.1016/j.niox.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Szabo C., Coletta C., Chao C., Módis K., Szczesny B., Papapetropoulos A. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Nat. Academ. Sci. U. S. A. 2013;110(30):12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurkowska H., Uchacz T., Roberts J., Wróbel M. Potential therapeutic advantage of ribose-cysteine in the inhibition of astrocytoma cell proliferation. Amino Acids. 2011;41(1):131–139. doi: 10.1007/s00726-010-0593-4. [DOI] [PubMed] [Google Scholar]
- 19.Takano N., Sarfraz Y., Gilkes D.M., Chaturvedi P., Xiang L., Suematsu M. Decreased expression of cystathionine β-synthase promotes glioma tumorigenesis. Mol. Cancer Res. : MCR. 2014;12(10):1398–1406. doi: 10.1158/1541-7786.MCR-14-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee Z.W., Teo X.Y., Tay E.Y., Tan C.H., Hagen T., Moore P.K. Utilizing hydrogen sulfide as a novel anti-cancer agent by targeting cancer glycolysis and pH imbalance. Br. J. Pharmacol. 2014;171(18):4322–4336. doi: 10.1111/bph.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baskar R., Li L., Moore P.K. Hydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. FASEB J. : Off.Pub. Fed. Am. Soc. Exp. Biol. 2007;21(1):247–255. doi: 10.1096/fj.06-6255com. [DOI] [PubMed] [Google Scholar]
- 22.Attene‐Ramos M.S., Nava G.M., Muellner M.G., Wagner E.D., Plewa M.J., Gaskins R.H. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ. Mol. Mutagen. 2010;51(4):304–314. doi: 10.1002/em.20546. [DOI] [PubMed] [Google Scholar]
- 23.Shen W., Li S., Chung S.H., Zhu L., Stayt J., Su T. Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-β1-induced permeability of centrally derived vascular endothelium. Eur. J. Cell Biol. 2011;90(4):323–332. doi: 10.1016/j.ejcb.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Hughes M.N., Centelles M.N., Moore K.P. Making and working with hydrogen sulfide: the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radical Biol. Med. 2009;47(10):1346–1353. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 25.Szabo C., Papapetropoulos A. International union of basic and clinical pharmacology. CII: pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol. Rev. 2017;69(4):497–564. doi: 10.1124/pr.117.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen X., Kolluru G.K., Yuan S., Kevil C.G. Measurement of H2S in vivo and in vitro by the monobromobimane method. Methods Enzymol. 2015;554:31–45. doi: 10.1016/bs.mie.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakl L., Lobachevsky P., Vokálová L., Durdík M., Marková E., Belyaev I. Validation of JCountPro software for efficient assessment of ionizing radiation-induced foci in human lymphocytes. Int. J. Radiat. Biol. 2016:1–8. doi: 10.1080/09553002.2016.1222093. [DOI] [PubMed] [Google Scholar]
- 28.Higgins J.A., Zainol M., Brown K., Jones G.D.D. Anthocyans as tertiary chemopreventive agents in bladder cancer: anti-oxidant mechanisms and interaction with mitomycin C. Mutagenesis. 2014;29(4):227–235. doi: 10.1093/mutage/geu009. [DOI] [PubMed] [Google Scholar]
- 29.Gyori B.M., Venkatachalam G., Thiagarajan P.S., Hsu D., Clement M.-V. OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2014;2:457–465. doi: 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B., Yee Aw T., Stokes K.Y. N-acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox biology. 2018;14:218–228. doi: 10.1016/j.redox.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson K.L., Lin W.-L.L., Miriyala S., Dayton R.D., Panchatcharam M., McCarthy K.J. p62 pathology model in the rat substantia nigra with filamentous inclusions and progressive neurodegeneration. PLoS One. 2017;12(1) doi: 10.1371/journal.pone.0169291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012;7(6):1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 33.Seluanov A., Mao Z., Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J. Vis. Exp. 2010;(43) doi: 10.3791/2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagel Z.D., Margulies C.M., Chaim I.A., McRee S.K., Mazzucato P., Ahmad A. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc. Nat. Academ. Sci. U. S. A. 2014;111(18):32. doi: 10.1073/pnas.1401182111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Attene-Ramos M.S., Wagner E.D., Plewa M.J., Gaskins H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. : MCR. 2006;4(1):9–14. doi: 10.1158/1541-7786.MCR-05-0126. [DOI] [PubMed] [Google Scholar]
- 36.Revet I., Feeney L., Bruguera S., Wilson W., Dong T.K., Oh D.H. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc. Nat. Academ. Sci. U. S. A. 2011;108(21):8663–8667. doi: 10.1073/pnas.1105866108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fortini P., Pascucci B., Parlanti E., D'Errico M., Simonelli V., Dogliotti E. 8-Oxoguanine DNA damage: at the crossroad of alternative repair pathways. Mutat. Res. 2003;531(1–2):127–139. doi: 10.1016/j.mrfmmm.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 38.Attene-Ramos M.S., Wagner E.D., Gaskins R.H., Plewa M.J. Hydrogen sulfide induces direct radical-associated DNA damage. Mol. Cancer Res. 2007;5(5):455–459. doi: 10.1158/1541-7786.MCR-06-0439. [DOI] [PubMed] [Google Scholar]
- 39.Turrens J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552(Pt 2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417(Pt 1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clayton D.A. Transcription and replication of mitochondrial DNA. Hum. Reprod. 2000;15(Suppl 2):11–17. doi: 10.1093/humrep/15.suppl_2.11. [DOI] [PubMed] [Google Scholar]
- 42.Paganetti H. Significance and implementation of RBE variations in proton beam therapy. Technol. Canc. Res. Treat. 2003;2(5):413–426. doi: 10.1177/153303460300200506. [DOI] [PubMed] [Google Scholar]
- 43.Sage E., Harrison L. Clustered DNA lesion repair in eukaryotes: relevance to mutagenesis and cell survival. Mutat. Res. 2011;711(1–2):123–133. doi: 10.1016/j.mrfmmm.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abe K., Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. : Off. J. Soc. Neurosci. 1996;16(3):1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fiorucci S., Antonelli E., Distrutti E., Rizzo G., Mencarelli A., Orlandi S. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129(4):1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 47.Kimura H. Metabolic turnover of hydrogen sulfide. Front. Physiol. 2012;3:101. doi: 10.3389/fphys.2012.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolluru G.K., Shen X., Bir S.C., Kevil C.G. Hydrogen sulfide chemical biology: pathophysiological roles and detection. Nitric Oxide : Biol. Chem. 2013;35:5–20. doi: 10.1016/j.niox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan S., Pardue S., Shen X., Alexander J.S., Orr A.W., Kevil C.G. Hydrogen sulfide metabolism regulates endothelial solute barrier function. Redox biology. 2016;9:157–166. doi: 10.1016/j.redox.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura Y., Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. : Off.Pub. Fed. Am. Soc. Exp. Biol. 2004;18(10):1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 51.Lu M., Hu L.-F., Hu G., Bian J.-S. Hydrogen sulfide protects astrocytes against H2O2-induced neural injury via enhancing glutamate uptake. Free Radic. Biol. Med. 2008;45(12):1705–1713. doi: 10.1016/j.freeradbiomed.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 52.Hoffman M., Rajapakse A., Shen X., Gates K.S. Generation of DNA-damaging reactive oxygen species via the autoxidation of hydrogen sulfide under physiologically relevant conditions: chemistry relevant to both the genotoxic and cell signaling properties of H(2)S. Chem. Res. Toxicol. 2012;25(8):1609–1615. doi: 10.1021/tx300066z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J., Shen X., Pardue S., Meram A.T., Rajendran S., Ghali G.E. The Ataxia telangiectasia-mutated and Rad3-related protein kinase regulates cellular hydrogen sulfide concentrations. DNA Repair. 2019;73:55–63. doi: 10.1016/j.dnarep.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szczesny B., Marcatti M., Zatarain J.R., Druzhyna N., Wiktorowicz J.E., Nagy P. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 2016;6(1):36125. doi: 10.1038/srep36125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao K., Ju Y., Li S., Altaany Z., Wang R., Yang G. S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 2014;15(7):792–800. doi: 10.1002/embr.201338213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szabo C., Ransy C., Módis K., Andriamihaja M., Murghes B., Coletta C. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014;171(8):2099–2122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kabil O., Banerjee R. Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 2010;285(29):21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collman J.P., Ghosh S., Dey A., Decréau R.A. Using a functional enzyme model to understand the chemistry behind hydrogen sulfide induced hibernation. Proc. Nat. Academ. Sci. U. S. A. 2009;106(52):22090–22095. doi: 10.1073/pnas.0904082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shifman J.M., Gibney B.R., Sharp R.E., Dutton P.L. Heme redox potential control in de novo designed four-alpha-helix bundle proteins. Biochemistry. 2000;39(48):14813–14821. doi: 10.1021/bi000927b. [DOI] [PubMed] [Google Scholar]
- 60.Kolling D.J., Brunzelle J.S., Lhee S., Crofts A.R., Nair S.K. Atomic resolution structures of rieske iron-sulfur protein: role of hydrogen bonds in tuning the redox potential of iron-sulfur clusters. Structure. 2007;15(1):29–38. doi: 10.1016/j.str.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Medvedev E.S., Couch V.A., Stuchebrukhov A.A. Determination of the intrinsic redox potentials of FeS centers of respiratory complex I from experimental titration curves. Biochim. Biophys. Acta. 2010;1797(9):1665–1671. doi: 10.1016/j.bbabio.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sarewicz M., Borek A., Cieluch E., wierczek M., Osyczka A. Discrimination between two possible reaction sequences that create potential risk of generation of deleterious radicals by cytochrome bc1: implications for the mechanism of superoxide production. Biochim. Biophys. Acta Bioenerg. 2010;1797(11):1820–1827. doi: 10.1016/j.bbabio.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 64.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Canc. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 65.Galadari S., Rahman A., Pallichankandy S., Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radical Biol. Med. 2017;104:144–164. doi: 10.1016/j.freeradbiomed.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 66.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 67.Cui L., Her S., Dunne M., Borst G.R., De Souza R., Bristow R.G. Significant radiation enhancement effects by gold nanoparticles in combination with cisplatin in triple negative breast cancer cells and tumor xenografts. Radiat. Res. 2017;187(2):147–160. doi: 10.1667/RR14578.1. [DOI] [PubMed] [Google Scholar]
- 68.Cotrim A.P., Yoshikawa M., Sunshine A.N., Zheng C., Sowers A.L., Thetford A.D. Pharmacological protection from radiation ± cisplatin-induced oral mucositis. Int. J. Radiat. Oncol. Biol. Phys. 2012;83(4):1284–1290. doi: 10.1016/j.ijrobp.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goodhead D.T. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int. J. Radiat. Biol. 1994;65(1):7–17. doi: 10.1080/09553009414550021. [DOI] [PubMed] [Google Scholar]
- 70.Harrison L., Malyarchuk S. Can DNA repair cause enhanced cell killing following treatment with ionizing radiation? Pathophysiology : Off. J. Int. Soc. 2002;8(3):149–159. doi: 10.1016/s0928-4680(01)00079-7. [DOI] [PubMed] [Google Scholar]
- 71.Malyarchuk S., Castore R., Harrison L. DNA repair of clustered lesions in mammalian cells: involvement of non-homologous end-joining. Nucleic Acids Res. 2008;36(15):4872–4882. doi: 10.1093/nar/gkn450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blaisdell J.O., Harrison L., Wallace S.S. Base excision repair processing of radiation-induced clustered DNA lesions. Radiat. Protect. Dosim. 2001;97(1):25–31. doi: 10.1093/oxfordjournals.rpd.a006634. [DOI] [PubMed] [Google Scholar]
- 73.Malyarchuk S., Castore R., Harrison L. Apex1 can cleave complex clustered DNA lesions in cells. DNA Repair. 2009;8(12):1343–1354. doi: 10.1016/j.dnarep.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]