

Abstract
Hematopoietic progenitor kinase 1 (HPK1 or MAP4K1) is a Ser/Thr kinase that operates via the c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) signaling pathways to dampen the T-cell response and antitumor immunity. Accordingly, selective HPK1 inhibition is considered a means to enhance antitumor immunity. Sunitinib, a multi-receptor tyrosine kinase (RTK) inhibitor approved for the management of gastrointestinal stromal tumors (GISTs), renal cell carcinoma (RCC), and pancreatic cancer, has been reported to inhibit HPK1 in vitro. In this report, we describe the crystal structures of the native HPK1 kinase domain in both nonphosphorylated and doubly phosphorylated states, in addition to a double phosphomimetic mutant (T165E,S171E), each complexed with sunitinib at 2.17–3.00-Å resolutions. The native nonphosphorylated cocrystal structure revealed an inactive dimer in which the activation loop of each monomer partially occupies the ATP- and substrate-binding sites of the partner monomer. In contrast, the structure of the protein with a doubly phosphorylated activation loop exhibited an active kinase conformation with a greatly reduced monomer–monomer interface. Conversely, the phosphomimetic mutant cocrystal structure disclosed an alternative arrangement in which the activation loops are in an extended domain-swapped configuration. These structural results indicate that HPK1 is a highly dynamic kinase that undergoes trans-regulation via dimer formation and extensive intramolecular and intermolecular remodeling of the activation segment.
Keywords: crystal structure, serine/threonine protein kinase, inhibitor, drug discovery, dimerization, conformational change, cancer, conformational plasticity, domain swapping, hematopoietic progenitor kinase 1 (HPK1), trans-regulation
Introduction
Hematopoietic progenitor kinase 1 (HPK12 or MAP4K1) comprises an N-terminal kinase domain, an intermediate region, and a C-terminal Citron homology domain (see Fig. 1A). In T-cells, HPK1 has been implicated as a negative regulator of signal transduction via phosphorylation and activation of the T-cell receptor adaptor protein SLP-76. Likewise, in B-cells, B-cell receptor signaling is down-regulated through HPK1-mediated phosphorylation and subsequent ubiquitination of activated B-cell linker protein (BLNK), an SLP-76 paralog (1). Upon activation and expansion of T-cells, caspase-mediated cleavage of HPK1 separates the kinase domain, HPK1-N, from the C terminus, HPK1-C. This cleavage event reverses the role of HPK1 from an activator of NF-κB into an inhibitor of NF-κB and leads to sensitization toward activation-induced cell death (2–5). Several in vivo studies of Hpk1−/− mice have provided genetic evidence for HPK1 as a negative regulator of the immune response and established that HPK1 functions as a potent immune response inhibitory kinase downstream of T-cell receptor–generated activation signals (6–8). Thus, HPK1 kinase has emerged as a potential target for immunotherapy by small-molecule inhibitors (9, 10).
Figure 1.

Domain architecture and structure of HPK1. A, schematic of primary sequence and domains of full-length HPK1 (top) and kinase domains used for crystallization (bottom). B, subunit structure of the HPK1+0P–sunitinib KD. N-lobe, cyan ribbon; C-lobe, green ribbon; AS, yellow; and P + 1 motif, orange. C, selected interactions at the HPK1+0P dimer interface. Chain B is colored slate. Dashed lines with numbers show distance (Å).
Sunitinib malate (SutentTM) is a multi-RTK inhibitor approved for the treatment of gastrointestinal stromal tumors, renal cell carcinoma, and pancreatic cancer. Although c-KIT, VEGFR, and platelet-derived growth factor receptor are its primary targets, sunitinib also binds to other kinases, including HPK1 (11, 12). Therefore, cocrystal structures of sunitinib bound to HPK1 are of interest as a starting point in the structure-based design of more potent and selective HPK1 inhibitors.
During our drug design campaign, we generated structures of the HPK1 kinase domain (KD) in complex with sunitinib and in a wide variety of conformations, including an inactive dimer (native nonphosphorylated kinase), an active dimer (native diphosphorylated kinase), and a three-dimensional (3D) domain-swapped dimer (phosphomimetic T165E,S171E mutant) in the inactive state. The diversity of conformational states observed, both in terms of the subunits and in distinct dimers, highlights the dynamic/flexible nature of the HPK1 kinase and suggests a role for dimerization as a mechanism for its regulation.
Results
In vitro inhibition of HPK1 activity by sunitinib and enhanced IL-2 production in sunitinib-treated T-cells
It has been previously shown in vitro that sunitinib can bind to the kinase domain of HPK1 with high affinity, with a dissociation constant (Kd) of 16 nm (12). To characterize binding of sunitinib to HPK1 in a construct most relevant to the cellular environment, we produced recombinant WT full-length HPK1 and preactivated the protein by in vitro autophosphorylation. The inhibition constant (Ki) for sunitinib against activated WT full-length HPK1 was measured to be about 10 nm (Fig. S1). Incubating sunitinib with Jurkat T-cells results in enhanced IL-2 production when these T-cells are stimulated with anti-CD3 and anti-CD28 monoclonal antibodies (Fig. S2). Therefore, the development of small molecules capable of inhibiting HPK1 kinase activity with exquisite specificity would benefit greatly from crystal structures of the HPK1 kinase domain in complex with inhibitor.
Structure of the inactive trans-inhibited HPK1–sunitinib complex
Initial attempts to express and purify WT HPK1 kinase domain (1–307 and 10–307) resulted in very low yields. To overcome this, we investigated the purification of the 10–307 construct in the presence of sunitinib and obtained sufficient amounts of the nonphosphorylated protein (HPK1+0P) for crystallization trials. Cocrystallization screening and crystal optimization (Fig. S3) generated a 2.17-Å-resolution structure of the HPK1+0P–sunitinib binary complex. The crystals belonged to the space group R32, and the HPK1+0P–sunitinib asymmetric unit (ASU) contained two molecules of the binary complex packed into a tight noncrystallographic dimer. Electron density for the protein backbone and side chains was of overall good to excellent quality. Structure refinement produced a well-validated atomic model with good refinement statistics of Rfree = 0.219 and Rwork = 0.192 (Table 1). The structure revealed the expected archetypal kinase fold: an N-terminal β-sheet–containing domain (N-lobe), a connecting hinge region, and a C-terminal α-helical domain (C-lobe) (Fig. 1B). As anticipated from previous structures of sunitinib bound to c-KIT (PDB ID code 3GOE) (13) and VEGFR2 (PDB ID code 4AGD) (14), sunitinib binds in the ATP site.
Table 1.
Crystallographic data collection and refinement statistics
r.m.s.d. is the root mean square deviation from ideal geometry.
| WT 10–307 +0P–sunitinib | WT 1–307 +2P–sunitinib | 1–307 T165E,S171E–sunitinib | |
|---|---|---|---|
| Data collection statistics | |||
| Wavelength (Å) | 1.00 | 1.00 | 1.00 |
| Resolution range (Å) | 107.95–2.17 | 74.97–2.97 | 60.18–2.03 |
| Space group | R32 | R32 | P1 |
| Cell edge a, b, c (Å); angle (°) | 165.91, 165.91, 163.58; 90.00, 90.00, 120.00 | 149.93, 149.93, 156.75; 90.00, 90.00, 120.00 | 55.81, 58.92, 60.93; 82.44, 82.31, 64.34 |
| Molecules per asymmetric unit | 2 | 2 | 2 |
| Total reflections (outer shell) | 454,280 (4,444) | 142,751 (1,488) | 155,437 (1,687) |
| Unique reflections (outer shell) | 46,182 (433) | 14,226 (149) | 43,684 (458) |
| Multiplicity (outer shell) | 9.8 (10.3) | 10.0 (10.0) | 3.6 (3.7) |
| Completeness (%) (outer shell) | 100.0 (99.3) | 100.0 (100.00) | 97.3 (95.8) |
| Mean I/σ(I) (outer shell) | 14.81 (2.33) | 12.5 (2.3) | 14.9 (2.10) |
| Wilson B-factor (Å2) | 44.31 | 55.04 | 42.36 |
| Rmerge (outer shell)a | 0.097 (1.210) | 0.196 (1.067) | 0.042 (0.620) |
| Rpimb | 0.033 (0.399) | 0.065 (0.356) | 0.026 (0.374) |
| CC1/2 (outer shell)c | 0.999 (0.701) | 0.996 (0.861) | 0.999 (0.874) |
| Refinement statistics | |||
| Resolution range (Å) | 17.78–2.17 | 74.97–2.97 | 27.69–2.03 |
| Rwork/Rfreed | 0.192/0.219 | 0.207/0.251 | 0.201/0.229 |
| Number of atoms (protein/ligand/water) | 4,463/58/412 | 4,225/58/16 | 4,332/58/320 |
| r.m.s.d. bond length (Å) | 0.014 | 0.056 | 0.013 |
| r.m.s.d. bond angle (°) | 1.71 | 1.00 | 1.57 |
| Ramachandran plot | |||
| Most favored region (%) | 98 | 95 | 96 |
| Allowed regions (%) | 2 | 5 | 4 |
| Disallowed regions (%) | 0 | 0 | 0 |
a Rmerge = ΣhklΣi|Ii − 〈I〉|/−ΣhklΣIi where Ii is the intensity of the ith observation, 〈I〉 is the mean intensity of the reflection, and the summations extend over all unique reflections (hkl) and all equivalents (i), respectively.
b Rpim is a measure of the quality of the data after averaging the multiple measurements, and Rpim = Σhkl[n/(n − 1)]1/2Σi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl) where n is the multiplicity and other variables are as defined for Rmerge (39).
c CC1/2 is the Pearson correlation coefficient.
d Rwork = Σ|Fo − Fc|/ΣFo where Fo and Fc are observed and calculated structure factors, respectively, Rfree was calculated from a randomly chosen 5% of reflections excluded from the refinement, and Rwork was calculated from the remaining 95% of reflections.
Two HPK1 kinase domains are arranged in a homodimer with the activation loop (AL) of each protomer coming into the interdomain cleft of the cognate protomer and partially occupying the ATP- and protein substrate–binding sites. Notably, a portion of the inhibitor-binding pocket is formed from AL residues of the adjacent protomer; Ala-167 Cβ is positioned ∼3 Å from the inhibitor F atom (Fig. 1C). In addition, the Arg-168 side chain from the noncrystallographic symmetry (NCS) molecule makes stabilizing interactions with its symmetry mate, including to the Oδ1 and Oδ2 atoms of Asp-155 in the DFG motif as well as the His-135 backbone O. This arrangement introduces considerable restraints on the DFG motif (residues 155–157), stabilizing it in the inactive orientation. Furthermore, the position of the NCS AL is incompatible with the αC-helix (αC) adopting an active, αC-in conformation. More specifically, Thr-175 from the AL of the opposite protomer is inserted between the catalytic lysine (Lys-46) and the conserved αC glutamate (Glu-62). These interactions stabilize the inactive αC-out conformation and exclude formation of the canonical active kinase Lys-46–Glu-62 salt bridge. The dimer is further stabilized by a β-sheet interaction where residues 171–173 of the 2-fold NCS monomers form four backbone hydrogen bonds (Fig. 2A). The AL is anchored by the DFG and P + 1 motifs, and the position of the P + 1 loop is typical with loop residues making only intramolecular contacts.
Figure 2.

Activation loops of the HPK1+0P and HPK1+2P dimers. A, the activation loops of the +0P dimer in ribbon representation with chain A in green and chain B in blue. Arrows show areas of β-strand. The DFG motif and phosphorylation sites are drawn as magenta and yellow sticks, respectively. Dashed lines indicate hydrogen bonds. B, the activation loops of the +2P dimer. The coloring scheme is as described in A. Dashed lines show interactions between protein and phosphate groups.
The tight subunit packing and high number of intermolecular interactions involving the active-site pocket and key regulatory motifs suggest a biologically relevant role for the dimer. To explore this further and quantitatively evaluate the crystal packing interface, we performed analysis of the structure using the Protein Interfaces and Surface Area (PISA) module in the CCP4 program suite (15). The analysis predicted the dimer to be stable in solution and revealed involvement of 62 residues in the dimer interface and 2253 Å2 of buried accessible surface area (Table S1 and Fig. S4). There is a significant ΔG of −22 kcal/mol for the dimer that includes 13 hydrogen bonds and 12 salt bridges in the interface.
Structure of the fully active diphosphorylated HPK1–sunitinib complex
Using the WT 1–307 construct purified in the presence of sunitinib, the cocrystal structure of the diphosphorylated HPK1–sunitinib complex (HPK1+2P) was obtained at 3.0-Å resolution. The crystals also belong to the space group R32 with two molecules in the ASU. However, the two molecules did not pack into a tight NCS dimer like the HPK1+0P structure. The two molecules in the ASU suggested a monomeric kinase in a nonphysiological dimer resulting from crystal packing. In contrast to the NCS dimer, PISA analysis predicted a distinct crystallographic dimer to be the only assembly stable in solution. The relative orientation of the two subunits identified by PISA was similar to that observed in the inactive HPK1+0P dimer; in each case, the subunits are assembled in a roughly parallel or “head-to-head” arrangement where the active sites are oriented to position sunitinib's terminal diethylamino group pointing away from the dimer interface and where the activation loops are arranged at the dimer interface in an overlapping antiparallel configuration (Fig. 3, A and B). Despite the similar arrangement of subunits in the dimer, the HPK1+2P structure is much more open with vastly reduced interface residues and buried surface area, a ΔG of only −9.4 kcal/mol, few hydrogen bonds, and no salt bridges, indicating a significantly weaker interaction (Table S1 and Fig. S4). Compared with the +0P structure, there are significant differences in the AL conformation and in the specific interactions made by AL residues (Fig. 2B). However, the DFG and P + 1 anchor points for the AL remain intact in both structures. From a purely structural standpoint, the interconversion of the inactive closed HPK1+0P dimer to the active open HPK1+2P dimer would require a relative subunit rotation of ∼61° and a displacement of ∼8 Å.
Figure 3.
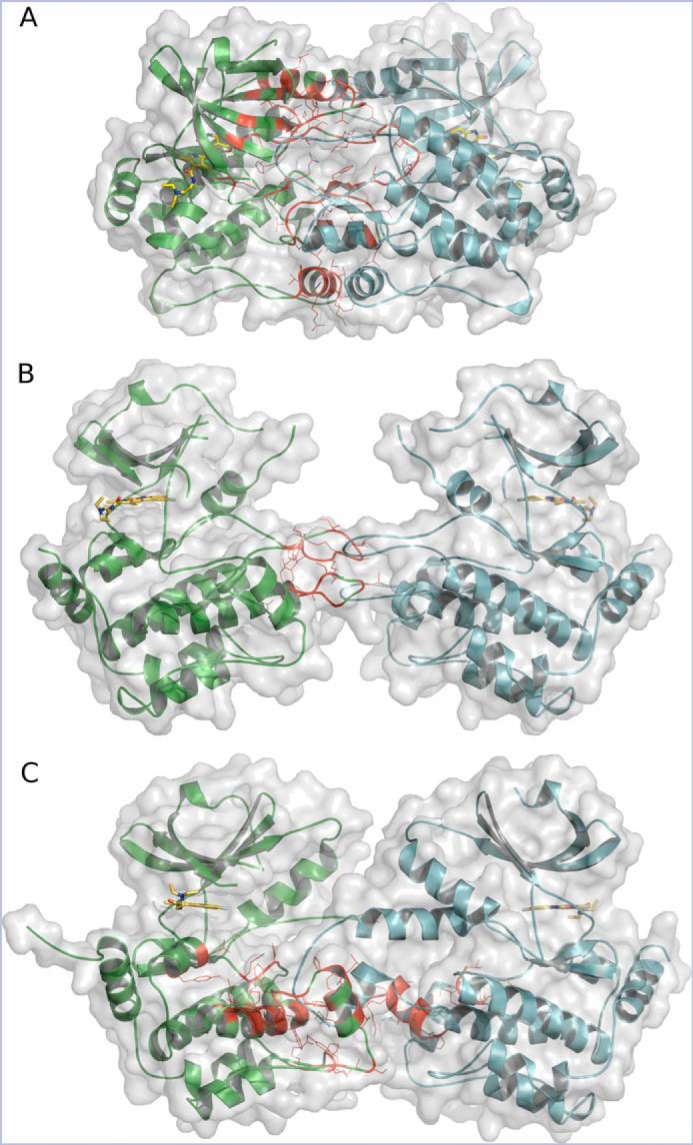
The configuration of HPK1–sunitinib dimers. A, HPK1+0P KD in a tightly closed head-to-head dimer. B, HPK1+2P KD in an open head-to-head dimer. C, HPK12PM KD in a head-to-head domain-swapped dimer. Chains A and B are colored green and cyan, respectively. Interface residues for chain A are shown as red lines, and inhibitor is drawn as yellow sticks.
At the level of the subunit structure, the active HPK1+2P and inactive HPK1+0P kinase domains have a similar overall fold. The most significant differences between the +0P and +2P structures are an inward movement of αC and a rearrangement of the AL starting at Asp-155 of the DFG and continuing to Thr-175 just preceding the P + 1 loop. The features of HPK1+2P that are characteristic of the active kinase fold include phosphorylation at Thr-165 and Ser-171, the αC-in position, the Lys-46–Glu-62 salt bridge and a DFG-in configuration. The phosphate oxygens of phospho-Ser-171 are stabilized by interactions with the sidechains of Arg-136 and Arg-168.
Comparing the positions of Thr-165 and Ser-171 in the +0P and +2P states, it is clear why phosphorylation is incompatible with the interface of the closed +0P dimer. As noted above, in the +0P complex, Thr-165 of one protomer is tightly packed against side chains from the N-terminal domain β-sheet and αC of the other protomer. A large, negatively charged phosphate group cannot be accommodated in this small hydrophobic space, and thus phosphorylation of Thr-165 would necessitate a remodeling of the AL at residues 163–167. Similarly, phosphorylation of Ser-171 would be incompatible with the dimeric β-strand interactions observed at residues 171–174 of the HPK1+0P dimer.
Structure of the phosphomimetic HPK1–sunitinib inhibitor complex in a domain-swapped dimer
Due to difficulties generating substantial amounts of the native +0P or +2P kinase domains, we investigated the production of the double phosphomimetic mutant (1–307 T165E,S171E, or HPK12PM). As validation, we compared enzyme kinetics and inhibition by sunitinib for HPK12PM and activated full-length WT HPK1. Although HPK12PM was found to be an order of magnitude less active than activated WT full-length HPK1, the apparent ATP Km values of HPK12PM and full-length WT HPK1 were found to be similar, 78 and 72 μm, respectively (Fig. S5). Sunitinib Ki values were about 6 and 10 nm, respectively (Fig. S1). Notably, HPK12PM could be purified in relatively high yield and in the absence of inhibitor and was amenable to the production of high-diffraction-quality crystals with a wide variety of inhibitor chemotypes using a single crystallization condition. The cocrystals obtained varied widely in their unit cell parameters and in the number of molecules in the ASU (2, 4, 8, and 12). Regardless of the crystallographic parameters, the structures invariably contained a dimer with a head-to-head subunit orientation and a 3D domain swap of the P + 1 and APE motifs. Reports of domain-swapped kinase structures are relatively rare but have been described for other kinases, including the closely related GLK (MAP4K3) (16), and Wu et al. (17) recently reported three crystal structures of the HPK1 KD that form domain-swapped dimers (PDB codes 6CQD, 6CQE, and 6CQF). 3D domain swapping is a well-established mechanism for assembling functional dimers or higher oligomers of proteins via replacement of identical structural elements (18–21). Here, we describe a dimeric 3D domain-swapped structure of HPK12PM–sunitinib in the inactive state.
With exception of the activation segment (AS), the overall subunit fold of the HPK12PM–sunitinib complex closely resembles that of the HPK1 native structures described above. The HPK12PM complex also assembles into a parallel head-to-head dimer where the arrangement of subunits is strikingly similar to the active HPK1+2P but with a much more extensive dimer interface (Fig. 3C). The most distinct feature of the HPK12PM structure is the presence of an extended AS, protruding across the dimer interface such that the P + 1 and APE motifs adopt a 3D domain-swapped configuration (Fig. 4A). The structure of the AS consists of a pair of antiparallel amphipathic α-helices (residues 159–171 and 180–189) connected by a well-ordered P + 1 loop. The residues of the P + 1 loop pack into an amphipathic pocket on the opposing HPK1 protomer and together with the APE motif define the domain swap (Fig. 3, B and C). In the domain-swapped dimer, the conserved APE glutamate, Glu-182, forms a bidentate ion pair with Arg-262 of the cognate protomer. This bidentate interaction is highly structurally conserved among kinases. The intermolecular pair is present in all reported domain-swapped kinases and is also observed as an intramolecular interaction in both HPK1 native structures described here.
Figure 4.

HPK1–sunitinib and comparable domain-swapped dimers from the PDB. A, the HPK12PM KD in a domain-swapped dimer. Chain A is shown as a green cartoon with transparent surface, and chain B is shown as a cyan cartoon. Inhibitor and WYMAPE residues are drawn as sticks. The active-site surface is colored yellow in subunit A. B, zoomed-in view of HPK12PM domain swap. Selected chain A side chains (green) surrounding YWMAPE residues from the P + 1 and APE motifs of chain B (cyan) are shown. C, equivalent view of HPK1+0P chain A (dark green) to illustrate the duplication of interactions in domain-swapped and nonswapped dimers. D, crystal structures of selected kinase domains from the PDB that assemble as domain-swapped dimers.
The dimer interface contains 53 residues, corresponding to 1997 Å2 of buried accessible surface area that includes four hydrogen bonds and nine salt bridges with a ΔG of −32.4 kcal/mol (Table S1 and Fig. S4), which is consistent with a physiologically relevant oligomer. In addition, the HPK12PM domain swap is very similar to published structures representing a diverse set of domain-swapped kinase domains ranging from less conserved members like CHK2 kinase to the closely related GLK (MAP4K3) (Fig. 4D).
Based on the observation of a DFG-out conformation, the αC-out position, and the absence of a Lys-46–Glu-62 salt bridge, the kinase subunits in the HPK12PM complex adopt the inactive fold. However, there is significant asymmetry within the HPK12PM ASU. For example, the AL appears much less ordered in chain A compared with chain B. Furthermore, the DFG conformation was intermediate for chain A but DFG-out for chain B. The position of αC was also ∼3 Å further out for chain A compared with chain B.
Sunitinib binding interactions
The electron density maps clearly indicate sunitinib bound in each of the complexes. Superposition of the structures shows that the kinase hinge region of residues 91–101 agrees well in the three complexes and that the oxindole pyrrole of sunitinib makes backbone hydrogen-bonding interactions with Cys-94 and Glu-92 (Fig. S6). Although sunitinib binding is stabilized via interactions with the hinge backbone, the overall position of sunitinib is affected by differences in the position of other residues, primarily in the G-loop and AL. For example, the oxindole ring is translated ∼0.9 Å farther into the binding pocket toward gatekeeper residue Met-91 in the +0P complex as compared with the +2P complex (Fig. S6C). The tertiary amine tail of sunitinib extends away from the kinase hinge into solvent, and its electron density was weak and ambiguous, indicating that the amine tail may occupy more than one preferred conformation.
Discussion
The crystal structures of the HPK1 KD described herein provide insight into the extraordinary structural plasticity of the HPK1 KD and its propensity for dimerization. The observation of the HPK1 KD in active and inactive kinase conformations as well as in both domain-swapped and nonswapped dimers offers a tantalizing clue for dimerization as a regulatory mechanism.
From a structural perspective, the inactive HPK1+0P dimer appears inhibited in trans; the intermolecular exchange of the AL stabilizes the kinase in an inactive state. The extent of the interactions at the dimer interface and the involvement of key regulatory motifs and residues argue in favor of a biologically relevant dimer. Perhaps paradoxically, this AL exchange positions the autophosphorylation residue, Thr-165, between the catalytic lysine (Lys-46) and the conserved αC glutamate (Glu-62) at the active site of the adjacent protomer (Fig. S7). Superposition of the structures of HPK1+0P and the insulin receptor kinase in complex with IRS2 KRLB peptide and ATP (PDB code 3BU5) positions the terminal phosphate of ATP within ∼8 Å of HPK1s' Thr-165 side-chain Oγ1 atom. Therefore, it seems plausible to speculate that conformational dynamics could allow for formation of the Asp-Lys-Thr catalytic triad and the autophosphorylation of Thr-165. Such a conformational change could be initiated by the trans-phosphorylation of Ser-171 by PRKD1, which would be consistent with the observation that trans-phosphorylation by PRKD1 and subsequent autophosphorylation of Thr-165 appear to be prerequisite for full activation of HPK1 (22). Biochemical and crystallographic studies of dimeric p21-activated kinase 2 (PAK2) and the homologous PAK1 (PDB ID code 1YHV) indicate that dimerization positions the AL of one protomer in the active site of the companion protomer, which is required for the trans-autophosphorylation reaction (23, 24).
The observation of the HPK1+2P in a head-to-head dimer is intriguing but possibly nonphysiological. Indeed, the ΔG of −9.4 kcal/mol predicted by PISA falls below the ΔG of >15–20 kcal/mol established as a range for reliable (1–2% errors) representation of complexes in crystals (25). Unfortunately, our efforts to determine the oligomeric state of the HPK1+0P and HPK1+2P in solution using size-exclusion chromatography with inline multiangle static light scattering (SEC-MALS) were inconclusive. In each case, the MALS analysis of the single peak eluting from the gel-filtration column predicted a mass in between those expected for the monomer and dimer (data not shown). In contrast, two distinct peaks were observed during SEC-MALS of the HPK12PM protein that corresponded well with the expected monomer/dimer masses (data not shown). These latter observations agree with the findings of Wu et al. (17). Considering the inconclusive SEC-MALS and PISA prediction for HPK1+2P, conclusions regarding the physiological relevance of the dimer require additional experimental evidence.
HPK1 now adds to the growing list of domain-swapped kinase structures that includes both active and inactive kinase folds from a diverse set of kinase family members, including CHK2 (PDB code 2CN5) (26), SLK (PDB code 2J51) (27), LOK/STK10 (PDB code 2J7T) (27), DAPK3 (PDB code 2J90) (27), OSR1 (PDB code 3DAK) (28), and GLK/MAP4K3 (PDB code 5J5T) (16). Moreover, Wu et al. (17) also reported the structure of a T165E,S171E double phosphomimetic mutant of HPK1 in complex with AMPPNP (PDB ID code 6CQF). Despite different crystallization conditions, different crystal parameters, and different ligands bound, the HPK12PM structure compares closely with the structure of Wu et al. (17). Superposition of the structures yields r.m.s.d. values of 1.564 Å for 482 Cα atoms and 0.429 Å for 202 Cα atoms for the dimer and monomer, respectively (Fig. S8).
The precise physiological role of the domain-swapped kinase remains unclear. However, the observation of kinases from diverse subfamilies assembled as domain-swapped dimers argues in favor of a physiologically relevant phenomenon. For example, structural studies of SLK, LOK, and DAPK3, each assembled as domain-swapped dimers, led the authors to propose a mechanism for kinase autophosphorylation of nonconsensus sites (27). It has also been suggested that dimerization of HPK1 may provide an additional layer of regulatory control over substrate selectivity or cis/trans-phosphorylation activity (29).
Inherent conformational plasticity is essential for the interconversion between active and inactive kinase states. In addition, biophysical studies have suggested that catalysis and conformational dynamics are linked, whereas NMR spectroscopy and molecular dynamics simulations have connected domain swapping to conformational flexibility in other protein molecules (30). Therefore, domain swapping is a likely consequence of the intrinsic conformational flexibility of HPK1 and other kinase domains. Additional structural and biochemical studies are needed to elucidate the functional roles of dimerization and domain swapping for HPK1 and other protein kinases.
Experimental procedures
Production of recombinant HPK1
HPK1 (GenBankTM NM_001042600.2) insect cell expression constructs were synthesized at GenScript and included a TEV-cleavable N-terminal poly-His purification tag. Recombinant baculoviruses were prepared by using the Bac-to-Bac method (Invitrogen) and were used to infect Sf21 insect cells. Recombinant HPK1 kinase domain was purified by double immobilized metal affinity chromatography following cell lysis in 50 mm Tris-HCl, pH 8.0, 250 mm NaCl, 0.25 mm TCEP, 10 μm leupeptin (Sigma), 10 μm E-64 (Sigma), and one EDTA-free protease inhibitor tablet (Roche Applied Science)/75 ml of buffer with tag removal between immobilized metal affinity chromatography steps followed by gel filtration over a Phenomenex 30 × 2.1-cm BioSep-s3000 size-exclusion column in 25 mm Tris-HCl, pH 8.0, 250 mm NaCl, 0.25 mm TCEP. Sunitinib was added to the nonmutant kinase domains for stabilization and concentrated to ∼20 mg/ml. The HPK1(1–307) T165E,S171E double mutant was prepared similarly but using 25 mm HEPES-NaOH, pH 7.5, 250 mm NaCl, 0.25 mm TCEP in the final step. Full-length HPK1 purification included 2 mm MgATP during the TEV cleavage dialysis step to allow for autophosphorylation.
Crystallization and crystallography
The sitting-drop vapor-diffusion method was used for crystallization. HPK1+0P crystals grew from 300 nl of protein (16 mg/ml) plus 300 nl of reservoir solution (4.19 m NaCl, 0.1 m HEPES, pH 6.5) at 21 °C. HPK1+2P crystals grew from 100 nl of protein (22 mg/ml) plus 100 nl of reservoir solution (0.2 m ammonium fluoride, 20% PEG 3350) at 13 °C. HPK12PM crystals grew from 200 nl of protein (15 mg/ml) plus 300 nl of reservoir solution (0.1 m Tris, pH 8, 20% 1,6-hexanediol, 10 mm magnesium sulfate, 24 mm barium acetate) at 13 °C. Crystals were cryoprotected in reservoir solution plus 20% glycerol and flash frozen in liquid N2.
X-ray diffraction data were collected at the Advanced Photon Source beamline 17-ID and processed with XDS (31) and POINTLESS, AIMLESS, or XSCALE via autoPROC from Global Phasing (32). Structure solution was by molecular replacement using Phaser (33). Model building was done in Coot (34) and refinement used REFMAC (35), PHENIX (36), or BUSTER (37). Structure validation was performed with MolProbity (38).
Author contributions
E. J., M. McTigue, R. A. G., T. W. J., T. S. F., R. K., S. S., S. B., and C. N. C. conceptualization; E. J. software; E. J., M. McTigue, S. T., M. Maestre, and C. N. C. formal analysis; E. J. validation; E. J., M. McTigue, and C. N. C. investigation; E. J., S. T., M. Maestre, S. S., and C. N. C. methodology; E. J., M. McTigue, and C. N. C. writing-original draft; E. J., M. McTigue, R. A. G., T. W. J., S. T., M. Maestre, S. S., and C. N. C. writing-review and editing; R. A. G., T. W. J., T. S. F., and R. K. project administration.
Supplementary Material
Acknowledgments
We thank Nicole Grable for insect cell culture and Oleg Brodsky for assistance with SEC-MALS analysis.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8 and Table S1.
The atomic coordinates and structure factors (codes 6NFY, 6NFZ, and 6NG0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HPK1
- hematopoietic progenitor kinase 1
- JNK
- c-Jun N-terminal kinase
- ERK
- extracellular signal-regulated kinase
- RTK
- receptor tyrosine kinase
- VEGFR
- vascular endothelial growth factor receptor
- KD
- kinase domain
- 3D
- three-dimensional
- ASU
- asymmetric unit
- PDB
- Protein Data Bank
- AL
- activation loop
- NCS
- noncrystallographic symmetry
- PISA
- Protein Interfaces and Surface Area
- GLK
- germinal center kinase–like kinase
- AS
- activation segment
- PAK
- p21-activated kinase
- SEC
- size-exclusion chromatography
- MALS
- multiangle static light scattering
- SLK
- STE20-like kinase
- LOK
- lymphocyte-oriented kinase
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate
- r.m.s.d.
- root mean square deviation
- TEV
- tobacco etch virus
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Wang X., Li J.-P., Kuo H.-K., Chiu L.-L., Dement G. A., Lan J.-L., Chen D.-Y., Yang C.-Y., Hu H., and Tan T.-H. (2012) Down-regulation of B cell receptor signaling by hematopoietic progenitor kinase 1 (HPK1)-mediated phosphorylation and ubiquitination of activated B cell linker protein (BLNK). J. Biol. Chem. 287, 11037–11048 10.1074/jbc.M111.310946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brenner D., Golks A., Becker M., Müller W., Frey C. R., Novak R., Melamed D., Kiefer F., Krammer P. H., and Arnold R. (2007) Caspase-cleaved HPK1 induces CD95L-independent activation-induced cell death in T and B lymphocytes. Blood 110, 3968–3977 10.1182/blood-2007-01-071167 [DOI] [PubMed] [Google Scholar]
- 3. Chen Y.-R., Meyer C. F., Ahmed B., Yao Z., and Tan T.-H. (1999) Caspase-mediated cleavage and functional changes of hematopoietic progenitor kinase 1 (HPK1). Oncogene 18, 7370–7377 10.1038/sj.onc.1203116 [DOI] [PubMed] [Google Scholar]
- 4. Arnold R., Liou J., Drexler H. C., Weiss A., and Kiefer F. (2001) Caspase-mediated cleavage of hematopoietic progenitor kinase 1 (HPK1) converts an activator of NFκB into an inhibitor of NFκB. J. Biol. Chem. 276, 14675–14684 10.1074/jbc.M008343200 [DOI] [PubMed] [Google Scholar]
- 5. Kiefer F., Tibbles L. A., Anafi M., Janssen A., Zanke B. W., Lassam N., Pawson T., Woodgett J. R., and Iscove N. N. (1996) HPK1, a hematopoietic protein kinase activating the SAPK/JNK pathway. EMBO J. 15, 7013–7025 10.1002/j.1460-2075.1996.tb01093.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alzabin S., Bhardwaj N., Kiefer F., Sawasdikosol S., and Burakoff S. (2009) Hematopoietic progenitor kinase 1 is a negative regulator of dendritic cell activation. J. Immunol. 182, 6187–6194 10.4049/jimmunol.0802631 [DOI] [PubMed] [Google Scholar]
- 7. Alzabin S., Pyarajan S., Yee H., Kiefer F., Suzuki A., Burakoff S., and Sawasdikosol S. (2010) Hematopoietic progenitor kinase 1 is a critical component of prostaglandin E2-mediated suppression of the anti-tumor immune response. Cancer Immunol. Immunother. 59, 419–429 10.1007/s00262-009-0761-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shui J.-W., Boomer J. S., Han J., Xu J., Dement G. A., Zhou G., and Tan T.-H. (2007) Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell–mediated immune responses. Nat. Immunol. 8, 84–91 10.1038/ni1416 [DOI] [PubMed] [Google Scholar]
- 9. Sawasdikosol S., Zha R., Yang B., and Burakoff S. (2012) HPK1 as a novel target for cancer immunotherapy. Immunol. Res. 54, 262–265 10.1007/s12026-012-8319-1 [DOI] [PubMed] [Google Scholar]
- 10. Liu J., Curtin J., You D., Hillerman S., Li-Wang B., Eraslan R., Xie J., Swanson J., Ho C.-P., Oppenheimer S., Warrack B. M., McNaney C. A., Nelson D. M., Blum J., Kim T., et al. (2019) Critical role of kinase activity of hematopoietic progenitor kinase 1 in anti-tumor immune surveillance. PLoS One 14, e0212670 10.1371/journal.pone.0212670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anastassiadis T., Deacon S. W., Devarajan K., Ma H., and Peterson J. R. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 10.1038/nbt.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., and Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 10.1038/nbt.1990 [DOI] [PubMed] [Google Scholar]
- 13. Gajiwala K. S., Wu J. C., Christensen J., Deshmukh G. D., Diehl W., DiNitto J. P., English J. M., Greig M. J., He Y.-A., Jacques S. L., Lunney E. A., McTigue M., Molina D., Quenzer T., Wells P. A., et al. (2009) KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. U.S.A. 106, 1542–1547 10.1073/pnas.0812413106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McTigue M., Murray B. W., Chen J. H., Deng Y.-L., Solowiej J., and Kania R. S. (2012) Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci U.S.A. 109, 18281–18289 10.1073/pnas.1207759109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 16. Marcotte D., Rushe M., Arduini M. R., Lukacs C., Atkins K., Sun X., Little K., Cullivan M., Paramasivam M., Patterson T. A., Hesson T., McKee D. T., May-Dracka T. L., Xin Z., Bertolotti-Ciarlet A., et al. (2017) Germinal-center kinase-like kinase co-crystal structure reveals a swapped activation loop and C-terminal extension. Protein Sci. 26, 152–162 10.1002/pro.3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu P., Sneeringer C. J., Pitts K. E., Day E. S., Chan B. K., Wei B., Lehoux I., Mortara K., Li H., Wu J., Franke Y., Moffat J. G., Grogan J. L., Heffron T. P., and Wang W. (2019) Hematopoietic progenitor kinase-1 structure in a domain-swapped dimer. Structure 27, 125–133.e4 10.1016/j.str.2018.10.025 [DOI] [PubMed] [Google Scholar]
- 18. Liu Y., and Eisenberg D. (2002) 3D domain swapping: as domains continue to swap. Protein Sci. 11, 1285–1299 10.1110/ps.0201402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bennett M. J., and Eisenberg D. (2004) The evolving role of 3D domain swapping in proteins. Structure 12, 1339–1341 10.1016/j.str.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 20. Bennett M. J., Schlunegger M. P., and Eisenberg D. (1995) 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 4, 2455–2468 10.1002/pro.5560041202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rousseau F., Schymkowitz J., and Itzhaki L. S. (2012) Implications of 3D domain swapping for protein folding, misfolding and function; in Protein Dimerization and Oligomerization in Biology (Matthews J. M., ed) pp. 137–152, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 22. Arnold R., Patzak I. M., Neuhaus B., Vancauwenbergh S., Veillette A., Van Lint J., and Kiefer F. (2005) Activation of hematopoietic progenitor kinase 1 involves relocation, autophosphorylation, and transphosphorylation by protein kinase D1. Mol. Cell. Biol. 25, 2364–2383 10.1128/MCB.25.6.2364-2383.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pirruccello M., Sondermann H., Pelton J. G., Pellicena P., Hoelz A., Chernoff J., Wemmer D. E., and Kuriyan J. (2006) A dimeric kinase assembly underlying autophosphorylation in the p21 activated kinases. J. Mol. Biol. 361, 312–326 10.1016/j.jmb.2006.06.017 [DOI] [PubMed] [Google Scholar]
- 24. Lei M., Robinson M. A., and Harrison S. C. (2005) The active conformation of the PAK1 kinase domain. Structure 13, 769–778 10.1016/j.str.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 25. Krissinel E. (2010) Crystal contacts as nature's docking solutions. J. Comput. Chem. 31, 133–143 10.1002/jcc.21303 [DOI] [PubMed] [Google Scholar]
- 26. Oliver A. W., Paul A., Boxall K. J., Barrie S. E., Aherne G. W., Garrett M. D., Mittnacht S., and Pearl L. H. (2006) Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J. 25, 3179–3190 10.1038/sj.emboj.7601209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oliver A. W., Knapp S., and Pearl L. H. (2007) Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem. Sci. 32, 351–356 10.1016/j.tibs.2007.06.004 [DOI] [PubMed] [Google Scholar]
- 28. Lee S.-J., Cobb M. H., and Goldsmith E. J. (2009) Crystal structure of domain-swapped STE20 OSR1 kinase domain. Protein Sci. 18, 304–313 10.1002/pro.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sawasdikosol S., and Burakoff S. (2019) The structure of HPK1 kinase domain: to boldly go where no immuno-oncology drugs have gone before. Structure 27, 1–3 10.1016/j.str.2018.12.009 [DOI] [PubMed] [Google Scholar]
- 30. Li Y., and Palmer A. G. III (2009) Domain swapping in the kinase superfamily: OSR1 joins the mix. Protein Sci. 18, 678–681 10.1002/pro.98 [DOI] [Google Scholar]
- 31. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., and Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 10.1107/S0907444911007773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 35. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 36. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O., and Vonrhein C. (2011) BUSTER, version 2.11. 2, Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 38. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weiss M. (2001) Global indicators of X-ray data quality. J. Appl. Crystallogr. 34, 130–135 10.1107/S0021889800018227 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.