

Abstract
Autophagy promotes cancer cell survival in response to p53 activation by the anticancer agent Nutlin-3a (Nutlin). We reported previously that Nutlin kills MDM2-amplified cancer cells and that this killing is associated with an inhibition of glucose metabolism, reduced α-ketoglutarate (α-KG) levels, and reduced autophagy. In the current report, using SJSA1, U2OS, A549, and MHM cells, we found that Nutlin alters histone methylation in an MDM2 proto-oncogene–dependent manner and that this, in turn, regulates autophagy-related gene (ATG) expression and cell death. In MDM2-amplified cells, Nutlin increased histone (H) 3 lysine (K) 9 and K36 trimethylation (me3) coincident with reduced autophagy and increased apoptosis. Blocking histone methylation restored autophagy and rescued these cells from Nutlin-induced killing. In MDM2-nonamplified cells, H3K9me3 and H3K36me3 levels were either reduced or not changed by the Nutlin treatment, and this coincided with increased autophagy and cell survival. Blocking histone demethylation reduced autophagy and sensitized these cells to Nutlin-induced killing. Further experiments suggested that MDM2 amplification increases histone methylation in Nutlin-treated cells by causing depletion of the histone demethylase Jumonji domain-containing protein 2B (JMJD2B). Finally, JMJD2B knockdown or inhibition increased H3K9/K36me3 levels, decreased ATG gene expression and autophagy, and sensitized MDM2-nonamplified cells to apoptosis. Together, these results support a model in which MDM2- and JMJD2B-regulated histone methylation levels modulate ATG gene expression, autophagy, and cell fate in response to the MDM2 antagonist Nutlin-3a.
Keywords: autophagy, p53, histone demethylase, mouse double minute 2 homolog (MDM2), histone methylation, ATG16L1, ULK1, Jumonji domain containing protein 2B (JMJD2B), nutlin, cancer, epigenetic regulation, chromatin regulation, MDM2 proto-oncogene
Introduction
WT p53 is a transcription factor and tumor suppressor that is activated in response to stress. Activated p53 can promote either transient cell cycle arrests that contribute to survival or permanent cell cycle arrest (senescence) or apoptosis that inhibits survival and contributes to tumor suppression. These effects are mediated by p53-responsive gene products including the cyclin-CDK inhibitor p21 or the apoptosis-inducing factors PUMA, Noxa, and Bax (1–4). The outcome of p53 activation (survival versus senescence/apoptosis) is believed to depend in part on the level of stress.
In addition to these canonical functions, p53 also has noncanonical functions that include its ability to regulate autophagy (5, 6). Autophagy is a process in which organelles, misfolded proteins, and other intracellular components are degraded in autophagolysosomes (7–9). Autophagy is a multistep process. A first step in autophagy is formation of phagophore membranes. This step is promoted by an autophagy initiating complex that includes the proteins ULK1 and ULK2. Subsequent steps are mediated in large part by the products of various autophagy-related genes (ATGs). These steps include elongation of phagophore membranes, capture of cargo (organelles, misfolded proteins, etc.) by phagophore membranes, conversion of phagophore membranes into autophagosomes, fusion of cargo-containing autophagosomes with lysosomes, and degradation of cargo in these autophagolysosomes by lysosomal enzymes. Autophagy occurs at low levels normally but is stimulated in response to metabolic stresses such as low nutrient levels. The degradation products of autophagy can be used to generate and maintain the levels of key nutrients and metabolites needed for survival. Autophagy in this case allows survival until the metabolic stress caused by low nutrient levels can be resolved. p53 has been reported to both promote and inhibit autophagy (6). p53 can promote autophagy, in part by activating ULK1 and also by binding the promoter regions of ULK1 and various ATG genes and promoting their expression (5, 10, 11). In contrast, Kroemer and colleagues (5) reported that cytoplasmic but not nuclear p53 can inhibit autophagy. There is some evidence that autophagy mediated by p53 increases survival. For example, treatment with the autophagy inhibitor bafilomycin A1 increased apoptosis in cells treated with the p53 activator Nutlin (12, 13).
p53 can also regulate cancer cell metabolism (14, 15). Cancer cells often have an altered metabolism that includes increased glucose uptake and glycolysis and reduced oxidative phosphorylation. p53 can inhibit glycolysis by repressing expression of glycolytic enzyme genes and promote oxidative phosphorylation by increasing expression of genes like SCO2 (15, 16). Most but not all MDM2-amplified cells undergo apoptosis in response to Nutlin treatment whereas most MDM2-nonamplified cells undergo cell cycle arrest with minimal apoptosis. We reported in MDM2-amplified cells that Nutlin treatment inhibits glucose metabolism and reduces α-ketoglutarate (α-KG)2 levels and that this is critical for Nutlin-induced apoptosis (12, 17, 18). In contrast, glucose metabolism and α-KG levels were maintained in MDM2-nonamplified cells treated with Nutlin. In these cells Nutlin increases autophagy that protects cells from apoptosis (12, 17). We also found the sensitivity of MDM2-amplified cells to Nutlin-induced apoptosis is due, in part, to MDM2-mediated down-regulation of SP1 and subsequent down-regulation of glycolytic genes (17).
Glycolysis promotes autophagy by, in some way, maintaining expression of various ATG genes in Nutlin-treated cells (12, 18), although the underlying mechanism for this is not known. Glycolytic metabolites are linked to histone modification that can regulate gene expression. Notably, α-KG is a metabolic intermediate of glucose. Recently we found that Nutlin suppresses α-KG and autophagy in MDM2-amplified cells while increasing α-KG and autophagy in MDM2-nonamplified cells (18). Importantly, α-KG is an activating cofactor for JMJD family histone lysine demethylases (19). These enzymes can regulate gene expression by altering the histone methylation status at gene promoters (20, 21). Histone methylation can regulate autophagy at gene expression levels. For example, Artal-Martinez de Narvajas et al. (22) reported the G9a histone methyltransferase inhibits autophagy by promoting H3K9me2 in the promoters of LC3 and other autophagy genes and repressing their expression. Histone methylations H3K27me3, H3K9me3, and H3K4me3 are found in LC3, ATG4b, and p62 gene promoters (23).
The JMJD2 (Jumonji C domain containing histone demethylase 2) family of proteins selectively demethylate H3K9me3 and H3K36me3. Among the JMJD2 family, JMJD2B is a p53 target gene (24). We envisioned that JMJD2B could be induced by Nutlin-mediated activation of p53 and then regulate histone methylation to affect ATG gene expression and autophagy. In the current report, we found JMJD2B-mediated histone demethylation promotes ATG gene expression, autophagy, and survival in MDM2-nonamplified cells treated with Nutlin. We also found that JMJD2B is depleted in MDM2-amplified cells treated with Nutlin in a manner that appears to be MDM2-dependent. The depletion of JMJD2B leads to increased histone methylation, reduced ATG gene expression and autophagy, and increased killing in MDM2-amplified cells.
Results
We previously showed glycolysis and α-KG can protect cells against Nutlin-induced apoptosis by in some way maintaining expression of ATG genes required for autophagy (12, 17, 18). α-KG is an intermediate metabolite of glucose and a cofactor for JMJD family histone lysine demethylases (19). Thus, we speculated JMJD histone demethylases could promote autophagy by regulating histone methylation and ATG gene expression (22). To begin to test this possibility, two MDM2-amplified cell lines (MHM and SJSA1) (17) and two MDM2-nonamplified cell lines (A549 and U2OS) were treated with Nutlin and the level of histone H3 lysine methylation determined by immunoblotting. As shown in Fig. 1A, H3K9me2/3 and H3K36me3 levels were decreased in Nutlin-treated A549 cells and unaffected in Nutlin-treated U2OS cells. In contrast, H3K9me2/3 and H3K36me3 levels were increased in Nutlin-treated MHM and SJSA1 cells. H3K4me3 levels were unaffected by Nutlin in all cells. H3K27me3 was barely detectable in U2OS, MHM, and SJSA1 cells compared with that in A549 cells. The results indicate H3K9me2/3 and H3K36me3 are increased in MDM2-amplified MHM and SJSA1 cells that are sensitive to Nutlin-induced apoptosis and either decreased or maintained in MDM2-nonamplified A549 and U2OS cells that are resistant to this apoptosis (17).
Figure 1.

Histone methylation status decides cell fate in Nutlin-treated cells. A, cells were treated 24 h with Nutlin (10 μm). Whole cell lysates were immunoblotted for the indicated proteins. B, cells were treated 24 h with vehicle or Nutlin (10 μm) and/or DZnep (10 μm). Whole cell lysates were immunoblotted for the indicated proteins. C, cells were treated 72 h with vehicle or Nutlin (10 μm) and/or DZnep (10 μm) or IOX-1 (0.1 mm) and then analyzed for sub-G1. % average sub-G1 cells (triplicate) is presented with S.D. indicated. The asterisk indicates there is a significant difference between Nutlin- and Nutlin plus DZnep–treated MHM (p < 0.01) and SJSA1 (p = 0.01). D and E, cells were treated 24 h with vehicle or Nutlin (10 μm) without or with IOX-1 (0.1 mm). Whole lysates were immunoblotted for the indicated proteins. F, cells were treated 72 h with vehicle or Nutlin (10 μm) and/or IOX-1 (0.1 mm) and then analyzed for sub-G1. Average % sub-G1 cells (triplicate) is presented with S.D. indicated. The asterisk indicates there is significant difference between Nutlin and Nutlin plus IOX-1 in A549 (p < 0.01) and U2OS (p < 0.01) cells.
Next we asked if increased histone methylation contributes to apoptosis. For this, apoptosis was monitored in MHM and SJSA1 cells co-treated with Nutlin and the pan-histone methylation inhibitor DZnep or IOX-1, a pan-inhibitor of JMJD2 histone demethylases. Co-treatment with DZnep blocked the increase in histone methylation seen in Nutlin-treated MHM cells (Fig. 1B) and reduced apoptosis in Nutlin-treated MHM and SJSA1 cells (Fig. 1C). In contrast, IOX-1 caused a slight increase in apoptosis in Nutlin-treated cells (Fig. 1C). These findings suggested that increased histone methylation contributes to Nutlin-induced apoptosis. To examine this further, we treated A549 and U2OS cells with Nutlin and IOX-1 and monitored histone methylation and apoptosis. Co-treatment with Nutlin and IOX-1 increased or maintained histone methylation in Nutlin-treated A549 and U2OS cells (Fig. 1, D and E) and sensitized these cells to apoptosis (Fig. 1F). Together, the results support the idea that increasing histone methylation sensitizes cells to Nutlin-induced apoptosis.
We next wished to ask if histone methylation regulates apoptosis in Nutlin-treated cells by affecting autophagy, because previous studies showed histone methylation can repress expression of LC3 and other autophagy genes (22). To examine this, we used autophagy flux as a measure of autophagy in Nutlin-treated cells. LC3-II is an autophagy protein that is degraded in autophagolysosomes. Bafilomycin A1 (BafA1) blocks autophagic degradation, including degradation of LC3-II. Thus, the extent to which LC3-II accumulates in BafA1-treated cells indicates the rate at which autophagic degradation is occurring, or autophagy “flux.” We first monitored LC3-II accumulation in MDM2-amplified MHM and SJSA1 cells treated with BafA1. As shown in Fig. 2, A and C, LC3-II accumulated in BafA1-treated MHM and SJSA1 cells, indicating autophagic degradation was occurring. LC3-II accumulated to a lesser extent in MHM and SJSA1 cells treated with BafA1 plus Nutlin versus cells treated with BafA1 alone (Fig. 2, A and C). This is consistent with our previous study (12) and indicates Nutlin inhibits autophagy flux in these cells. Importantly, LC3-II accumulated to a greater extent in cells co-treated with BafA1 plus Nutlin and DZNep compared with cells treated with BafA1 plus DZNep only (Fig. 2, A and C). This indicates Nutlin treatment promotes autophagy flux when histone methylation is inhibited by DZnep. Monodansylcadaverine (MDC) is a fluorescent marker of intact autophagolysosomes, and MDC labeling is a common measure of autophagy (26). We monitored MDC labeling of autophagic vesicles in MHM and SJSA1 cells treated with Nutlin alone or Nutlin plus DZnep. As shown in Fig. 2, B and D, MDC labeling was reduced in Nutlin-treated MHM and SJSA1 cells, consistent with reduced autophagy. However, Nutlin increased MDC labeling when histone methylation was inhibited by co-treatment with DZnep. Altogether, the results indicate decreasing histone methylation can restore or increase autophagy in Nutlin-treated MHM and SJSA1 cells.
Figure 2.
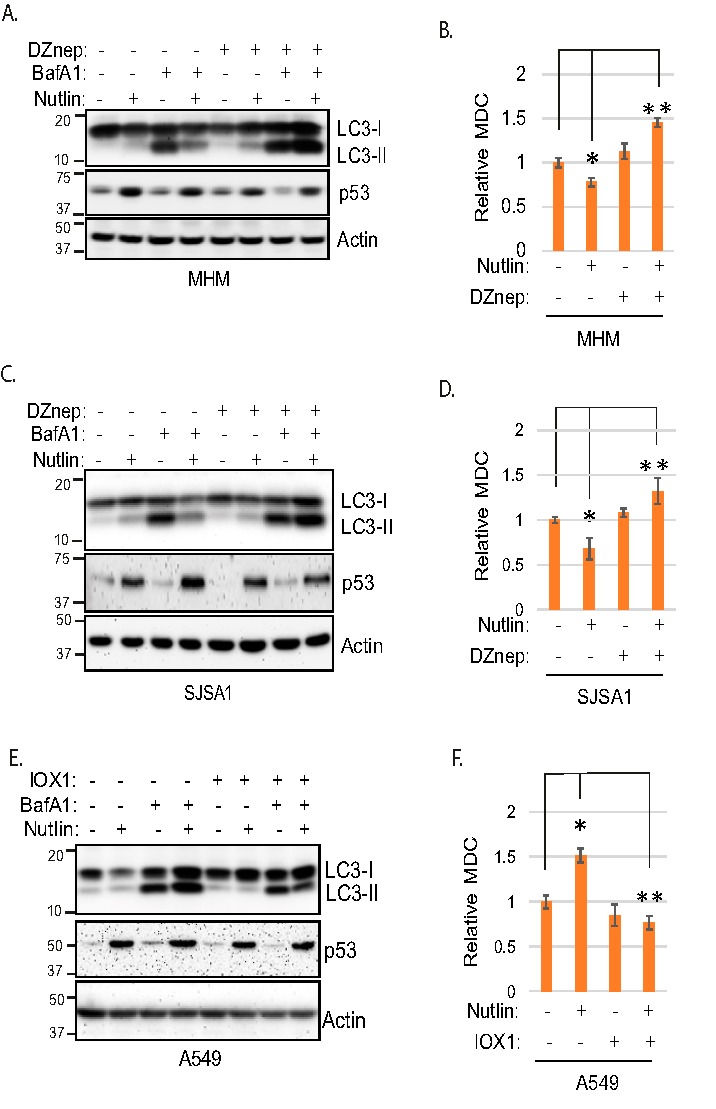
Histone methylation suppresses autophagy. A and C, MHM and SJSA1 cells were treated 24 h with Nutlin (10 μm) and/or DZnep (10 μm) in the presence or absence of BafA1 (10 nm); lysates were immunoblotted for the indicated proteins. B and D, MHM and SJSA1 cells were treated 24 h with Nutlin (10 μm) and/or DZnep (10 μm) and then analyzed for MDC sequestration, average relative MDC (triplicate) is presented with S.D. indicated. The asterisk indicates there is a significant difference between vehicle and Nutlin (p < 0.05) as well as Nutlin and Nutlin plus DZnep (p < 0.01) in MHM and SJSA1 cells. E, A549 cells were treated 24 h with Nutlin (10 μm) and/or IOX-1 (0.1 mm) in the presence or absence of BafA1(10 nm); lysates were immunoblotted for the indicated proteins. F, A549 cells were treated 24 h with Nutlin (10 μm) and/or IOX-1 (0.1 mm) and then analyzed for MDC sequestration, average relative MDC (triplicate) is presented with S.D. indicated. The asterisk indicates there is a significant difference between vehicle and Nutlin (p < 0.01) as well as Nutlin and Nutlin plus IOX-1 (p < 0.01) cells.
We next asked if increasing histone methylation by IOX1 treatment could reduce autophagy flux and MDC labeling in MDM2-nonamplified A549 cells. As shown in Fig. 2E, LC3-II accumulated to a greater extent in A549 cells co-treated with Nutlin plus BafA1 compared with cells treated with BafA1 alone (Fig. 2E). This is consistent with our previous studies and indicates Nutlin can increase autophagy flux in these cells. However, LC3-II accumulated to a lesser extent in cells co-treated with BafA1 plus Nutlin and IOX1 compared with cells treated with BafA1 plus Nutlin alone (Fig. 2E). This result suggests increasing histone methylation by IOX-1 treatment reduces autophagy flux. As shown in Fig. 2F, MDC labeling was increased by Nutlin treatment in A549 cells, and this effect was blocked by IOX1 (Fig. 2F). In total, these results suggest increasing histone methylation in Nutlin-treated A549 cells reduces autophagy.
The above results suggest histone methylation, specifically H3K9/K36me3, is differentially regulated in MDM2-amplified (MHM and SJSA1) cells versus MDM2-nonamplified (A549 and U2OS) cells and plays a repressive role in autophagy. JMJD2 family proteins promote demethylation of H3K9/K36me3, and JMJD2B is a p53 target gene (24, 27). We speculated JMJD2B may regulate histone methylation and autophagy in response to p53 activation by Nutlin. To test this possibility, we first monitored JMJD2B gene and protein expression in Nutlin-treated MHM, SJSA1, A549, and U2OS cells. As shown in Fig. 3A, JMJD2B mRNA levels were increased in all four cell lines treated with Nutlin. In U2OS and A549 cells, JMJD2B protein was also increased in response to Nutlin (Fig. 3B), consistent with the mRNA increase. However, JMJD2B protein was markedly decreased in Nutlin-treated MHM and SJSA1 cells (Fig. 3B). Notably, MDM2 levels were highly induced in MHM/SJSA1 cells compared with that in A549/U2OS cells (Fig. 3B). Co-treatment with the proteasome inhibitor MG132 restored JMJD2B protein levels in MHM and SJSA1 cells treated with Nutlin (Fig. 3C), at least partially, but only slightly increased JMJD2B protein levels in U2OS and A549 cells (Fig. 3D). These results suggest the decrease in JMJD2B protein levels in MDM2-amplified MHM/SJSA1 cells is because of increased protein degradation.
Figure 3.

Nutlin up-regulates JMJD2B proteins in U2OS and A549 but down-regulates in MHM and SJSA1 cells. A, cells were treated 24 h with vehicle or Nutlin (10 μm). JMJD2B mRNA were determined and presented (average from triplicate, S.D. is too small to show). B, cells were treated 24 h with vehicle or Nutlin (10 μm). Lysates were immunoblotted for the indicated proteins. C and D, cells were treated 24 h with Nutlin (10 μm) and then with MG132 (10 μm) for 4 h. Lysates were immunoblotted for the indicated proteins.
Results in Fig. 3 suggested that reduced levels of JMJD2B may cause increased histone methylation and, subsequently, reduced autophagy and increased death in MDM2-amplified cells treated with Nutlin. If this were true, we reasoned knockdown of JMJD2B would also increase histone methylation in MDM2-nonamplified cells treated with Nutlin, and this would coincide with reduced autophagy and increased cell death. To test this, A549 cells were transfected with control siRNA or JMJD2B siRNA and then treated with vehicle or Nutlin. As shown in Fig. 4A, H3K9me3 and H3K36me3 levels were increased in JMJD2B knockdown cells treated with Nutlin but not in control cells. Immunoblotting confirmed JMJD2B levels were depleted by the JMJD2B siRNA. We again used LC3 accumulation in BafA1-treated cells as a measure of autophagy flux. As shown in Fig. 4, B and D, LC3-II accumulated to a greater extent in control A549 and U2OS cells treated with Nutlin plus BafA1 compared with BafA1 alone, indicating Nutlin promotes autophagy flux in these cells. However, this effect was lost when JMJD2B was depleted by siRNA. Similarly, Nutlin-treatment increased MDC labeling in control A549 and U2OS cells but not in cells where JMJD2B was depleted by siRNA (Fig. 4, C and E). These results indicate that depletion of JMJD2B blocks or reduces autophagy in Nutlin-treated A549 and U2OS cells. Finally, we monitored apoptosis (% sub-G1 cells) in response to Nutlin in either control A549 and U2OS cells or cells in which JMJD2B was depleted by siRNA. As shown in Fig. 4, C and E, Nutlin promoted apoptosis only in cells where JMJD2B was depleted but not in control cells. These results support a role for JMJD2B in suppressing histone methylation to maintain or increase autophagy and survival in MDM2-nonamplified cells treated with Nutlin.
Figure 4.

Knockdown of JMJD2B suppresses autophagy and sensitizes cells to Nutlin. A, A549 cells were transfected with control siRNA or JMJD2B siRNA and then treated 24 h with Nutlin (10 μm); lysates were immunoblotted for the indicated proteins. B and D, A549 and U2OS cells were transfected with control siRNA or JMJD2B siRNA and then treated 24 h with Nutlin (10 μm) and/or BafA1 (10 nm). Whole cell lysates were immunoblotted for the indicated proteins. Note: A and B are from the same experiment. In B, the first four lanes of the JMJD2B blot are reused from A to illustrate differences in JMJD2B expression in the absence and presence of BafA1. C, left panel, A549 cells were transfected with control siRNA or JMJD2B siRNA and then treated 24 h with vehicle or Nutlin (10 μm). MDC sequestration was determined and average relative MDC (triplicate) is presented with S.D. indicated (left panel). The asterisk indicates there is a significant difference between vehicle-treated and Nutlin-treated conditions in control siRNA (p < 0.01) and as well as Nutlin-treated control siRNA and JMJD2B siRNA (p < 0.01) cells. C, right panel, cells were treated similarly for 72 h and then analyzed for sub-G1. % sub-G1 cell were presented with S.D. indicated. There is a significant difference between Nutlin-treated control siRNA and JMJD2B siRNA (p < 0.01) cells. E, left panel, U2OS cells were transfected with control siRNA or JMJD2B siRNA and then treated 24 h with vehicle or Nutlin (10 μm). MDC sequestration was determined and average relative MDC (triplicate) is presented with S.D. indicated. The asterisk indicates there is a significant difference between vehicle-treated and Nutlin-treated conditions in control siRNA (p < 0.05) as well as Nutlin-treated control siRNA and JMJD2B siRNA (p < 0.05) cells. E, right panel, cells were treated similarly for 72 h and analyzed for sub-G1. % sub-G1 cells were presented with S.D. indicated. There is a significant difference between Nutlin-treated control siRNA and JMJD2B siRNA (p < 0.01) cells.
MDM2 is an ubiquitin E3-ligase that can bind and promote the degradation of p53 and other proteins. The finding that JMJD2B was depleted in MDM2-amplified cells treated with Nutlin but not in MDM2-nonamplified cells raised the possibility that MDM2 may bind and promote JMJD2B degradation in these cells. We carried out co-immunoprecipitation studies in SJSA1 cells to ask if MDM2 and JMJD2B can bind each other. In first experiments, to avoid JMJD2B degradation, cells were treated with Nutlin for only 6 h. This treatment length increases MDM2 levels but does not yet cause depletion of JMJD2B. As shown in Fig. 5A, JMJD2B co-immunoprecipitated with MDM2 in SJSA1 cells treated with Nutlin for 6 h. We carried out similar experiments in which SJSA1 cells were treated with Nutlin for 24 h followed by treatment with MG132 for 4 h. As shown in Fig. 5B, JMJD2B levels were reduced with 24-h Nutlin treatment but restored by MG132. Further, MDM2 was co-immunoprecipitated with JMJD2B in SJSA1 cells treated with Nutlin for 24 h and subsequently treated with MG132 (Fig. 5B). The results demonstrate JMJD2B and MDM2 can interact. Because MDM2 is ubiquitin ligase, we tested if MDM2 down-regulates JMJD2B via ubiquitination by co-expression of MDM2 and JMJD2B in p53-null H1299 cells. However, we failed to see JMJD2B ubiquitination by MDM2 (data not shown). Thus, we did not gain any evidence for JMJD2B degradation directly regulated by MDM2. Nutlin increases MDM2 protein levels but also inhibits the ability of MDM2 to bind and degrade p53 and other proteins that interact with MDM2 in the p53-binding domain. We speculated that if Nutlin reduces JMJD2B protein levels and blocks autophagy in MDM2-amplified cells by inhibiting MDM2, then depletion/knockdown of MDM2 would have the same effect as Nutlin. To examine this, we monitored JMJD2B protein levels, histone methylation, and autophagy in SJSA1 cells that were either treated with Nutlin or where MDM2 was depleted by siRNA and/or treated with Nutlin. We choose SJSA1 for this experiment because we have found MDM2 can be more efficiently knocked down with transfection of MDM2 siRNA compared with MHM cells. As shown in Fig. 5C, both knockdown of MDM2 and Nutlin treatment increased p53 levels. However, only Nutlin treatment but not MDM2 knockdown caused depletion of JMJD2B and increased histone methylation (H3K9me3). Moreover, MDM2 knockdown blocked the Nutlin-induced increase in H3K9me3 and decrease in JMJD2B. Further, Nutlin treatment reduced MDC labeling (Fig. 5D) and reduced autophagy flux (reduced the accumulation of LC3 by BafA1) (Fig. 5E) which was reversed by MDM2 knockdown. Finally, Nutlin treatment caused abundant apoptosis in SJSA1 cells which was also reversed by MDM2 knockdown (Fig. 5F). These results indicate the depletion of JMJD2B and inhibition of autophagy in MDM2-amplified cells treated with Nutlin is MDM2-dependent.
Figure 5.

MDM2 associates with and mediates down-regulation of JMJD2B. A, SJSA1 cells were treated 6 h with Nutlin (10 μm) and MG132 (10 μm). Lysates were immunoprecipitated (IP) with anti-MDM2 antibody and blotted for JMJD2B and MDM2. B, SJSA1 cells were treated 24 h with Nutlin (10 μm) and then 4 h with MG132 (10 μm). Lysates were immunoprecipitated (IP) with anti-JMJD2B antibody and blotted for MDM2 and JMJD2B. C, SJSA1 cells were transfected with control siRNA or MDM2 siRNA and then treated with vehicle or Nutlin (10 μm) for 24 h. Whole cell lysates were immunoblotted for the indicated proteins. D, control siRNA and MDM2 siRNA cells were treated with vehicle or Nutlin (10 μm) for 24 h. MDC sequestration was determined and average relative MDC (triplicate) is presented with S.D. indicated. The asterisk indicates there is a significant difference between vehicle and Nutlin (p < 0.05) in control cells and between Nutlin-treated control cells and Nutlin-treated MDM2 knockdown cells (p < 0.01). E, control siRNA and MDM2 siRNA cells were treated 24 h with vehicle or Nutlin (10 μm) in the presence or absence of BafA1(10 nm). Cell lysates were immunoblotted for the indicated proteins. F, control siRNA and MDM2siRNA cells were treated with vehicle or Nutlin (10 μm) for 72 h and then analyzed for sub-G1. Average % sub-G1 cells were presented with S.D. indicated. The asterisk indicates there is significant differences between vehicle and Nutlin cells (p < 0.01) as well as Nutlin-treated control siRNA cells and Nutlin-treated MDM2 siRNA cells (p < 0.01).
We reported previously that ULK1, ATG3, ATG7, and ATG16L1 are down-regulated by Nutlin in MHM and SJSA1 cells and this is reversed by α-KG (18). Because α-KG is a cofactor for JMJD2B, it may up-regulate ATG genes via JMJD2B-mediated histone demethylation. Because JMJD2B levels are increased by Nutlin in A549 and U2OS cells, we speculated JMJD2B may promote ATG gene expression in these cells. To test this, we first compared ATG gene expression in response to Nutlin in MHM, SJSA1, A549, and U2OS cells. The results show that ULK1, ATG3, ATG5, ATG10, and ATG16L1 are down-regulated by Nutlin in MHM and SJSA1 cells (Fig. 6A). However, these genes are not down-regulated by Nutlin in A549 and U2OS cells and, in fact, ULK1 and ATG16L1 are up-regulated by Nutlin in these cells (Fig. 6B).
Figure 6.

JMJD2B promotes ULK1 and ATG16L1 expression. A and B, MHM and SJSA1 cells (A) and A549 and U2OS cells (B). mRNA for the indicated ATG genes were determined and presented as graphs (average from triplicate, S.D. is too small to show). C and E, A549 and U2OS cells were transfected with control siRNA or JMJD2B siRNA and treated with vehicle or Nutlin for 24 h. mRNA for ULK1 and ATG16L1 were determined and presented as graphs with S.D. indicated. The asterisk indicates there are significant differences between vehicle and Nutlin treatment in control A549 and U2OS cells (p < 0.05), and between Nutlin-treated control cells and Nutlin-treated JMJD2B knockdown cells (p < 0.01). D and F, whole cell lysates were immunoblotted for the indicated proteins.
The opposite effect of Nutlin on ULK1 and ATG16L1 expression in MHM/SJSA1 versus A549/U2OS cells may be regulated by JMJD2B and histone methylation. If so, then knockdown of JMJD2B should decrease ULK1 and ATG16L1 expression in A549 and U2OS cells whereas inhibition of histone methylation should increase their expression in MHM and SJSA1 cells. To test this, we first knocked down JMJD2B with siRNA in A549 and U2OS cells and monitored ULK1 and ATG16L1 expression. The results showed that ULK1 and ATG16L1 mRNA levels were increased by Nutlin in control cells. However, this Nutlin-induced increase in ULK1 and ATG16L1 mRNA was prevented by JMJD2B knockdown (Fig. 6, C and E). Immunoblotting also showed that Nutlin induced an increase in ULK1 and ATG16L1 proteins that was blocked by knockdown of JMJD2B (Fig. 6, D and F). These results show that JMJD2B promotes ULK1 and ATG16L1 expression in Nutlin-treated A549 and U2OS cells. Finally, we sought further evidence that histone methylation regulates ATG gene expression in MDM2-amplified cells treated with Nutlin. To this end, we inhibited histone methylation in MHM cells by DZnep treatment and analyzed expression of ULK1 and ATG16L1 in response to Nutlin. Consistent with Fig. 6A and our previous report (18), Nutlin treatment decreased ULK1 and ATG16L1 expression in MHM and SJSA1 cells (Fig. 7A). Importantly, however, co-treatment with DZnep blocked the decrease caused by Nutlin. Immunoblotting showed that ULK1 and ATG16L1 proteins were decreased by Nutlin and this effect was blocked or reversed by DZnep (Fig. 7B).
Figure 7.

Histone methylation represses ULK1 and ATG16L1 expression. A, MHM and SJSA1 were treated 24 h with vehicle or Nutlin (10 μm) in the presence or absence of DZnep (10 μm). mRNA for ULK1 and ATG16L1 were determined and presented as graphs with S.D. indicated (triplicate). The asterisk indicates there are significant differences between vehicle and Nutlin treatment in control MHM and SJSA1 cells (p < 0.05), and between Nutlin-treated and Nutlin plus DZnep-treated cells (p < 0.01). B, whole cell lysates were immunoblotted for the indicated proteins. The double asterisk indicates there are significant differences between Nutlin treated and Nultin plus DZnep treated cells (p < 0.01).
Altogether, the results suggest p53-mediated up-regulation of JMJD2B and subsequent histone demethylation promote ULK1 and ATG16L1 expression and autophagy in MDM2-nonamplified cells treated with Nutlin. However, in MDM2-amplified cells treated with Nutlin, JMJD2B protein is down-regulated in a MDM2-dependent manner, leading to increased histone methylation, repression of ULK1 and ATG16L1 expression, reduced autophagy, and increased apoptosis.
Discussion
MDM2 antagonists like Nutlin activate WT p53 and are being developed as therapeutic agents for p53 WT cancers. We reported that p53 activated by Nutlin treatment can either promote or inhibit autophagy dependent on the MDM2 gene amplification status in cells. Results from the current study indicate the histone demethylase JMJD2B promotes autophagy via up-regulating autophagy (ATG) genes and determines cell fate in response to Nutlin. JMJD2B belongs to the JMJD2 family of proteins that removes methyl groups specifically from histone H3 lysine 9 and lysine 36 (19, 20). The JMJD2B gene is also transcriptionally activated by p53 (24). We treated different cell lines with Nutlin to activate p53 and then monitored histone methylation and autophagy. In the MDM2-amplified cell lines MHM and SJSA1, Nutlin treatment increased H3K9me2/3 and H3K36me3 levels. This increased histone methylation coincided with reduced expression of ULK1 and ATG16L1, reduced autophagy flux, and apoptosis. Importantly, decreasing histone methylation by DZnep restored ULK1 and ATG16L1 expression and autophagy and rescued these cells from Nutlin-induced killing. In MDM2-nonamplified cell lines U2OS and A549, Nutlin treatment decreased H3K9me2/3 and H3K36me3 levels. This decrease in histone methylation coincided with increased autophagy and increased expression ULK1 and ATG16L1. Increasing histone methylation through knockdown of JMJD2B or use of a JMJD2 inhibitor IOX1 reduced ULK1 and ATG16L1 expression and autophagy in these cells and sensitized the cells to apoptosis by Nutlin. The results support a model in which p53-regulated autophagy and cell fate in response to Nutlin treatment is controlled by JMJD2-dependent histone demethylation.
It has been reported that p53 and MDM2 can bind and/or regulate the activity and expression of different histone methylation and demethylation enzymes, and that they have opposing effects on histone methylation. For example, Zheng et al. (27) reported p53 promotes expression of the JMJD2B histone demethylase while reducing expression of the histone methyltransferase SUV39H1. The authors found that this results in reduced H3K9me3 levels, relaxed chromatin structure, and increased DNA repair. In contrast, MDM2 was reported to increase H3K9 methylation in p53 target genes, due most likely to the ability of MDM2 to bind and recruit SUV39H1 and EHMT1 (another histone methyltransferase) to p53-responsive genes (28). Tang et al. reported p53 can repress expression of the histone methyltransferase EZH2, which normally promotes H3K27 trimethylation (29). By contrast, Wienken et al. (30) reported that MDM2 can increase H3K27me3 by binding the EZH2-containing polycomb repressor complex 2 (PRC2). Importantly, p53 has also been reported to interact with SUV39H1, SETD2, and JMJD2 proteins to regulate target gene expression (31, 32). Results from the current study suggest another mechanism by which p53 and MDM2 can regulate histone methylation that involves changes in JMJD2B protein levels. Specifically, we found Nutlin increased H3K9me2/3 and H3K36me3 levels in MDM2-amplified MHM and SJSA1 cells, but either did not alter or reduced the levels of these methylations in MDM2-nonamplified U2OS and A549 cells. JMJD2 family proteins remove methyl groups specifically from H3K9 and H3K36 (19, 20). Given the effect of Nutlin treatment on JMJD2B levels in these different cells, we speculated changes in histone methylation could result from changes in JMJD2B protein level. Knockdown of JMJD2B or inhibition of JMJD2 by IOX1 increased histone methylation in Nutlin-treated A549 and U2OS cells. This supports the idea that reduced histone methylation in Nutlin-treated A549 and U2OS cells requires JMJD2 demethylase activity. Together, these data indicate histone methylation in Nutlin-treated cells is regulated, at least in part, by JMJD2 histone demethylases.
We found JMJD2B mRNA and protein expression in response to Nutlin is different in MDM2-amplified versus MDM2-nonamplified cells. JMJD2B mRNA was increased by Nutlin in all the cell lines, consistent with reports that JMJD2B is transcriptionally activated by p53. JMJD2B protein was increased by Nutlin treatment in A549 and U2OS cells. However, JMJD2B protein levels were markedly decreased by Nutlin treatment in the MDM2-amplified cells MHM and SJSA1. This decrease was because of enhanced protein degradation because it was largely prevented by the proteasome inhibitor MG132. Nutlin binds MDM2 and increases MDM2 protein levels, but also inhibits MDM2 from functionally interacting with p53 and other proteins that bind the MDM2 N terminus. We therefore asked if inhibiting MDM2 in MDM2-amplified cells (by siRNA knockdown) could mimic the effects of Nutlin. However, MDM2 knockdown did not reduce JMJD2B protein levels and also did not reduce ULK1 and ATG16L gene expression and autophagy. Moreover, knockdown of MDM2 blocked Nutlin-induced loss of JMJD2B, inhibition of autophagy, and apoptosis. These results indicate the ability of Nutlin to reduce JMJD2B levels and reduce ULK1/ATG16L expression and autophagy is not because of inhibition of MDM2 activity. Given that Nutlin binds MDM2, the results suggest the ability of Nutlin to reduce JMJD2B levels in MDM2-amplified cells and cause a subsequent reduction in ULK1/ATG16L expression and autophagy requires MDM2 protein. Because MDM2 is an E3 ubiquitin ligase, we speculated the very high MDM2 levels may bind and promote JMJD2B degradation in MDM2-amplified cells treated with Nutlin. Indeed, we observed MDM2 and JMJD2B co-immunoprecipitate with each other in MDM2-amplified cells treated with Nutlin and MG132. However, in subsequent and ongoing studies we have gained no evidence that MDM2 can promote the ubiquitination or degradation of JMJD2B in transfected cells. Further studies are required to determine how MDM2 affects JMJD2B protein levels in Nutlin-treated, MDM2-amplified cells.
Previous studies showed histone methylation can repress various genes known to participate in autophagy. For example, Artal-Martinez de Narvajas et al. (22) reported the G9a histone methyltransferase inhibits autophagy by promoting H3K9me2 in the promoters of LC3 and other autophagy genes and repressing their expression. p53 has been reported to promote or inhibit autophagy dependent on its subcellular localization. For example, nuclear p53 can promote autophagy by binding the promoters of ULK1, DRAM1, and various ATG genes and promoting their expression. In contrast, cytoplasmic p53 has been reported to inhibit autophagy. The JMJD2B gene is transcriptionally activated by p53. In the current report JMJD2B mRNA and protein levels were increased in A549 and U2OS cells treated with Nutlin, most likely because of p53-mediated activation of the JMJD2B gene. Further, increased expression of JMJD2B was necessary for increased ULK1 and ATG16L expression and autophagy in these cells. These results indicate yet another way in which p53 can promote autophagy is by increasing JMJD2B expression and causing a subsequent increase in ULK1 and ATG16L. A question that arises is how JMJD2B promotes ULK1/ATG16L expression and autophagy. H3K9me3 and H3K36me3 are substrates for demethylation by JMJD2B and both are associated with condensed chromatin and repression of gene transcription. Thus, one possibility is that JMJD2B increases ULK1 and ATG16L gene expression by reducing H3K9me3 and/or H3K36me3 levels in the ULK1 and ATG16L gene promoters. It is also possible that JMJD2B modulates expression of other proteins that, in turn, increase expression of ULK1 and ATG16L.
MDM2-amplified cancer cells are, in most cases, especially sensitive to Nutlin-induced apoptosis. The molecular basis for this is incompletely understood. Results from the current study indicate the heightened sensitivity of MDM2-amplified cells to Nutlin-induced apoptosis can be explained in part by reduced JMJD2B, increased histone methylation, and subsequent loss of ATG gene expression and autophagy (see proposed model in Fig. 8). Further, the results suggest JMJD2 histone demethylases could be targeted to increase cancer cell sensitivity to MDM2 antagonists like Nutlin.
Figure 8.
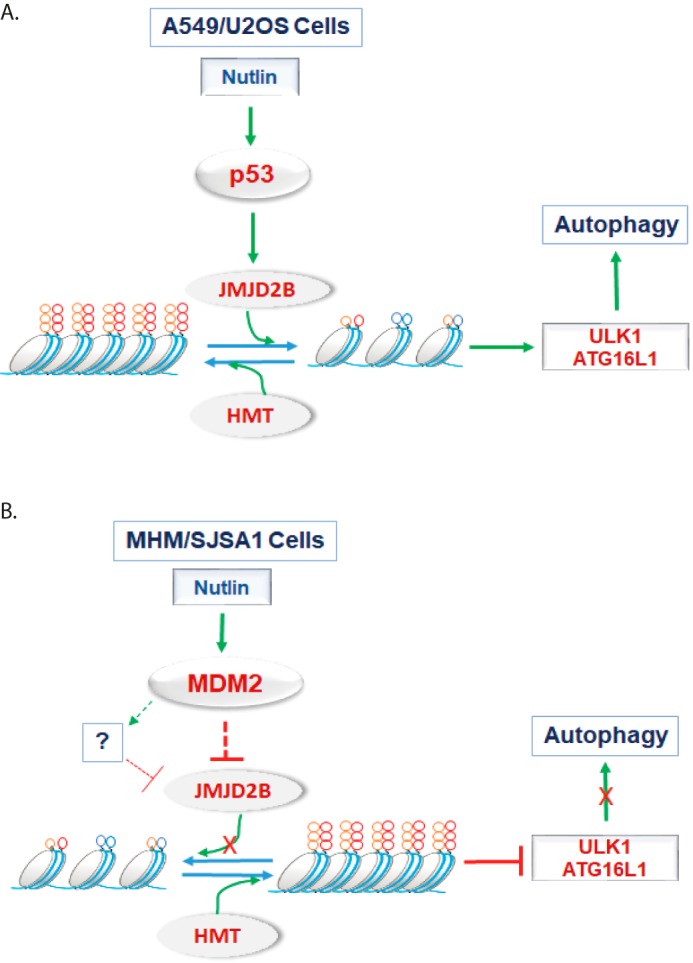
Proposed model. A, in MDM2-nonamplified A549 and U2OS cells, Nutlin activates p53 that up-regulates JMJD2B. JMJD2B demethylates H3K9me3 and H3K36me3 that are promoted by histone methyltransferases (HMT). Histone demethylation leads to transactivation of ULK1 and ATG16L1 genes that are critical for p53-mediated autophagy. B, in MDM2-amplified MHM and SJSA1 cells, Nutlin-induced p53 increases MDM2 to very high levels. High levels of MDM2 lead to degradation of JMJD2B through an unknown mechanism. Loss of JMJD2B results in increased HMT-mediated histone methylation that represses ULK1 and ATG16L1 genes leading to suppression of p53-mediated autophagy.
Experimental procedures
Cells and reagents
SJSA1, U2OS, A549 cells were from ATCC. MHM cells were kindly provided by Dr. Ola Myklebost, Norwegian Radium Hospital. All cell lines were grown in RPMI medium with 10% fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml). Cells were plated 48 h before treatment. Nutlin, MDC, dimethyl ketoglutarate (DMKG), BIX-01294, and BafA1 were obtained from Sigma-Aldrich. DZnep was from EMD Millipore (Darmstadt, Germany). IOX1 was from Selleck Chemicals (Houston, TX).
Immunoblotting
Whole cell extracts were prepared by scraping cells in RIPA buffer, resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Thermo Fisher Scientific). Antibodies to ATG16L1, H3K4me3, H3K9me2, H3K9me3, H3K27me3, H3K36me3, H3, and OGDH were from Cell Signaling Technology (Danvers, MA); LC3B and ULK1 antibodies were from Abcam (Cambridge, MA). p62, β-actin, and p53 (DO-1) antibodies were from Santa Cruz Biotechnology (Dallas, TX). Primary antibodies were detected with goat anti-mouse or goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase was from Invitrogen, using Clarity chemiluminescence from Bio-Rad.
Flow cytometry
For cell cycle analysis, cells were harvested and fixed in 25% ethanol overnight. The cells were then stained with propidium iodide (25 μg/ml, Calbiochem). Flow cytometry analysis was performed on a GalliosTM Flow Cytometer (Beckman Coulter), analyzed with FlowJo 10 (Treestar Inc.). For each sample, 10,000 events were collected.
siRNA-mediated transient knockdown
JMJD2B siRNA (ON-TARGETplus SMARTpool) and Control siRNA (ON-TARGETplus siControl Non-targeting Pool) were purchased from GE Dharmacon (Lafayette, CO) and were transfected according to the manufacturer's guidelines using DharmaFECT 1 reagent.
RNA isolation and real-time quantitative PCR analysis
Total RNA was prepared using Total RNA Mini Kit (IBI Scientific, Dubuque, IA); the first cDNA strand was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA). Manufacturers' protocols were followed in each case. The PCR primers for ULK1, ATG3, 4a, 7, 10, 12, 16L1, LC3, and β-actin genes were described previously (12, 18). SYBR Green PCR kit (Midwest Scientific, St. Louis, MO) was used according to the manufacturer's instructions. AB7300 system was used as follows: Activation at 95 °C; 2 min, 40 cycles of denaturation at 95 °C; 15 s and annealing/extension at 60 °C; 60 s, followed by melt analysis ramping from 60 °C to 95 °C. Relative gene expression was determined by the ΔΔCt method using β-actin to normalize.
Quantitative analysis of autophagosomes and autolysosomes
For analysis of autophagosome/autolysosomes, MDC sequestration was conducted as described previously (25).
Statistical analysis
One-way analysis of variance (ANOVA) was used to determine the statistical significance of differences among experimental groups. Student's t test was used to determine the statistical significance between experimental groups.
Author contributions
L. D. and C. G. M. conceptualization; L. D., R. E. P., and C. G. M. data curation; L. D. and C. G. M. formal analysis; L. D. investigation; L. D. writing-original draft; L. D. and C. G. M. project administration; R. E. P., X. L., and L. C. methodology; C. G. M. supervision; C. G. M. funding acquisition; C. G. M. writing-review and editing.
This work was supported by National Cancer Institute, National Institutes of Health Grant 1 R21 CA185036–01A1 (to C. G. M) and by a Swim Across America Foundation Grant (to C. G. M). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- α-KG
- α-ketoglutarate
- BafA1
- bafilomycin A1
- MDC
- monodansylcadaverine
- TCA
- tricarboxylic acid.
References
- 1. Schuler M., and Green D. R. (2001) Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans. 29, 684–688 10.1042/bst0290684 [DOI] [PubMed] [Google Scholar]
- 2. Yu J., and Zhang L. (2005) The transcriptional targets of p53 in apoptosis control. Biochem. Biophys. Res. Commun. 331, 851–858 10.1016/j.bbrc.2005.03.189 [DOI] [PubMed] [Google Scholar]
- 3. Gartel A. L., Serfas M. S., and Tyner A. L. (1996) p21–negative regulator of the cell cycle. Proc. Soc. Exp. Biol. Med. 213, 138–149 10.3181/00379727-213-44046 [DOI] [PubMed] [Google Scholar]
- 4. Riley T., Sontag E., Chen P., and Levine A. (2008) Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 10.1038/nrm2395 [DOI] [PubMed] [Google Scholar]
- 5. Maiuri M. C., Galluzzi L., Morselli E., Kepp O., Malik S. A., and Kroemer G. (2010) Autophagy regulation by p53. Curr. Opin. Cell Biol. 22, 181–185 10.1016/j.ceb.2009.12.001 [DOI] [PubMed] [Google Scholar]
- 6. Levine B., and Abrams J. (2008) p53: The Janus of autophagy? Nat. Cell Biol. 10, 637–639 10.1038/ncb0608-637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rubinsztein D. C. (2012) Autophagy–alias self-eating–appetite and ageing. EMBO Rep. 13, 173–174 10.1038/embor.2012.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wesselborg S., and Stork B. (2015) Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell Mol. Life Sci. 72, 4721–4757 10.1007/s00018-015-2034-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White E., Mehnert J. M., and Chan C. S. (2015) Autophagy, metabolism, and cancer. Clin. Cancer Res. 21, 5037–5046 10.1158/1078-0432.CCR-15-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kenzelmann Broz D., and Attardi L. D. (2013) TRP53 activates a global autophagy program to promote tumor suppression. Autophagy 9, 1440–1442 10.4161/auto.25833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ci Y., Shi K., An J., Yang Y., Hui K., Wu P., Shi L., and Xu C. (2014) ROS inhibit autophagy by downregulating ULK1 mediated by the phosphorylation of p53 in selenite-treated NB4 cells. Cell Death Dis. 5, e1542 10.1038/cddis.2014.506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duan L., Perez R. E., Davaadelger B., Dedkova E. N., Blatter L. A., and Maki C. G. (2015) p53-regulated autophagy is controlled by glycolysis and determines cell fate. Oncotarget 6, 23135–23156 10.18632/oncotarget.5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fitzwalter B. E., Towers C. G., Sullivan K. D., Andrysik Z., Hoh M., Ludwig M., O'Prey J., Ryan K. M., Espinosa J. M., Morgan M. J., and Thorburn A. (2018) Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev. Cell 44, 555–565.e3 10.1016/j.devcel.2018.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gottlieb E., and Vousden K. H. (2010) p53 regulation of metabolic pathways. Cold Spring Harbor Perspect. Biol. 2, a001040 10.1101/cshperspect.a001040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vousden K. H., and Ryan K. M. (2009) p53 and metabolism. Nat. Rev. Cancer 9, 691–700 10.1038/nrc2715 [DOI] [PubMed] [Google Scholar]
- 16. Puzio-Kuter A. M. (2011) The role of p53 in metabolic regulation. Genes Cancer 2, 385–391 10.1177/1947601911409738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duan L., Perez R. E., Chen L., Blatter L. A., and Maki C. G. (2018) p53 promotes AKT and SP1-dependent metabolism through the pentose phosphate pathway that inhibits apoptosis in response to Nutlin-3a. J. Mol. Cell Biol. 10, 331–340 10.1093/jmcb/mjx051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duan L., Perez R. E., and Maki C. G. (2018) Alpha ketoglutarate levels, regulated by p53 and OGDH, determine autophagy and cell fate/apoptosis in response to Nutlin-3a. Cancer Biol. Ther. 20, 252–260 10.1080/15384047.2018.1523858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu C., and Thompson C. B. (2012) Metabolic regulation of epigenetics. Cell Metab. 16, 9–17 10.1016/j.cmet.2012.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kooistra S. M., and Helin K. (2012) Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 10.1038/nrm3327 [DOI] [PubMed] [Google Scholar]
- 21. Johansson C., Tumber A., Che K., Cain P., Nowak R., Gileadi C., and Oppermann U. (2014) The roles of Jumonji-type oxygenases in human disease. Epigenomics 6, 89–120 10.2217/epi.13.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Artal-Martinez de Narvajas A., Gomez T. S., Zhang J. S., Mann A. O., Taoda Y., Gorman J. A., Herreros-Villanueva M., Gress T. M., Ellenrieder V., Bujanda L., Kim D. H., Kozikowski A. P., Koenig A., and Billadeau D. D. (2013) Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell Biol. 33, 3983–3993 10.1128/MCB.00813-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biga P. R., Latimer M. N., Froehlich J. M., Gabillard J. C., and Seiliez I. (2017) Distribution of H3K27me3, H3K9me3, and H3K4me3 along autophagy-related genes highly expressed in starved zebrafish myotubes. Biol. Open 6, 1720–1725 10.1242/bio.029090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castellini L., Moon E. J., Razorenova O. V., Krieg A. J., von Eyben R., and Giaccia A. J. (2017) KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 45, 3674–3692 10.1093/nar/gkw1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duan L., Danzer B., Levenson V. V., and Maki C. G. (2014) Critical roles for nitric oxide and ERK in the completion of prosurvival autophagy in 4OHTAM-treated estrogen receptor-positive breast cancer cells. Cancer Lett. 353, 290–300 10.1016/j.canlet.2014.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vázquez C. L., and Colombo M. I. (2009) Assays to assess autophagy induction and fusion of autophagic vacuoles with a degradative compartment, using monodansylcadaverine (MDC) and DQ-BSA. Methods Enzymol. 452, 85–95 10.1016/S0076-6879(08)03606-9 [DOI] [PubMed] [Google Scholar]
- 27. Zheng H., Chen L., Pledger W. J., Fang J., and Chen J. (2014) p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene 33, 734–744 10.1038/onc.2013.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen L., Li Z., Zwolinska A. K., Smith M. A., Cross B., Koomen J., Yuan Z. M., Jenuwein T., Marine J. C., Wright K. L., and Chen J. (2010) MDM2 recruitment of lysine methyltransferases regulates p53 transcriptional output. EMBO J. 29, 2538–2552 10.1038/emboj.2010.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang X., Milyavsky M., Shats I., Erez N., Goldfinger N., and Rotter V. (2004) Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene 23, 5759–5769 10.1038/sj.onc.1207706 [DOI] [PubMed] [Google Scholar]
- 30. Wienken M., Dickmanns A., Nemajerova A., Kramer D., Najafova Z., Weiss M., Karpiuk O., Kassem M., Zhang Y., Lozano G., Johnsen S. A., Moll U. M., Zhang X., and Dobbelstein M. (2016) MDM2 associates with polycomb repressor complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol. Cell 61, 68–83 10.1016/j.molcel.2015.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xie P., Tian C., An L., Nie J., Lu K., Xing G., Zhang L., and He F. (2008) Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell. Signal. 20, 1671–1678 10.1016/j.cellsig.2008.05.012 [DOI] [PubMed] [Google Scholar]
- 32. Kim T. D., Shin S., Berry W. L., Oh S., and Janknecht R. (2012) The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J. Cell. Biochem. 113, 1368–1376 10.1002/jcb.24009 [DOI] [PubMed] [Google Scholar]