

Abstract
Colorectal cancer is one of the most common malignancies worldwide, as it is often diagnosed at an advanced stage. Chimeric antigen receptor (CAR) T cell therapy has demonstrated remarkable success and emerged as one of the most promising therapeutic strategies in multiple malignancies. The purpose of this study was to investigate the anti-tumor activity of NKG2D CAR-T cells against human colorectal cancer cells. A non-viral third-generation NKG2D CAR was constructed, and subsequently transduced into T cells to obtain the NKG2D CAR-T cells. In vitro, NKG2D CAR-T cells showed cytotoxicity against human colorectal cancer cells in a dose-dependent manner compared with untransduced T cells. In addition, IL-2 and IFN-γ secreted by these cells were significantly higher than those by untransduced T cells. In vivo, NKG2D CAR-T cells significantly suppressed tumor growth, reduced tumor sizes and extended overall survival of mice in a xenograft model of HCT-116 cells. Furthermore, human NKG2D-positive lymphocytes infiltration could be found in the tumor sections of NKG2D CAR-T cells-treated mice. There were no severe pathological changes found in vital organs in any of the treatment groups. NKG2D CAR-T cells showed excellent killing effect and represented a promising immunotherapeutic strategy against human colorectal cancer.
Keywords: Chimeric antigen receptor, NKG2D, minicircle DNA, colorectal cancer
Introduction
Colorectal cancer is one of the most common malignancies among both men and women worldwide, with about 1.4 million new cases and almost 700,000 deaths in 2012. In 2015, 191,000 deaths (111,100 male and 80,000 female) were ascribed to this disease and 376,300 new cases (215,700 male and 160,600 female) were diagnosed in China [1]. By 2030, the global burden of colorectal cancer is expected to increase by 60% to more than 2.2 million new cases and 1.1 million deaths [2]. However, the majority of patients are diagnosed with an advanced stage of the disease due to insidious onset and lack of early onset specific symptoms. In the United States, localized stage of colorectal cancer is diagnosed in 39% of patients, for which the 5-year survival rate is 90%. The survival rate declines to 71% and 14% for patients diagnosed with regional and distant-stage disease, respectively [3]. Therefore, powerful therapeutic strategies for the treatment of colorectal cancer are urgently needed.
Chimeric antigen receptor (CAR) T cell therapy, as a novel adoptive cellular immunotherapy, has recently provided promising treatment modality for various malignancies. Generally, CARs are synthetic receptors that combine an extracellular ligand-specific recognition domain, typically a single-chain variable fragment, with a transmembrane domain and one or several intracellular signaling domains. When expressed in T cells using viral-based and non-viral approaches, CARs will provide both antigen-binding and T cell activating functions in a major histocompatibility complex (MHC)-independent manner. A number of CARs have been reported over the past decade, targeting an array of cell surface tumor antigens [4]. Early trials using CAR-T cells have showed promising results in the treatment of patients with advanced hematologic cancers, including relapsed or refractory acute lymphoblastic leukemia, chronic lymphocytic leukemia, and non-Hodgkin lymphoma [5]. Recently, two CD19 CAR-T cell therapies (KymriahTM & YescartaTM) have received FDA approval for the treatment of B-cell malignancies [6]. The clinical progress obtained in the treatment of hematological cancers with CAR-T cell therapies has not been recapitulated in the treatment of solid tumors, mainly due to the inefficient T cell trafficking, immunosuppressive tumor microenvironment, suboptimal antigen recognition specificity, and lack of safety control [7]. Furthermore, virtually all CARs are transduced into the T cells using viral vectors, which may contribute to clonal expansion, oncogenic transformation, variegated transgene expression and transcriptional silencing [8].
Natural killer group 2, member D (NKG2D) is an important activating receptor expressed on the surface of human natural killer (NK) cells, invariant natural killer T (NKT) cells, γδ T cells, CD8+ T cells, and some autoreactive or immunosuppressive CD4+ T cells [9]. In humans, NKG2D ligands (NKG2DLs) include major histocompatibility complex (MHC) class I-related chain A and B (MICA and MICB, respectively) and six unique long 16 binding protein (ULBP1-6). These molecules are present at low or undetectable levels on normal cells but rapidly appear on the surface of stressed, malignant transformed, and infected cells [10]. Upon ligands binding, NKG2D activates immune cells through the adaptor molecule DAP10, thereby triggering cellular proliferation, pro-inflammatory cytokines production, and target cell elimination. These functions emphasize the importance of the receptor NKG2D in immunotherapy for cancer. Currently, several preclinical studies and clinical trials applying NKG2D CAR-T cells in cancer immunotherapy have been performed [11-14].
In this study, a non-viral third-generation NKG2D CAR was constructed, and subsequently transduced into T cells to obtain the NKG2D CAR-T cells. NKG2D CAR-T cells demonstrate a specific and efficient cytotoxicity against human colorectal cancer cells both in vitro and in vivo.
Materials and methods
Cell lines and reagents
Two human colorectal cancer cells (LS174T and HCT-116) were obtained from American Tissue Culture Collection (ATCC, USA) and cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 1% streptomycin-penicillin mixture in a 95% air and 5% CO2 atmosphere at 37°C.
NKG2D CAR construction
NKG2D CAR consists of the CD8α signal sequence, the extracellular portion of human NKG2D receptor (amino acids 82-216), the hinge region of CD8α, the transmembrane and intracellular domain of CD28, the intracellular signaling domains of human 4-1BB and CD3ζ. The above sequences were all referred to NCBI GeneBank based on previous reports and optimized suitable for expression. EcoRI and BamHI sites were included at both ends. The sequence was synthesized by DetaiBio Co., Ltd (Nanjing, China) and confirmed by gene sequencing (Life Technologies, China). The entire sequence was cloned into the parental minicircle plasmid pMC.CMV-MCS-EF1-GFP-SV40polyA (System Biosciences, Palo Alto, CA, USA) according to the manufacturer’s instructions. This parental minicircle plasmid was transformed into ZYCY10P3S2T minicircle production E. coli to generate minicircle vector. The new minicircle DNA vector was named NKG2D CAR minicircle DNA vector (Figure 1).
Figure 1.
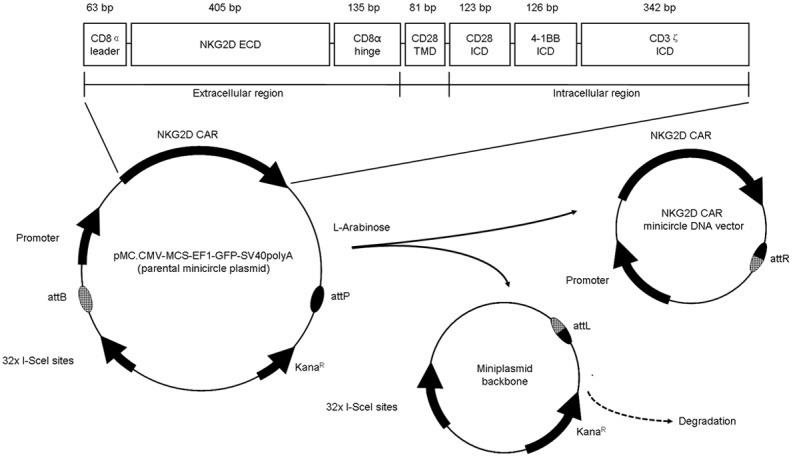
Schema of NKG2D CAR minicircle DAN non-viral vector construction. NKG2D CAR contains a CD8α leader signal sequence sequence, an extracellular domain (ECD) of human NKG2D receptor, a hinge region of CD8α, a transmembrane domain (TMD) of CD28, and an intracellular domain (ICD) of CD28, 4-1BB and CD3ζ. This gene fragment was cloned into the parental cloning vector pMC.CMV-MCS-EF1-GFP-SV40polyA. After transformation into ZYCY10P3S2T minicircle production E. coli, the parental cloning vector underwent a recombination event between the PhiC321 attB and attP sites, resulting in two products --- the NKG2D CAR minicircle DNA vector and the miniplasmid backbone. In the process, L-arabinose was added into the media to induce the I-SceI endonuclease expression, resulting in degradation of the miniplasmid backbone.
CAR-T cells preparation
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by Ficoll density gradient centrifugation (Tianjin Haoyang Biological Manufacture Co., Ltd., China). Isolated PBMCs were cultured and expanded in RPMI 1640 (Gibco, Life Technologies) supplemented with 10% fetal bovine serum (Gibco, Life Technologies) in a 95% air and 5% CO2 atmosphere at 37°C on day 1. Human recombinant interleukin-2 (IL-2; PeproTech, Rocky Hill, NJ, USA) was added every 2 days to a 300 IU/ml final concentration, and a cell density of 0.5-2 × 106 cells/ml was maintained. Three days after isolation, 5 × 106 PBMCs were electroporated with 10 µg NKG2D CAR minicircle DNA vector or empty minicircle DNA vector using the P3 Primary Cell 4D-Nucleofector X Kit (Lonza, Switzerland) following the manufacturer’s instructions. Subsequently, the cells were activated with Human T-Activator CD3/CD28 Dynabeads (Thermo Fisher Scientific, Waltham, MA) for 2 days. After 48 h, cells were collected for further transfection efficiency and cytotoxicity analysis.
Flow cytometric analysis
Cell phenotypes were analyzed by flow cytometry. NKG2DLs expressions of two human colorectal cancer cells (LS174T and HCT-116) were detected with human MICA PE-conjugated antibody, human MICB PE-conjugated antibody, human ULBP-1 PE-conjugated antibody, human ULBP-2/5/6 PE-conjugated antibody, and human ULBP-3 PE-conjugated antibody (all R&D Systems, Inc., Minneapolis, MN, USA). The expression of NKG2D, CD4 and CD8 on CAR-T cells was directly detected with human NKG2D/CD314 APC-conjugated antibody, CD4 PerCP-Cy5.5-conjugated antibody and CD8 PerCP-Cy5.5-conjugated antibody (all BD, San Diego, CA, USA). 1 × 105 cells were incubated with saturating amounts of antibodies in the dark for 30 min at room temperature, washed in PBS, and then resuspended in a proper volume of PBS. Flow cytometry acquisition was performed with a FACSCalibur Flow Cytometer (BD Immunocytometry Systems, Franklin Lakes, NJ). FlowJoTM 7.6.5 software (TreeStar, San Carlos, USA) was used for data analysis and calculation of the geometric mean fluorescence intensity (MFI). Isotype-matched antibodies were used in all analyses.
Western blot analysis
After treatment, the cells were washed twice with ice cold PBS, lysed in protein lysis buffer for 30 min on ice, and centrifuged at 12,000 rpm at 4°C for 20 min. Protein concentration was quantified using the BCA protein assay Kit with BSA as the standard. Protein samples were separated with SDS-PAGE, and transferred to PVDF membranes, which were then blocked with 5% (v/v) non-fat milk at 37°C for 2 h. The membranes were subsequently incubated overnight at 4°C with anti-CD3ζ antibody (1:1000) (Santa Cruz Biotechnology, CA), and then with secondary antibody (1:5000) for 1 h at room temperature. Imaging was performed using the Odyssey Fc System (LICOR, USA). The densitometry of the bands was analyzed using NIH ImageJ software.
Cytotoxicity assays
According to the manufacturer’s instructions, the cytotoxicity and specificity of engineered T cells was evaluated by lactate dehydrogenase (LDH) release assay kit (Biyotime, China), which was a nonradioactive alternative to the [51Cr] and [3H]-thymidine release assay. Briefly, colorectal cancer cells were plated in 96-well plates at a density of 1 × 105 cells per well. Following overnight incubation, the medium was replaced with serum free medium. Then, control T cells, or NKG2D CAR-T cells were co-incubated with target cells at different effector: target cell (E:T) ratios of 5:1, 10:1, 20:1 for 24 h. Following this, the plates were centrifuged for 5 min at 400 × g. 120 μl of supernatant from each well was transferred to a new 96-well plates and mixed with 60 μl of lactic acid dehydrogenase substrate mixture. To determine the maximal LDH release, 20 μl LDH release solution was added to the maximum release control 1 h prior to harvesting supernatant. After incubation at room temperature for 30 min, the absorbance was measured at 490 nm using a BioTek’s Microplate spectrophotometer (Gene Company Limited, USA). The percentage of cytotoxicity was calculated using the following formula: Cytotoxicity (%) = (experimental LDH release - spontaneous LDH release)/(maximal LDH release - spontaneous LDH release) × 100. All conditions were carried out in triplicate, and the experiments were replicated three times.
Cytokine release assays
T cells and target cells were cocultured at a 10:1 E:T ratio for 24 hours (not supplemented with IL-2). Cell supernatants were harvested for cytokine measurements of IL-2 and interferon (IFN)-γ concentrations using enzyme-linked immunosorbent assay (ELISA) kits (eBioscience, San Diego, CA, USA), according to the manufacturer’s protocols.
Mice and in vivo experiments
Four to six-week-old male NOD/SCID mice were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and housed in the specific-pathogen-free (SPF) conditions with clean sawdust bedding. All procedures involving mice were reviewed and approved by the Ethics Committee of Hebei Medical University. HCT-116-Luc human colorectal cancer cells were purchased from China Infrastructure of Cell Line Resources (Beijing, China) and cultured in RPMI-1640, supplemented with 10% fetal bovine serum, 1% streptomycin-penicillin mixture in a 95% air and 5% CO2 atmosphere at 37°C. To establish the tumor model, each mouse was inoculated subcutaneously with 1 × 106 HCT-116-Luc cells in 200 μl phosphate buffered saline (PBS) on the right flank. When the tumor burden reached about 150-250 mm3 after tumor cell inoculation, the mice were randomly divided into three groups (5 in each group). On day 0 and 7, tumor-bearing mice were treated by the tail vein injection with PBS (control group), 1 × 107 untransduced T cells (untransduced T cells group), or 1 × 107 NKG2D CAR-T cells (NKG2D CAR-T cells group), respectively. The body weight and tumor volume of mice was recorded every 5 days. Tumor growth was also monitored by luciferase activity on day 20 using the In Vivo Imaging System (Berthold Technologies, German) 10 min after subcutaneous injection with 150 mg/kg D-luciferin (Solarbio, Beijing, China). On day 25, blood samples were collected from mice. Samples were centrifuged and serum was used for cytokine detection assay as described previously. Subsequently, the animals were sacrificed and the heart, liver, lungs, spleen, and kidneys were harvested for further analysis.
Immunohistochemistry
For immunohistochemical analysis, all samples were fixed in 4% paraformaldehyde and embedded in paraffin. Paraffin sections were cut at 4 mm and deparaffinized with xylene and rehydrated in graded ethanol. Internal peroxidase was inactivated with 3% hydrogen peroxide in 100% methanol for 30 min. Antigen retrieval was performed by microwave oven heating in 10 mM citrate buffer for 15 min. 10% normal goat serum in PBS was then added to the sections for 30 min at room temperature to block nonspecific antibody binding. The sections were then incubated with primary antibodies for human NKG2D (diluted 1:300) overnight at 4°C. After rinses in PBS, the sections were then incubated with biotinylated secondary antibody and horseradish peroxidase-conjugated streptavidin. Labeling was visualized with diaminobenzidine (DAB) to produce a brown color, and sections were counterstained with hematoxylin. For negative controls, the primary antibody was omitted.
Statistical analysis
Data are expressed as means ± standard deviation (SD). Statistical analysis was performed by one-way ANOVA. Statistical significance was defined as P < 0.05.
Results
Expression of NKG2DLs on human colorectal cancer cells
The surface expression of ligands for NKG2D receptor on two human colorectal cancer cells (LS174T and HCT-116) was assayed by flow cytometry. As shown in Figure 2, LS174T cells expressed high levels of MICA and ULBP-3 but low levels of MICB, ULBP1, and ULBP-2/5/6 on their surface. HCT-116 cells expressed high levels of MICA, MICB, ULBP-2/5/6 and ULBP-3. No detectable levels of surface ULBP-1 was found in HCT-116 cells. These data were consistent with previous reports [15,16].
Figure 2.

Detection of NKG2DLs expression on two human colorectal cancer cells. LS174T and HCT-116 cells were stained with the specific antibody (human MICA PE-conjugated antibody, human MICB PE-conjugated antibody, human ULBP-1 PE-conjugated antibody, human ULBP-2/5/6 PE-conjugated antibody, and human ULBP-3 PE-conjugated antibody, filled histogram) or isotype control antibody (mouse IgG2B PE-conjugated antibody and mouse IgG2A PE-conjugated antibody, open histogram). MFI ratios of specific staining versus staining of isotype control are detailed in the histograms.
Construction of NKG2D CAR and generation of NKG2D CAR-T cells
To generate NKG2D CAR-expressed T cells in vitro, we constructed a minicircle DNA vector encoding a non-viral third-generation NKG2D CAR. The minicircle DNA vector incorporated a green fluorescent protein (GFP) sequence which was driven by the moderate EF1α promoter to facilitate identification of transfectants via GFP imaging. CD3/CD28-activated T cells from healthy donors were electroporated with the NKG2D CAR minicircle DNA vector, and expanded in the presence of IL-2 for further analysis of transfection efficiency. The surface expression of NKG2D on T cells was detected by flow cytometric analysis using human NKG2D/CD314 APC-conjugated antibody at days 2 after electroporation. As shown in Figure 3A, the proportion of cells expressing NKG2D was much higher in NKG2D CAR-T cells than that in control T cells. GFP expression detected by flow cytometry also confirmed the successful generation of NKG2D CAR-T cells.
Figure 3.
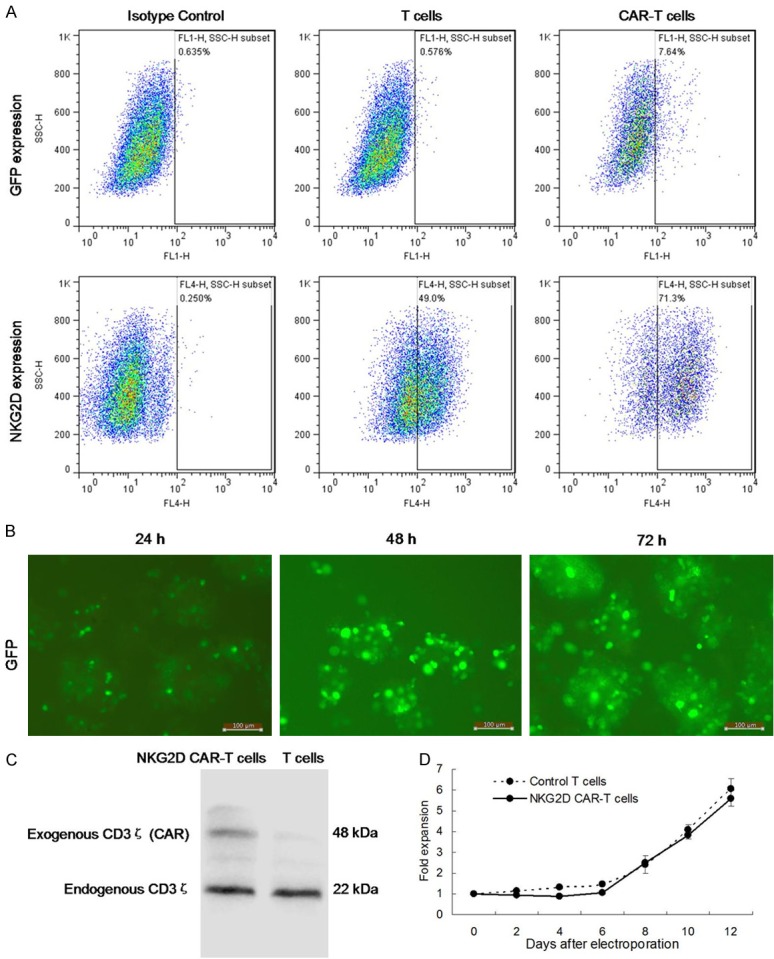
Characterization of NKG2D-specific CAR-T cells. A. Expression of NKG2D CAR and GFP on T cells or NKG2D CAR-T cells detected by flow cytometry. Left, isotype control. Middle, T cells control. Right, NKG2D CAR-T cells. B. Fluorescence microscopy images of GFP expression in CAR-T cells were observed by fluorescence microscopy for 24, 48, or 72 hours (Magnification × 200). C. The expression level of exogenous CD3ζ was analyzed by Western blotting. D. In vitro fold expansion of T cells following stimulation of Human T-Activator CD3/CD28 Dynabeads was assayed using cell counting assays.
As shown in Figure 3B, fluorescence microscopy images showed that GFP expression was observed at 24 hours after the minicircle DNA vector NKG2D CAR transfection, and sustained to at least 72 hours. As shown in Figure 3C, the expression level of exogenous CD3ζ was notably upregulated in NKG2D CAR-T cells than that in control T cells. In addition, as shown in Figure 3D, control T cells and NKG2D CAR-T cells displayed a similar expansion rate (5-6 folds) after 12 days of culture, despite a significant decrease in NKG2D CAR-T cells count on day 2 to 4 possibly due to the cells injury caused by electroporation.
Expression of lymphocytic phenotypes on NKG2D CAR-T cells
In healthy donors, peripheral T cells are usually either CD4+ or CD8+ with a small percentage of CD4+ CD8+ double positive cells (< 5%) [17]. CD4+ T cells are crucial for CD8+ T cell responses. In the absence of CD4+ help, primed CD8+ T cells never develop the enhanced capacity to divide and secrete cytokines characteristic of CD8+ memory cells [18]. Alteration in the T-cell composition and phenotype is related to the production of an effective immune response. Therefore, we determined the frequencies and phenotypes of CD4+ and CD8+ cells subsets among the T cells or NKG2D CAR-T cells by flow cytometry. As shown in Figure 4A, in the NKG2D CAR-T cells, there were a slightly larger proportion of CD4+ cells compared to T cells after electroporation and expansion. As shown in Figure 4B, the increased abundance of CD4+ cells was accompanied by a decreased percentage of CD8+ cells in NKG2D CAR-T cells.
Figure 4.
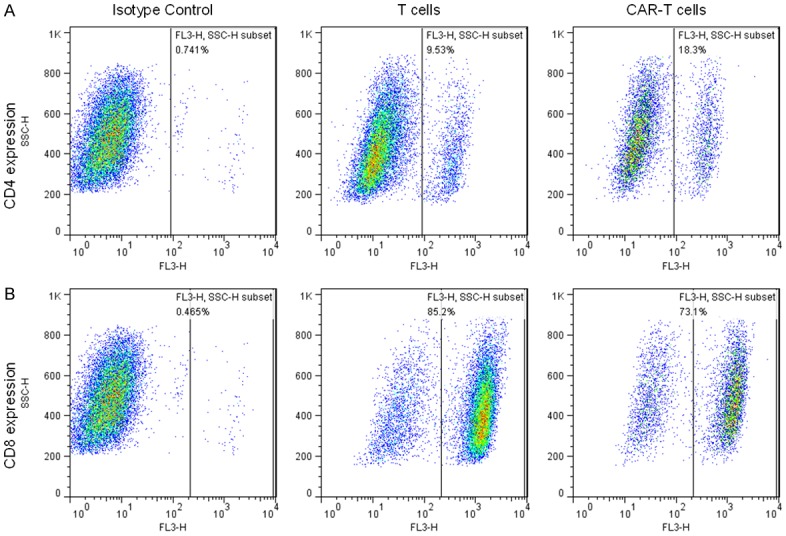
Phenotype Changes of NKG2D-specific CAR-T cells. A. Expression of CD4 on T cells or NKG2D CAR-T cells detected by flow cytometry. B. Expression of CD8 on T cells or NKG2D CAR-T cells detected by flow cytometry. Left, isotype control. Middle, T cells control. Right, NKG2D CAR-T cells.
Anti-tumor capacity of NKG2D CAR-T cells in vitro
To determine the cytotoxic activity, NKG2D CAR-T cells were co-cultured with LS174T or HCT-116 cells at the E:T ratios of 5:1, 10:1, 20:1 for 24 hours in 96-well-plate triplicately. Target cell death and cell lysis were analyzed based on the measurement of LDH activity released from the cytosol of damaged cells into the supernatant. As shown in Figure 5A, compared with untransduced T cells, NKG2D CAR-T cells showed stronger cytolytic effect on both of LS174T and HCT-116 cells at each E:T ratio. The cytotoxicity against LS174T and HCT-116 cells exceeded 47% and 52% at 20:1 of E:T ratio, respectively. Furthermore, to determine the T cell activation, NKG2D CAR-T cells were co-cultured with LS174T or HCT-116 cells at the E:T ratios of 10:1 for 24 hours in 96-well-plate triplicately. Cell-free medium was collected and analyzed for IL-2 and IFN-γ production by ELISA. As shown in Figure 5B, compared with untransduced T cells, NKG2D CAR-T cells secreted a substantial amount of IL-2 and IFN-γ when they were co-cultured with LS174T or HCT-116 cells.
Figure 5.
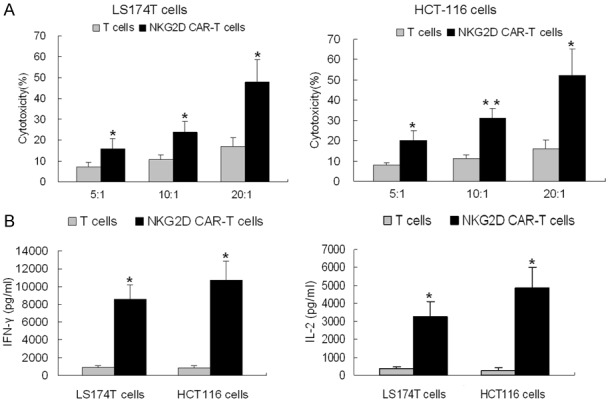
In vitro anti-tumor capacity of NKG2D CAR-T cells. A. The cytotoxicity of T cells (black line) and NKG2D CAR-T cells (gray line), against LS174T (left) or HCT-116 (right) was analyzed by LDH release assay at the indicated E:T ratios. B. Levels of IL-2 (left) and IFN-γ (right) secreted by T cells and NKG2D CAR-T cells were analyzed by ELISA after co-cultured with LS174T or HCT-116 cells for 24 hours. Results are expressed as means ± SD of triplicate experiments. Differences between the T cells and NKG2D CAR-T cells at each E:T ratio were evaluated using Student’s t-test. *P < 0.05 vs. T cells. **P < 0.01 vs. T cells.
Anti-tumor capacity of NKG2D CAR-T cells in vivo
To further confirm the anti-tumor effect of NKG2D CAR-T cells in vivo, we established xenograft models by intravenously inoculating HCT-116-Luc human colorectal cancer cells into NOD/SCID mice. A schema of the experimental protocol for this in vivo model was provided in Figure 6A. As shown in Figure 6B, on day 20, the in vivo bioluminescence imaging results showed that tumor growth was significantly suppressed in the mice treated with NKG2D CAR-T cells, as compared with those treated with PBS or untransduced T cells. As shown in Figure 6C, the tumor volumes were consistent with the corresponding results obtained by in vivo bioluminescence imaging. Moreover, better survival (Figure 6D) were observed in the mice treated with NKG2D CAR-T cells, compared with those treated with PBS or untransduced T cells. In order to track the tumor homing of CAR-T cells, we investigated NKG2D expression by immunohistochemistry in tissue samples from mice. As shown in Figure 6E, human NKG2D-positive lymphocytes infiltration was found in the tumor sections of NKG2D CAR-T cells-treated animals.
Figure 6.
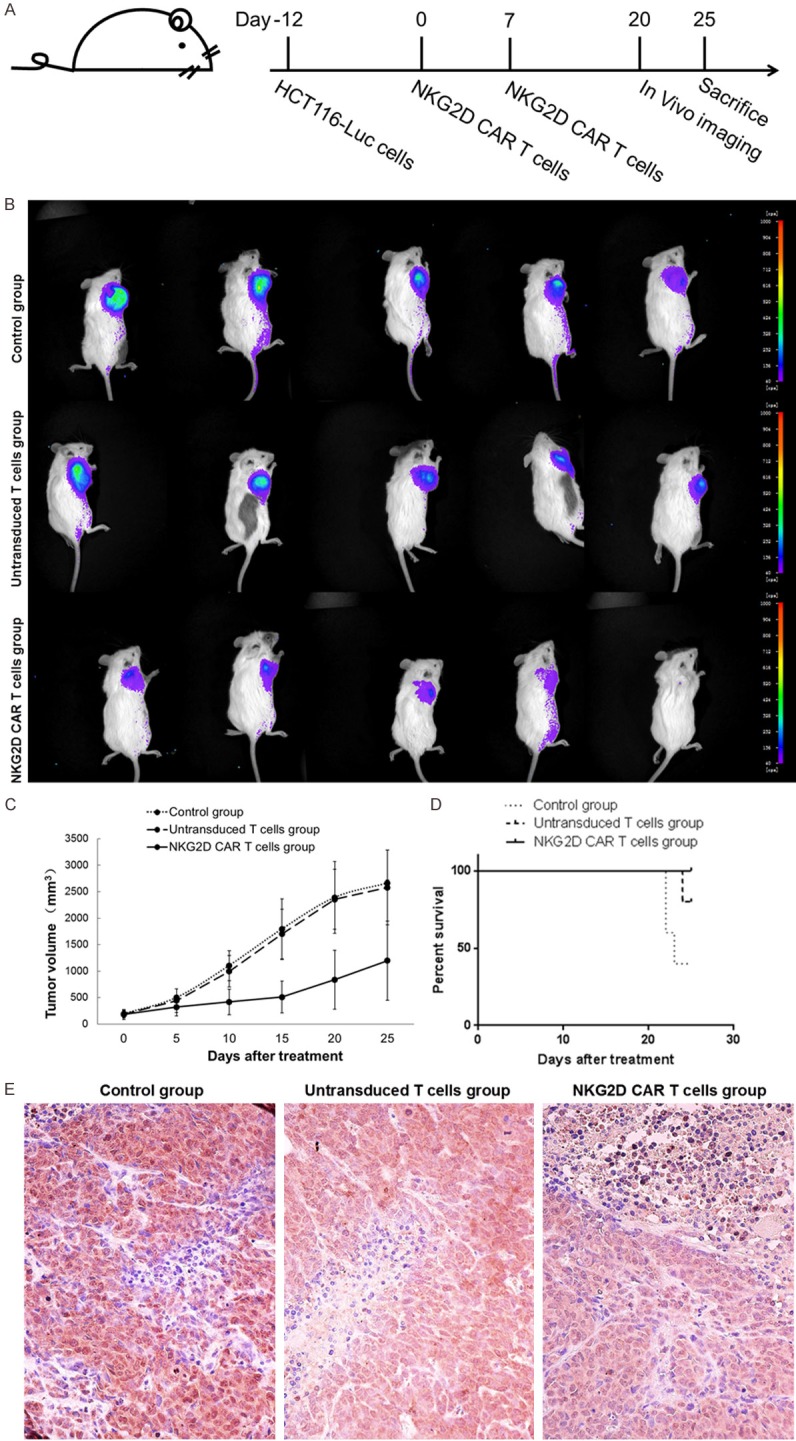
Anti-tumor capacities of NKG2D CAR-T cells in vivo. A. Schematic of the experimental protocol. NOD/SCID mice were inoculated subcutaneously with 1 × 106 HCT-116-Luc cells on day-12, and subsequently treated by the tail vein injection with 1 × 107 NKG2D CAR-T cells (NKG2D CAR-T cells group) on day 0 and 7. B. In vivo bioluminescence imaging was used to measure tumor growth of mice on day 20. C. Tumor volumes were measured by calipers every 5 days. Data are expressed as means ± SD. D. Overall survival of mice was presented in Kaplan-Meier curves. E. Tumor sections were stained with anti-human NKG2D specific antibody for detection of T cells (Magnification × 400).
Safety of NKG2D CAR-T cells in vivo
We further confirm the safety of NKG2D CAR-T cells in xenograft models. Despite a gradual loss of body weight (Figure 7A) in the mice treated with NKG2D CAR-T cells, there was no pronounced fever, loss of appetite, fatigue, vomiting, diarrhea, seizures, tremor, or other severe cytokine release syndrome observed in any of three groups. To further evaluate the organ toxicities in vivo, we performed pathological inspections of vital organs to investigate pathological changes by hematoxylin-eosin (HE) staining. As shown in Figure 7B, except for obvious congestion and inflammation (especially in the lungs) in the mice treated with PBS, no other obvious pathological changes were found in the heart, liver, lungs, spleen, and kidney sections in any of the treatment groups.
Figure 7.
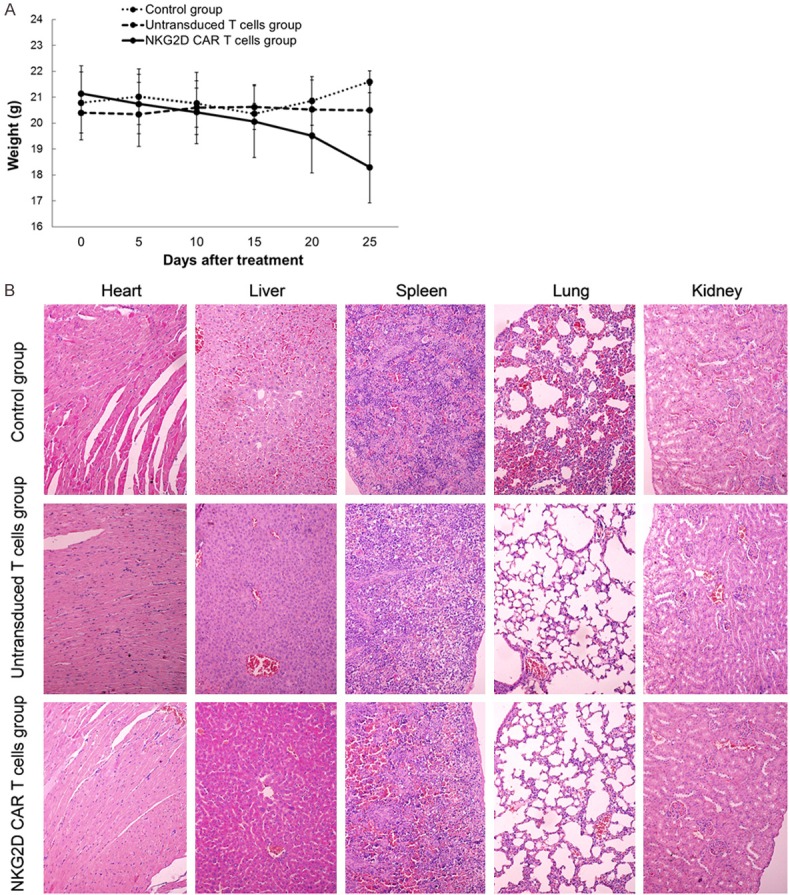
Safety of NKG2D CAR-T cells in vivo. A. Body weight was measured using an electronic scale every 5 days. Data are expressed as means ± SD. B. HE staining analysis was performed to examine the pathological changes of the heart, liver, lungs, spleen, and kidneys of the mice. (Magnification × 200).
Discussion
Globally, colorectal cancer is one of the common causes of cancer related death. Despite great advances in the diagnosis and treatment over the past decade, colorectal cancer continues to have a high incidence rate and mortality rate [1]. Therefore, it is of great significance to develop effective targeted drugs for the treatment of colorectal cancer. In recent years, numerous studies have highlighted CAR-T cell therapy as a promising therapeutic strategy to redirect the immune system to selectively attack tumor cells. The anti-tumor effect of CAR-T therapy has been evaluated in a variety of preclinical tumor models and clinical trials for the treatment of hematopoietic malignancies and solid tumors, including lymphoma, multiple myeloma, osteosarcoma, head/neck cancer, breast cancer, glioblastoma, pancreatic cancer, neuroblastoma, ovarian cancer and so on [19,20]. Moreover, exciting progresses have been made as evidenced by the two CAR-T therapies against the CD19 protein approved for clinical use by the U.S. FDA in 2017 [6].
Specially, several published studies have shown promising results of CAR-T therapy in the treatment of colorectal cancer. In 2000, Daly et al. reported that human lymphocytes transduced with chimeric GA733 specifically recognized colon carcinoma cells in cytokine release assays and lysed EGP40-expressing tumor cells [21]. In 2012, Tamada et al. engineered an anti-FITC CAR-T cells resulting in delayed growth of colon cancer but unexpectedly led to the outgrowth of EGF receptor (EGFR)-negative tumor cells [22]. In 2014, Blat et al. reported that CEA-specific CAR regulatory T cells (Tregs) significantly decreased the lung tumor burden in the azoxymethane-dextran sodium sulfate model [23]. In 2016, Katz et al. reported that pegional intraperitoneal infusion of CAR-T resulted in superior protection against carcinoembryonic antigen (CEA+) murine colon cancer cells compared with systemically infused CAR-Ts [24]. In 2017, Ang et al. reported the effectiveness of anti-epithelial cell adhesion molecule (EpCAM) CAR-expressing T cells for the local treatment of rectal cancer in mice [25]. In 2018, Magee et al. reported GUCY2C murine CAR-T cells recognized and killed human colorectal cancer cells endogenously expressing GUCY2C, providing durable survival in a human xenograft model in immunodeficient mice [26]. In addition to these preclinical studies, several clinical trials have confirmed the safety and efficacy of this approach in the treatment of colorectal cancer [27,28], although not in all studies [29]. Therefore, CAR-T therapy may represent a novel therapeutic strategy for patients with colorectal cancer.
Target antigen selection is a critical factor for a successful CAR-T therapy since it determines the off-target toxicity, specificity, and efficacy. In contrast to haematopoietic malignancies, many tumor antigens of epithelial cancer origin are not only expressed on tumors but also on other normal tissues [30]. To some extent, solid tumors may not be an ideal target for immune cells. Therefore, it would be necessary to explore highly specific CARs target antigens. To date, several antigens have been studied as targets for CAR-T therapies in solid tumors, such as CD70, CD171, EGFRvIII, ErbB, GPC3, PSCA, ROR1 [31].
NKG2D is an important activating receptor encoded by the killer cell lectin-like receptor subfamily K member 1 (Klrk1) gene [32]. It is mainly present on NK cells in both mice and humans, whereas it is constitutively expressed on CD8+ T cells in humans but only expressed upon T-cell activation in mice [32]. Human NKG2D receptor recognizes numerous and highly diversified ligands, including MICA, MICB, ULBP1, ULBP-2, ULBP-3, ULBP-4, ULBP-5, and ULBP6 [10]. Generally, NKG2DLs are absent on normal cells, but can be induced by a variety of stresses. Upon binding to NKG2DLs, NKG2D can modulate cell activity and survival. On tumor cell, NKG2DLs can participate in immune cell-mediated destruction by engaging NKG2D to activate NK cells and costimulate effector T cells [33]. In this study, we analyzed the surface expression of NKG2DLs on two human colorectal cancer cells, LS174T and HCT-116 cells. Consistent with other reports [15,16], LS174T cells expressed high levels of MICA and ULBP-3, and HCT-116 cells expressed high levels of MICA, MICB, ULBP-2/5/6 and ULBP-3. These results suggest that LS174T and HCT-116 cells are potential targets for immunotherapeutic approaches.
Thus, we designed and constructed a non-viral vector encoding NKG2D CAR by using minicircle DNA. Generally, most researchers employed lentiviral or retroviral vector systems for transduction of T cells with CAR. These viral vector systems can efficiently and lastingly transfect T cells. The transfection process itself is usually a simple addition of the vector reagent to the culture vessel [34]. Nevertheless, host immune response and insertional mutagenesis remain the major obstacles in their safe and broad application. To bypass these obstacles, Sleeping Beauty transposon system, recombinant eukaryotic expression vector and, in vitro transcription (IVT) mRNA vector were also used as vector systems for transduction of T cells with CAR [20]. In this study, we constructed a minicircle DNA vector encoding a non-viral third-generation NKG2D CAR. Non-viral vectors present certain advantages over viral vector, such as simple large scale production and low host immunogenicity. We found that NKG2D CAR could be properly folded and specifically bind to the T cell membrane, which was determined by flow cytometry, fluorescence microscopy, and western blotting. Frankly, we found some limitations in our study. One problem is that the NKG2D and GFP expression were apparently time-dependent, and persistence was relatively short. Another problem is that transfection efficiency of the minicircle DNA vector was particular low by using Lipofectamine reagent. Electroporation seems to be optimal for higher transfection efficiency but reduced viability. Despite these issues, in vitro and in vivo anti-tumor capacity of NKG2D CAR-T cells seems not to be influenced.
In our study, NKG2D CAR contains the CD8α signal sequence, the extracellular domain of human NKG2D receptor, the hinge region of CD8α, the transmembrane and intracellular domain of CD28, the intracellular signaling domains of human 4-1BB and CD3ζ. Classical CARs are classified into three or even more generations, differing on the intracellular signaling domain number of T cell receptors [35]. Compared with the first generation of CARs, the second and third generation of CARs incorporated activation domain and costimulatory domain for the expansion, prolonged antitumor activity, and cytokine secretion (such as IL-2, TNFα, and IFN-γ) of T cells [36]. On this basis, CAR-modified T cells therapy has emerged as a promising strategy for cancer immunotherapy. Indeed, consistent with previous studies, our findings suggested that NKG2D CAR-T cells had stronger cytolytic effect and secreted increased levels of IL-2 and IFN-γ against LS174T and HCT-116 cells in vitro.
To further confirm the anti-tumor effect of NKG2D CAR-T cells in vivo, we established xenograft models by intravenously inoculating human colorectal cancer cells into NOD/SCID mice. Considering that HCT-116 cells are more sensitive than LS174T cells to NKG2D CAR-T cells, we established HCT-116 xenograft models into NOD/SCID mice. As expected, the results obtained by in vivo demonstrated that NKG2D CAR-T cells significantly reduced tumor growth and extended overall survival of mice. Surprisingly, a mouse of NKG2D CAR-T cells group displayed an extraordinary effect of tumor suppression. The underlying mechanism remains to be further explored. Moreover, the mice treated with NKG2D CAR-T cells showed a gradual loss of body weight. We presumed that the suppressed tumor growth might be the main cause of this symptom. Nevertheless, there was no dramatical fever, loss of appetite, fatigue, vomiting, diarrhea, seizures, tremor, or other severe cytokine release syndrome observed in any of three groups. Additionally, NKG2D CAR-T cells have not shown toxicity against healthy tissues in animal models.
Taken together, the present study described the first report of antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo by using a non-virus vector. Our study demonstrated that the NKG2D CAR-T cells exhibited specific cytotoxicity against LS174T and HCT-116 cells in an NKG2D specific manner in vitro, particularly for HCT-116 cells. In vivo treatment of NKG2D CAR-T cells significantly reduced tumor growth and extended overall survival of HCT-116 xenograft models. In conclusion, NKG2D CAR-T cells showed excellent killing effect and represents a promising immunotherapeutic strategy against human colorectal cancer.
Acknowledgements
The Key projects in medical science research of Hebei Health and Family Planning Commision (20180153).
Disclosure of conflict of interest
None.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–91. doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, Jemal A. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–93. doi: 10.3322/caac.21395. [DOI] [PubMed] [Google Scholar]
- 4.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–98. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric antigen receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. 2017;8:1850. doi: 10.3389/fimmu.2017.01850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arabi F, Torabi-Rahvar M, Shariati A, Ahmadbeigi N, Naderi M. Antigenic targets of CAR T cell therapy. A retrospective view on clinical trials. Exp Cell Res. 2018;369:1–10. doi: 10.1016/j.yexcr.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol. 2018;11:22. doi: 10.1186/s13045-018-0568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–7. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, Cerboni C, Santoni A. NKG2D and its ligands: “One for All, All for One”. Front Immunol. 2018;9:476. doi: 10.3389/fimmu.2018.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonangeli F, Soriani A, Cerboni C, Sciumè G, Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front Immunol. 2017;8:1583. doi: 10.3389/fimmu.2017.01583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song DG, Ye Q, Santoro S, Fang C, Best A, Powell DJ Jr. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum Gene Ther. 2013;24:295–305. doi: 10.1089/hum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Sun M, Yu S, Liu K, Li X, Shi H. Potential therapeutic strategy for gastric cancer peritoneal metastasis by NKG2D ligands-specific T cells. Onco Targets Ther. 2015;8:3095–104. doi: 10.2147/OTT.S91122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernández L, Metais JY, Escudero A, Vela M, Valentín J, Vallcorba I, Leivas A, Torres J, Valeri A, Patiño-García A, Martínez J, Leung W, Pérez-Martínez A. Memory T cells expressing an NKG2D-CAR efficiently target osteosarcoma cells. Clin Cancer Res. 2017;23:5824–35. doi: 10.1158/1078-0432.CCR-17-0075. [DOI] [PubMed] [Google Scholar]
- 14.Lonez C, Hendlisz A, Shaza L, Aftimos P, Vouche M, Donckier V, Machiels JH, Van Den Eynde M, Canon JL, Carrasco J, Odunsi K, Sahebjam S, Rottey S, Braun N, Verma B, Gilham DE, Lehmann FF. Celyad’s novel CAR T-cell therapy for solid malignancies. Curr Res Transl Med. 2018;66:53–6. doi: 10.1016/j.retram.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Sers C, Kuner R, Falk CS, Lund P, Sueltmann H, Braun M, Buness A, Ruschhaupt M, Conrad J, Mang-Fatehi S, Stelniec I, Krapfenbauer U, Poustka A, Schäfer R. Down-regulation of HLA Class I and NKG2D ligands through a concerted action of MAPK and DNA methyltransferases in colorectal cancer cells. Int J Cancer. 2009;125:1626–39. doi: 10.1002/ijc.24557. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H, Wang F, Ju X, Kong L, An T, Zhao Z, Liu J, Li Y. Aurovertin B sensitizes colorectal cancer cells to NK cell recognition and lysis. Biochem Biophys Res Commun. 2018;503:3057–63. doi: 10.1016/j.bbrc.2018.08.093. [DOI] [PubMed] [Google Scholar]
- 17.Menard LC, Fischer P, Kakrecha B, Linsley PS, Wambre E, Liu MC, Rust BJ, Lee D, Penhallow B, Manjarrez Orduno N, Nadler SG. Renal cell carcinoma (RCC) tumors display large expansion of double positive (DP) CD4+ CD8+ T cells with expression of exhaustion markers. Front Immunol. 2018;9:2728. doi: 10.3389/fimmu.2018.02728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barinov A, Galgano A, Krenn G, Tanchot C, Vasseur F, Rocha B. CD4/CD8/Dendritic cell complexes in the spleen: CD8+ T cells can directly bind CD4+ T cells and modulate their response. PLoS One. 2017;12:e0180644. doi: 10.1371/journal.pone.0180644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han S, Latchoumanin O, Wu G, Zhou G, Hebbard L, George J, Qiao L. Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett. 2017;390:188–200. doi: 10.1016/j.canlet.2016.12.037. [DOI] [PubMed] [Google Scholar]
- 20.Kim MG, Kim D, Suh SK, Park Z, Choi MJ, Oh YK. Current status and regulatory perspective of chimeric antigen receptor-modified T cell therapeutics. Arch Pharm Res. 2016;39:437–52. doi: 10.1007/s12272-016-0719-7. [DOI] [PubMed] [Google Scholar]
- 21.Daly T, Royal RE, Kershaw MH, Treisman J, Wang G, Li W, Herlyn D, Eshhar Z, Hwu P. Recognition of human colon cancer by T cells transduced with a chimeric receptor gene. Cancer Gene Ther. 2000;7:284–91. doi: 10.1038/sj.cgt.7700121. [DOI] [PubMed] [Google Scholar]
- 22.Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18:6436–45. doi: 10.1158/1078-0432.CCR-12-1449. [DOI] [PubMed] [Google Scholar]
- 23.Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. 2014;22:1018–28. doi: 10.1038/mt.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, Boutros C, Hanna N, Junghans RP. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23:142–8. doi: 10.1038/cgt.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ang WX, Li Z, Chi Z, Du SH, Chen C, Tay JC, Toh HC, Connolly JE, Xu XH, Wang S. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget. 2017;8:13545–59. doi: 10.18632/oncotarget.14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magee MS, Abraham TS, Baybutt TR, Flickinger JC Jr, Ridge NA, Marszalowicz GP, Prajapati P, Hersperger AR, Waldman SA, Snook AE. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res. 2018;6:509–16. doi: 10.1158/2326-6066.CIR-16-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, Zhang R, Xiong Z, Wei Z, Shen J, Luo Y, Zhang Q, Liu L, Qin H, Liu W, Wu F, Chen W, Pan F, Zhang X, Bie P, Liang H, Pecher G, Qian C. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ metastatic colorectal cancers. Mol Ther. 2017;25:1248–58. doi: 10.1016/j.ymthe.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH, Sherwin SA. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22. doi: 10.1186/s40425-017-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, Byatte AJ, Kirillova N, Valle JW, Sharma SK, Chester KA, Westwood NB, Halford SER, Nabarro S, Wan S, Austin E, Hawkins RE. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66:1425–36. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutting T, Burgermeister E, Härtel N, Ebert MP. Checkpoints and beyond - immunotherapy in colorectal cancer. Semin Cancer Biol. 2018 doi: 10.1016/j.semcancer.2018.04.003. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 31.Schmidts A, Maus MV. Making CAR T cells a solid option for solid tumors. Front Immunol. 2018;9:2593. doi: 10.3389/fimmu.2018.02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prajapati K, Perez C, Rojas LBP, Burke B, Guevara-Patino JA. Functions of NKG2D in CD8+ T cells: an opportunity for immunotherapy. Cell Mol Immunol. 2018;15:470–9. doi: 10.1038/cmi.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. 2018;51:55–61. doi: 10.1016/j.coi.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer RK, Bowles PA, Kim H, Dulgar-Tulloch A. Industrializing autologous adoptive immunotherapies: manufacturing advances and challenges. Front Med (Lausanne) 2018;5:150. doi: 10.3389/fmed.2018.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allegra A, Innao V, Gerace D, Vaddinelli D, Musolino C. Adoptive immunotherapy for hematological malignancies: current status and new insights in chimeric antigen receptor T cells. Blood Cells Mol Dis. 2016;62:49–63. doi: 10.1016/j.bcmd.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Yu S, Li A, Liu Q, Li T, Yuan X, Han X, Wu K. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017;10:78. doi: 10.1186/s13045-017-0444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]