

Abstract
All-trans retinoic acid (ATRA) resistance continues to be a critical problem in acute promyelocytic leukemia (APL)-relapsed patients. In this study, a clinically achievable concentration of enzastaurin synergized with ATRA to induce differentiation and apoptosis in ATRA-resistant APL cell lines, NB4-R1 and NB4-R2. Mechanistically, although enzastaurin is a protein kinase Cβ (PKCβ) inhibitor, PKCβ may not be required since the activity of PKCβ was not suppressed by enzastaurin-ATRA (enz-ATRA) co-treatment, and another PKCβ-selective inhibitor did not mimic the effects of enzastaurin. An MEK inhibitor but not a RAF-1 inhibitor suppressed enz-ATRA treatment-triggered differentiation, activation of MEK/ERK and up-regulation of CCAAT/enhancer binding protein β (C/EBPβ) and/or PU.1. Therefore, RAF-1-independent MEK/ERK signaling was required for enz-ATRA treatment-induced differentiation via modulation of the protein levels of C/EBPβ and/or PU.1. Enz-ATRA treatment collapsed mitochondrial transmembrane potential without the activation of caspase-3, -6 and -7. Moreover, caspase-3/7- and caspase-6-specific inhibitors had no inhibitory effect on enz-ATRA treatment-triggered apoptosis. Therefore, enz-ATRA treatment-induced apoptosis was mitochondria-dependent but caspase-independent. Enz-ATRA treatment degraded PML-RARα, which may be involved in enz-ATRA treatment-induced dual effects and may also be beneficial for APL eradication. These findings may provide a potential therapy for ATRA-resistant APL patients.
Keywords: Acute promyelocytic leukemia, all-trans retinoic acid, apoptosis, differentiation, enzastaurin
Introduction
Since the introduction of all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) in the conventional chemotherapy of acute promyelocytic leukemia (APL), the remission and overall survival have been dramatically improved to exceptional rates [1]. Nevertheless, there are still 5%-10% patients who eventually relapse and/or become resistant to ATRA [2]. Until now, ATO has been the first choice for ATRA-resistant relapsed APL patients [3]. However, as ATO therapy is more and more widely adopted in APL treatment, acquired resistance to ATO therapy has been recognized in clinical practice [4]. Therefore, the development of novel approaches to avoid or reverse ATRA resistance continues to be a goal in the treatment of this disease. Gemtuzumab ozogamicin (GO) has been successfully used in combination with ATRA and ATO for newly diagnosed APL patients, as well as used as a single agent for patients with molecular relapsed APL [5-9]. However, no large randomized study of GO for APL treatment has been performed. LG-362B was demonstrated to overcome ATRA resistance in vitro and in vivo via targeting of PML-RARα [2]. However, the clinical applicability of LG-362B remains to be determined. Other agents, such as cAMP, STI571, granulocyte colony-stimulating factor, tumor necrosis factor, oridonin, dasatinib, matrine and interferon-γ have been shown to synergize with ATRA to induce differentiation in ATRA-resistant APL cells [10-17]. Clinical trials are urgently needed to verify their efficacy.
Protein kinase C (PKC) is a family of serine/threonine kinases, which consists of 13 isozymes that are involved in proliferation, differentiation, apoptosis, cell migration and gene expression. Intensive studies have explored the role of PKC in carcinogenesis and have rendered it as an attractive target for cancer therapy. PKCα is specifically down-regulated during human neutrophil terminal differentiation, suggesting its negative role in neutrophil differentiation [18]. Although PKC activity has been confirmed to be increased by ATRA treatment, both in the APL cell line-NB4 and in APL primary cells, its role in ATRA-induced granulocytic differentiation has been controversial [19-22]. A structural-biology study showed that ATRA competed with a PKC activator to bind to the C2-domian of PKCα and may thereby modulate PKCα activity [23]. Interestingly, PKCα and PKCγ are able to phosphorylate retinoic acid receptor α (RARα) at S157 in vitro and subsequently disrupt the formation of RARα/retinoid X receptor (RXR) heterodimer, resulting in decreased transcriptional activity [24]. Therefore, there is interference between retinoic acid (RA)-signaling and PKC-signaling pathways. Moreover, PKCδ contributes to ATRA resistance by overexpression of topoisomerase II β [19]. However, activated PKCδ has also been demonstrated to be required for ATRA-induced differentiation in APL cells [22]. Therefore, the role of PKCδ in ATRA-induced differentiation in APL cells has been disputed.
Enzastaurin is an isoenzyme-specific derivative of PKC pan-inhibitor staurosporine. It was designed to suppress the activation of PKCβ by inhibiting the binding of ATP. Unlike the unacceptable toxicity of staurosporine, enzastaurin has been demonstrated to be safe and well tolerated in multiple clinical trials. Moreover, it has exhibited promising anti-cancer activity in a variety of preclinical studies [25].
For hematological malignances, enzastaurin either as a single agent or in combination with other medicines exerts anti-cancer activity in acute myeloid leukemia, lymphoma and multiple myeloma cells by inhibiting proliferation or promoting apoptosis [25]. However, to our knowledge, enzastaurin has not yet been reported to induce/enhance differentiation. As mentioned above, since PKC may be one of the mediators of ATRA resistance in APL-relapsed patients and may also be the negative regulator of neutrophil-terminal differentiation, these phenomena prompted us to investigate whether enzastaurin could restore ATRA sensitivity in ATRA-resistant APL cell lines. This study used clinically achievable concentrations of enzastaurin. Unexpectedly, the combination of enzastaurin and ATRA (enz-ATRA) induced both terminal granulocytic differentiation and apoptosis in ATRA-resistant APL cell lines, NB4-R1 and NB4-R2, in a dose-dependent manner. Further study showed that the enz-ATRA combination-overcoming differentiation block required MEK/ERK-mediated modulation of the protein levels of CCAAT/enhancer-binding protein β (C/EBPβ) and/or PU.1. Additionally, the enz-ATRA combination-induced apoptosis was mitochondria-dependent but caspase-independent. Enzastaurin also synergized with ATRA to degrade PML-RARα, the pathogenic protein of APL.
Material and methods
Reagents
ATRA was purchased from Sigma-Aldrich (St Louis, MO, USA). Enzastaurin and sorafenib tosylate were purchased from Selleckchem Chemicals (Houston, TX, USA). U0126 and Z-DEVD-FMK were obtained from EMD Chemicals (San Diego, CA, USA). Z-VEID-FMK was purchased from R&D systems (Minneapolis, MN, USA). A PKCβ inhibitor was obtained from Merck (Darmstadt, Germany). All reagents were dissolved in dimethyl sulfoxide (DMSO).
Cell culture, cell viability and cell proliferation
The ATRA-resistant cell lines, NB4-R1 and NB4-R2 (kindly gifted from Dr Michel Lanotte, Hopital Saint Louis, Paris, France), were cultured in RPMI-1640, supplemented with 10% fetal calf serum (Thermo Fisher Scientific Inc, Waltham, MA, USA) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Trypan-blue exclusion was used to evaluate cell viability.
Cell differentiation assays
Cell maturation was evaluated by cellular morphology, nitroblue tetrazolium (NBT) reduction assay and the content of cell surface differentiation-related antigen CD11b. Morphology was determined with May-Grunwald-Giemsa’s staining and viewed at 1000× magnification. For NBT reduction, 1×106 cells were collected and incubated with 1 mg/mL NBT (Sigma-Aldrich) solution containing 10 μM phorbol 12-myristate 13-acetate (Sigma-Aldrich) at 37°C for 1 h. Cells were lysed by 10% sodium dodecyl sulfate (SDS) and 0.04 M hydrochloric acid. The absorbance at O.D 570 nm was detected by spectrophotometer (Beckman Coulter, Brea, CA, USA). The expression of cell surface differentiation-related antigen CD11b (Coulter, Marseilles, France), was determined via flow cytometry (EPICS XL, Coulter, Hialeah, FL, USA).
Annexin-V analysis
Annexin-V assay was performed according to instructions provided in the Annexin V-7AAD Apoptosis Detection Kit (BD Biosciences Pharmingen, San Diego, CA, USA). Briefly, 5×105 cells were harvested and washed with binding buffer provided in the kit. Then, cells were incubated with 5 μL annexin-V and 5 μL 7-Amino-Actinomycin at room temperature in the dark for 15 min. Fluorescent intensities were determined via flow cytometry (Coulter).
Determination of mitochondrial transmenbrane potentials (DYm) on flow cytometry
After washing twice with PBS, about 1×106 cells were incubated (37°C, 30 min) with 10 mg/mL rhodamine 123 (Rh123). Then, 50 mg/mL propidium iodide (PI) was added to cells. Fluorescent intensities were determined via flow cytometry (Coulter).
Western-blotting analysis
Cells were washed with phosphate-buffered saline (PBS) and lysed with RIPA buffer (Sigma-Aldrich). Cell lysates were centrifuged at 13,000 rpm for 10 min at 4°C. Supernants were collected and quantified by Bio-Rad Dc protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Nest, 20 or 50 μg protein extracts were loaded onto 8% SDS-polyacrylamide gel, subjected to electrophoresis, and were then transferred to polyvinylidene difluoride membranes (GE Healthcare UK Ltd, Buckinghamshire, UK). After blocking with 5% nonfat milk or BSA in PBS, the membranes were probed with the following primary antibodies: RARα, C/EBPβ, C/EBPε, PU.1 from Santa Cruz Biotech (Santa Cruz, CA, USA); phospho-p44/42 Erk1/2 (Thr202/Try204), phospho-MEK1/2 (Ser218/222), Phospho-PKC (pan) (βII Ser660), Phospho-PKCα/βII (Thr638/641) from Cell Signaling Technology (Beverly, MA, USA); β-actin from Sigma-Aldrich. Subsequently, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (GE Healthcare UK Ltd). Immunocomplexes were visualized with chemiluminescence kit (GE Healthcare UK Ltd) according to the manufacturer’s instr-uctions. To detect Erk1/2, MEK1/2 and PKCβ, the same membrane incubated with the antibodies to phosphorylated Erk1/2, MEK1/2 or PKCβ was stripped with stripping buffer (2% SDS, 100 mM beta-mercaptoethanol, 50 mM Tris, pH 6.8), followed by blocking and probing with anti-Erk1/2 (Cell Signaling Technology), anti-MEK1/2 (Cell Signaling Technology) or anti-PKCβ (Santa Cruz Biotech).
Immunofluorescent analysis for PML/PML-RARa proteins
Cells were centrifuged onto slides and fixed with 4% paraformaldehyde. After blocking with 5% BSA in PBS, cells were incubated with anti-PML monoclonal antibody (Santa Cruz Biotech) for 1 h at room temperature. Then, cells were washed with PBS and incubated with FITC-conjugated anti-mouse IgG (Sigma). Subsequently, cells were washed with PBS and covered with mounting medium (Agilent Technologies, Santa Clara, CA, USA). Slides were viewed on a Zeiss LSM870 confocal fluorescent microscope (Carl Zeiss Microscopy GmbH, Jena, Germany).
Statistical analysis
For NBT reduction, a two-tailed paired Student’s t test was used (n = 3). The flow-cytometric analysis of CD11b was analyzed by chi-square test (n = 20,000).
Results
The combination of enzastaurin and ATRA induces terminal granulocytic differentiation and apoptosis of NB4-R1 and NB4-R2 cells in a dose-dependent manner
A phase I clinical trial using oral enzastaurin (500 mg QD or 250 mg BID) showed that the average drug concentration of enzastaurin and its active metabolite under steady-state conditions (Cav, ss) is between 1120-2000 nM [26]. For feasibility in future clinical applications, this study used a clinically achievable concentration of 2 μM as the maximum concentration of enzastaurin in both NB4-R1 and NB4-R2 cells. As illustrated in Figures 1A, 1B, 2A and 2B, a low concentration (1 μM) of enzastaurin treatment alone for four days only suppressed proliferation, while a high concentration (2 μM) of enzastaurin inhibited cell growth and also reduced cell viability in both cell lines. The combination of 1 μM enzastaurin and ATRA attenuated proliferation and slightly decreased cell viability in both cell lines. However, the combination of 2 μM enzastaurin and ATRA induced significant growth inhibition and cell-viability reduction in both cell lines. Therefore, the enz-ATRA combination inhibited proliferation and triggered cell death in both cell lines in a dose-dependent manner.
Figure 1.

Effects of enz-ATRA treatment on cell growth, survival, apoptosis and differentiation in NB4-R1 cells. NB4-R1 cells were treated with 1 μM (1EN), 2 μM enzastaurin (2EN), 1 μM ATRA (RA) and in enz-ATRA combination (EN+RA) for four days. One representative experiment of cell growth (A) and cell viability (B) is shown. Each value represents the mean ± SD of triplicate samples. Similar results were obtained in three independent experiments. Representative morphology of NB4-R1 cells treated with the indicated drugs for four days (C). Scale bar represents 5 μm and the magnification is 1,000. Similar results were obtained in three independent experiments. Annexin-V assay of NB4-R1 cells treated with enzastaurin or/and ATRA for four days (D). The percentages of Annexin V+ cells are shown in the corresponding panels. Results were representative among three independent experiments. Differentiation was also evaluated by NBT-reduction assay (E) and flow-cytometric analysis of CD11b expression in NB4-R1 cells (F) with the indicated treatment for four days. For NBT-reduction assay, one representative experiment is shown. Each value represents the mean ± SD of triplicate samples. Similar results were obtained in three independent experiments. For flow-cytometric analysis of CD11b expression, each value represents the mean ± SD of three independent measurements. *P<0.05, **P<0.01, ***P<0.001 versus DMSO treated cells. ##P<0.01, ###P<0.001, versus ATRA treated cells. &&P<0.01, &&&P<0.001, as compared with 1EN+RA in NB4-R1 cells. The representative histogram of flow-cytometric analysis of CD11b expression in NB4-R1 cells with the indicated treatment for four days is also shown (G). The percentages of CD11b+ cells are shown in the corresponding panels.
Figure 2.

Effects of enz-ATRA treatment on cell growth, survival, apoptosis and differentiation in NB4-R2 cells. NB4-R2 cells were treated with 1 μM (1EN), 2 μM enzastaurin (2EN), 1 μM ATRA (RA) and in enz-ATRA combination (EN+RA) for four days. One representative experiment of cell growth (A) and cell viability (B) is shown. Each value represents the mean ± SD of triplicate samples. Similar results were obtained in three independent experiments. Representative morphology of NB4-R2 cells treated with the indicated drugs for four days (C). Scale bar represents 5 μm and the magnification is 1,000. Similar results were obtained in three independent experiments. Annexin-V assay of NB4-R2 cells treated with enzastaurin or/and ATRA for four days (D). The percentages of Annexin V+ cells are shown in the corresponding panels. Results were representative among three independent experiments. Differentiation was also assessed by NBT-reduction assay (E) and flow-cytometric analysis of CD11b expression in NB4-R2 cells (F) with the indicated treatment for four days. For NBT-reduction assay, one representative experiment is shown. Each value represents the mean ± SD of triplicate samples. Similar results were obtained in three independent experiments. For flow-cytometric analysis of CD11b expression, each value represents the mean ± SD of three independent measurements. **P<0.01, ***P<0.001 versus DMSO treated cells. ##P<0.01, ###P<0.001, versus ATRA treated cells. &&P<0.01, &&&P<0.001, as compared with 1EN+RA in NB4-R2 cells. The representative histograms of flow-cytometric analysis of CD11b expression in NB4-R2 cells with the indicated treatment for four days are also shown (G). The percentages of CD11b+ cells are shown in the corresponding panels.
Morphologically, a slightly decreased nuclear/cytoplasm ratio was observed in both cell lines treated with 1 μM enzastaurin for four days and in NB4-R1 cells treated with 1 μM ATRA for the same period (Figures 1C and 2C). After treatment with 2 μM enzastaurin for four days in both cell lines, some cells presented a decreased nuclear/cytoplasm ratio with kidney-shape nuclei, while some displayed nuclear fragmentation that was the characteristic of apoptosis (Figures 1C and 2C). More mature morphology was observed in both cell lines treated with any concentration of enzastaurin and ATRA co-treatment (Figures 1C and 2C). Moreover, with the combination of 2 μM enzastaurin and ATRA for four days, fully granulocytic-differentiated cells, such as lobed nuclei accompanied by markedly decreased nuclear/cytoplasm ratio were presented in both cell lines. Compared with 1 μM enzastaurin and ATRA co-treatment, more apoptotic cells were observed in both cell lines with 2 μM enzastaurin and ATRA co-treatment (Figures 1C and 2C). In both cell lines, the content of Annexin V+ cells was increased with 2 μM enzastaurin or the combination of any concentration of enzastaurin and ATRA for four days. The combination of 2 μM enzastaurin and ATRA had the highest content of Annexin V+ cells and showed synergistic effect (Figures 1D and 2D). Thus, apoptosis was induced by enz-ATRA combined treatment or by 2 μM enzastaurin in both cell lines. Consistent with the morphological results, enz-ATRA treatment for four days also enhanced NBT reduction in a dose-dependent manner in both cell lines (Figures 1E and 2E). Moreover, a synergistic effect of enzastaurin and ATRA on the content of CD11b+ cells was also observed in a dose-dependent manner in both cell lines (Figures 1F, 1G, 2F and 2G). Therefore, these results demonstrate that the combination of enzastaurin and ATRA exerts dual effects, triggering differentiation and apoptosis in a dose-dependent manner.
PKCβ is not involved in enz-ATRA treatment-triggered differentiation and apoptosis
To further investigate the mechanisms of enz-ATRA treatment-triggered differentiation and apoptosis, 2 μM enzastaurin was used in the subsequent studies. Since enzastaurin was designed to inhibit PKCβ, we first examined the role of PKCβ in enz-ATRA treatment-triggered differentiation and apoptosis. Phosphorylation of Ser660 or Thr641 controls PKCβ activity [27]. Phosphorylation of Thr641 but not that of Ser660 could be detected. With any treatment for 3 h, the phosphorylated level of this site remained unchanged in NB4-R2 cells and was elevated in NB4-R1 cells (Figure 3A). Thus, enzastaurin alone or in combination may not inhibit PKCβ activity. To further confirm the role of PKCβ, another PKCβ inhibitor was combined with ATRA to test whether such combination could mimic the effect of enzastaurin and ATRA co-treatment. According to manufacturer’s instructions, the IC50 to inhibit PKCβI and PKCβII of this PKCβ inhibitor is 21 nM and 5 nM, respectively. 200 nM PKCβ inhibitor was used. There were no typical fully differentiated cells or apoptotic cells present with PKCβ inhibitor-ATRA co-treatment in both cell lines (Figure 3B and 3C). Meanwhile, the content of CD11b+ cells was only slightly enhanced with PKCβ inhibitor-ATRA co-treatment compared with ATRA, but did not lead to a similar increase of CD11b+ cells by enz-ATRA co-treatment (Figure 3D-F). The content of Annexin V+ cells was hardly increased with PKCβ inhibitor-ATRA co-treatment in both cell lines when compared with ATRA (Figure 3G and 3H). Therefore, enz-ATRA treatment did not inhibit the activity of PKCβ and PKCβ may not be required for enz-ATRA treatment-triggered differentiation and apoptosis.
Figure 3.

PKCβ may not be involved in enz-ATRA treatment-induced differentiation and apoptosis. (A) NB4-R1 (left panel) and NB4-R2 (right panel) cells were treated with 2 μM enzastaurin (ENZA), 1 μM ATRA (RA) alone and in combination (ENZA+RA) for 3 h. The activation of PKCβ was measured by Western-blotting analysis of phosphorylated PKCβ at serine 641. The same membrane incubated with anti-phospho-PKCβ (Ser 641) was stripped and followed by detection of PKCβ. The expression of β-actin was evaluated as internal control. The morphology of NB4-R1 (B) and NB4-R2 (C) cells treated with 200 nM PKCβ inhibitor and/or 1 μM ATRA (RA) for four days. Scale bar represents 5 μm and the magnification is 1,000. One representative experiment among three independent assays is shown. Differentiation was also assessed by flow-cytometric analysis of CD11b expression (D), and each value represents the mean ± SD of three independent measurements. ##P<0.01, versus ATRA treated cells. The representative histograms of flow-cytometric analysis of CD11b expression in NB4-R1 (E) and NB4-R2 (F) cells with the indicated treatment for four days are also shown. Apoptosis was evaluated by flow-cytometric analysis of Annexin-V in NB4-R1 (G) and NB4-R2 (H) cells with the indicated treatment for four days. The percentages of CD11b+ cells or Annexin-V+ cells are shown in the corresponding panels. Similar results were obtained in three independent experiments.
Enz-ATRA treatment degrades PML-RARα, elevates the protein levels of C/EBPβ and PU.1, and activates MEK/ERK pathway
PML-RARa is not only a molecular marker of APL but has also been demonstrated to play an important role in the pathogenesis of APL [1]. ATRA and ATO are both regarded as successful examples of targeted therapy for malignance due to their ability to degrade PML-RARα. As shown in Figure 4A, ATRA treatment alone for 72 h decreased the protein level of PML-RARα in NB4-R1 cells, but not NB4-R2 cells, while enz-ATRA treatment promoted the degradation of PML-RARα in both cell lines. PML-RARα sequesters PML from PML nuclear body (PML-NB) and disrupts PML-NB, whereas the degradation of PML-RARα by ATRA treatment restores PML-NB [3]. PML-NB was visualized as a doughnut-shaped macromolecular structure by immunofluorescent analysis with an anti-PML antibody. In APL cells, PML-RARα delocalized PML into aberrant micro-speckled nuclear structures. As shown in Figure 4B, in NB4-R1 and NB4-R2 cells, hundreds of very small intranuclear dots were detected. With enz-ATRA treatment for 72 h, the micro-punctate nuclear dots were replaced with a few large nuclear dots in some NB4-R1 and NB4-R2 cells. This result suggests that PML-NB was restored in some cells with enz-ATRA treatment via degradation of PML-RARα.
Figure 4.
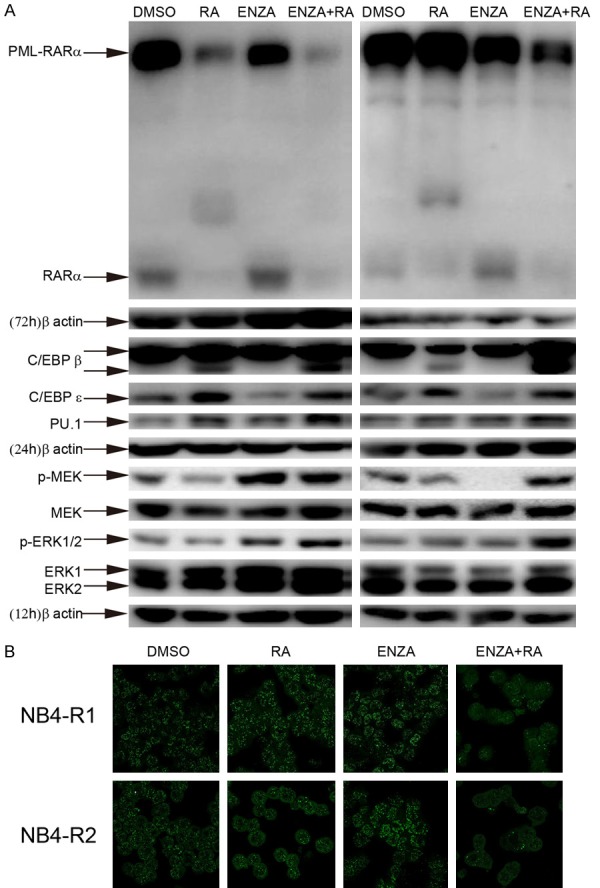
Enz-ATRA treatment degrades PML-RARa, enhances the protein levels of C/EBPβ and PU.1 and activates MEK/ERK pathway. A. NB4-R1 (left panel) and NB4-R2 (right panel) cells were treated with 2 μM enzastaurin (ENZA), 1 μM ATRA (RA) alone and in combination (ENZA+RA) for 12 h, 24 h or 72 h. The same membrane incubated with the antibodies to phosphorylated Erk1/2 or MEK1/2 was stripped and followed by detection of MEK and ERK1/2. Each time point has the corresponding expression of β-actin as internal control. Similar results were obtained in three independent experiments. B. Subcellular localization of PML/PML-RARa was analyzed by immunofluorescence with the indicated treatments for 72 h. One representative experiment among three independent assays is shown.
Next, we explored the mechanisms of enz-ATRA treatment-induced differentiation. Since RA is a natural differentiation inducer of granulocytes and the addition of enzastaurin overcame ATRA resistance in APL cells, we investigated several proteins and signal pathways involved in ATRA-induced differentiation in APL cells. C/EBPβ, C/EBPε and PU.1 (a member of the ets family) play a crucial role in the maturation of the myeloid lineages [28]. In addition, their expression was reported to be induced by ATRA, and they were all required for ATRA-promoted differentiation in APL cells [29-31]. In this study, with enz-ATRA treatment for 24 h, the protein levels of PU.1 and C/EBPβ, especially the short form but not C/EBPε, was remarkable elevated in both cell lines (Figure 4A).
The mounting literature has highlighted the important role of MEK/ERK pathway in certain cytokine-induced myeloid differentiation, as well as in ATRA-triggered differentiation of APL cells [32-34]. As shown in Figure 4A, 12 h earlier than the induction of C/EBPβ and PU.1, the phosphorylation of MEK and ERK was enhanced significantly with enz-ATRA treatment in both cell lines. Thus, the enz-ATRA combination activated MEK/ERK pathway earlier than its modulation of C/EBPβ and PU.1.
Enz-ATRA treatment induces differentiation through RAF-1-independent MEK/ERK modulation of the protein levels of C/EBPβ and/or PU.1
To further investigate the role of MEK/ERK pathway in enz-ATRA treatment-triggered differentiation and apoptosis, cells were pretreated with 0.5 μM U0126, which is a specific inhibitor of MEK. Of note, this concentration of U0126 was confirmed to be the highest that had no effect on cell viability when added to enz-ATRA treatment. U0126 attenuated MEK activity in both cell lines, as determined by Western blotting of phosphorylated ERK1/2 (Figure 5A and 5B). With U0126 pretreatment, fully differentiated cells observed in enz-ATRA treatment were replaced by primary cells with a large nuclear/cytoplasm ratio and round nuclei in both cell lines. However, typical apoptotic cells were still present (Figure 5C and 5D). The contents of CD11b+ cells were also significantly suppressed by U0126 in both cell lines (Figure 5F-H). Meanwhile, Annexin V+ cells were partially inhibited by U0126 pretreatment (Figure 5E). Since there is post-maturation apoptosis, U0126 partially inhibiting apoptosis might be the result of the suppression of differentiation. To clarify the source of apoptotic cells inhibited by U0126, cells were double-stained by CD11b and Annexin V. About half of differentiated cells (CD11b+ cells) underwent apoptosis in NB4-R1 and NB4-R2 cells (Annexin V+/CD11b+ cells compared with CD11b+ cells, 34.7±2.1% vs. 74.8±3.3% in NB4-R1 cells and 44.6±2.6% vs. 79.3±4.5% in NB4-R2 cells, Figure 5F, 5I and 5J). Moreover, most of the cells inhibited by U0126 were CD11b+ cells in both cell lines (U+En+RA compared with EN+RA, Annexin V+/CD11b+ cells in NB4-R1 were 26.4±1.3% vs. 34.7±2.1%, Annexin V+/CD11b- cells in NB4-R1 were 4.8±0.7% vs. 5.6±0.6%, Annexin V+/CD11b+ cells in NB4-R2 were 24.3±1.6% vs. 44.6±2.6%, Annexin V+/CD11b- cells in NB4-R2 were 9.4±1.7% vs. 14.2±2.3%, Figure 5I and 5J). Therefore, about half of differentiated cells underwent apoptosis and U0126 mainly suppressed post-maturation apoptosis. These results suggest that the U0126 inhibitory effect on apoptosis might be the result of its suppression of differentiation. In the presence of U0126, enz-ATRA treatment-enhanced protein level of PU.1 was remarkably decreased in both cell lines while the protein level of C/EBPβ was suppressed only in NB4-R2 cells (Figure 5K and 5L). Taken together, these results demonstrate enz-ATRA treatment-induced differentiation via MEK/ERK modulation of the protein levels of C/EBPβ and/or PU.1, while MEK/ERK pathway may have only a limited effect on enz-ATRA combination-induced apoptosis.
Figure 5.

MEK inhibition primarily suppresses differentiation and restores the protein levels of C/EBPβ or PU.1. Cells were exposed to 0.5 μM U0126 for 1 h prior to other treatment. The attenuation of MEK activation by U0126 (U) was detected by Western-blotting analysis of phosphorylated ERK1/2 in NB4-R1 (A) and NB4-R2 cells (B) with indicated treatments for 12 h and 36 h, respectively. The same membrane incubated with the antibody to phosphorylated Erk1/2 was stripped and followed by detection of ERK1/2. Similar results were obtained in three independent experiments. Effect of U0126 on morphology in NB4-R1 (C) and NB4-R2 cells (D) incubated with the indicated drugs for four days. Scale bar represents 5 μm and the magnification is 1,000. One representative experiment among three independent assays is shown. Similar results were obtained in three independent experiments. The effect of U0126 on apoptosis and differentiation was also confirmed by Annexin-V assay (E) and flow-cytometric analysis of CD11b expression (F) in NB4-R1 and NB4-R2 cells with the indicated treatments for four days. Each value represented the mean ± SD of three independent measurements. ###P<0.001 versus ENZA+RA. The representative histograms of flow-cytometric analysis of CD11b expression in NB4-R1 (G) and NB4-R2 cells (H) with the indicated drugs for four days are also shown. The percentages of CD11b+ cells are shown in the corresponding panels. The column graph of CD11b and Annexin V double staining in NB4-R1 (I) and NB4-R2 cells (J) with the indicated drugs for four days. Results were representative among three independent experiments. The protein levels of C/EBPβ and PU.1 in NB4-R1 (K) and NB4-R2 (L) cells with the indicated drugs for 48 h was determined by Western-blotting analysis. Expression of β-actin was assessed as internal control. Similar results were obtained in three independent experiments.
RAF-1 is a classical upstream regulator of MEK/ERK pathway. To clarify whether RAF-1 was required for enz-ATRA activation of MEK/ERK pathway, cells were pretreated with 0.5 μM sorafenib tosylate, a specific inhibitor of RAF-1 for 1 h. However, sorafenib tosylate did not attenuate the phosphorylation of MEK in both cell lines (Figure 6A and 6B). Meanwhile, sorafenib tosylate did not inhibit enz-ATRA treatment-induced differentiation, as determined by morphology (Figure 6C and 6D) and CD11b expression (Figure 6E-G). Thus, enz-ATRA combination-induced differentiation and activation of MEK/ERK pathway may be independent of RAF-1.
Figure 6.

RAF-1 is not required for enz-ATRA combination-activated MEK/ERK pathway and differentiation. NB4-R1 and NB4-R2 cells were pretreated with 0.5 μM sorafenib tosylate (SORA) for 1 h. The effect of SORA on MEK activation was measured by phosphorylated MEK with the indicated treatments for 12 h and 36 h respectively in NB4-R1 (A) and NB4-R2 cells (B). The same membrane incubated with the antibody to phosphorylated MEK was stripped and followed by detection of MEK. Expression of β-actin was assessed as internal control. Similar results were obtained in three independent experiments. The effect of SORA on enz-ATRA treatment-induced differentiation for four days was observed by morphologic changes in NB4-R1 (C) and NB4-R2 cells (D). Scale bar represents 5 μm and the magnification is 1,000. One representative experiment among three independent assays is shown. The effect of SORA on enz-ATRA treatment-induced differentiation was also confirmed by flow-cytometric analysis of CD11b expression (E). Each value represents the mean ± SD of three independent measurements. ###P<0.001 versus ENZA+RA. The representative histograms of flow-cytometric analysis of CD11b expression in NB4-R1 (F) and NB4-R2 cells (G) with the indicated drugs for four days are also shown. The percentages of CD11b+ cells are shown in the corresponding panels.
Enz-ATRA treatment-triggered apoptosis is mitochondria-dependent but caspase-independent
Apoptosis can be classified into extrinsic apoptosis which is death receptor-dependent, and intrinsic apoptosis which is mitochondria-dependent [35]. The mitochondrial transmembrane potential (Δψm) was assessed by uptake of rhodamine 123 (Rh123). With double staining of Rh123 and PI, most untreated and ATRA-treated NB4-R1 and NB4-R2 cells were PI negative and Rh123 positive. With enzastaurin treatment for four days, Rh123 negative cells were presented and enz-ATRA treatment remarkably elevated this cell population in both cell lines (Figure 7A and 7B). Thus, enz-ATRA treatment-triggered apoptosis acted through an intrinsic apoptotic pathway. Apoptosis is mainly mediated by caspases, which are subdivided into initiators and executors, the latter of which include caspase-3, -6 and -7. Caspases normally exist as inactive zymogens and are cleaved into large and small subunits when being activated [35]. As shown in Figure 7C and 7D, caspase-3 and -7 remained intact with any treatment for four days in both cell lines, while caspase-6 was not detected. Further studies showed that different concentrations of DEVD (caspase-3/7 inhibitor) or VEID (caspase-6 inhibitor) did not suppress enz-ATRA treatment-promoted apoptosis in either cell lines (Figure 7E and 7F). Therefore, enz-ATRA treatment-triggered apoptosis may be caspase-independent.
Figure 7.

Enz-ATRA treatment-triggered apoptosis is mitochondria-dependent but caspase-independent. NB4-R1 (A) and NB4-R2 (B) cells were treated with 2 μM enzastaurin (EN) and/or 1 μM ATRA (RA) for four days. One representative scatter plots of flow-cytometric analysis of mitochondrial transmembrane potential (assessed by uptake of rhodamine 123 [Rh123]) is shown. The percentages of Rh123- cells are shown in the corresponding panels. Western-blotting analysis of caspase-3, caspase-6 and caspase-7 in NB4-R1 (C) and NB4-R2 (D) cells treated with 2 μM enzastaurin and/or 1 μM ATRA for 24 h. Expression of β-actin was assessed as internal control. Similar results were obtained in three independent experiments. NB4-R1 and NB4-R2 cells were pretreated with different concentrations of (1, 2, 4 μM) DEVD (caspase-3/7 inhibitor) or different concentrations of (1, 5, 10 μM) VEID (caspase-6 inhibitor) for 1 h prior to enz-ATRA treatment for four days. The effect of DEVD or VEID on enz-ATRA treatment-triggered apoptosis in NB4-R1 (E) and NB4-R2 (F) cells was determined by Annexin V analysis. The percentages of Annexin V+ cells are shown in the corresponding panels. Results were representative among three independent experiments.
Discussion
In this study, the combination of enzastaurin and ATRA exerted dual effects, triggering differentiation and apoptosis in a dose-dependent manner in ATRA-resistant APL cell lines. Enzastaurin has been reported to promote apoptosis or inhibit proliferation in a variety of malignances [25]. To our knowledge, this is the first study to demonstrate enzastaurin’s differentiation-enhancing and ATRA-resistance-reversing effects at a clinically achievable concentration. Hence, the efficacy of enz-ATRA combination on the primary cells from APL-relapsed patients merits further investigation.
Although enzastaurin was designed to target PKCβ, the activity of PKCβ was not inhibited either by enzastaurin alone or with enz-ATRA treatment. Moreover, another PKCβ specific inhibitor could not mimic the effect of enzastaurin in its combination with ATRA. Thus, PKCβ might not involve in enz-ATRA combination-induced differentiation and apoptosis. PKCβ-independent effect of enzastaurin has also been reported in other cell lines [36-38]. Enzastaurin inhibits PKCβ at lower concentrations, while it also inhibits other PKC isozymes at higher concentrations reached in clinical trials (1-4 μM) [36]. Thus, we still cannot exclude the involvement of other PKC isozymes in enz-ATRA treatment-induced dual effects.
In this study, further experiments showed that MEK/ERK pathway was activated, and that the protein levels of C/EBPβ and PU.1 were elevated remarkably with enz-ATRA treatment. An MEK specific inhibitor, U0126, suppressed enz-ATRA treatment-induced differentiation and restored protein levels of C/EBPβ and/or PU.1. Although U0126 partially inhibited enz-ATRA combination-triggered apoptosis, it was confirmed that the inhibitory effect of U0126 on apoptosis was the result of its suppression of differentiation. Unexpectedly, a specific inhibitor of RAF-1, sorafenib tosylate, neither attenuated the phosphorylation of MEK nor inhibited enz-ATRA treatment-induced differentiation. Hence, enz-ATRA combination-induced differentiation was controlled by RAF-1-independent MEK/ERK-mediated modulation of the protein levels of C/EBPβ and/or PU.1. Mounting evidence has indicated a critical role of MEK/ERK signaling in myeloid differentiation and its induction of C/EBPβ and PU.1 expression [32-34,39-41]. However, the mechanisms of how MEK/ERK signaling regulates the expression of C/EBPβ and PU.1 remain unknown. Since PKC, phosphatidylinositol-3 kinase (PI3K), protein kinase A (PKA) and MEK kinase (MEKK) are all known upstream kinases to activate MEK, RAF-1-independent MEK/ERK activation is not rare [42-45].
The degradation of PML-RARα fusion protein, the main driver of APL, by two active drugs, ATO and ATRA has been regarded as the key mechanism of successful therapy for APL. In the present study, the addition of enzastaurin was shown to promote the degradation of PML-RARα in both cell lines. The mechanism of enzastaurin accelerating the degradation of PML-RARα by ATRA remains to be elucidated. However, the MEK/ERK pathway might not be involved since ATRA has been demonstrated to degrade PML-RARα by a caspase-3-like proteasome, and the role of MEK/ERK in PML-RARα destruction has been ruled out in ATRA-induced differentiation in APL cells [34,46]. Although other agents in combination with ATRA have been shown to induce differentiation in ATRA-resistant APL cell lines, they have failed to degrade PML-RARα [10-12,14,16]. It is noteworthy that loss of PML-RARα has been demonstrated to be essential for APL clearance [46]. It has been suggested that, similar to ATRA or ATO, the combination of enzastaurin and ATRA might eradicate APL due to abolishing leukemia-initiating activity of PML-RARα. Whether enz-ATRA treatment has the same efficacy as ATRA and/or ATO needs to be further surveyed. In ATRA-treated APL cells, the degradation of PML-RARα could reinitiate myeloid differentiation by forming RA-bound RARα complex [3]. The degradation of PML-RARα by enz-ATRA combination might also trigger differentiation in a similar manner.
Due to the collapse of Δψm by enz-ATRA treatment, the combination of enzastaurin and ATRA-triggered apoptosis was mitochondrial-dependent. However, enz-ATRA treatment did not activate any of known executor casapses and the inhibitors of these caspases could not suppress enz-ATRA treatment-promoted apoptosis. Thus, enz-ATRA treatment-triggered apoptosis was caspase-independent. Consistent with our study, caspase has also not been required for enzastaurin-induced apoptosis in multiple myeloma cell line [37]. Besides executor casapses, nuclear translocation of apoptosis-inducing factor (AIF), apoptosis-inducing factor-homologous mitochondrion-associated inducer of death (AMID) or endonuclease G (endo G) promotes caspase-independent apoptosis [47-49]. Whether these proteins were involved in enz-ATRA treatment-triggered apoptosis remains to be elucidated. Moreover, PML-RARa fusion protein could disrupt PML-NB and interfere with the normal function of PML as a growth suppression and apoptotic activator by forming heterodimers with PML [46]. Since PML-NB was shown to be recovered with enz-ATRA treatment in some cells, the destruction of PML-RARα fusion protein might also contribute to enz-ATRA treatment-triggered apoptosis by restoring the normal function of PML.
Taken together, a clinically achievable concentration of enzastaurin synergizes with ATRA to induce apoptosis and differentiation in ATRA-resistant APL cell lines. Mechanistically, enz-ATRA combination-induced differentiation is controlled by RAF-1-independent MEK/ERK-mediated modulation of the protein levels of C/EBPβ and/or PU.1, while enz-ATRA combination-mediated apoptosis is mitochondrial-dependent but caspase-independent. Moreover, the degradation of PML-RARα by enz-ATRA treatment may be involved in enz-ATRA treatment-induced dual effects and may also be beneficial to APL eradication. These findings may provide a potential therapy for ATRA-resistant APL patients.
Acknowledgements
This work is supported by the Natural Science Foundation of Shanghai (17ZR1417100).
Disclosure of conflict of interest
None.
References
- 1.Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, Shi JY, Zheng PZ, Yan H, Liu YF, Chen Y, Shen Y, Wu W, Tang W, Waxman S, de The H, Wang ZY, Chen SJ, Chen Z. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci. 2004;101:5328–5335. doi: 10.1073/pnas.0400053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Lin Q, Lv F, Liu N, Xu Y, Liu M, Chen Y, Yi Z. LG-362B targets PML-RARα and blocks ATRA resistance of acute promyelocytic leukemia. Leukemia. 2016;30:1465–1474. doi: 10.1038/leu.2016.50. [DOI] [PubMed] [Google Scholar]
- 3.McCulloch D, Brown C, Iland H. Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: current perspectives. Onco Targets Ther. 2017;10:1585–1601. doi: 10.2147/OTT.S100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lou Y, Ma Y, Sun J, Ye X, Pan H, Wang Y, Qian W, Meng H, Mai W, He JS, Tong H, Jin J. Evaluating frequency of PML-RARA mutations and conferring resistance to arsenic trioxide-based therapy in relapsed acute promyelocytic leukemia patients. Ann Hematol. 2015;94:1829–1837. doi: 10.1007/s00277-015-2477-x. [DOI] [PubMed] [Google Scholar]
- 5.Lo-coco F, Cimino G, Breccia M, Noguera I, Diverio D, Finolezzi E, Pogliani EM, Di Bona E, Micalizzi C, Kropp M, Venditti A, Tafuri A, Mandelli F. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood. 2004;104:1995–1999. doi: 10.1182/blood-2004-04-1550. [DOI] [PubMed] [Google Scholar]
- 6.Godwin CD, Gale RP, Walter RB. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia. 2017;31:1855–1868. doi: 10.1038/leu.2017.187. [DOI] [PubMed] [Google Scholar]
- 7.Aribi A, Kantarjian HM, Estey EH, Koller CA, Thomas DA, Kornblau SM, Faderl SH, Laddie NM, Garcia-Manero G, Cortes JE. Combination therapy with arsenic trioxide, all-trans retinoic acid, and gemtuzumab ozogamicin in recurrent acute promyelocytic leukemia. Cancer. 2007;109:1355–1359. doi: 10.1002/cncr.22524. [DOI] [PubMed] [Google Scholar]
- 8.Ravandi F, Estey E, Jones D, Faderl S, O’Brien S, Fiorentino J, Pierce S, Blamble D, Estrov Z, Wierda W, Ferrajoli A, Verstovsek S, Garcia-Manero G, Cortes J, Kantarjian H. Effective treatment of acute promyelocytic leukemia with all-Trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J. Clin. Oncol. 2009;27:504–510. doi: 10.1200/JCO.2008.18.6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abaza Y, Kantarjian H, Garcia-Manero G, Estey E, Borthakur G, Jabbour E, Faderl S, O’Brien S, Wierda W, Pierce S, Brandt M, McCue D, Luthra R, Patel K, Kornblau S, Kadia T, Daver N, DiNardo C, Jain N, Verstovsek S, Ferrajoli A, Andreeff M, Konopleva M, Estrov Z, Foudray M, McCue D, Cortes J, Ravandil F. Long-term outcome of acute promyelocytic leukemia treated with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab. Blood. 2017;129:1275–1283. doi: 10.1182/blood-2016-09-736686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higuchi T, Kizaki M, Omine M. Induction of differentiation of retinoic acid-resistant acute promyelocytic leukemia cells by the combination of all-trans retinoic acid and granulocyte colony-stimulating factor. Leuk Res. 2004;28:525–532. doi: 10.1016/j.leukres.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 11.Witcher M, Hoi YS, Guo Q, Miller WH. Combination of retinoic acid and tumor necrosis factor overcomes the maturation block in a variety of retinoic acid-resistant acute promyelocytic leukemia cells. Blood. 2004;104:3335–3342. doi: 10.1182/blood-2004-01-0023. [DOI] [PubMed] [Google Scholar]
- 12.Gao F, Tang Q, Yang P, Fang Y, Li W, Wu Y. Apoptosis inducing and differentiation enhancement effect of oridonin on the all-trans-retinoic acid-sensitive and -resistant acute promyelocytic leukemia cells. Int J Lab Hematol. 2010;32:114–122. doi: 10.1111/j.1751-553X.2009.01147.x. [DOI] [PubMed] [Google Scholar]
- 13.He P, Liu Y, Zhang M, Wang X, Xi J, Wu D, Li J, Cao Y. Interferon-γ enhances promyelocytic leukemia protein expression in acute promyelocytic cells and cooperates with all-trans-retinoic acid to induce maturation of NB4 and NB4-R1 cells. Exp Ther Med. 2012;3:776–780. doi: 10.3892/etm.2012.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding M, Weng XQ, Sheng Y, Wu J, Liang C, Cai X. Dasatinib synergizes with ATRA to trigger granulocytic differentiation in ATRA resistant acute promyelocytic leukemia cell lines via Lyn inhibition-mediated activation of RAF-1/MEK/ERK. Food Chem Toxicol. 2018;119:464–478. doi: 10.1016/j.fct.2017.10.053. [DOI] [PubMed] [Google Scholar]
- 15.Wu D, Shao K, Sun J, Zhu F, Ye B, Liu T, Shen Y, Huang H, Zhou Y. Matrine cooperates with all-trans retinoic acid on differentiation induction of all-trans retinoic acid-resistant acute promyelocytic leukemia cells (NB4-LR1): possible mechanisms. Planta Med. 2014;80:399–408. doi: 10.1055/s-0034-1368183. [DOI] [PubMed] [Google Scholar]
- 16.Gianni’ M, Kalaç Y, Ponzanelli I, Rambaldi A, Terao M, Garattini E. Tyrosine kinase inhibitor STI571 potentiates the pharmacologic activity of retinoic acid in acute promyelocytic leukemia cells: effects on the degradation of RARα and PML-RARα. Blood. 2001;97:3234–3243. doi: 10.1182/blood.v97.10.3234. [DOI] [PubMed] [Google Scholar]
- 17.Guillemin MC, Raffoux E, Vitoux D, Kogan S, Soilihi H, Lallemand-Breitenbach V, Zhu J, Janin A, Daniel MT, Gourmel B, Degos L, Dombret H, Lanotte M, de Thé H. In vivo activation of cAMP signaling induces growth arrest and differentiation in acute promyelocytic leukemia. J Exp Med. 2002;196:1373–1380. doi: 10.1084/jem.20021129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devalia V, Thomas NS, Roberts PJ, Jones HM, Linch DC. Down-regulation of human protein kinase C alpha is associated with terminal neutrophil differentiation. Blood. 1992;80:68–76. [PubMed] [Google Scholar]
- 19.McNamara S, Nichol JN, Wang H, Miller WH. Targeting PKCδ-mediated topoisomerase IIΒ overexpression subverts the differentiation block in a retinoic acid-resistant APL cell line. Leukemia. 2010;24:729–739. doi: 10.1038/leu.2010.27. [DOI] [PubMed] [Google Scholar]
- 20.Zhao KW, Li X, Zhao Q, Huang Y, Li D, Peng ZG, Shen WZ, Zhao J, Zhou Q, Chen Z, Sims PJ, Wiedmer T, Chen GQ. Protein kinase Cdelta mediates retinoic acid and phorbol myristate acetate-induced phospholipid. Blood. 2004;104:3731. doi: 10.1182/blood-2004-04-1630. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Shao G, Chen S, Wang X, Wang ZY. Studies on the relationship between protein kinase C and differentiation of human promyelocytic leukemia cells induced by retinoic acid. Leuk Res. 1989;13:869–874. doi: 10.1016/0145-2126(89)90039-8. [DOI] [PubMed] [Google Scholar]
- 22.Kambhampati S, Li Y, Verma A, Sassano A, Majchrzak B, Deb DK, Parmar S, Giafis N, Kalvakolanu DV, Rahman A, Uddin S, Minucci S, Tallman MS, Fish EN, Platanias LC. Activation of protein kinase C delta by all-trans-retinoic acid. J Biol Chem. 2003;278:32544–32551. doi: 10.1074/jbc.M301523200. [DOI] [PubMed] [Google Scholar]
- 23.Ochoa WF, Torrecillas A, Fita I, Verdaguer N, Corbalán-García S, Gomez-Fernandez JC. Retinoic acid binds to the C2-domain of protein kinase Cα. Biochemistry. 2003;42:8774–8779. doi: 10.1021/bi034713g. [DOI] [PubMed] [Google Scholar]
- 24.Delmotte H, Lefebvre P. Serine 157, a retinoic acid receptor a residue phosphorylated by protein kinase C. Mol Biol. 1999;274:38225–38231. doi: 10.1074/jbc.274.53.38225. [DOI] [PubMed] [Google Scholar]
- 25.Bourhill T, Narendran A, Johnston RN. Enzastaurin: a lesson in drug development. Crit Rev Oncol Hematol. 2017;112:72–79. doi: 10.1016/j.critrevonc.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Kreisl TN, Kim L, Moore K, Duic P, Kotliarova S, Walling J, Musib L, Thornton D, Albert PS, Fine HA. A phase I trial of enzastaurin in patients with recurrent gliomas. Clin Cancer Res. 2009;15:3617–3623. doi: 10.1158/1078-0432.CCR-08-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keranen LM, Dutil EM, Newton AC. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol. 1995;5:1394–1403. doi: 10.1016/s0960-9822(95)00277-6. [DOI] [PubMed] [Google Scholar]
- 28.Lekstrom-Himes JA. The role of C/EBPε in the terminal stages of granulocyte differentiation. Stem Cells. 2001;19:125–133. doi: 10.1634/stemcells.19-2-125. [DOI] [PubMed] [Google Scholar]
- 29.Duprez E, Wagner K, Koch H, Tenen DG. C/EBPβ: a major PML-RARA-responsive gene in retinoic acid-induced differentiation of APL cells. EMBO J. 2003;22:5806–5816. doi: 10.1093/emboj/cdg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller B, Pabst T, Fos J. ATRA resolves the differentiation block in t(15;17) acute myeloid leukemia by restoring PU.1 expression. Blood. 2006;107:3330–3338. doi: 10.1182/blood-2005-07-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park DJ, Chumakov AM, Vuong PT, Chih DY, Gombart AF, Miller WH, Koeffler HP. CCAAT/enhancer binding protein e is a potential target gene in acute promyelocytic leukemia treatment. J Clin Invest. 1999;103:1399–1408. doi: 10.1172/JCI2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miranda MB, McGuire TF, Johnson DE. Importance of MEK-1/2 signaling in monocytic and granulocytic differentiation of myeloid cell lines. Leukemia. 2002;16:683–692. doi: 10.1038/sj.leu.2402400. [DOI] [PubMed] [Google Scholar]
- 33.Miranda MB, Xu H, Torchia JA, Johnson DE. Cytokine-induced myeloid differentiation is dependent on activation of the MEK/ERK pathway. Leuk Res. 2005;29:1293–1306. doi: 10.1016/j.leukres.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 34.Weng XQ, Sheng Y, Ge DZ, Wu J, Shi L, Cai X. RAF-1/MEK/ERK pathway regulates ATRA-induced differentiation in acute promyelocytic leukemia cells through C/EBPβ, C/EBPε and PU.1. Leuk Res. 2016;45:68–74. doi: 10.1016/j.leukres.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 2016;8:603–619. doi: 10.18632/aging.100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Querfeld C, Rizvi MA, Kuzel TM, Guitart J, Rademaker A, Sabharwal SS, Krett NL, Rosen ST. The selective protein kinase Cβ inhibitor enzastaurin induces apoptosis in cutaneous T-cell lymphoma cell lines through the AKT pathway. J Invest Dermatol. 2006;126:1641–1647. doi: 10.1038/sj.jid.5700322. [DOI] [PubMed] [Google Scholar]
- 37.Rizvi MA, Ghias K, Davies KM, Ma C, Weinberg F, Munshi HG, Krett NL, Rosen ST. Enzastaurin (LY317615), a protein kinase Cbeta inhibitor, inhibits the AKT pathway and induces apoptosis in multiple myeloma cell lines. Mol Cancer Ther. 2006;5:1783–1789. doi: 10.1158/1535-7163.MCT-05-0465. [DOI] [PubMed] [Google Scholar]
- 38.Ruvolo PP, Zhou L, Watt JC, Ruvolo VR, Burks JK, Jiffar T, Kornblau S, Konopleva M, Andreeff M. Targeting PKC-mediated signal transduction pathways using enzastaurin to promote apoptosis in acute myeloid leukemia-derived cell lines and blast cells. J Cell Biochem. 2011;112:1696–1707. doi: 10.1002/jcb.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Harrison JS, Studzinski GP. Isoforms of p38MAPK gamma and delta contribute to differentiation of human AML cells induced by 1,25-dihydroxyvitamin D3. Exp Cell Res. 2011;317:117–130. doi: 10.1016/j.yexcr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu J, Wu DM, Zheng YL, Hu B, Cheng W, Zhang ZF, Li MQ. Troxerutin counteracts domoic acid-induced memory deficits in mice by inhibiting CCAAT/enhancer binding protein -mediated inflammatory response and oxidative stress. J Immunol. 2013;190:3466–3479. doi: 10.4049/jimmunol.1202862. [DOI] [PubMed] [Google Scholar]
- 41.Kusuyama J, Komorizono A, Bandow K, Ohnishi T, Matsuguchi T. CXCL3 positively regulates adipogenic differentiation. J Lipid Res. 2016;57:1806–1820. doi: 10.1194/jlr.M067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin JL, Chen HC, Fang HI, Robinson D, Kung HJ, Shih HM. MST4, a new Ste20-related kinase that mediates cell growth and transformation via modulating ERK pathway. Oncogene. 2001;20:6559–6569. doi: 10.1038/sj.onc.1204818. [DOI] [PubMed] [Google Scholar]
- 43.Cerioni L, Palomba L, Cantoni O. The Raf/MEK inhibitor PD98059 enhances ERK1/2 phosphorylation mediated by peroxynitrite via enforced mitochondrial formation of reactive oxygen species. FEBS Lett. 2003;547:92–96. doi: 10.1016/s0014-5793(03)00675-6. [DOI] [PubMed] [Google Scholar]
- 44.Winston BW, Lange-Carter CA, Gardner AM, Johnson GL, Riches DW. Tumor necrosis factor alpha rapidly activates the mitogen-activated protein kinase (MAPK) cascade in a MAPK kinase kinase-dependent, c-Raf-1-independent fashion in mouse macrophages. Proc Natl Acad Sci U S A. 1995;92:1614–1618. doi: 10.1073/pnas.92.5.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J Neurochem. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 46.Ablain J, Leiva M, Peres L, Fonsart J, Anthony E, de Thé H. Uncoupling RARA transcriptional activation and degradation clarifies the bases for APL response to therapies. J Exp Med. 2013;210:647–653. doi: 10.1084/jem.20122337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cande C. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002;115:4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 48.Kaku Y, Tsuchiya A, Kanno T, Nishizaki T. HUHS1015 induces necroptosis and caspase-independent apoptosis of MKN28 human gastric cancer cells in association with AMID accumulation in the nucleus. Anticancer Agents Med Chem. 2015;15:242–247. doi: 10.2174/1871520614666140922122700. [DOI] [PubMed] [Google Scholar]
- 49.Ren SX, Cheng AS, To KF, Tong JH, Li MS, Shen J, Wong CC, Zhang L, Chan RL, Wang XJ, Ng SS, Chiu LC, Marquez VE, Gallo RL, Chan FK, Yu J, Sung JJ, Wu WK, Cho CH. Host immune defense peptide LL-37 activates caspase-independent apoptosis and suppresses colon cancer. Cancer Res. 2012;72:6512–6523. doi: 10.1158/0008-5472.CAN-12-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]