

Abstract
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors worldwide, characterized by a high degree of malignancy, a poor prognosis, and chemotherapy resistance. The anticancer activities of cantharidin and its derivatives have been widely reported. Sodium demethylcantharidate is a derivative of cantharidin that shows excellent anticancer activity against multiple types of cancer, including HCC. However, its mechanism of action remains unclear. We evaluated the anticancer activities of sodium demethylcantharidate in SMMC-7721 and Bel-7402 HCC cells in the current study and demonstrated that sodium demethylcantharidate effectively inhibited the proliferation of SMMC-7721 and Bel-7402 HCC cells in a dose- and time-dependent manner. Flow cytometry analysis showed that sodium demethylcantharidate could induce apoptosis. Western blotting revealed that endoplasmic reticulum (ER) stress-related proteins (p-IRE1, GRP78/BiP, CHOP, the spliced form of XBP1, and caspase-12) were upregulated in SMMC-7721 and Bel-7402 cells after exposure to sodium demethylcantharidate. Based on those results, we confirmed that sodium demethylcantharidate could induce apoptosis via ER stress. Moreover, a significant attenuation of SMMC-7721 cell tumorigenesis was observed after sodium demethylcantharidate treatment in a nude mouse xenograft model. These findings elucidate one mechanism underlying the anticancer activity of sodium demethylcantharidate against HCC.
Keywords: Sodium demethylcantharidate, hepatocellular carcinoma, ER stress, apoptosis
Introduction
According to GLOBOCAN 2018, liver cancer may be the sixth most common cancer and fourth lethal cause in cancer worldwide, with approximately 841,000 new liver cancer cases and 782,000 deaths annually [1]. The most common type of primary liver cancer is hepatocellular carcinoma (HCC), comprising 75%-85% of all cases [1,2]. Infection by hepatitis B or C viruses, heavy alcohol consumption, obesity, and aflatoxin B1 contribute to the high burden in Asia and Eastern Africa, especially in Mongolia [1,3]. Currently, surgical resection, liver transplantation, chemotherapy, radiotherapy, and targeted therapy are the standard treatments for HCC patients [4]. However, HCC still shows a poor prognosis, with a 5-year survival rate of less than 12% due to relapse and metastasis [5]. Moreover, HCC is characterized by chemotherapy resistance. Therefore, a better understanding of the mechanistic bases of HCC is urgently required to design more effective therapy for HCC [6].
One recent anticancer strategy is to cause apoptosis by promoting endoplasmic reticulum (ER) stress. The ER is primarily responsible for the correct folding and post-translational modification of membrane proteins and secretory proteins [7]. ER stress, a reaction caused by ER dysfunction and disturbance of the internal environment, is characterized by misfolded and unfolded protein aggregation and Ca2+ balance disorder in the ER lumen. To eliminate the abnormal proteins, the unfolded protein response (UPR) is triggered and mediated by inositol-requiring enzyme 1 (IRE1)-dependent splicing of the mRNA encoding X-box binding protein 1 (XBP1), cleavage and translocation of activating transcription factor 6 (ATF6), and phosphorylation of PKR-like ER kinase (PERK) [8]. When ER stress is prolonged or too severe to be resolved, cell apoptosis will be initiated by caspase-12 [9], CHOP/GADD153 [10], IRE1/ASK1/JNK [11], and p53 [12] signal pathways. Subsequently, cytochrome C is released from mitochondria, promoting the formation of the apoptosome and resulting in the cleavage of caspase-9 and caspase-3/6/7 and subsequent induction of cell death [13]. Causing ER stress with drugs such as bortezomib, nelfinavir, and tanespinmycin can promote apoptosis and successfully treat cancers [14-17]. Thus, searching for other ER-stress inducers is a meaningful direction for cancer treatment development.
Cantharis vesicatoria, a traditional Chinese folk drug from blister beetles, has a history of more than 2,000 years and is used to treat furuncles, ulcers, and foot warts [18]. Cantharidin was proven to be the main active compound isolated from Cantharis vesicatoria. Much literature has reported that cantharidin and its derivatives are effective in treating ovarian cancer, lung cancer, colon cancer, and breast cancer, among other cancer types [19-22]. Sodium demethylcantharidate is a derivative of cantharidin that shows excellent anticancer activity against many types of cancer, including HCC. However, the mechanism of action remains unclear [23-25]. In the current study, we illuminated the anticancer mechanism of sodium demethylcantharidate in two HCC cell lines (SMMC-7721 and Bel-7402) and a mouse xenograft tumor model.
Materials and methods
Materials
Sodium demethylcantharidate was purchased from Targetmol (Shanghai, China). Sulforhodamine B (SRB) was purchased from Sigma (Shanghai, China). FITC Annexin V Apoptosis Detection Kit I was purchased from Becton Dickinson Biosciences (Franklin Lakes, NJ, USA). The antibodies used in this study, Bcl-2, Bax, cleaved caspase-3, cleaved caspase-9, GRP78/BiP, CHOP, and HRP-conjugated anti-rabbit and anti-mouse secondary antibodies, were from Cell Signaling Technology (Beverly, MA, USA). Phosphorylated IRE1(p-IRE1), caspase-12, and XBP1s were from abcam (Cambridge, MA, USA). GAPDH was from TDY Bio (Beijing, China).
Cell lines and culture conditions
The hepatocellular carcinoma cell lines SMMC-7721 and Bel-7402 were purchased from Genechem Co., LTD (Shanghai, China). Both cell lines were cultured in RPMI-1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Biological Industries, LTD, Beit Haemek, Israel) and 1% penicillin/streptomycin (Solarbio, Beijing, China) and were maintained at 37°C in a humidified incubator containing 5% CO2.
Cell viability assay
Cells were seeded in 96-well plates at a density of 8×103 cells/well and were cultured for 24 h to allow adherence. Next, the cells were treated with sodium demethylcantharidate at final concentrations of 0, 6.25, 12.5, 25, 50, and 100 μM. After the cells were incubated for 24 or 48 h, SRB colorimetry was performed. The absorbance of each well was measured at 530 nm using a SpectraMax i3X plate reader (Molecular Devices, San Jose, CA, USA). The assays were performed at least three times.
Colony formation assay
Cells were seeded in 6-well plates at a density of 500 cells/well and were cultured for 24 h. Next, various concentrations of sodium demethylcantharidate (0, 9, 18, or 36 µM) were added for 24 h. Fourteen days later, the cells were washed with phosphate-buffered solution (PBS), fixed with methanol for 5 min, and stained with Giemsa solution (Solarbio, Beijing, China). Finally, clones were counted to evaluate cell proliferation.
Fluorescent staining
Cells grown in 96-well plates were treated with sodium demethylcantharidate (0, 9, 18, or 36 µM) for 24 h and fixed with 4% paraformaldehyde for 25 min and then were washed with PBS and stained with Hoechst 33342 (Beyotime, Shanghai, China) for 30 min. Fluorescence images were captured using a fluorescence microscope (Olympus, Tokyo, Japan).
Cell apoptosis analysis
Cells were plated in 6-well plates for 24 h and then were treated with sodium demethylcantharidate (0, 9, 18, or 36 µM) for 24 h. Cells were washed with PBS and harvested with trypsin. The collected cell samples were incubated with Annexin V for 15 min in the dark and then were incubated with PI for 5 min, followed by evaluation for apoptosis using a BD Accuri C6 Plus Flow Cytometer.
Western blot assay
Cells were plated in 6-well plates for 24 h and then were treated with sodium demethylcantharidate (0, 9, 18, or 36 µM) for 24 h. Cells were lysed with RIPA lysis buffer and protein concentration was quantified using the BCA assay. Proteins were resolved (50 µg per lane) by 8%, 10%, or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Beyotime, Shanghai, China), followed by transfer onto polyvinylidene fluoride membranes. After blocking with western blocking buffer (Beyotime, Shanghai, China), the membrane was incubated with specific primary antibodies overnight at 4°C, followed by incubation with the corresponding secondary antibodies for 1.5 h at room temperature. The membranes were incubated with Immobilon Western Chemiluminescent HRP Substrate (Millipore, MA, USA) and the protein bands were visualized using a BIO-RAD ChemiDoc Imaging System (Hercules, CA, USA).
Tumor xenograft assay in vivo
Suspensions of SMMC-7721 cells (1×107) were injected subcutaneously into the back of each 4-week-old male (n=12) BALB/c nude mouse (Beijing Huahengkang Biological Technology, Beijing, China) and were maintained in specific pathogen-free (SPF) conditions. One week later, when the mean volume of the tumors reached approximately 70 mm3, the mice were randomized to either the vehicle group (n=6) or sodium demethylcantharidate treatment group (n=6). Sodium demethylcantharidate was dissolved in normal saline, and intraperitoneal injection was performed every other day at a dose of 4.3 mg/kg. The control group was treated with only normal saline at the same volume. The nude mice were weighed every other day. The tumor volume was measured every other day with a caliper and was calculated as length × width2/2 [26].
Statistical analysis
All assays were conducted as three independent experiments (n=3). The data were expressed as means ± SD. All statistical analyses were conducted using GraphPad Prism 6.01 from Graphpad Software (La Jolla, CA, USA). One-way ANOVA and Student’s t-test were employed to analyze the differences between sets of data. A p value < 0.05 was considered statistically significant.
Results
Suppression of HCC cell proliferation by sodium demethylcantharidate
We examined the effect of sodium demethylcantharidate on the cell viability of SMMC-7721 and Bel-7402 cells for 24 and 48 hours by SRB assay. SRB colorimetry showed that sodium demethylcantharidate treatment significantly decreased the viability of SMMC-7721 and Bel-7402 cells in a dose- and time-dependent manner (Figure 1A and 1B). The colony-forming assay revealed that the number of colonies formed in both SMMC-7721 and Bel-7402 cells was reduced obviously under the influence of sodium demethylcantharidate treatment in a dose-dependent manner (Figure 1C and 1D). With an increasing concentration of sodium demethylcantharidate, more chromatin underwent condensation and polynucleosomal fragmentation, and nuclear shrinking occurred in Hoechst dye-stained slices (Figure 1E). These results confirmed the anti-proliferative activity of sodium demethylcantharidate in the two HCC cell lines.
Figure 1.
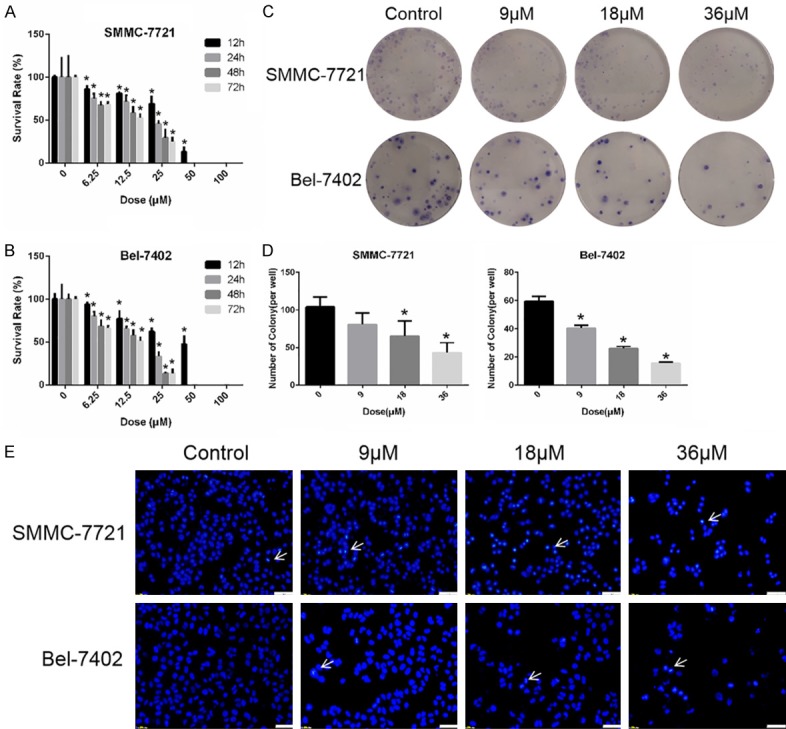
Sodium demethylcantharidate effectively suppressed the proliferation of HCC Cells. (A) SMMC-7721 and (B) Bel-7402 cells were treated with various doses of sodium cantharidinate for 0, 12, 24, 48 or 72 h respectively. Cell viability was determined by SRB colorimetry. (C and D) SMMC-7721 and Bel-7402 cells were treated with sodium demethylcantharidate (0, 9, 18 or 36 µM) for 24 h. The colony formation assay was then performed to detect proliferation. (E) SMMC-7721 and Bel-7402 cells were treated with sodium demethylcantharidate (0, 9, 18 or 36 µM) for 24 h. The apoptosis of SMMC-7721 and Bel-7402 cells was detected by Hoechst staining. Bar: 50 μm. The Hoechst-positive cells (white arrows pointed) were recognized as apoptotic. The experiments were repeated at least three times. The data are expressed as the means ± SD of three experiments (*P<0.05 vs. control).
Apoptosis induction by sodium demethylcantharidate in HCC cells
Flow cytometry was used to evaluate sodium demethylcantharidate-induced cell death. The pro-apoptotic effect of sodium demethylcantharidate was concentrationdependent (Figures 2A, 2B and S1). To further study the mechanism of sodium demethylcantharidate-induced apoptosis, we examined the caspase activity in sodium demethylcantharidate-treated cells. Western blot analysis indicated that the levels of cleaved caspase-3, cleaved caspase-9, and Bax/Bcl-2 were increased by sodium demethylcantharidate dose dependently (Figure 2C). These results suggested that sodium demethylcantharidate treatment could induce apoptosis in HCC cells via the intrinsic pathway.
Figure 2.

Sodium demethylcantharidate induced apoptosis in HCC cells. (A) SMMC-7721 and (B) Bel-7402 cells were treated with sodium demethylcantharidate (0, 9, 18 or 36 µM) for 24 h. Apoptosis was measured via Annexin V/propidiumiodide (PI) staining and quantified by flow cytometry. (C) Apoptosis proteins including Bax, Bcl-2, cleaved caspase-3 and cleaved caspase-9 were analyzed by Western blotting. The experiments were repeated at least three times. The data are expressed as the means ± SD of three experiments (*P<0.05 vs. control).
ER stress induction by sodium demethylcantharidate in HCC cells
ER stress plays an important role in the initiation of agent-induced apoptosis [27,28]. Additionally, the expression of Bcl-2 is inhibited by sodium demethylcantharidate dose dependently as shown above. CHOP, which is highly expressed when ER stress occurs, is involved in regulating apoptosis-related proteins, such as down-regulating the expression of Bcl-2 [29]. Therefore, we hypothesized that the exacerbation of ER stress contributed to HCC cell apoptosis by sodium demethylcantharidate treatment. Next, we examined the expression of ER stress-related proteins. Western blot analysis indicated that p-IRE1, GRP78/BiP, XBP1s, caspase-12, and CHOP levels were higher in SMMC-7721 and Bel-7402 cells exposed to sodium demethylcantharidate (0, 9, 18, or 36 µM) than in control cells (Figure 3). These results indicated that ER stress is an important feature of sodium demethylcantharidate-induced apoptosis in HCC cells.
Figure 3.
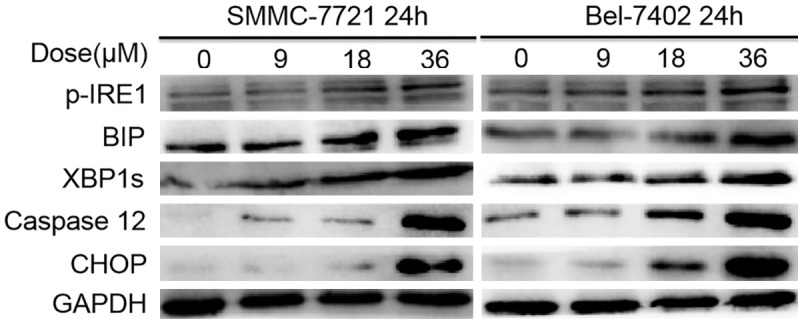
Sodium demethylcantharidate induced ER stress in HHC cells. ER stress markers, including p-IRE1, GRP78/BiP, XBP1s, Caspase 12 and CHOP were analyzed in concentration course manner by Western blotting.
Sodium demethylcantharidate significantly decrease the tumorigenicity of SMMC-7721 cells in vivo
To evaluate the anticancer effect of sodium demethylcantharidate in vivo, nude mice were inoculated with SMMC-7721 cells, and tumor tumorigenesis was monitored. Compared with vehicle controls, the tumor mass and volume were significantly reduced following sodium demethylcantharidate treatment (Figure 4A, 4C, 4D). Western blot analysis indicated that p-IRE1, GRP78/BiP, XBP1s, caspase-12, and CHOP levels were higher in dissected tumors of the xenograft model exposed to sodium demethylcantharidate (Figure 4B). The body weights of the two groups were similar; thus, the differences in the tumor mass and volume were not attributed to a loss of weight (Figure 4E). These data suggested that sodium demethylcantharidate could suppress hepatocellular carcinoma tumorigenesis in vivo.
Figure 4.
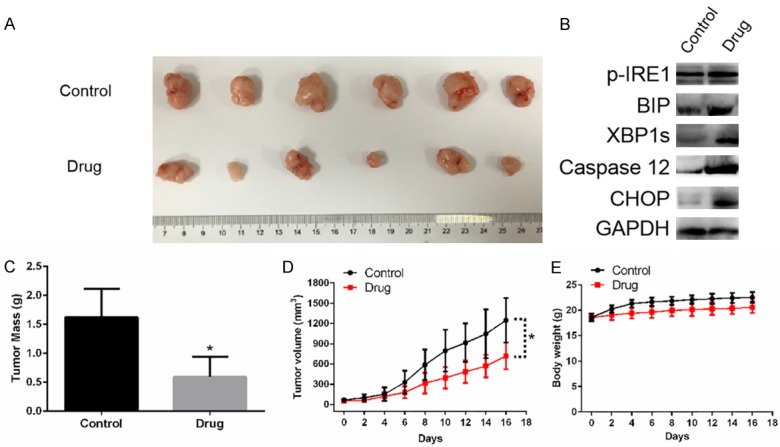
Drug (Sodium demethylcantharidate) significantly decrease the tumorigenicity of SMMC-7721 cells in a nude mouse xenograft model. A. The dissected tumors from each group of mice 16 days after inoculation with SMMC-7721 cells. B. Drug induced ER stress in dissected tumors of the xenograft model. C. Mass of the dissected tumors for the two groups. Each histogram represents mean ± SD of six mice. D. Tumor volumes were measured every two days. E. Body weight of nude mice were measured every two days. There were no obvious change between the two group, indicating the decrease of tumor mass and volume were not caused by a loss of body weight. Data are represented as means ± SD. *P<0.05 vs. control.
Discussion
HCC is one of the most fatal cancers worldwide, and its incidence is increasing. Surgical resection, radiotherapy, and systemic chemotherapy are the main approaches for HCC treatment [1]. However, these available strategies are not effective enough to eliminate its poor prognosis. Thus, it is necessary to identify other curative treatments for HCC.
Sodium demethylcantharidate is a novel derivative of cantharidin and has been reported to have good anticancer activity against multiple cancers [23-25]. In the present study, we found that sodium demethylcantharidate significantly decreased the viability and proliferation of SMMC-7721 and Bel-7402 cells in a dose- and time-dependent manner via induction of apoptosis. Apoptosis is a common mechanism of cancer cell death after chemical treatment. We observed elevation of Bax/Bcl-2 levels and elevated cleavage of caspase-9 and caspase-3. Thus, sodium demethylcantharidate treatment could stimulate the apoptosis of HCC cells via the intrinsic pathway. Compared with the increase in Bax expression, the suppression of Bcl-2 was more obvious, especially in the Bel-7402 cell line (Figure 2C). Activating CHOP, which triggers cell growth arrest and ER stress-related apoptosis, is known to inhibit the expression of Bcl-2 [29]. Thus, we hypothesized that sodium demethylcantharidate induced cell death due to ER stress.
Our observation of increased autophosphorylation of IRE1 (Figures 3 and 4B), a key molecule that functions as a rheostat capable of regulating cell fate [30], suggests this might initiate the apoptotic signal. Consistent with this, BIP acts as a master regulator for UPR [31], and phosphorylated IRE1 cleaves XBP1 mRNA at an intron, allowing the production of XBP1 protein. In addition, IRE1 activates the ASK1/JNK signal that then upregulates and activates CHOP and Bax [26]. As Bax/Bcl-2 is elevated, cytochrome C is released from mitochondria, which forms the apoptosome. Finally, the apoptosome activates and cleaves caspase-9 and then caspase-3, leading to apoptosis.
In summary, we demonstrated that the apoptosis of HCC cells after sodium demethylcantharidate exposure can be caused by ER stress.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81660611, 81760746, 81560669), Science & Technology Foundation for the Excellent Youth Scholars of Guizhou Province (KY[2015]12), Science & Technology Talent Support Project of the Educational Department of Guizhou Province (KY [2018]055), Guizhou Provincial Science & Technology Program (QKH[2019]1346), Guizhou Provincial Science & Technology “125-Plan” Major Project ([2015]039), Science & Technology Plan of Zunyi (2018[03]), Science & Technology Plan Project of Huichuan District in Zunyi (201630), and the start-up Foundation of Zunyi Medical University (F-800, F-906).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace MC, Preen D, Jeffrey GP, Adams LA. The evolving epidemiology of hepatocellular carcinoma: a global perspective. Expert Rev Gastroenterol Hepatol. 2015;9:765–779. doi: 10.1586/17474124.2015.1028363. [DOI] [PubMed] [Google Scholar]
- 4.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 5.Vande Lune P, Abdel Aal AK, Klimkowski S, Zarzour JG, Gunn AJ. Hepatocellular carcinoma: diagnosis, treatment algorithms, and imaging appearance after transarterial chemoembolization. J Clin Transl Hepatol. 2018;6:175–188. doi: 10.14218/JCTH.2017.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun CY, Zhu Y, Li XF, Tang LP, Su ZQ, Wang XQ, Li CY, Yang HM, Zheng GJ, Feng B. Norcantharidin alone or in combination with crizotinib induces autophagic cell death in hepatocellular carcinoma by repressing c-Met-mTOR signaling. Oncotarget. 2017;8:114945–114955. doi: 10.18632/oncotarget.22935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan J, Jiang X, Yin G, He L, Liu J, Long Z, Jiang Z, Yao K. Anacardic acid induces cell apoptosis of prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling pathway. Oncol Rep. 2017;38:1373–1382. doi: 10.3892/or.2017.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Kaufman R. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 13.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 14.Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 2004;24:9695–9704. doi: 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner ER, Danish M, Hollander MC, Kawabata S, Tsokos M, Figg WD, Steeg PS, Dennis PA. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13:5183–5194. doi: 10.1158/1078-0432.CCR-07-0161. [DOI] [PubMed] [Google Scholar]
- 17.Pyrko P, Kardosh A, Wang W, Xiong W, Schonthal AH, Chen TC. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007;67:10920–10928. doi: 10.1158/0008-5472.CAN-07-0796. [DOI] [PubMed] [Google Scholar]
- 18.Puerto Galvis CE, Vargas Mendez LY, Kouznetsov VV. Cantharidin-based small molecules as potential therapeutic agents. Chem Biol Drug Des. 2013;82:477–499. doi: 10.1111/cbdd.12180. [DOI] [PubMed] [Google Scholar]
- 19.Hart ME, Chamberlin AR, Walkom C, Sakoff JA, McCluskey A. Modified norcantharidins; synthesis, protein phosphatases 1 and 2A inhibition, and anticancer activity. Bioorg Med Chem Lett. 2004;14:1969–1973. doi: 10.1016/j.bmcl.2004.01.093. [DOI] [PubMed] [Google Scholar]
- 20.Tarleton M, Gilbert J, Sakoff JA, McCluskey A. Synthesis and anticancer activity of a series of norcantharidin analogues. Eur J Med Chem. 2012;54:573–581. doi: 10.1016/j.ejmech.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Hill TA, Stewart SG, Ackland SP, Gilbert J, Sauer B, Sakoff JA, McCluskey A. Norcantharimides, synthesis and anticancer activity: synthesis of new norcantharidin analogues and their anticancer evaluation. Bioorg Med Chem. 2007;15:6126–6134. doi: 10.1016/j.bmc.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 22.Thaqi A, Scott JL, Gilbert J, Sakoff JA, McCluskey A. Synthesis and biological activity of Delta-5,6-norcantharimides: importance of the 5,6-bridge. Eur J Med Chem. 2010;45:1717–1723. doi: 10.1016/j.ejmech.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Hua J. The research on liver targeting delivery system of sodium norcantharidate liposome modiffied by galactolipid. Journal of Mathematical Medicine. 2009 [Google Scholar]
- 24.You-Suo XU, Meng QS, Feng SU, Zhao TB. Clinical study of sodium norcantharidin combined with transcatheter hepatic arterial chemoembolization as treatment for advanced liver cancer. Medical Recapitulate. 2011 [Google Scholar]
- 25.Ke H, Li X, Wang X. Treatment of primary hepatocellular carcinoma with iodine 125 particle implantation combined with norcantharidin sodium and the changes of serum VEGF and bFGF. Gansu Medical Journal. 2017 [Google Scholar]
- 26.Qiu C, Zhang T, Zhang W, Zhou L, Yu B, Wang W, Yang Z, Liu Z, Zou P, Liang G. Licochalcone a inhibits the proliferation of human lung cancer cell lines A549 and H460 by inducing G2/M cell cycle arrest and ER stress. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18081761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou P, Chen M, Ji J, Chen W, Chen X, Ying S, Zhang J, Zhang Z, Liu Z, Yang S, Liang G. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget. 2015;6:36505–36521. doi: 10.18632/oncotarget.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewy TG, Grabowski JM, Bloom ME. BiP: master regulator of the unfolded protein response and crucial factor in flavivirus biology. Yale J Biol Med. 2017;90:291–300. [PMC free article] [PubMed] [Google Scholar]
- 31.Ron D, Hubbard SR. How IRE1 reacts to ER stress. Cell. 2008;132:24–26. doi: 10.1016/j.cell.2007.12.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.