

Abstract
Background
Glioblastoma (GBM) is a lethal, heterogeneous human brain tumor, with regulatory mechanisms that have yet to be fully characterized. Previous studies have indicated that the transcriptional repressor REST (repressor element-1 silencing transcription factor) regulates the oncogenic potential of GBM stem cells (GSCs) based on level of expression. However, how REST performs its regulatory role is not well understood.
Methods
We examined 2 independent high REST (HR) GSC lines using genome-wide assays, biochemical validations, gene knockdown analysis, and mouse tumor models. We analyzed in-house patient tumors and patient data present in The Cancer Genome Atlas (TCGA).
Results
Genome-wide transcriptome and DNA-binding analyses suggested the dopamine receptor D2 (DRD2) gene, a dominant regulator of neurotransmitter signaling, as a direct target of REST. Biochemical analyses and mouse intracranial tumor models using knockdown of REST and double knockdown of REST and DRD2 validated this target and suggested that DRD2 is a downstream target of REST regulating tumorigenesis, at least in part, through controlling invasion and apoptosis. Further, TCGA GBM data support the presence of the REST-DRD2 axis and reveal that high REST/low DRD2 (HRLD) and low REST/high DRD2 (LRHD) tumors are specific subtypes, are molecularly different from the known GBM subtypes, and represent functional groups with distinctive patterns of enrichment of gene sets and biological pathways. The inverse HRLD/LRHD expression pattern is also seen in in-house GBM tumors.
Conclusions
These findings suggest that REST regulates neurotransmitter signaling pathways through DRD2 in HR-GSCs to impact tumorigenesis. They further suggest that the REST-DRD2 mechanism forms distinct subtypes of GBM.
Keywords: REST, DRD2, glioblastoma subtypes
Key Points.
REST-DRD2 mechanism identifies unique subtypes of GBM.
Potential neurotransmitter signaling in REST-DRD2 subtypes.
Importance of the Study
In the present work, we identify the REST-DRD2 regulatory axis as a mechanism in a class of GSCs. Our results suggest that REST represses DRD2 and that REST-DRD2 forms a mechanistic axis in the process of tumorigenesis. This axis can be used to classify GBM tumors into subtypes that are distinct from the known GBM subtypes. Thus, we describe a mechanism that contributes to the GSC-mediated tumorigenesis potentially via neurotransmitter signaling pathways. Based on our findings, we propose that REST-DRD2 status be considered in potential mechanism-based therapeutic approaches.
Glioblastoma (GBM) is a highly heterogeneous and lethal human brain tumor.1–3 Ongoing research in the GBM field has provided promising approaches to GBM therapy.4,5 GBM was also found to be made up of multiple molecular subtypes, indicating that targeted therapy would be effective.6–9 However, all newly diagnosed GBM patients are treated with a similar therapeutic regimen, resulting in poor patient outcomes. Thus, there is an urgent need for precision medicine approaches that target specific molecular mechanisms.
GBM tumors contain stemlike cells (GSCs) that contribute to tumor initiation, development, growth, and resistance to conventional therapies that leads to relapse.10–13 Accordingly, GSCs present an excellent experimentation-amenable system for the development of targeted therapeutic approaches to GBM.
Repressor element-1 silencing transcription factor (REST) is a transcriptional repressor and a major epigenetic modifier. It was originally found to repress neurogenesis by repressing the terminal neuronal differentiation genes.14,15 A conditional full-length knock-out mouse model confirmed this role.16 REST has been found to regulate many biological processes.17–21 Building on our studies of REST in medulloblastoma,22–24 we and others recently discovered that REST plays a role in GBM and that tumors with GSCs that have high levels of REST expression (HR-GSCs) have different properties than do tumors with GSCs that have low levels (LR-GSCs).25–30 Indeed, findings of The Human Protein Atlas (www.proteinatlas.org) supported such conclusions. However, it is not known how REST performs its oncogenic role, which is a gap in basic knowledge and a barrier to the therapeutic targeting of HR-GSCs.
Our findings described here suggest that a major mechanism of REST-mediated tumorigenesis in HR-GSCs is direct repression of the DRD2 gene, a dominant regulator of neurotransmitter signaling31 that positively regulates some GBM tumors and GSCs32 and has cell- and context-dependent oncogenic or tumor suppressor activity.32,33 This impact on tumorigenicity is attributable, at least in part, to its role in controlling invasion and apoptosis. We analyzed data from The Cancer Genome Atlas (TCGA) GBM database to determine whether DRD2 has both oncogenic and tumor suppressor activity in GBM, depending on the GBM subtype. The results suggested that DRD2 is expressed at varying levels and that tumors can be stratified into distinct high REST/low DRD2 (HRLD) and low REST/high DRD2 (LRHD) subtypes. We validated this classification using orthotopic mouse tumor models derived from HRLD and LRHD GSCs. The results supported the view that the 2 subtypes represent different oncogenic potential. Intriguingly, our analyses of the TCGA GBM data also indicated a significant difference in the median patient age when patients were stratified by this mechanism and that this difference is primarily due to the presence of isocitrate dehydrogenase (IDH) mutant tumors.
Materials and Methods
Details of methods are provided in the Supplementary material. Patient-derived GSCs were isolated with patient consent according to the protocol approved by the institutional review board and cultured as we described previously.25,30 Mouse intracranial tumorigenic analyses were performed following the approved protocol of the Institutional Animal Care and Use Committee as we described previously.25,30 Performed were immunofluorescence assays (using anti-REST antibody: Sigma-Aldrich, HPA006079; anti-DRD2 antibody: Santa Cruz Biotechnology, sc-5303; ki67 antibody: Dako), cell transductions for overexpression or knockdown (using lentiviruses from Sigma-Aldrich or Open Biosystems), RNA preparation using TRIzol reagent (Invitrogen), quantitative reverse transcription polymerase chain reaction (qRT-PCR; using the reverse transcription kit from Life Technologies and performed on an ABI7900 detection system [Applied Biosystems]), western blotting and chromatin immunoprecipitation (ChIP) assays (using anti-REST antibody; Upstate), genome-wide ChIP sequencing (ChIP-seq) analyses (using KAPA Biosystems Library), cell apoptosis assay (terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling [TUNEL]) (Roche), and in vitro invasion assay (BD Biosciences). These procedures were previously described.25,30
Experimental Design and Statistical Analysis
An unpaired 2-tailed Student’s t-test was performed to determine the differences between the control and experimental groups using GraphPad Prism software. A P-value of <0.05 was considered statistically significant. The results of statistical significance tests are described in each Fig.. All bar graph data represent at least 3 independent experiments. All image data represent at least 3 independent areas. Microarray data were analyzed with Agilent GeneSpring GX11.5 and Ingenuity Pathway Analysis software. Mouse survival analyses were determined using the Kaplan–Meier method with a 2-sided log-rank test.
Results
REST Represses DRD2 Expression in GSCs
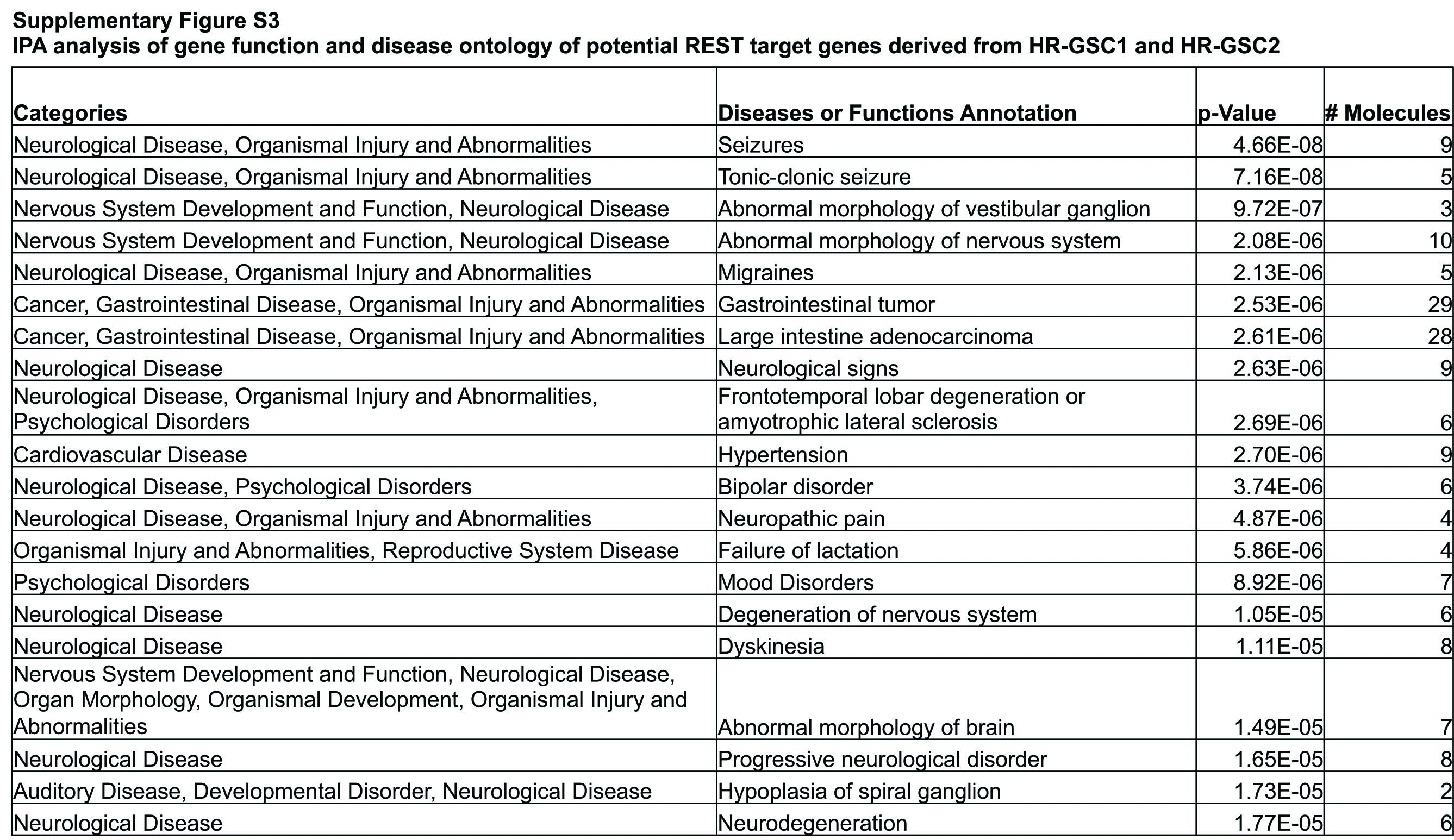
We and others had previously found that GSCs can be classified as HR-GSCs and LR-GSCs based on the level of REST expression25,26 (see Supplementary Fig. 1). To determine the downstream targets of REST in HR-GSCs, we analyzed our previously published transcriptome profiles of the 2 HR-GSC lines with loss-of-REST function and 1 LR-GSC line with gain-of-REST function, plus the corresponding control lines.25,30 For the initial bioinformatic screening of upregulated transcripts at the gene probe level upon short hairpin (sh)REST treatment (compared with short hairpin non-targeting [shNT] controls) in HR-GSC1 and HR-GSC2, we selected genes that were simply upregulated by at least 1.25-fold in either HR-GSC1 cells (2664 genes) or HR-GSC2 cells (3628 genes). We then took the genes that were common in both the cells (703 genes). We further filtered the genes by 2 published REST target lists (by Sun et al34 and Mortazavi et al35). This resulted in 31 potential REST target genes common in both the cell types (Supplementary Fig. 2; genes are sorted by the fold change in the HR-GSC1 and HR-GSC2 cells). We then used Ingenuity Pathway Analyses to analyze these 31 genes to determine gene function and disease ontologies (Supplementary Fig. 3). Neurological disease was identified among the top categories.
The 31-gene target list contains many previously validated REST targets, such as GRIA2,36CHGB,37NTRK3,38GABRB3,39VGF,40 and SNAP25.16,37 Interestingly, only 2 of the genes, NTRK3 and DRD2, were common REST targets in both the Sun and Mortazavi lists (Supplementary Fig. 2). Because Sun et al used a system of neuronal differentiation of embryonic stem cells and Mortazavi et al used Jurkat cells to screen for the potential REST targets, our finding of only 2 targets in our system that are common to both lists supports the previously held idea that REST function is context dependent, and the specific REST target list for bioinformatic uses should be chosen keeping this in mind.
The list also included DRD2, a dominant regulator of neurotransmitter signaling and a recently discovered regulator of GBM and GSCs.32 We were particularly interested in DRD2: A previous genome-wide REST DNA-binding screen had identified DRD2 as a potential REST target34 because of its critical role in neurotransmitter signaling,31,41 and because of its involvement in GBM.32
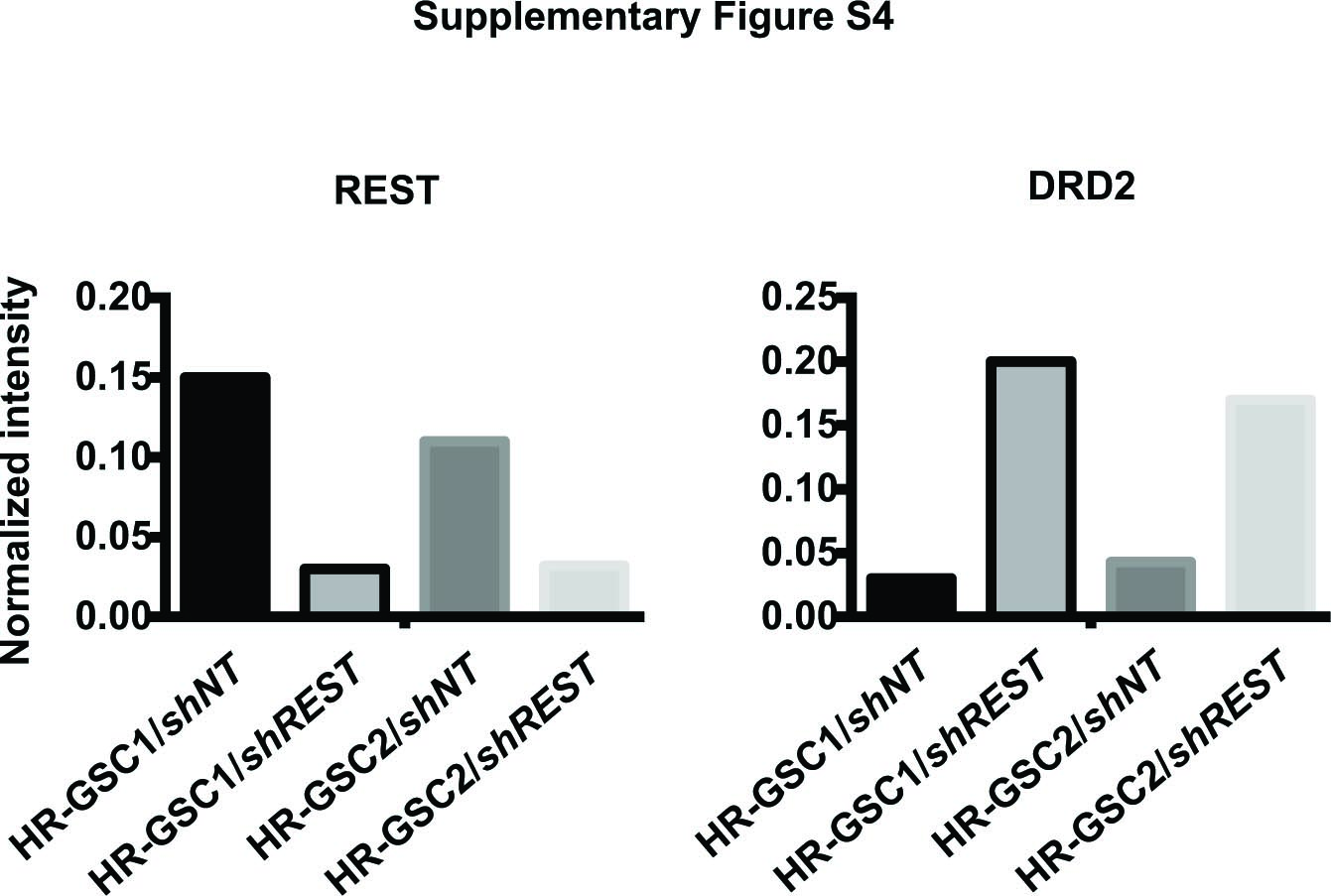
To biochemically validate our bioinformatic results, we assayed for DRD2 expression using qRT-PCR in the 2 HR-GSC lines with loss-of-REST function using shREST. As shown, knockdown of REST increased DRD2 transcript levels by 65-fold in HR-GSC1 cells and by 10-fold in HR-GSC2 cells compared with the shNT controls (Fig. 1A, B). We used western blotting assays to analyze changes in the protein expression levels. As shown, knockdown of REST caused an increase in DRD2 protein levels in both HR-GSC lines (Fig. 1C; quantification of the western blotting data is shown in Supplementary Fig. 4). To confirm the REST loss-of-function results, we performed gain-of-function experiments. We overexpressed REST in the LR-GSC line, which we had studied previously,25 using an exogenous expression vector. As shown, overexpression of REST caused a decrease in DRD2 transcript levels (Fig. 1D), supporting the data obtained with HR-GSC1 and HR-GSC2. However, to obtain a clean, mechanistic approach, we excluded LR-GSCs from this work for further tumorigenic assays, since the tumorigenicities of these cells are not regulated by REST as previously shown.25
Fig. 1.

REST suppresses DRD2 gene expression in HR-GSCs. (A, B) Loss-of-function and rescue experiments. Knockdown of REST using shREST lentiviruses, compared with lentiviruses carrying the shNT control, increased DRD2 transcript levels by 65-fold in HR-GSC1 cells (A) and by 10-fold in HR-GSC2 cells (B). (C) Protein extracts from cells generated in (A) and (B) were subjected to western blot analysis. (D) Overexpression of REST in LR-GSC causes decreased DRD2 transcript levels. (E) REST binds to the DRD2 gene chromatin, as determined by unbiased genome-wide ChIP-seq assays. REST binds to a genomic region spanning 5000–5200 bp upstream of the DRD2 transcription start site (arrowheads). IgG control did not show such binding. (F) Validation of REST binding to the DRD2 gene chromatin by quantitative ChIP. REST loss-of-function HR-GSC1 and HR-GSC2 cells and their corresponding control cells as described in (A) above were processed for ChIP. REST binding to the DRD2 chromatin is shown as fold enrichment. Primer sets corresponding to the control non-binding site 4400–4600 (P1) and another set corresponding to the REST binding site 5000–5200 (P2) on the DRD2 chromatin region are shown; *P < 0.05. (G) REST-DRD2 inverse relationship exists at the protein level in our in-house GBM patient tumors. One HR- and one LR-GBM tumor specimen were analyzed by double immunofluorescence using anti-REST and anti-DRD2 antibodies. Results suggest distinct HRLD and LRHD subtypes.
To determine whether the expression of DRD2 in HR-GSC cell lines was regulated by REST-mediated repression of DRD2 gene chromatin, we performed an unbiased genome-wide ChIP-seq analysis using anti-REST antibody and HR-GSC2 cells. As shown, REST was found to bind to a region spanning 5000 to 5020 bp upstream of the transcription start site (Fig. 1E). In contrast, control immunoglobulin (Ig)G did not reveal any binding in this region. Taken together, these results indicate that REST regulated DRD2 expression in HR-GSCs.
To further validate our REST ChIP-seq results, we performed quantitative ChIP assays using anti-REST antibody in cell extracts obtained from HR-GSC1 and HR-GSC2 cells treated with either shREST or shNT control. We used 2 primer sets: one corresponding to a control non-binding site 4400–4600 bp upstream of the transcription start site (P1) and another corresponding to the DRD2 chromatin region 5000–5200 bp upstream of the transcription start site (P2). As shown, REST bound to the DRD2 gene chromatin at the 5000–5200 site in both cell lines when they were treated with shNT compared with IgG control (Fig. 1F). In addition, the binding was reduced in both cell lines when REST was knocked down with shREST. The negative control site (4400–4600) produced signals similar to those of IgG control.
To validate the existence of the HRLD and LRHD subtypes at the protein level in our GBM patient specimens, we obtained HR and LR specimens from our previous study25 and performed double immunofluorescence analyses using antibodies against REST and DRD2. As shown in Fig. 1G, REST and DRD2 expression levels were inversely correlated.
DRD2 Is a Downstream Target in REST-Mediated GSC Tumorigenesis
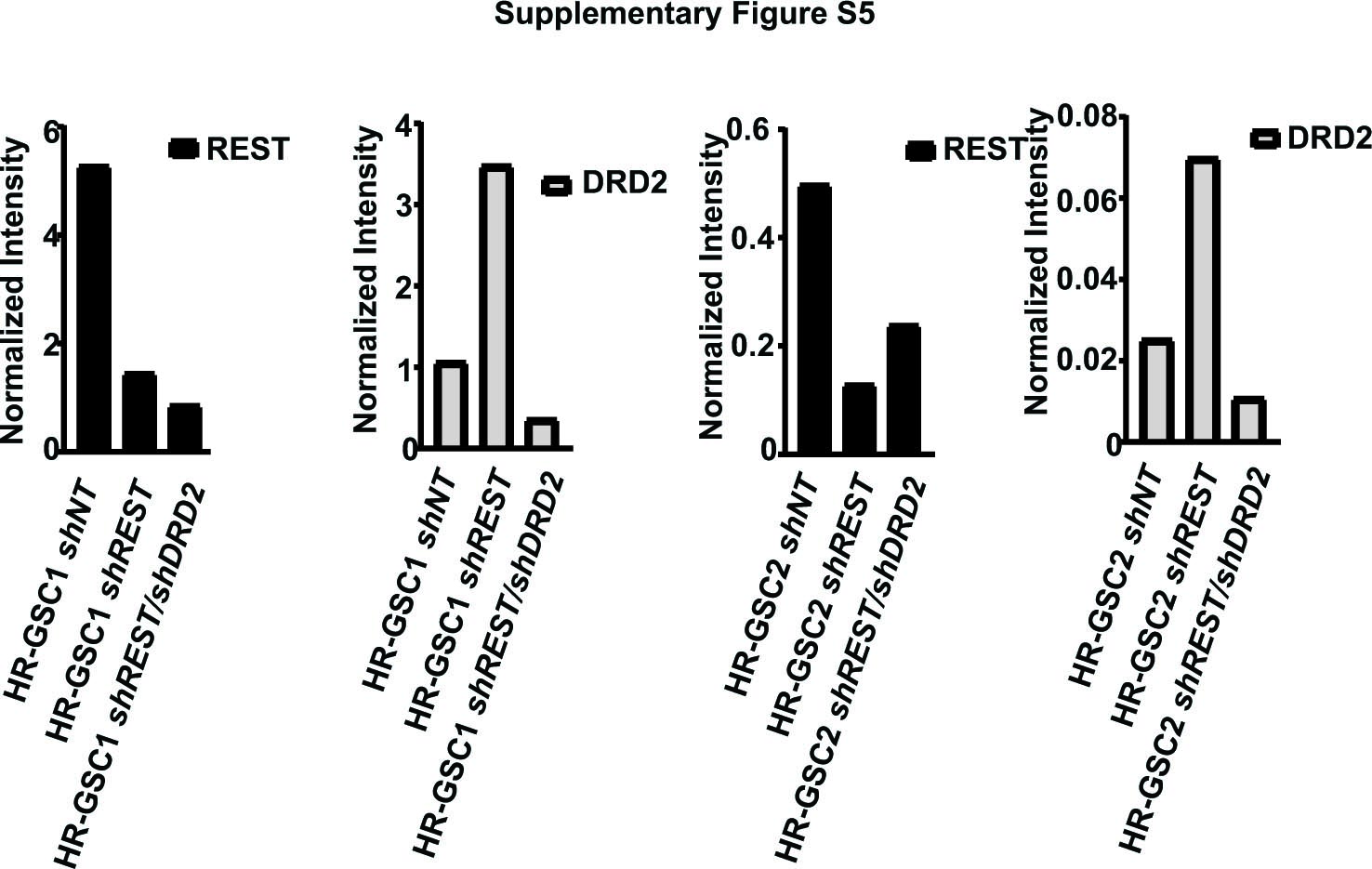
To determine the impact of the REST-DRD2 relationship on tumor formation, we determined whether the REST-DRD2 pathway regulated GSC-mediated tumorigenesis. Because GBM is a highly heterogeneous tumor, resulting in many patient-specific genomic alterations, it is important to examine the causal relationship between REST and DRD2 and tumorigenesis using isogenic GSC cells. We used western blotting to confirm the knockdown of REST in HR-GSC1 cells that had been infected with lentiviruses containing shREST or shNT control (Fig. 2A; Supplementary Fig. 5). We transplanted these cells into the brains of nude mice, as described previously, using a screw-guided system, which aids in determining the location of the injection site and further analysis.25,30 We then performed Kaplan–Meier survival analyses. As shown in Fig. 2B and Supplementary Fig. 4, knockdown of REST using shREST caused a longer survival duration compared with shNT control, similar to what we had observed previously.25,30
Fig. 2.

DRD2 is a downstream target in REST-mediated GSC tumorigenesis. (A, B) Knockdown of REST in HR-GSC1 (A) and HR-GSC2 (B) cells using shREST decreased REST protein levels compared with the shNT control, as expected. Double knockdown of REST and DRD2 (shREST/shDRD2) in these cells showed lowered levels of both REST and DRD2 proteins. (C, D) REST-DRD2 pathway regulates survival of mice. While shREST increased mouse survival compared with shNT control (red arrows), shREST/shDRD2 double knockdown in these isogenic cells reversed the effect of shREST (green arrows) (C: HR-GSC1; D: HR-GSC2). N = number of mice.
To determine whether this increase was due to the increase in DRD2 expression, we further knocked down DRD2 in these cells using shDRD2, resulting in isogenic cells with double knockdown of REST and DRD2 (HR-GSC1/shREST/shDRD2) and confirmed the results by western blot analysis (Fig. 2A). As expected, double knockdown caused decreased DRD2 expression, but the REST levels remained similar to those seen after single shREST knockdown, suggesting that DRD2 is downstream of the REST signaling pathway. We then transplanted the double knockdown cells into the brains of nude mice and measured survival using Kaplan–Meier analysis. As shown in Fig. 2B, shDRD2 strongly reversed the effects of shREST in the isogenic GSC lines and shortened the survival duration of the mice. Similar results were obtained when we used HR-GSC2 cells (Fig. 2C, D).
In a different set of experiments, we transplanted the cells into the brains of nude mice as described above but sacrificed all mice at day 50 posttransplantation. We excised the mouse tumors and analyzed the cut sections (using the bolt site as a reference point) using hematoxylin and eosin staining. As shown, tumors resulting from shNT-treated HR-GSCs had a high degree of invasion of cells into the adjacent hemispheres (Fig. 3A, HR-GSC1). When the same cells were treated with shREST, the resulting tumors were less invasive. These results not only were consistent across all animals tested but also recapitulated the behavior of HR-GSC tumors that we had observed previously.25,30 In contrast, tumors arising from the shREST/shDRD2 double knockdown isogenic GSCs showed a strong rescue of the effect of shREST and increased invasion, with tumor cells migrating to the adjacent hemisphere. Similar results were also observed when we used the HR-GSC2 cells (Fig. 3B). Thus, the REST-DRD2 pathway appears to affect GSC tumor invasion. This effect was also seen when the experimental cells were tested in in vitro invasion assays (Supplementary Fig. 6).
Fig. 3.

REST-DRD2 pathway regulates invasion and apoptosis in REST-mediated GSC tumorigenesis. (A, B) REST-DRD2 pathway regulates invasion of GSC-mediated brain tumors. Mouse brain tumor sections were examined. Tumors resulting from shNT-treated HR-GSC1 (A) and HR-GSC2 (B) cells had a high degree of invasion, with cells invading to the adjacent hemispheres as shown previously.27,31 While shREST decreased tumor invasion compared with shNT control, shREST/shDRD2 double knockdown reversed the effect of shREST. (C, D) REST-DRD2 pathway regulates apoptosis of GSC-mediated brain tumors. Mouse brain tumors were also examined for apoptosis using TUNEL assays. While shREST caused an increase in tumor apoptosis, shREST/shDRD2 double knockdown reversed this effect (C: HR-GSC1; D: HR-GSC2). *P-value is <0.05. (E, F) REST-DRD2 pathway does not cause significant cell proliferation. We examined the mouse tumor sections for cell proliferation using ki67 labeling as described previously.31 Although there was a tendency of increased cell proliferation upon REST knockdown and its reversal by REST and DRD2 double knockdown in these cells, the P-value was not significant (E: HR-GSC1; F: HR-GSC2).
We examined apoptosis and cell proliferation in similar tumor sections using TUNEL assays and ki67 labeling, respectively, and immunofluorescence. As shown, shREST treatment led to increased tumor apoptosis, while shREST/shDRD2 double knockdown reversed the effects of shREST and decreased tumor apoptosis (Fig. 3C, HR-GSC1; Fig. 3D, HR-GSC2). In contrast, cell proliferation was not significantly affected by the REST-DRD2 pathway (Fig. 3E, HR-GSC1; Fig. 3F, HR-GSC2). Thus, taken together, these results suggest that DRD2 is a downstream effector of REST-mediated GSC tumorigenesis.
HRLD and LRHD Subtypes Represent Distinct Functional Groups and Distinct Subtypes, with Enrichment of Specific Gene Sets and Biological Pathways in the Dataset from TCGA
Because DRD2 was found to positively regulate some GBM tumors and GSCs,32 we determined whether DRD2 expression varies in GBM patients and whether HRLD and LRHD also result in specific subtypes. We accessed level 1 TCGA human genome RNA-seq data for GBM through the cgHub portal (cghub.ucsc.edu). After excluding the relapse tumors, the database includes 154 cases with RNA-seq values.
We first performed a gene differential expression analysis using the DESeq2 Bioconductor package. To stratify all patients, we used a median expression cutoff method.42,43 In this approach, half of all patient samples were classified as “high” and the other half were classified as “low” for REST (and, separately, for DRD2) based on the median expression level. We then classified the patients on the basis of combined REST-DRD2 status. Four possible groups emerged: HRLD (39 patients), LRHD (39 patients), high REST/high DRD2 (HRHD, 38 patients), and low REST/low DRD2 (LRLD, 38 patients). Using the REST-DRD2 regulatory axis to classify patients in this manner, we found that about half of all patients were classified as belonging to either the HRLD or LRHD subtypes. The third and fourth subtypes (HRHD and LRLD) are likely impacted by a different mechanism.
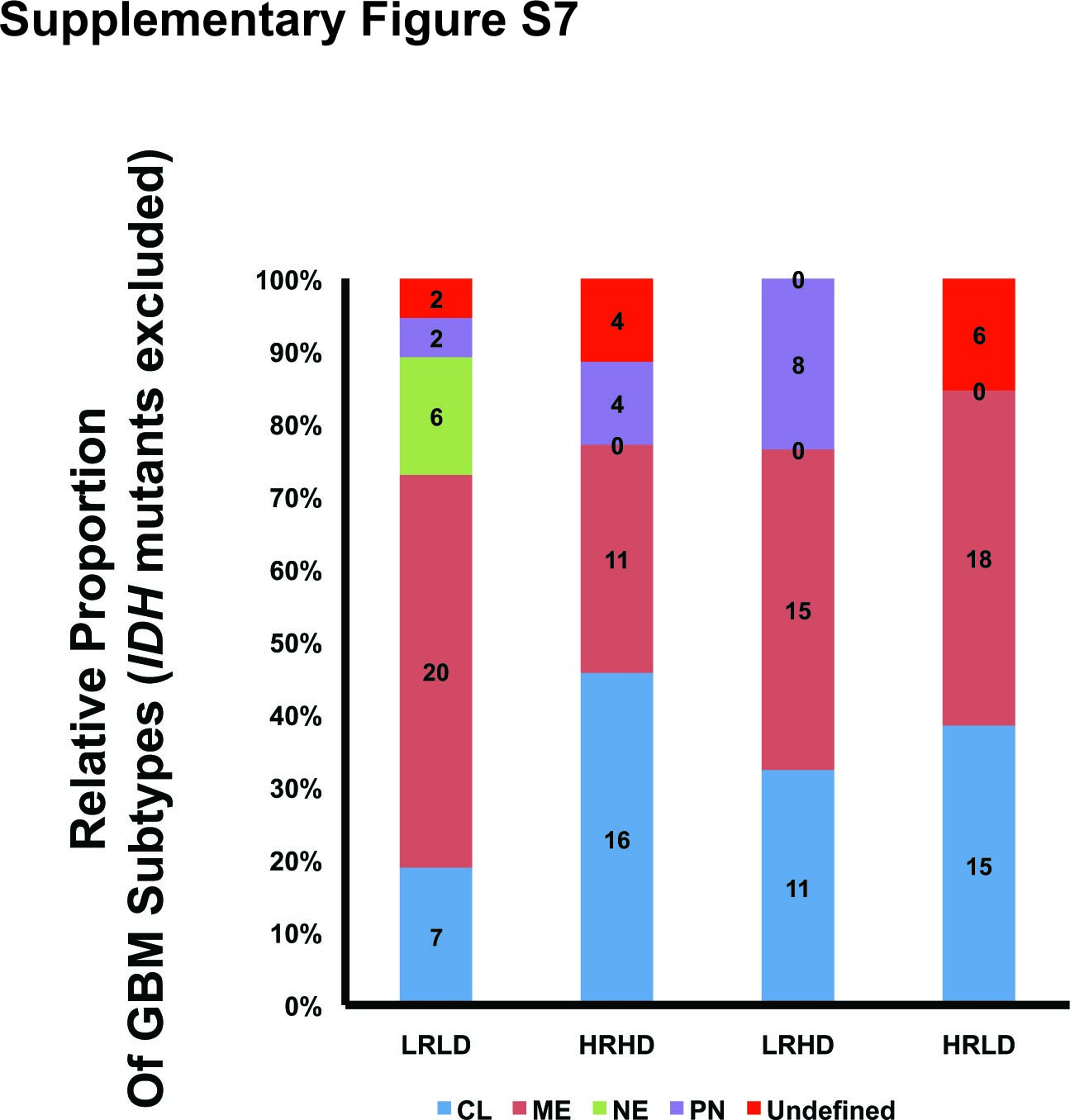
We wanted to determine whether the subtypes identified on the basis of the REST-DRD2 mechanism were distinct from the 4 known GBM subtypes (neural, proneural, mesenchymal, and classical). We performed a direct comparison of known molecular subtype markers to the gene expression patterns present in HRLD, LRHD, HRHD, and LRLD subtypes using the GBM database of TCGA. Of the 154 patients, 16 did not have known subtype markers (n.a. or undefined). The results in Fig. 4A indicate that none of our subtypes corresponded completely to any of the known subtype markers. They were composed of a mixture of the known markers. In particular, HRLD was composed of a mixture of classical and mesenchymal markers, whereas LRHD was composed of a mixture of classical, mesenchymal, and proneural markers. HRHD was composed of classical, mesenchymal, and proneural markers. LRLD was composed of all 4 markers, whereas none of the other subtypes contained neural markers. Interestingly, HRLD did not contain any proneural subtype cases, whereas LRHD did. This might have been anticipated because HRLD contains cases of high REST, which is a repressor of neurogenesis. We further determined the impact of IDH mutants on our classification (number of IDH mutant cases: LRLD = 1, HRHD = 1, LRHD = 3, and HRLD = 0). Even when IDH mutants were removed (Supplementary Fig. 7), the mechanism based on REST-DRD2 classification was distinct from the known GBM subtypes. Taken together, these results suggest that HRLD and LRHD form GBM subtypes that are distinct from any of the known GBM subtypes.
Fig. 4.

HRLD and LRHD correspond to distinct molecular subtypes in TCGA dataset. (A) REST-DRD2 mechanism-based classifications of TCGA GBM tumors into HRLD, LRHD, HRHD, and LRLD subtypes were distinct from the 4 known GBM subtypes (neural [NE], proneural [PN], mesenchymal [ME] and classical [CL]). Tumors with no known subtype markers are also shown (undefined). (B, C) HRLD and LRHD form molecularly distinct subtypes. Clustering analyses of the 5731 DE signature genes in HRLD and LRHD tumors suggest distinct molecular subtypes (B). Clustering analyses of the same DE signature genes using all patients, including HRLD, LRHD, HRHD, and LRLD suggest that whereas HRLD and LRHD subtypes formed distinct, robust clusters, HRHD and LRLD did not (C). (D) GSEA using TCGA expression data for HRLD and LRHD tumors suggests distinct molecular stratification. The gene sets were based on the biological processes from gene ontology.
To determine whether HRLD and LRHD form molecularly distinct subtypes, we utilized the DESeq2 package to perform differential expression (DE) analysis. The analysis revealed 5731 DE signature genes between HRLD and LRHD. We used Pearson distance and complete linkage in the clustering analysis. The results of a chi-square test (P < 0.05) suggested that HRLD and LRHD form 2 molecularly distinct subtypes (Fig. 4B). When we added the 4 “normal” samples available in TCGA in the analyses (Supplementary Fig. 8), the normal samples clearly clustered into a distinct group from HRLD and LRHD samples. This observation further suggests that the DE genes are uniquely represented by HRLD and LRHD samples.
We then determined whether the 5731 DE signature genes were specific in clustering HRLD and LRHD subtypes by considering all patients, including HRLD, LRHD, HRHD, and LRLD. The results in Fig. 4C, obtained with adjusted P-value <0.05, indicate that whereas HRLD and LRHD subtypes form distinct robust clusters, HRHD and LRLD did not, supporting the view that HRLD and LRHD form distinct subtypes. The expression difference between HRLD and LRHD may imply functional divergence between these 2 subtypes.
To determine whether the subtypes correspond to different functional groups, we performed a gene set enrichment analysis (GSEA) using the tool developed by the Broad Institute (www.broadinstitute.org/gsea). GSEA uses a collection of differentially expressed gene sets annotated for the gene ontology biological process from the Molecular Signatures Database. We performed a further clustering analysis using the top 20 gene sets from each subtype (total 300 genes) with R programming packages that visualized the clustering result using a heatmap that classified patients into groups (Fig. 4D). The results of the chi-square test (P < 0.05) suggest that the HRLD and LRHD subtypes represent distinct functional groups with distinctive patterns of enrichment of specific gene sets and biological pathways.
With this refined data, we further computed the top 20 gene sets expressed in HRLD versus LRHD. The top gene sets in LRHD (Fig. 5A) were dominated by neuronal differentiation and development (synaptic signaling, regulation of postsynaptic membrane potential, regulation of neurotransmitter levels, neurotransmitter transport, neuropeptide signaling). This was expected because REST is a repressor of neuronal differentiation genes, and neural cells with low REST, such as the LRHD subtype, would likely show expression of neuronal differentiation genes; the results confirmed the robustness of the subtype classification. In contrast, the top gene sets in HRLD (Fig. 5B) were dominated by cilium assembly and organization (axoneme assembly, microtubule bundle formation, cilium organization, cilium morphogenesis). The corresponding top enrichment plots for LRHD and HRLD are shown in Fig. 5C and D, respectively. Taken together, these data suggest that HRLD and LRHD represent distinct subtypes in the GBM patient database of TCGA.
Fig. 5.

HRLD and LRHD subtypes represent distinct functional groups with distinctive patterns of enrichment of specific gene sets and biological pathways in the dataset of TCGA. (A, B) Top 20 gene sets that are enriched in LRHD (A) and HRLD (B) tumors (false discovery rate, 0.01). (C, D) GSEA enrichment plots showing that the gene sets related to synaptic signaling (enriched in LRHD, C) and axoneme assembly (enriched in HRLD, D).
We then determined the overall survival (OS) and progression-free survival (PFS) of HRLD and LRHD patients using TCGA. As shown in Fig. 6A (OS) and 6B (PFS), there was no significant difference between the 2 subtypes for either OS or PFS. We then compared OS of all patients classified by the REST-DRD2 mechanism. As shown in Supplementary Fig. 9, no survival advantage was seen even when all patients were considered. Interestingly, the 4 established molecular subtypes of GBM also do not show any survival differences.7 Thus, our results are similar to the published survival outcomes of the known GBM subtypes and suggest that although our subtypes represent molecularly and functionally distinct groups, they do not vary in patient survival outcomes with current, non-targeted approaches to treatment.
Fig. 6.

Survival analyses and the median patient age at diagnosis between HRLD and LRHD patients using TCGA. (A, B) There is no significant difference between the HRLD and LRHD subtypes for either overall survival (A) or progression-free survival (B). (C) Differential impact of age on HRLD versus LRHD subtypes in the dataset of TCGA. Analyses of TCGA GBM dataset indicates that there is a significant difference in median patient age between HRLD and LRHD subtypes, but not between HR and LR subtypes. (D) The difference in the median patient age between HRLD and LRHD subtypes appears to be due to the presence of IDH mutants in the LRHD group. There is no significant difference in the median age between the LRHD and HRLD patients when IDH mutants are excluded.
We further analyzed the dataset of TCGA in terms of the median patient age at diagnosis between HRLD and LRHD subtypes. There are 153 patient cases (one patient’s age is not available). When all patients are included, HR contained 77 patients, LR contained 76 patients, HRLD contained 39 patients, and LRHD contained 39 patients. As shown in Fig. 6C, the median patient age significantly differed between the HRLD and LRHD subtypes: 61.6 years for HRLD and 54.6 years for LRHD (P = 0.033). There was no significant difference when patients were stratified by HR and LR alone. Because patients with IDH mutant tumors are generally younger, we considered the IDH mutants in the database. There are 3 IDH mutants in HR, 6 in LR, 0 in HRLD, and 5 in LRHD subtypes. Thus, when the IDH mutants were excluded from the analyses (Fig. 6D), there are a total of 144 cases: 74 for HR, 70 for LR, 39 for HRLD, and 34 for LRHD. Excluding the IDH mutants from the LRHD group, there was no significant difference in the median age between the LRHD and HRLD patients. Therefore, the difference in the median patient age between HRLD and LRHD appears to be due to the presence of IDH mutants in the LRHD group.
Discussion
Our results described here suggest that REST represses the DRD2 gene and that the REST-DRD2 mechanism can be used to stratify GBM into distinct subtypes: HRLD and LRHD. We believe that the work presented here opens a field in which this mechanism can be further studied to determine patient-specific, mechanism-based therapeutic approaches. Interestingly, DRD2 encodes a critical nigrostriatal receptor involved in regulating many physiological processes, including locomotor activity.31,41 Whether there is a mechanistic role of the REST-DRD2 mechanism in controlling movement disorder in GBM patients is unknown. We are currently examining such a possibility.
Although many studies have linked REST overexpression to both medulloblastoma and GBM, REST-specific pharmacological inhibitors are not part of standard therapy. Various approaches to block REST function have been undertaken in experimental models, such as histone deacetylase inhibitors44 and small-molecule inhibitors of REST.45 In an attempt to block REST-mediated repression and activate its target genes, we previously generated a recombinant form of REST, REST-VP16, by replacing the 2-repressor domain of REST with the strong activation domain VP16.46 REST-VP16 converted neural stem cells into differentiated neurons47 and myoblasts into a functional neuronal phenotype.48,49 Delivery of REST-VP16 through viral vectors also blocked REST-mediated medulloblastoma tumor formation in orthotopic mouse models.47 Whether such a gene therapy approach could be used to block HR-GBM tumors using REST-VP16 is still unknown.
Finally, our results further indicate that REST represses the DRD2 gene and suggest that the REST-DRD2 mechanism can be used to stratify GBM into distinct subtypes: HRLD and LRHD. Whereas a previously published study suggested that DRD2 provides oncogenic activity in some GBM,32 DRD2 exhibits tumor suppressor activity in HRLD. Thus, the role of DRD2 in GBM is context dependent. This context-dependent function of molecules in tumorigenesis is not restricted to DRD2. Overexpression of REST has been found to positively regulate the oncogenic properties of many medulloblastoma, GBM, and neuroblastoma models.22–30,44,50 In contrast, deletion of REST and p53 led to GBM proneural subtype–like tumors in adult mice,16 suggesting that the function of REST in GBM tumors is also dependent on context. In the case of REST, while the oncogenic function depends on its role in regulating properties such as maintenance of stemness, cellular invasion, and apoptosis,25,30 its tumor suppressor function depends on its role in maintaining genomic integrity.16 In the p53-null background, an additional lack of REST in p53-null/REST-null mice results in cells with DNA damage that cannot be eliminated through p53-mediated apoptosis, resulting in a tumor phenotype. Thus, the oncogenic and tumor suppressor functions of molecules likely depend on the mechanism involved.
Funding
This work was partially supported by the National Institutes of Health grants (CA97124 and NS81684; S.M.), B*CURED Brain Cancer Research grant (S.M.), MD Anderson Institutional Research Grant (S.M.), and CPRIT grant (RP170005; C.C.).
Conflict of interest statement. The authors declare no conflict of interest.
Authorship statement. LL, BLV, BL, CC, ALM, MMK, DHK, JG, VH, GNF, and SM designed this work and analyzed the data; LL, BLV, BL, CC, ALM, MMK, DHK, KI, YL, JG, VH, AP-H, GR, VB, FFL, and GNF helped or performed experiments and analyses; LL, BLV, BL, CC, ALM, MMK, DHK, and GNF helped in preparation of the manuscript; SM wrote the manuscript.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 2. Wen PY, Reardon DA. Neuro-oncology in 2015: Progress in glioma diagnosis, classification and treatment. Nat Rev Neurol. 2016;12(2):69–70. [DOI] [PubMed] [Google Scholar]
- 3. Gladson CL, Prayson RA, Liu WM. The pathobiology of glioma tumors. Annu Rev Pathol. 2010;5:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grauwet K, Chiocca EA. Glioma and microglia, a double entendre. Nat Immunol. 2016;17(11):1240–1242. [DOI] [PubMed] [Google Scholar]
- 5. Weller M, Roth P, Preusser M, et al. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat Rev Neurol. 2017;13(6):363–374. [DOI] [PubMed] [Google Scholar]
- 6. Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. [DOI] [PubMed] [Google Scholar]
- 7. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Noushmehr H, Weisenberger DJ, Diefes K, et al. ; Cancer Genome Atlas Research Network Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brennan CW, Verhaak RG, McKenna A, et al. ; TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dirks PB. Brain tumor stem cells: the cancer stem cell hypothesis writ large. Mol Oncol. 2010;4(5):420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kahlert UD, Cheng M, Koch K, et al. Alterations in cellular metabolome after pharmacological inhibition of Notch in glioblastoma cells. Int J Cancer. 2016;138(5):1246–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chong JA, Tapia-Ramírez J, Kim S, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80(6):949–957. [DOI] [PubMed] [Google Scholar]
- 15. Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267(5202):1360–1363. [DOI] [PubMed] [Google Scholar]
- 16. Nechiporuk T, McGann J, Mullendorff K, et al. The REST remodeling complex protects genomic integrity during embryonic neurogenesis. Elife. 2016;5:e09584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol. 2005;15(5):500–506. [DOI] [PubMed] [Google Scholar]
- 18. Kagalwala MN, Singh SK, Majumder S. Stemness is only a state of the cell. Cold Spring Harb Symp Quant Biol. 2008;73:227–234. [DOI] [PubMed] [Google Scholar]
- 19. Roopra A, Dingledine R, Hsieh J. Epigenetics and epilepsy. Epilepsia. 2012;53(Suppl 9):2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang JY, Zukin RS. REST, a master transcriptional regulator in neurodegenerative disease. Curr Opin Neurobiol. 2018;48:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baldelli P, Meldolesi J. The transcription repressor REST in adult neurons: physiology, pathology, and diseases. eNeuro. 2015;2(4):0010–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lawinger P, Venugopal R, Guo ZS, et al. The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells. Nat Med. 2000;6(7):826–831. [DOI] [PubMed] [Google Scholar]
- 23. Fuller GN, Su X, Price RE, et al. Many human medulloblastoma tumors overexpress repressor element-1 silencing transcription (REST)/neuron-restrictive silencer factor, which can be functionally countered by REST-VP16. Mol Cancer Ther. 2005;4(3):343–349. [DOI] [PubMed] [Google Scholar]
- 24. Su X, Gopalakrishnan V, Stearns D, et al. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol Cell Biol. 2006;26(5):1666–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kamal MM, Sathyan P, Singh SK, et al. REST regulates oncogenic properties of glioblastoma stem cells. Stem Cells. 2012;30(3):405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Conti L, Crisafulli L, Caldera V, et al. REST controls self-renewal and tumorigenic competence of human glioblastoma cells. PLoS One. 2012;7(6):e38486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang J, Meng Q, Zhao W, et al. An expression based REST signature predicts patient survival and therapeutic response for glioblastoma multiforme. Sci Rep. 2016;6:34556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wagoner MP, Roopra A. A REST derived gene signature stratifies glioblastomas into chemotherapy resistant and responsive disease. BMC Genomics. 2012;13:686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bai Y, Lathia JD, Zhang P, Flavahan W, Rich JN, Mattson MP. Molecular targeting of TRF2 suppresses the growth and tumorigenesis of glioblastoma stem cells. Glia. 2014;62(10):1687–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marisetty AL, Singh SK, Nguyen TN, Coarfa C, Liu B, Majumder S. REST represses miR-124 and miR-203 to regulate distinct oncogenic properties of glioblastoma stem cells. Neuro Oncol. 2017;19(4):514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217. [DOI] [PubMed] [Google Scholar]
- 32. Li J, Zhu S, Kozono D, et al. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget. 2014;5(4):882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang H, Wu K, Ma J, Du Y, Cao C, Nie Y. Dopamine D2 receptor suppresses gastric cancer cell invasion and migration via inhibition of EGFR/AKT/MMP-13 pathway. Int Immunopharmacol. 2016;39:113–120. [DOI] [PubMed] [Google Scholar]
- 34. Sun YM, Greenway DJ, Johnson R, et al. Distinct profiles of REST interactions with its target genes at different stages of neuronal development. Mol Biol Cell. 2005;16(12):5630–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mortazavi A, Leeper Thompson EC, Garcia ST, Myers RM, Wold B. Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire. Genome Res. 2006;16(10):1208–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Noh KM, Hwang JY, Follenzi A, et al. Repressor element-1 silencing transcription factor (REST)–dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A. 2012;109(16):E962–E971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klajn A, Ferrai C, Stucchi L, et al. The rest repression of the neurosecretory phenotype is negatively modulated by BHC80, a protein of the BRAF/HDAC complex. J Neurosci. 2009;29(19):6296–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Epping MT, Lunardi A, Nachmani D, et al. TSPYL2 is an essential component of the REST/NRSF transcriptional complex for TGFβ signaling activation. Cell Death Differ. 2015;22(8):1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tanaka M, Bailey JN, Bai D, Ishikawa-Brush Y, Delgado-Escueta AV, Olsen RW. Effects on promoter activity of common SNPs in 5′ region of GABRB3 exon 1A. Epilepsia. 2012;53(8):1450–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moss AC, Jacobson GM, Walker LE, Blake NW, Marshall E, Coulson JM. SCG3 transcript in peripheral blood is a prognostic biomarker for REST-deficient small cell lung cancer. Clin Cancer Res. 2009;15(1): 274–283. [DOI] [PubMed] [Google Scholar]
- 41. Li L, Marisetty A, Kamal MM, et al. REST overexpresison in mice causes deficits in spontaneous locomotion. Sci Rep. 2018. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bredel M, Scholtens DM, Yadav AK, et al. NFKBIA deletion in glioblastomas. N Engl J Med. 2011;364(7):627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sathyan P, Zinn PO, Marisetty AL, et al. Mir-21-Sox2 axis delineates glioblastoma subtypes with prognostic impact. J Neurosci. 2015;35(45):15097–15112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor P, Fangusaro J, Rajaram V, et al. REST is a novel prognostic factor and therapeutic target for medulloblastoma. Mol Cancer Ther. 2012;11(8):1713–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Conforti P, Zuccato C, Gaudenzi G, et al. Binding of the repressor complex REST-mSIN3b by small molecules restores neuronal gene transcription in Huntington’s disease models. J Neurochem. 2013;127(1):22–35. [DOI] [PubMed] [Google Scholar]
- 46. Immaneni A, Lawinger P, Zhao Z, et al. REST-VP16 activates multiple neuronal differentiation genes in human NT2 cells. Nucleic Acids Res. 2000;28(17):3403–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Su X, Kameoka S, Lentz S, Majumder S. Activation of REST/NRSF target genes in neural stem cells is sufficient to cause neuronal differentiation. Mol Cell Biol. 2004;24(18):8018–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watanabe Y, Kameoka S, Gopalakrishnan V, et al. Conversion of myoblasts to physiologically active neuronal phenotype. Genes Dev. 2004;18(8):889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gopalakrishnan V, Bie B, Sinnappah-Kang ND, et al. Myoblast-derived neuronal cells form glutamatergic neurons in the mouse cerebellum. Stem Cells. 2010;28(10):1839–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singh A, Rokes C, Gireud M, et al. Retinoic acid induces REST degradation and neuronal differentiation by modulating the expression of SCF(β-TRCP) in neuroblastoma cells. Cancer. 2011;117(22):5189–5202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.