

Abstract
Background
Upregulation of staphylococcal nuclease domain-containing protein 1 (SND1) is a common phenomenon in different human malignant tissues. However, little information is available on the underlying mechanisms through which SND1 affects glioma cell proliferation and invasion.
Methods
SND1, Ras homolog family member A (RhoA), and marker of proliferation Ki-67 (MKI67) were analyzed in 187 gliomas by immunostaining. The correlation between those markers and patients’ prognoses was assessed using the Kaplan–Meier estimator. Gene Ontology, chromatin immunoprecipitation, electrophoretic mobility shift assay, and chromosome conformation capture were applied to identify SND1-activated target genes. We also used MTT, colony formation, transwell and orthotopic implantation assays to investigate SND1 function in glioma cell proliferative and invasive activity.
Results
We identified SND1 and RhoA as independent predictors of poor prognosis in glioma patients. SND1 knockdown significantly suppressed the proliferation and invasion of glioma cells. Mechanistically, we discovered that SND1 facilitated malignant glioma phenotypes by epigenetically inducing chromatin topological interaction, which activated downstream RhoA transcription. RhoA sequentially regulated expression of CCND1, CCNE1, CDK4, and CDKN1B and accelerated G1/S phase transition in glioma cell proliferation.
Conclusions
Our findings identify SND1 as a novel chromatin architectural modifier and promising prognostic indicator for glioma classification and treatment.
Keywords: chromatin conformation remodeling, glioma, malignance, prognostic biomarker, SND1
Key Points.
1. SND1 is an independent predictor for golima prognosis.
2. SND1 is a novel chromatin architectural modifier and facilitates proliferation, migration and invasion of glioma cells.
Importance of the Study
Our present results identify SND1 as an independent predictor and a novel chromatin architectural modifier. SND1 can recruit GCN5 to certain DNA loci. By inducing histone acetylation and remodeling the chromatin conformation of the RhoA promoter, SND1 can directly upregulate the transcription of RhoA. The novel SND1/GCN5/RhoA axis further triggers the cyclin/CDK signaling pathway and facilitates proliferation, migration, and invasion of glioma cells. Therefore, our study provides new insights into the diagnostic and prognostic determination as well as therapeutic intervention for gliomas based on SND1 functions.
Glioma has the highest incidence among primary brain tumors.1 The high-grade malignant glioblastoma (GBM) is easy to relapse and is lethal. Due to the diverse intragroup survival, variations exist among individual patients,2,3 current glioma histopathology diagnostic criteria has deficiency on comprehensive evaluation of glioma prognosis.4 Therefore, exploring novel prognostic biomarkers can improve the subclassification and prognostic evaluation for gliomas.
Overexpression of staphylococcal nuclease domain-containing 1 (SND1) has been detected in several human carcinogenesis processes. SND1 facilitates carcinogenesis by enhancing the RNA-induced silencing complex function in breast,5 colon,6 and hepatocellular carcinoma.7 SND1 may regulate the radioresistance of tumor cells.8 SND1 also accelerates breast cancer metastasis via Smurf19 and the SMAD pathway.10 However, the effects of SND1 overexpression on gliomagenesis, progression, and the underlying mechanisms thereof remain poorly understood.
Our results reveal that SND1 facilitates the proliferation and invasion of glioma cells. We identified RhoA, a small GTPase implicating cancer proliferation, apoptosis, angiogenesis, migration, and invasion,11–14 as a potential target of SND1 through human GBM mRNA profiling and bioinformatics analyses. The regulatory mechanism and biologic effects of SND1 and RhoA were also determined. We further investigated the expression patterns and prognostic values of these 2 genes in 187 glioma tissue specimens. Our findings indicate that SND1 and RhoA have the potential to be prognostic markers and therapeutic targets for malignant gliomas.
Materials and Methods
Tissue Collection and Clinical Information
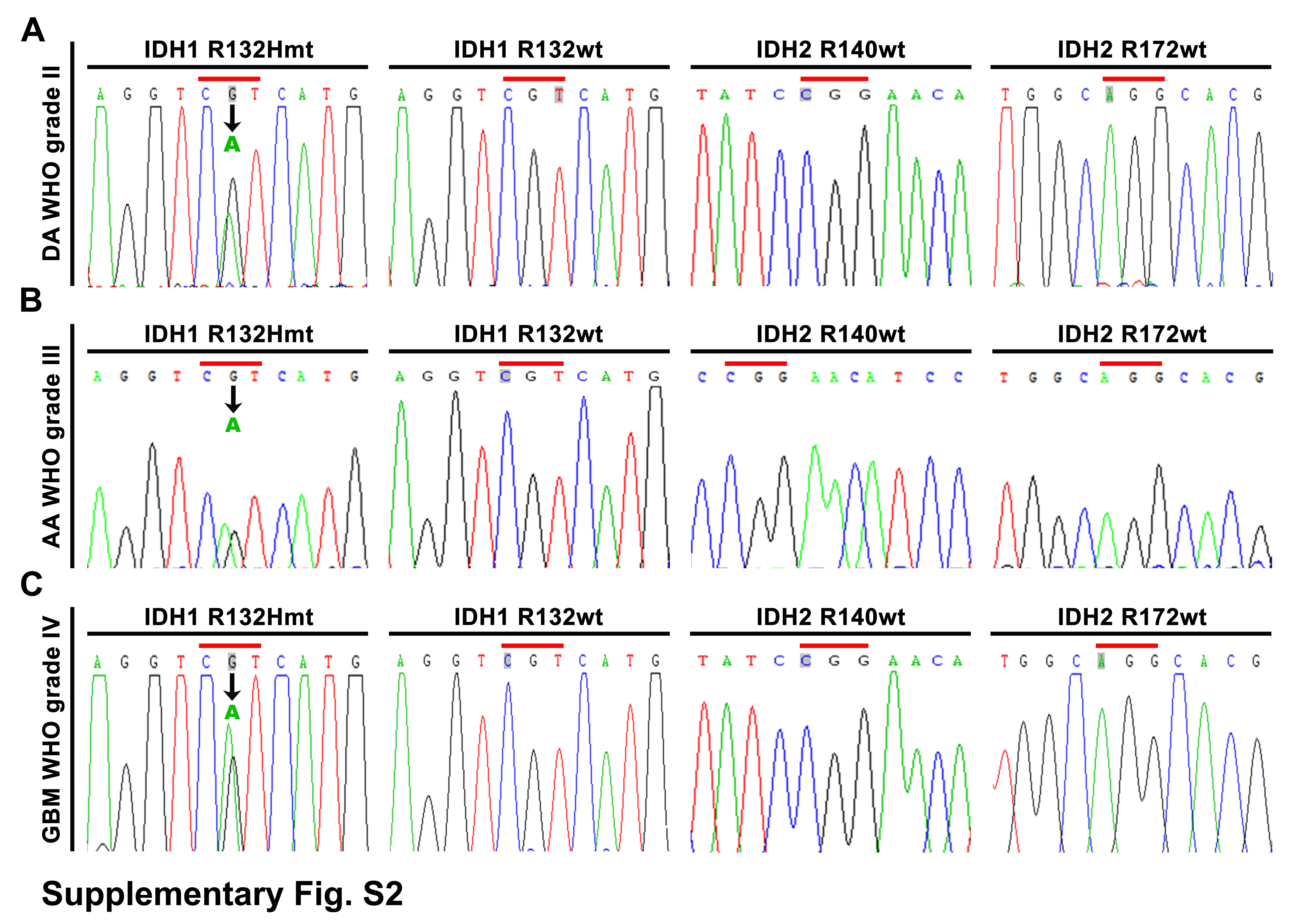
The tissue samples of 187 astrocytic gliomas and 20 control brain tissues from patients without neoplasm were collected at Tianjin Medical University General Hospital (TMUGH) with informed consent. All the data involving patients were handled under the guidance of the Declaration of Helsinki and the principles of the Ethics Committee of TMUGH. The tissues were formalin fixed and paraffin embedded (FFPE). Histopathological diagnoses were independently made by 2 neuropathologists according to the 2016 World Health Organization (WHO) classification of central nervous system tumors.15 The genetic types were ascertained by Sanger sequencing of the gene mutations of isocitrate dehydrogenase 1 and 2 (IDH1/2) and by comparative genomic hybridization (CGH) or fluorescence in situ hybridization detecting 1p/19q status. No 1p/19q codeletion was found in them, thus excluding the possibility of oligodendroglial tumors. The clinical and pathological features of the 187 gliomas are shown in Supplementary Table 1. The clinical features and gene expression information of the 468 glioma samples in the low-grade glioma (LGG) + GBM dataset of The Cancer Genome Atlas (TCGA) (https://cancergenome.nih.gov/) were applied to validate patients’ prognostic values and the correlation of SND1 and RhoA expressions.
Comparative Genomic Hybridization Assay
The SND1 locus gain were detected by CGH; see details in Supplementary Methods.
IDH Mutation Assays
The gene mutations of IDH1/2 were detected using Sanger sequencing, as described in the Supplementary Methods.
Immunohistochemistry
Immunohistochemistry (IHC) staining was applied according to a previously described procedure.16 The antibodies used in IHC are listed in Supplementary Table 12. The proteins were then visualized by a Vectastain ABC (avidin-biotin complex) Detection System (including the biotinylated secondary antibody and ABC complex). The 10 fields (×400) from highly expressing areas (hotspots) were selected under a Leica DM6000B microscope. Image Pro Plus 5.0 software (Media Cybernetics) was used to count the positive cell number and total cell number in the 10 fields and calculate the percentage of positive cells in all cells (labeling index [%], LI).
Isolation and Maintenance of Cells
The patient-derived primary GBM cells were isolated from fresh human GBM tissues as described in the Supplementary Methods. The U118MG cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (Gibco) as described in the Supplementary Methods.
Lentivirus and Stable Subcell Line Construction
Empty vector lentiviruses (vector), lentiviruses expressing RhoA protein (RhoA) with blasticidin resistance, lentiviruses expressing SND1 protein (SND1), SND1 short hairpin (sh)RNAs (SND1-sh1: 5′-GGTACCATCCTTCATCCAAAT-3′; SND1-sh2: 5′-GGACAAGGCCGGCA ACTTTAT-3′) and scrambled shRNA (scramble: 5′-TTCTCCGAACGTGTCACGT-3′) with puromycin resistance were obtained from GenePharma (pGLVU6/Puro vector). The U118MG and primary GBM cells were cultured in 6 cm dishes and treated with 50 μL of the virus. After 48 hours, puromycin (8 μg/mL) or blasticidin (14 μg/mL) was added to the cells to stably select subcell lines (scramble/vector, SND1-sh1/Vector, SND1-sh2/vector, SND1-sh1/SND1, SND1-sh2/SND1, scramble/RhoA, SND1-sh1/RhoA, SND1-sh2/RhoA). The above cells were then transfected by lentiviruses expressing firefly luciferase with neomycin resistance (pCMV-Luci, Clontech) for in vivo imaging. After 48 hours, neomycin (200 μg/mL) was used to select stable cell lines.
Orthotopic Implantation of the GBM Cell Lines
The U118MG and primary GBM cells expressing luciferase were implanted (7.5 × 104 cells) into the brains of 6-week-old non-obese diabetic, severe combined immunodeficiency (NOD-SCID) mice (Charles River Laboratories). Details of experiments are shown in the Supplementary Methods. All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University.
DNA Electrophoretic Mobility Shift Assay
Supplementary Table 2 lists all the RhoA promoter probes used in this study (biotin-labeled, unlabeled, and mutated, Beyotime). Details are shown in the Supplementary Methods.
Complementary DNA Microarray and Data Analysis
The transcription profiles of U118 and primary GBM cell with different treatments were detected by cDNA microarray (n = 3/group). See details in Supplementary Methods.
Chromatin Immunoprecipitation–Chip and ChIP-qPCR Assays
An SND1 antibody (8 μg/mL; Abcam ab65078) and immunoglobulin G antibody (8 μg/mL; Millipore) were used to identify the binding loci of SND1. Supplementary Table 3 lists all the primers for quantitative (q)PCR. The experimental details are outlined in the Supplementary Methods.
Quantitative Reverse Transcription PCR and Western Blot
The qRT-PCR primers are listed in Supplementary Table 3, the antibodies used for western blot are listed in Supplementary Table 12. See details in Supplementary Methods.
Chromosome Conformation Capture
Scramble and SND1 knockdown (SND1-sh1/2) U118MG and primary GBM cells were used for chromosome conformation capture (3C) assay; the experimental details are outlined in the Supplementary Methods.
Flow Cytometry Assay
The U118MG and primary GBM cells were stained with propidium iodide and analyzed by flow cytometers; the experimental details are outlined in the Supplementary Methods.
Colony Formation Assay
The colonies of U118MG cells were counted 10 days after seeding, the details are shown in the Supplementary Methods.
Invasion Assays In Vitro
U118MG and primary GBM cells were seeded in transwell inserts (24-well, 8-μm pore size; Millipore). The invasion cells were counted after 24 hours. The details are listed in the Supplementary Methods.
MTT Proliferation Assay
The U118MG and primary GBM cells were seeded into 96-well plates, then the cells were stained by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) and the absorbance of each well at 490 nm was measured to detect the proliferation condition. See the details in the Supplementary Methods.
Statistical Methods
The data are displayed as the mean ± SD. The cell line–based experiments were repeated in triplicate (n = 3/group), and the in vivo experiments were performed in quintuplicate (n = 5/group). Statistical significance was set as P < 0.05. The details are summarized in the Supplementary Methods.
Results
SND1 Correlates with Glioma Grades and Proliferation and Predicts Poor Patient Outcome
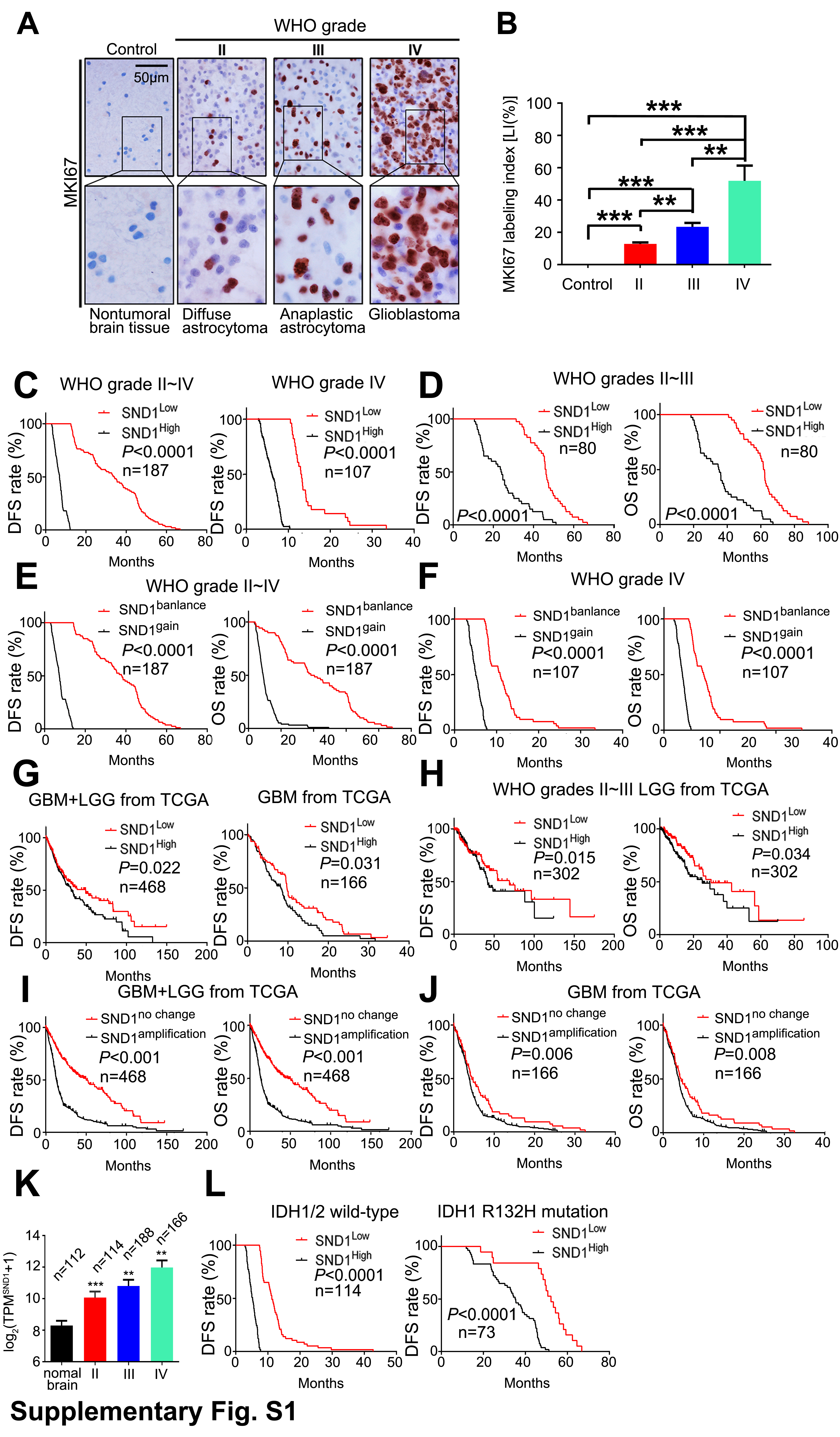
On the SND1 locus, both incidences of the CGH genomic DNA gain from our WHO grades II–IV glioma samples and the gene copy amplification from TCGA GBM+LGG dataset are correlated with glioma grade (Fig. 1A). IHC was applied to detect SND1 and the marker of proliferation Ki-67 (MKI67) in the FFPE specimens of 187 cases of gliomas with different WHO grades (diffuse astrocytoma, grade II, 40 cases; anaplastic astrocytoma, grade III, 40 cases; and GBM, grade IV, 107 cases) and 20 control brain tissue samples. SND1 expression was dramatically increased in high-grade glioma (Fig. 1B–C). The SND1 mRNA levels from the aforementioned TCGA dataset also increase as glioma grade rises (Supplementary Figure 1K). MKI67 expression showed a similar trend (Supplementary Figure 1A–B) and was positively related to SND1 (Fig. 1C right). Then we use Kaplan–Meier (KM) survival analysis to evaluate the relation of SND1 expression and its locus condition to survival of WHO grades II–IV gliomas and grade IV GBM. Patients had shorter survival time when they had higher SND1 expression levels and SND1 locus DNA gain (Fig. 1D, Supplementary Figure 1C, E, and F). The above results were further verified by the diffuse astrocytoma, anaplastic astrocytoma, and GBM cases from the dataset of TCGA (Fig. 1E, Supplementary Figure 1G, I, and J). In our grades II–III glioma samples and TCGA LGG dataset (grades II–III), a higher SND1 level could predict shorter survival time (Supplementary Figure 1D and H). In our 187 samples, 73 cases bore the IDH1 R132H mutation (Supplementary Figure 2). We found that higher SND1 levels were associated with shorter disease-free survival and overall survival (OS) in both IDH1/2 wild-type and mutation groups (Fig. 1F and Supplementary Figure 1L). The survival data were analyzed by univariate and multivariate statistics, and the results showed that amplification and overexpression of the SND1 gene could independently indicate poor prognosis for both the glioma patients and TCGA samples (Supplementary Tables 5–8).
Fig. 1.

SND1 levels correlate with WHO grades and prognosis of glioma. (A) CGH results of WHO grades III–IV gliomas (left and upper right) and SND1 gene amplifications condition from TCGA (lower-right). (B) IHC staining of SND1. Scale bar, 50 μm. (C) Comparison of SND1 protein level (LI [%]) among 187 glioma samples and 20 control brain samples (left), and the linear regression compares the expression of SND1 and MKI67 in 187 gliomas (right). (D) KM analysis of the outcomes of WHO grades II-IV glioma (left) and grade IV GBM (right) patients stratified by SND1 LI (OS). (E) KM analysis of the effect of SND1 mRNA level on GBM+LGG and GBM survival from TCGA (OS; GBM+LGG, n = 468, left; GBM, n = 166, right). (F) Survival analysis of the relation between SND1 protein expression and patients’ outcome for all wild-type IDH1/2 glioma (left, n = 114) and IDH1 R132H mutation-bearing (right, n = 73) glioma patients (OS). Patients were stratified by median SND1 levels. **P < 0.01; ***P < 0.001.
SND1 Promotes the Proliferative and Invasive Ability of GBM Cells
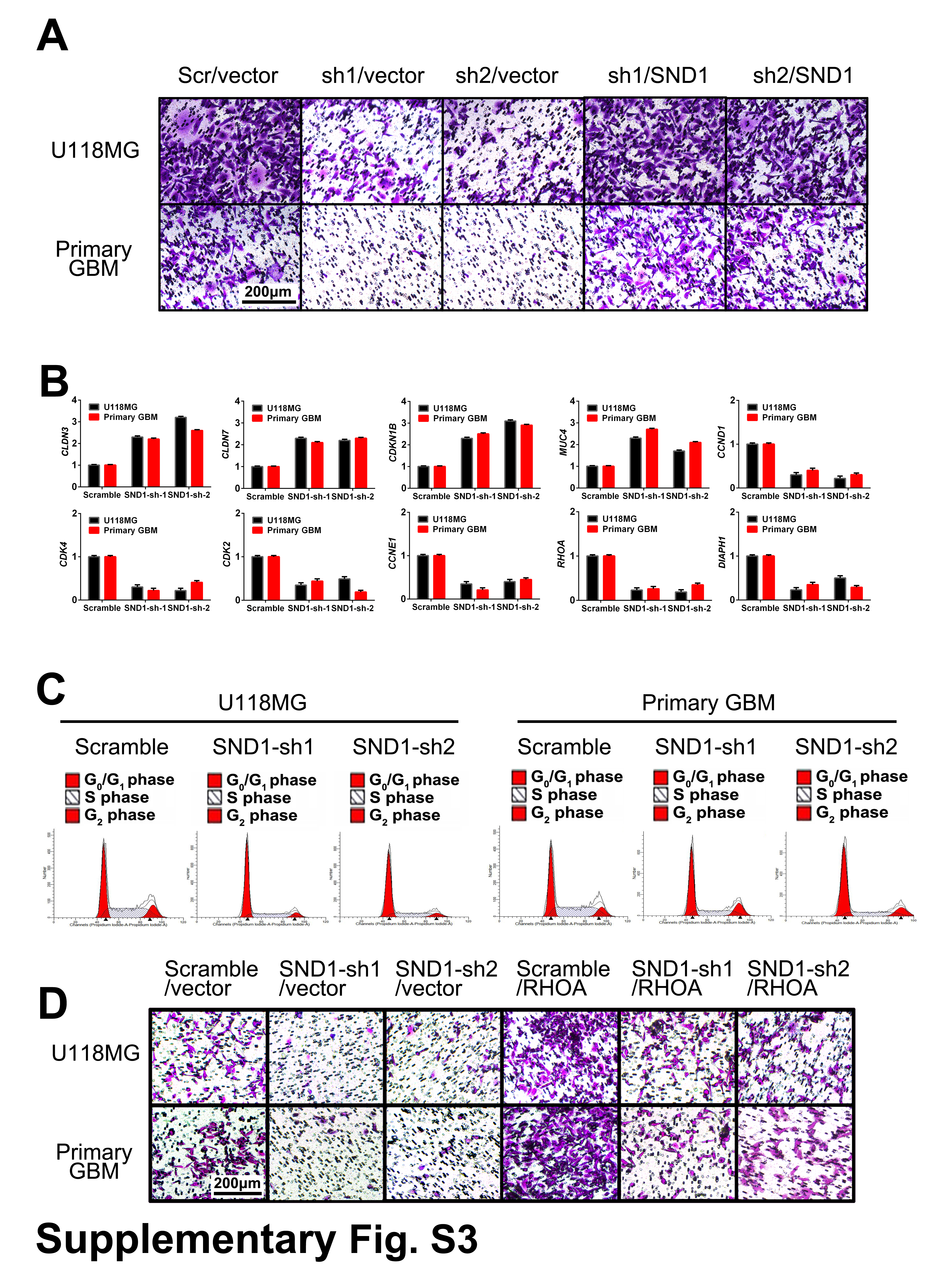
To assess the effects of SND1 on GBM cell proliferation and invasion, we performed transwell, MTT, colony formation, and orthotopic tumor implantation assays. We found that SND1 knockdown (SND1-sh1/vector, sh2/vector) dramatically inhibited the invasion and proliferation of U118MG and primary GBM cells, whereas SND1 rescue expression (SND1-sh1/SND1, sh2/SND1) significantly facilitated invasion and proliferation (Fig. 2A–C, Supplementary Figure 3A). The inhibitory effect of SND1 knockdown on GBM cell proliferation was also verified by the colony formation assays (Fig. 2D). We then assessed the in vivo role of SND1 in GBM cell proliferation. U118MG and patient-derived primary GBM cells expressing firefly luciferase were cotransfected with viruses containing control RNA (scramble) or SND1 shRNAs (SND1-sh1, sh2) and performed in a xenograft model. The above cells were orthotopically transplanted into 6-week-old NOD-SCID mice (n = 5). The bioluminescence images showed that mice injected with SND1 knockdown cells bore smaller tumors (Fig. 2E). The IHC results of the tissue samples of xenograft tumors showed the MKI67 levels were also decreased in the SND1 knockdown groups (Fig. 2F, upper panel, MKI67). Hematoxylin and eosin (HE) staining showed that obvious invasion occurred in the scramble group (the red arrow showed the invasion distance), while no significant invasion was found in the SND1-sh groups (Fig. 2F, middle and bottom panel, HE). KM results showed that mice in the SND1 knockdown group survived longer than the mice in the scramble control group (Fig. 2G). Our results reveal that SND1 overexpression is an important factor in promoting the invasion and proliferation of GBM cells.
Fig. 2.

SND1 facilitates GBM cell invasion and proliferation. (A) Western blotting results of the SND1 level in U118MG and primary GBM cells of control (scramble/vector), SND1-knockdown (SND1-sh1/vector, SND1-sh2/vector), and SND1 rescue expression (SND1-sh1/SND1, SND1-sh2/SND1) groups. (B) The transwell results of aforementioned U118MG and primary GBM cells. (C) MTT proliferation assays of aforementioned cells. (D) Colony formation of U118MG cells expressing control shRNA (scramble) or SND1-shRNAs (SND1-sh1, sh2). *P < 0.05, **P < 0.01. (E) U118MG and primary GBM cells bearing control shRNA (scramble) or shRNAs against SND1 (SND1-sh1, sh2) were inoculated orthotopically into NOD-SCID mice (n = 5). Tumor volumes were measured by the ISVS system after 28 days (U118MG) or 21 days (primary GBM) of initial implantation. (F) IHC staining for MKI67 (upper panel) in a xenograft tumor from mice transplanted with U118MG and primary GBM cells bearing control shRNA (scramble) or shRNAs against SND1 (SND1-sh1, sh2). Representative images of the invasion condition in HE stained tissue from the scramble, SND1-sh1, and SND1-sh2 (middle and bottom panel) xenografts. The red dashed line marks the tumor border, and the red arrow displays the invasion distance. (G) Survival results analyzed by the KM estimator showed that mice bearing SND1-sh1 and sh2 GBM cells survived longer than the control mice (scramble, P < 0.001).
SND1 Activates a Cluster of Genes Related to Cancer Biology in GBM Cells
To further investigate the function of SND1 in GBM cells, we knocked down SND1 in U118MG and patient-derived primary GBM cells; we then used cDNA microarrays to screen the downstream gene transcription profile. Functional annotation showed significantly enriched differentially expressed oncologic genes at low false discovery rates (data were presented as mean of each group, false discovery rate < 0.05, fold change > 2 in either direction, n = 3 for each group). Forty-nine genes that were downregulated and 32 genes that were upregulated upon SND1 knockdown were enriched (Fig. 3A, Supplementary Table 9). Among the 81 genes, cyclin-dependent kinase (CDK) 4, cyclin (CCN) E1, RhoA, CCND1, CDK2, DIAPH1 (diaphanous related formin 1), and SMAD2 were the most downregulated; CDK inhibitor 1B (CDKN1B), claudin (CLDN) 7, CLDN3, and MUC4 (mucin 4, cell surface associated) were the most upregulated. The functional groups identified for the most commonly downregulated genes involved the regulation of cell cycle arrest and cell proliferation and motility, while the most upregulated genes regulated cell adhesion and the DNA damage checkpoint (Fig. 3B). Supplementary Table 10 lists all the individual SND1-targeted genes and their related molecular pathways. The 5 most important pathways involved with SND1-sh are the mitotic DNA damage checkpoint, cell motility, cell adhesion, regulation of cell proliferation, regulation of cell cycle arrest and G1/S transition. The microarray results were confirmed by qRT-PCR in U118 and primary GBM cells (Supplementary Figure 3B). SND1 upregulates multiple target genes via transcriptional activation. We used chromatin immunoprecipitation (ChIP)-chip to detect the SND1-bound loci in U118MG cells and found that SND1 bound to the promoters of 2505 genes. By Pearson correlation analysis of the mRNA expressions in 166 TCGA GBM, we screened 4637 genes whose mRNAs were coexpressed with SND1 (Pearson’s R > 0.3, P < 0.05). After combining them with the 81 genes from the microarray data above, 4 potential direct targets (red) and 19 indirect targets (green) overlapping among the 3 groups were revealed (Fig. 3C, Supplementary Table 11). Furthermore, we used the GeneMANIA package17 of Cytoscape software to analyze and generate the SND1 regulative network. The result showed that SND1 could upregulate RhoA and other indirect target genes in gliomas (Fig. 3D).
Fig. 3.

SND1 knockdown represses a cluster of cell proliferation and motility genes. (A) Functional profiling of differentially expressed genes among U118MG and primary GBM cells with scrambled shRNAs (scramble) or SND1 shRNAs (SND1-sh1 and sh2) transfection (n = 3 for each group). The 2 arrowheads label 32 upregulated genes and 49 downregulated genes after SND1 knockdown (SND1-sh1, sh2). Representative SND1-sh promoted (red) and SND1-sh inhibited genes (green) are shown (left) based on their function (right). (B) The 5 most important pathways enriched among the SND1 candidate regulatory targets are shown in the red box. See also Supplementary Table 10. (C) Venn diagram shows SND1-activated downstream target genes. (D) Network plot illustrating relationships among SND1, RhoA (bold red circle), and downstream pathway members (bold black circle).
SND1 Upregulates RhoA Transcription by Inducing Long-Range Chromatin Conformation Remodeling at the RhoA Promoter in GBM Cells
PCR and western blotting showed that shRNA SND1 knockdown (SND1-sh1, sh2) dramatically reduced the RhoA mRNA and RhoA protein in U118MG and primary GBM cells, whereas they were significantly recovered by SND1 rescue expression (sh1/SND1, sh2/SND1) (Supplementary Figure 4A). Bioinformatics analysis revealed that there were 2 potential SND1-binding sites in the RhoA promoter (Fig. 4A top; R1 and R2). ChIP-qPCR verified that SND1 bound to both SND1-binding sites in the RhoA promoter. After SND1 knockdown (SND1-sh1, sh2), the enrichment at R1 and R2 was significantly decreased, which further confirms that the enrichment of SND1 at the RhoA promoter is specific (Fig. 4A). Electrophoretic mobility shift assay (EMSA) showed that the nuclear extract formed complexes with labeled probes containing SND1-binding sites in the RhoA promoter (Fig. 4B; Lane 2) and that the complex could bind to anti-SND1 antibody and form a supershift band (Fig. 4B; Lane 5).
Fig. 4.

SND1 upregulates RhoA transcription by inducing long-range chromatin conformation remodeling on the RhoA promoter. (A) SND1-binding sites predicted in the RhoA promoter (top figure) and ChIP-qPCR analyses of the capacities for them to bind with SND1. (B) Binding between SND1 and the RhoA promoter verified by EMSA (Lanes 2, 4 and 5). (C and D) 3C results showed the cross-linking efficiency between SND1 binding sites R1 and R2 in both U118MG and primary GBM cells with or without SND1 knockdown (R1 as the anchor, C; R2 as the anchor, D). DpnII digestion sites are labeled by the vertical lines, and the arrows represent the individual PCR primers. **P < 0.01. (E) Schematic displaying the assembling process of the transcription activation complex on the RhoA promoter.
Histone lysine residue acetylation is a milestone event in transcription initiation, which also induces chromatin architectural alternation to allow long-range transcription factor recruitment. Our results showed that SND1 could bind to histone acetyltransferase GCN5.10 To confirm the ability of SND1 and GCN5 to colocalize on the RhoA promoter, we performed ChIP-qPCR targeting histone H3K9-Ac and GCN5 in U118MG and primary GBM cells in which endogenous SND1 was silenced (SND1-sh1, sh2) and with scramble shRNA (scramble). GCN5 bound to the SND1 binding sites (R1 and R2) of the RhoA promoter in scramble cells. Interestingly, GCN5 lost the ability to localize on R1 and R2 in SND1-sh1 and sh2 cells (Supplementary Figure 4B). The histone H3K9-Ac modification condition shared a similar pattern with GCN5 recruitment (Supplementary Figure 4C). Our results confirm that SND1 can upregulate RhoA gene transcription by recruiting GCN5 to the RhoA promoter and inducing histone acetylation.
Immunoprecipitation results showed that SND1 could physically interact with GCN5 in U118MG and primary GBM cells, which further confirms that SND1 is the recognition partner of GCN5 on the RhoA promoter (Supplementary Figure 4D). The luciferase reporter assay showed that the full-length RhoA promoter could be activated by SND1, while deletion of the R1 or R2 conserved motif would repress RhoA promoter activity, suggesting that both R1 and R2 regions are essential for SND1 to regulate RhoA transcription (Supplementary Figure 4E). Chromatin topological alternation requires long-distance interaction between cis-transcriptional elements and the transcription factors located on them. Experiments by 3C were used to detect the spatial propinquity between the conserved motifs R1 and R2. When the R1 region was used as a PCR anchor, the strongest long-range chromatin associations were detected between R1 and R2 in U118MG and primary GBM cells, while the associations were dramatically decreased after SND1 knockdown in both cells (Fig. 4C; SND1-sh1, sh2). When the R2 region was set as the PCR anchor, a similar cross-linking signal between R1 and R2 was detected (Fig. 4D). These data confirmed the hypothesis that SND1, inducing long-range chromatin cross-linking, plays an important role in RhoA transcription activation. SND1 recruits GCN5 to R1 and R2 and induces histone acetylation and chromatin conformation remodeling, which ultimately activates RhoA transcription (Fig. 4E).
SND1 Accelerates the Cell Cycle and Proliferation of GBM Cells via Upregulating RhoA
To identify the underlying mechanisms by which SND1 promotes GBM cell proliferation, we focused on RhoA and G1/S-phase checkpoint regulators (ie, CCND1, CCNE1, CDK4, and CDKN1B [p27Kip1]), to explore their function in the SND1-driven proliferation of GBM cells. The endogenous SND1 knockdown (SND1-sh1, sh2) in U118MG and primary GBM cells could significantly reduce RhoA, CCND1, CCNE1, and CDK4 and increase CDKN1B (Fig. 5A). Concordantly, flow cytometry results indicated that SND1 shRNA transfection significantly suppressed G1/S-phase cell cycle transition in GBM cells (Fig. 5B, Supplementary Figure 3C).
Fig. 5.

SND1 accelerates the GBM cell cycle, proliferation, and invasion in vitro via upregulating RhoA. (A) Western blot results of SND1, RhoA, CCND1, CCNE1, CDK4, and CDKN1B in U118MG and primary GBM cells of the control (scramble) and SND1-knockdown (SND1-sh1, sh2) groups. (B) Cell cycle analyses by flow cytometry of the cells described above. (C) Quantitative RT-PCR detection of SND1 and RhoA mRNA levels in U118MG and primary GBM cells of the scramble/vector, SND1-sh1/vector, SND1-sh2/vector, scramble/RhoA, SND1-sh1/ RhoA, and SND1-sh2/ RhoA groups. (D) Western blot results of SND1, RhoA, CCND1, CCNE1, CDK4, and CDKN1B in the cells described in (C). (E) Results of transwell invasive cell counts of the cells as indicated in (C). (F) MTT proliferation assay of the cells described above. *P < 0.05, **P < 0.01.
We used rescued RhoA expression to further determine whether RhoA mediates SND1-facilitated GBM cell proliferation. Subsequently, U118MG and primary GBM cells were stable-transfected with scrambled shRNA virus/empty vector virus (scramble/vector; control), SND1-shRNA viruses/empty vector virus (SND1-sh1, sh2/vector; endogenous SND1 knockdown), scrambled shRNA virus/RhoA expression virus (scramble/RhoA; exogenous RhoA overexpression), or SND1-shRNA/RhoA co-expression (SND1-sh1, sh2/RhoA; RhoA rescue expression). RhoA mRNA and protein levels were significantly decreased in SND1-sh1, sh2/vector cells but increased in scramble/RhoA cells, while the RhoA mRNA and protein in SND1-sh1, sh2/RhoA groups were kept at basal levels matching the controls (Fig. 5C and D). Moreover, RhoA depletion induced by the SND1 knockdown distinctly decreased CCND1, CCNE1, and CDK4 expression and increased CDKN1B expression in GBM cells (Fig. 5D), thereby inhibiting GBM cell proliferation and invasion (Fig. 5E, F and Supplementary Figure 3D). The RhoA rescue transfection not only effectively blocked the negative or positive regulation of SND1 knockdown on CCND1, CCNE1, CDK4, and CDKN1B (Fig. 5D) but also prevented the inhibition of invasion and proliferation of GBM cells (Fig. 5E, F, Supplementary Figure 3D). The result is consistent with the effect of SND1 knockdown. Our findings uncover a new pathway in which RhoA functions as a signal relay for SND1 to promote GBM cell proliferation and invasion by upregulating CCND1, CCNE1, and CDK4 and downregulating CDKN1B.
SND1 Induces GBM Malignant Phenotype via RhoA In Vivo
To confirm the role of RhoA in SND1 inducing GBM cell proliferation in vivo, we transduced U118MG and primary GBM cells with lentiviruses expressing SND1 shRNAs (SND1-sh1, sh2) or co-transduced the SND1-sh1 or sh2 plus a lentivirus expressing exogenous RhoA (SND1-sh1, sh2/RhoA) in a rescue assay. The results of U118MG and primary GBM cell xenografting and bioluminescence imaging detection verified that the SND1 knockdown significantly decreased transplant tumor sizes, whereas the RhoA rescue expression neutralized the above effect of the SND1 knockdown (Fig. 6A and B). KM results indicated that SND1 knockdown groups (SND1-sh1, sh2/vector) survived significantly longer than the control (scramble/vector) and the RhoA ectopic expression (scramble/RhoA) groups, while the RhoA rescue groups (SND1-sh1, sh2/RhoA) had similar survival time to the scramble/vector groups (Fig. 6C). The tissue samples of xenografted tumors in vivo showed that RhoA upregulation (scramble/RhoA) observably facilitated tumor invasion, whereas the SND1 knockdown (SND1-sh1, sh2/vector) effectively inhibited tumor invasion. Furthermore, the RhoA rescue expression (SND1-sh1, sh2/RhoA) restored the invasion ability of GBM cells (Fig. 6D).
Fig. 6.

SND1 accelerates GBM in vivo growth via RhoA; the RhoA levels indicate high glioma grades and poor prognoses. (A) U118MG and primary GBM cells with empty vector plus scramble control shRNA (scramble/vector) or SND1 shRNA (SND1-sh1/vector, SND1-sh2/vector), or ectopic RhoA plus scrambled shRNA (scramble/RhoA) or SND1 shRNA (SND1-sh1/RhoA, SND1-sh2/RhoA) were inoculated orthotopically into NOD-SCID mice (n = 5). (B) Tumors were measured by the IVIS imaging system. (C) Survival analysis results of the tumor transplant experiment. (D) Representative HE-stained images of the invasion condition among the cells described in (A). Scale bar, 100 μm. (E) IHC results of RhoA expression. Scale bar, 50 μm. (F) Comparing the RhoA LIs among different WHO grades in 187 gliomas and 20 normal brain tissue samples. (G) Linear regression results showed the correlation between SND1 and RhoA in 187 gliomas. (H) Survival analysis results by RhoA LI in WHO grades II–IV glioma samples (left) and grade IV GBM samples (right).
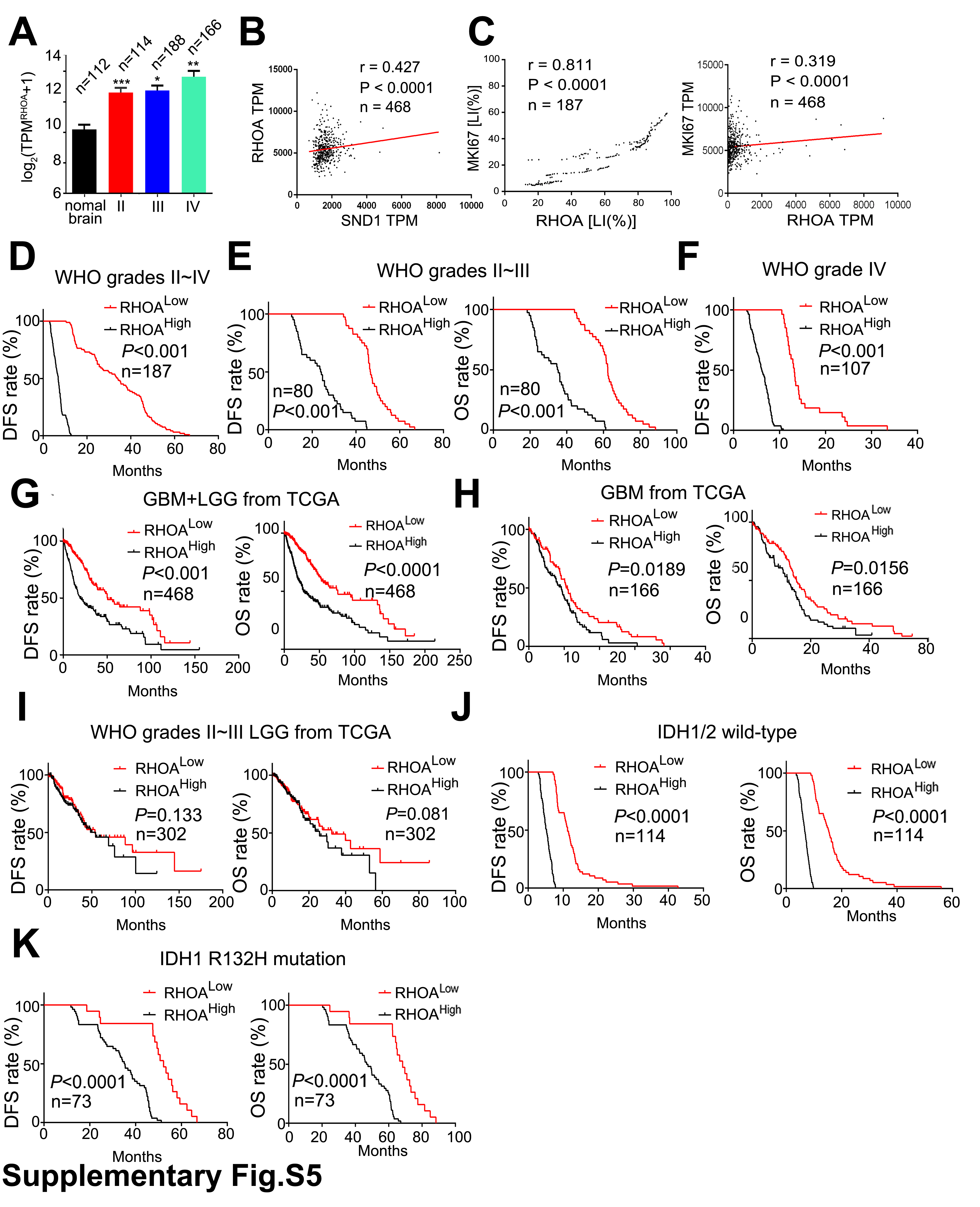
We performed IHC staining in our FFPE samples to detect RhoA level. Compared with nontumoral brain tissues, RhoA expression was elevated in gliomas and was positively correlated with the WHO grade of the glioma, being highest in grade IV GBM (Fig. 6E and F). The above result was also verified by the GBM+LGG profiles from TCGA. The RhoA level correlated with glioma grade (Supplementary Figure 5A). Moreover, SND1 expression was positively correlated with RhoA expression (Fig. 6G), which was further verified by the mRNA data in 468 GBM+LGG from TCGA (Supplementary Figure 5B). Furthermore, the RhoA mRNA and protein were also positively correlated with those of MKI67 in our samples and the GBMs from TCGA (Supplementary Figure 5C). Higher expression of RhoA predicted shorter survival time in our glioma and GBM patients (Fig. 6H and Supplementary Figure 5D–F). RhoA level also predicted shorter survival time in TCGA GBM and GBM+LGG datasets; however, RhoA has no statically significant effect on the survival times of TCGA grades II‒III LGG (Supplementary Figure 5G–I). A high level of RhoA predicted shorter survival time in both the IDH1/2 wild-type and IDH1 R132H mutation groups (Supplementary Figure 5J–K). Similar to SND1, RhoA has the potential to be an independent glioma prognosis indicator, as suggested by multivariate and univariate analyses in our samples and TCGA GBM+LGG dataset (Supplementary Tables 5–8). The results further confirm the upregulating role of SND1 on RhoA and its effects on the biological behavior of malignant gliomas.
Discussion
It is well documented that SND1 is overexpressed in different malignant tissues.7,18,19 In this study, we confirmed that anaplastic astrocytoma and GBM had gain and amplification on SND1 locus, which may upregulate SND1 expression. Previous results showed the suppression of upstream miRNAs could also upregulate SND1.20,21 Then we identified SND1 as a glioma promoter facilitating cell proliferation and invasion, as well as a potential specific biomarker for prognosis-based glioma subclassification. We further verified that 4 SND1-activated genes were closely related to cancer biology, and RhoA is the most relevant to SND1 among those 4 genes. In this study, our results confirmed that RhoA is the direct downstream target gene of SND1. As a transcription coactivator, SND1 binds to the conserved sites of the RhoA promoter and induces RhoA transcription.
Our study showed that SND1 may induce RhoA transcription by recruiting GCN5 and inducing H3K9 acetylation on the RhoA promoter.10 GCN5 is a highly evolutionarily conserved lysine acetyltransferase (KAT) that can catalyze histone acetylation on most gene promoters.22,23 The regulation of the GCN5 catalytic activity is controlled by different partner subunits.24,25 Our results verified that SND1 could induce long-range interactions between 2 SND1 binding sites (R1 and R2) in the RhoA promoter, which linked the histone H3K9 acetylation with special chromatin conformation remodeling. Our results indicate a novel function of SND1 as a chromatin architecture regulator that regulates gene expression by promoting transcriptional cis-acting element interaction.
It is known that RhoA promotes glioma cell invasion by inducing actin rearrangement and enhancing amoeboid migration.26,27 However, the upstream and downstream mechanisms underlying RhoA-stimulated glioma cell proliferation remain to be further elucidated. The complexes of CCND1/CDK4 and CCNE1/CDK2 are key drivers of cell G1/S phase transition and proliferation,28–31 whereas CDKN1B is an inhibitor of the 2 complexes.32 In this study, we found that SND1 could increase expression of CCND1, CCNE1, and CDK4 and decrease expression of CDKN1B by directly upregulating RhoA, thereby promoting glioma cell proliferation by accelerating the transition from the G1 phase of the cell cycle to the S phase.
Overexpression of RhoA has been found in many malignant tumors.33 Our results suggest that RhoA is a potential biomarker distinguishing glioma grades. Furthermore, the combined results of the KM and Cox analyses verified that RhoA was also an independent survival predictor for glioma patients. Since SND1 had multiple other target genes and regulatory approaches, the prognostic value of RhoA was less than that of SND1 in gliomas. Furthermore, SND1 and RhoA stably were expressed in FFPE tissue sections as detected by IHC staining. Thus, they might be promising novel molecular markers for diagnostic and classification purposes in human gliomas.
In summary, we revealed novel mechanisms of gliomagenesis and malignant progression driven by SND1 via directly activating RhoA transcription by inducing histone acetylation and remodeling the chromatin conformation of the RhoA promoter. By upregulating RhoA, SND1 promotes glioma cell proliferation and invasion, which provided a strong rationale for targeting SND1 and RhoA in malignant glioma therapy. Moreover, we linked SND1-induced long-range chromatin communication remodeling with poor prognoses in glioma patients.
Funding
This research was supported by the National Natural Science Foundation of China (81672592, 81502166 and 81872061), the Program of Science and Technology of Tianjin Municipality (16JCQNJC13400 and 17JCYBJC27100), and the Program of Tianjin Municipal Health Bureau (15KJ147).
Conflict of interest statement. All authors declare no conflict of interest pertaining to this work.
Authorship statement: Conception and design: Lin Yu, Shizhu Yu, Development of methodology: Lin Yu, Jinling Xu, Jing Liu, Acquisition of data (acquired and managed patients, provided facilities, etc): Huibian Zhang, Cuiyun Sun, Cuijuan Shi, Xuexia Zhou, Analysis and interpretation of data (eg, statistical analysis, biostatistics, computational analysis): Qian Wang, Dan Hua, Wenjun Luo, Writing, review, and/or revision of the manuscript: Lin Yu, Xiuwu Bian, Shizhu Yu, Administrative, technical, or material support (ie, reporting or organizing data, constructing databases): Shizhu Yu, Study supervision: Shizhu Yu
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Brodbelt A, Greenberg D, Winters T, Williams M, Vernon S, Collins VP; (UK) National Cancer Information Network Brain Tumor Group Glioblastoma in England: 2007-2011. Eur J Cancer. 2015;51(4):533–542. [DOI] [PubMed] [Google Scholar]
- 2. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109(1):93–108. [DOI] [PubMed] [Google Scholar]
- 3. Gorlia T, van den Bent MJ, Hegi ME, et al. . Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol. 2008;9(1):29–38. [DOI] [PubMed] [Google Scholar]
- 4. Pal S, Bi Y, Macyszyn L, Showe LC, O’Rourke DM, Davuluri RV. Isoform-level gene signature improves prognostic stratification and accurately classifies glioblastoma subtypes. Nucleic Acids Res. 2014;42(8):e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blanco MA, Alečković M, Hua Y, et al. . Identification of staphylococcal nuclease domain-containing 1 (SND1) as a Metadherin-interacting protein with metastasis-promoting functions. J Biol Chem. 2011;286(22):19982–19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang N, Du X, Zang L, et al. . Prognostic impact of Metadherin-SND1 interaction in colon cancer. Mol Biol Rep. 2012;39(12):10497–10504. [DOI] [PubMed] [Google Scholar]
- 7. Jariwala N, Rajasekaran D, Mendoza RG, et al. . Oncogenic role of SND1 in development and progression of hepatocellular carcinoma. Cancer Res. 2017;77(12):3306–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu X, Zhang C, Meng H, et al. . Oncoprotein Tudor-SN is a key determinant providing survival advantage under DNA damaging stress. Cell Death Differ. 2018;25(9):1625–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu L, Liu X, Cui K, et al. . SND1 acts downstream of TGFβ1 and upstream of Smurf1 to promote breast cancer metastasis. Cancer Res. 2015;75(7):1275–1286. [DOI] [PubMed] [Google Scholar]
- 10. Yu L, Di Y, Xin L, et al. . SND1 acts as a novel gene transcription activator recognizing the conserved Motif domains of Smad promoters, inducing TGFβ1 response and breast cancer metastasis. Oncogene. 2017;36(27):3903–3914. [DOI] [PubMed] [Google Scholar]
- 11. González-González A, Muñoz-Muela E, Marchal JA, et al. . Activating transcription factor 4 modulates TGFβ-induced aggressiveness in triple-negative breast cancer via SMAD2/3/4 and mTORC2 signaling. Clin Cancer Res. 2018;24(22):5697–5709. [DOI] [PubMed] [Google Scholar]
- 12. Yu OM, Benitez JA, Plouffe SW, et al. . YAP and MRTF-A, transcriptional co-activators of RhoA-mediated gene expression, are critical for glioblastoma tumorigenicity. Oncogene. 2018;37(41):5492–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zuo Y, Ulu A, Chang JT, Frost JA. Contributions of the RhoA guanine nucleotide exchange factor Net1 to polyoma middle T antigen-mediated mammary gland tumorigenesis and metastasis. Breast Cancer Res. 2018;20(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chillà A, Margheri F, Biagioni A, Del Rosso M, Fibbi G, Laurenzana A. Mature and progenitor endothelial cells perform angiogenesis also under protease inhibition: the amoeboid angiogenesis. J Exp Clin Cancer Res. 2018;37(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Louis DN, Perry A, Reifenberger G, et al. . The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 16. Liu J, Xu J, Li H, et al. . miR-146b-5p functions as a tumor suppressor by targeting TRAF6 and predicts the prognosis of human gliomas. Oncotarget. 2015;6(30):29129–29142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warde-Farley D, Donaldson SL, Comes O, et al. . The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–W220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rajasekaran D, Jariwala N, Mendoza RG, et al. . Staphylococcal nuclease and tudor domain containing 1 (SND1 Protein) promotes hepatocarcinogenesis by inhibiting monoglyceride lipase (MGLL). J Biol Chem. 2016;291(20):10736–10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arretxe E, Armengol S, Mula S, Chico Y, Ochoa B, Martínez MJ. Profiling of promoter occupancy by the SND1 transcriptional coactivator identifies downstream glycerolipid metabolic genes involved in TNFα response in human hepatoma cells. Nucleic Acids Res. 2015;43(22):10673–10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emdad L, Janjic A, Alzubi MA, et al. . Suppression of miR-184 in malignant gliomas upregulates SND1 and promotes tumor aggressiveness. Neuro Oncol. 2015;17(3):419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu J, Yang J, Yu L, et al. . miR-361-5p inhibits glioma migration and invasion by targeting SND1. Onco Targets Ther. 2018:11: 5239–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vermeulen M, Eberl HC, Matarese F, et al. . Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142(6):967–980. [DOI] [PubMed] [Google Scholar]
- 23. Chang P, Fan X, Chen J. Function and subcellular localization of Gcn5, a histone acetyltransferase in Candida albicans. Fungal Genet Biol. 2015;81:132–141. [DOI] [PubMed] [Google Scholar]
- 24. Ringel AE, Cieniewicz AM, Taverna SD, Wolberger C. Nucleosome competition reveals processive acetylation by the SAGA HAT module. Proc Natl Acad Sci U S A. 2015;112(40):E5461–E5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee D, Goldberg AL. Muscle Wasting in Fasting Requires Activation of NF-κB and Inhibition of AKT/Mechanistic Target of Rapamycin (mTOR) by the Protein Acetylase, GCN5. J Biol Chem. 2015;290(51):30269–30279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimizu A, Nakayama H, Wang P, et al. . Netrin-1 promotes glioblastoma cell invasiveness and angiogenesis by multiple pathways including activation of RhoA, cathepsin B, and cAMP-response element-binding protein. J Biol Chem. 2013;288(4):2210–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Talamillo A, Grande L, Ruiz-Ontañon P, et al. . ODZ1 allows glioblastoma to sustain invasiveness through a Myc-dependent transcriptional upregulation of RhoA. Oncogene. 2017;36(12):1733–1744. [DOI] [PubMed] [Google Scholar]
- 28. Peurala E, Koivunen P, Haapasaari KM, Bloigu R, Jukkola-Vuorinen A. The prognostic significance and value of cyclin D1, CDK4 and p16 in human breast cancer. Breast Cancer Res. 2013;15(1):R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duong MT, Akli S, Macalou S, et al. . Hbo1 is a cyclin E/CDK2 substrate that enriches breast cancer stem-like cells. Cancer Res. 2013;73(17):5556–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee Y, Dominy JE, Choi YJ, et al. . Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510(7506):547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun L, Huang Y, Wei Q, et al. . Cyclin E-CDK2 protein phosphorylates plant homeodomain finger protein 8 (PHF8) and regulates its function in the cell cycle. J Biol Chem. 2015;290(7):4075–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wan C, Hou S, Ni R, et al. . MIF4G domain containing protein regulates cell cycle and hepatic carcinogenesis by antagonizing CDK2-dependent p27 stability. Oncogene. 2015;34(2):237–245. [DOI] [PubMed] [Google Scholar]
- 33. Orgaz JL, Herraiz C, Sanz-Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases. 2014;5: e29019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.