

Abstract
The unicellular green microalga Chlamydomonas reinhardtii is a powerful photosynthetic model organism which is capable of heterotrophic growth on acetate as a sole carbon source. This capacity has enabled its use for investigations of perturbations in photosynthetic machinery as mutants can be recovered heterotrophically. Fixation of acetate into cellular carbon metabolism occurs first by its conversion into acetyl‐CoA by a respective synthase and the generation of succinate by the glyoxylate cycle. These metabolic steps have been recently determined to largely occur in the peroxisomes of this alga; however, little is known about the trafficking and import of acetate or its subcellular compartmentalization. Recently, the genes of five proteins belonging to the GPR1/FUN34/YaaH (GFY) superfamily were observed to exhibit increased expression in C. reinhardtii upon acetate addition, however, no further characterization has been reported. Here, we provide several lines of evidence to implicate Cr GFY1–5 as channels which share structural homology with bacterial succinate‐acetate channels and specifically localize to microbodies, which are surprisingly distinct from the glyoxylate cycle‐containing peroxisomes. We demonstrate structural models, gene expression profiling, and in vivo fluorescence localization of all five isoforms in the algal cell to further support this role.
Keywords: acetate transporter, Chlamydomonas, GFY protein, membrane channel, microbodies
1. INTRODUCTION
The green microalga Chlamydomonas reinhardtii is a unique model organism for many areas of fundamental and applied research. Its utility as a model organism arises from the capacity for heterotrophic growth on acetate as a carbon source in addition to oxygenic photosynthesis. This has enabled the generation of mutants perturbed in photosynthesis that can nevertheless be regenerated, maintained, and investigated to understand the dynamics of light‐driven metabolism (Rochaix, 2002). This alga can also combine these two metabolic capacities, resulting in more rapid growth under mixotrophic conditions for biotechnological applications (Lauersen, Baier, et al., 2016). Acetate is a two‐carbon (C2) molecule that is assimilated for heterotrophic growth in C. reinhardtii by the glyoxylate cycle. This pathway is a modified version of the mitochondrial tricarboxylic acid (mTCA) cycle and yields net carbon gain from exogenous acetate (Kornberg & Krebs, 1957; Kornberg & Madsen, 1957). The reactions of isocitrate lyase (ICL1) and malate synthase (MAS1) bypass the substrate decarboxylation steps of mTCA to produce intermediates and succinate as the net C4 product for gluconeogenesis, mTCA cycle, or cytosolic reactions. In eukaryotic cells, the enzymatic reactions of glyoxylate cycle are distributed between the peroxisomal matrix and the cytosol (Kunze & Hartig, 2013; Kunze, Pracharoenwattana, Smith, & Hartig, 2006). All enzymes of the glyoxylate cycle in C. reinhardtii have been localized to algal peroxisomes including a single acetyl‐CoA synthase (ACS3), with the sole exception of ICL1, which is cytosolic (Lauersen, Willamme, et al., 2016). A mutant deficient in ICL1 was unable to utilize acetate heterotrophically and exhibited reduced enzyme abundances of other glyoxylate cycle genes (Plancke et al., 2014).
Highly compartmentalized metabolic processes are separated in eukaryotic cells by microenvironments mediated by organellar membranes. These structures require specific intracellular transport proteins dedicated to facilitating and regulating the movement of metabolites which cannot pass by diffusion. In C. reinhardtii, carbon metabolism has been extensively studied (Johnson & Alric, 2013), although little has been reported regarding proteins involved in metabolite transport. According to TransportDB2 (Elbourne, Tetu, Hassan, & Paulsen, 2017), there are at least 519 predicted membrane proteins related to transport in the genome of C. reinhardtii (v5.5) (~2.9% of total encoded proteins). However, none has been extensively studied for their role in acetate transport.
It currently remains unclear whether uptake of acetate by the cell occurs by diffusion or active transport. The mechanisms of intracellular acetate transport are also unclear. Members of GPR1/FUN34/YaaH superfamily have been identified in all domains of life but are more commonly found in bacteria, archaea, and fungi (Goodenough et al., 2014; Ribas et al., 2019). These small molecule protein channels were originally discovered in mutants of the yeast Yarrowia lipolytica, which were unable to grow on acetate. The involvement of GFY membrane proteins in acetate uptake has been demonstrated in Escherichia coli (enzyme: SatP_Ec or YaaH) (Sá‐Pessoa et al., 2013; Sun et al., 2018), Citrobacter koseri (SatP_Ck) (Qiu et al., 2018), Methanosarcina acetivorans (AceP) (Ribas et al., 2019), Saccharomyces cerevisiae (Ady2) (Paiva, Devaux, Barbosa, Jacq, & Casal, 2004), and Aspergillus nidulans (AcpA) (Robellet, Flipphi, Pégot, MacCabe, & Vélot, 2008). GFY proteins have also been implicated in acetate sensing for the yeast Y. lipolytica (Gpr1) (Augstein, Barth, Gentsch, Kohlwein, & Barth, 2003; Tzschoppe, Augstein, Bauer, Kohlwein, & Barth, 1999). Hallmarks of GFY proteins are six transmembrane domains and at least two distinct conserved amino acid signatures which have been determined to be directly involved in transport mechanisms by substrate interaction studies (Qiu et al., 2018; Ribas et al., 2019). Two recently resolved crystal structures of bacterial GFYs include the SatP_Ec from E. coli (Sá‐Pessoa et al., 2013; Sun et al., 2018) and SatP_Ck from Citrobacter koserii (Qiu et al., 2018). In C. reinhardtii, five GFY genes emerged as candidate acetate permeases in clusters of transcripts responsive to acetic acid (Goodenough et al., 2014).
Here, we suggest that these five CrGFY isoforms are channel proteins which likely permit acetate transport specifically across microbody or intracellular organellar membranes. Based on the strong structural identity and conserved key amino acid positions, we demonstrate that CrGFY1–5 proteins have fundamental features in common with the acetate‐succinate channels SatP_Ck, and SatP_Ec. These models support an analogous selective transport mechanism for the CrGFY proteins in C. reinhardtii microbodies. In vivo protein localization was used here to determine the subcellular localization of each of the five protein isoforms because distinct isoform involvement in different physiological states may be suggested by variable expression levels. Their characterization sheds light on the mechanisms employed to enable acetate assimilation as a carbon source in green algae. However, their localizations present a perplexing and nonintuitive localization which also opens new questions of intracellular metabolism and transport in this alga.
2. MATERIALS AND METHODS
2.1. Algal strains, cultivation and transformation
Chlamydomonas reinhardtii cell wall‐less strain CC‐4533 (cw15 mt‐ nit1‐) was used as a wild‐type strain for physiological, PCR, and RT‐qPCR analyses. The derived insertional mutants for the genes GFY1 (gfy1, strain code: LMJ.RY0402.062945), GFY2 (gfy2‐1: LMJ.RY0402.153639; gfy2‐2: LMJ.RY0402.239051), and GFY3 (gfy3: LMJ.RY0402.178275) were obtained from the Chlamydomonas Library Project (CLiP) (Li et al., 2016), carrying insertion conferring paromomycin antibiotic resistance (AphVIII gene, ParR) (Sizova, Fuhrmann, & Hegemann, 2001). For each strain, the position of inserted AphVIII gene was verified by PCR protocol on single colonies with primers listed in Table S4 and mapped to the gene sequences (Figure 2d). Lack of the corresponding transcript for the knocked‐out GFY genes was verified by RT‐qPCR as described in the section “Nucleic acid extractions and RT‐qPCR analyses.” Wild‐type CC‐4533 and insertional mutants were backcrossed with the isogenic wild‐type strain CC‐4402 (isoloP mt+) until a 50 ParR:50 ParS segregation progeny was obtained. The UVM4 strain (kindly provided by Ralph Bock to the Algae Biotechnology and Bioenergy Group at the CeBiTec, Bielefeld University) was used as a recipient strain for the protein localization experiments (Neupert, Karcher, & Bock, 2009). The algal cultures were grown in Tris acetate phosphate (TAP) or Tris minimum phosphate (TMP) (Harris, 1989) under continuous light, on an agar plate or shaken in Erlenmeyer flasks. Light intensity for strain maintenance was 8–15 μmol photons m−2 s−1 and about 80 μmol photons m−2 s−1 for growth. Anaerobic adapted algal suspensions were prepared as follows: TAP‐cultured cells were grown until exponential phase, washed three times, and concentrated in TMP medium to 2 × 108 cells/mL. Suspensions were flushed with N2 for 10 min in a sealed Falcon tube using a long needle and incubated in the dark under gentle shaking. After 6 h, cells were collected by centrifugation at 4500 g for 5 min and the supernatant subject to additional centrifugation (14,000 g for 10 min) prior to analysis. Where specified, antibiotics were added at 5 mg/L for paromomycin and 10 mg/L for hygromycin B. Transformation procedure was performed as previously reported (Shimogawara, Fujiwara, Grossman, & Usuda, 1998) with ~500 ng of KpnI‐XbaI linearized plasmid vectors. Electroporated cells were recovered under dim light in 2 ml of liquid TAP medium overnight and spread onto TAP agar plates supplemented with appropriate antibiotics. Plates were incubated under the light intensity of ~100 μmol photons m−2 s−1 until the colonies reached around 1 mm of diameter, then picked by sterile toothpick, and transferred to 96‐colony grids on square TAP agar plates. Cell number was determined using a Z‐series Coulter Counter set in 3–11 μm size range (Beckman Coulter).
2.2. Measurement of fermentative products
l‐Lactic acid, formate, acetic acid, and ethanol were quantified by high‐performance liquid chromatography (Shimadzu) using a Supercogel C610‐H ion exchange column (Sigma‐Aldrich) kept at 35°C, using H3PO4 0.1% as mobile phase in isocratic mode (0.5 mL/min). Twenty microliters of supernatant was injected onto the column, retention peaks were recorded using a refractive index detector (RID‐20A, Shimadzu), and quantification was performed by comparison with known amounts of standards (Roth) in LabSolution software (Shimadzu). The cell dry weight used for data normalization was obtained by sample lyophilization in a benchtop freeze dryer (Labconco Corporation).
2.3. DNA construction
In order to determine in vivo protein localizations of CrGFY1–5, the gene coding regions from start codon until the last amino acid before the stop codon of CrGFY1–5 genes were individually PCR amplified from genomic DNA and cloned into a pOpt_mVenus_Paro backbone vector (Lauersen, Kruse, & Mussgnug, 2015) in‐frame with the YFP variant mVenus (hereafter YFP). Genes of interest were cloned to generate fusions with YFP in either N‐ or C‐terminal orientations using the unique restriction sites of the pOpt vector concept (Lauersen et al., 2015). PCRs were performed using Q5 Polymerase and GC enhancer solution (New England Biolabs) following manufacturer's protocols with genomic DNA as a template; all primers used are listed in Table S4. In detail, for cloning GFY genes to have a C‐terminal YFP fusion, the stop codons were substituted with the triplet GGT or ACC, both coding for the amino acid glycine. Correct sizes of the amplifications were verified in 1.2% agarose gel stained with Midori Green (Nippongenetics). Each ORF sequence was amplified with the combination of NdeI and/or BglII or EcoRI and/or EcoRV endonuclease restriction sites added to the 5′ of oligonucleotides to enable cloning into the pOpt vector based on the naturally occurring sites in the genomic sequences. Unless otherwise mentioned, Fastdigest restriction enzymes (Thermo Scientific) were used in this work. PCR fragments and plasmid backbone were digested with the respective restriction enzymes followed by vector dephosphorylation (FastAP, Thermo Scientific), subsequent fragment ligation using T4 DNA ligase (New England Biolabs), and transformation by heat shock into chemically competent E. coli DH5α cells. Bacterial transformant colonies were selected on Luria broth (LB) medium plates and grown in liquid medium for plasmid isolation, using 150 mg/L of ampicillin for selection. The plasmid insertions were verified by colony PCR in selected bacterial clones with the mVup or mVdw primers pairs (Table S4). Hereafter, the resulting DNA constructions obtained are represented in Figure 4, from i to x. Vectors a and c are pOpt_mVenus_Paro and pOpt_mCerulean3_Hyg, respectively, from Lauersen et al., 2015. Vector b was previously developed by our group (Lauersen, Willamme, et al., 2016). This vector expresses a modified mVenus (YFP) reporter with the C‐terminal amino acids of C. reinhardtii malate synthase 1 gene HIVTKTPSRM*, which is a confirmed peroxisomal targeting signal type 1 (PTS1) sequence (Lauersen, Willamme, et al., 2016). Vector d (Figure 4) was generated by digestion of the pOpt_cCA_mCerulean3_Hyg vector (Lauersen et al., 2015) with EcoRV‐EcoRI restriction endonucleases and ligation of a DNA fragment built by annealing the P‐5′‐AAGGACGAGCTGTAAG‐3′‐ and P‐5′‐AATTCTTACAGCTCGTCCTT‐3′ oligonucleotide pair. The resulting protein is targeted to the ER and retained there by the C‐terminal KDEL* ER‐retention signal. In both vectors b and d, a stop codon was introduced after each signal peptide in order to prevent translation of the strepII tag of the pOpt vector backbone. Vector e (Figure 5) was generated by adding the N‐terminal amino acids of citrate synthase 2 (CIS2) gene (MSSRLDVLSRQMCGMQLSPTAGEEE) to vector c as described in Lauersen, Willamme, et al., (2016). CIS2 contains a confirmed PTS2 targeting peptide and is a key enzyme in the glyoxylate cycle of the peroxisomal microbodies (Lauersen, Willamme, et al., 2016). Prior to algal transformation, the amplification products were verified by sequencing (Sanger sequencing service GENEWIZ, United Kingdom) and linearized with XbaI‐KpnI.
2.4. In silico and database analyses
Protein and genomic sequences were obtained from Phytozome database v.12 ( http://phytozome.jgi.doe.gov/pz/-portal.html), C. reinhardtii genome v.5.5, UniProt protein references are indicated between brackets. GFY1–5 gene sequences have the following accession numbers: GFY1, Cre17.g700450 (A8IQI1); GFY2, Cre17.g700650 (A8IQH3); GFY3, Cre17.g700750 (A8IQG4); GFY4, Cre17.g702900 (A8IQ56), and GFY5, Cre17.g702950 (A8IQ53). ClustalOmega ( https://www.ebi.ac.uk/Tools/msa/clustalo/) and T‐COFFEE ( http://tcoffee.crg.cat/apps/tcoffee/index.html) were used for multiple sequence alignment analyses and to generate percent identity matrixes.
2.5. Transformant screening, identification, and single‐cell fluorescence microscopy
Initial screening of C. reinhardtii transformants was performed on colonies at the agar plate level for YFP fluorescence corresponding to GFY fusion protein expression as previously described (Lauersen et al., 2015; Lauersen, Willamme, et al., 2016). Colonies exhibiting fluorescence were selected and cultivated in 1 ml TAP in 24‐well microtiter plates with ~100 μmol photons m−2 s−1 light intensity overnight prior to single‐cell fluorescence microscopy. Single‐cell fluorescence was captured as previously described (Lauersen, Baier, et al., 2016).
2.6. Nucleic acid extraction and RT‐qPCR analyses
Nucleic acids were extracted with phenol chloroform procedure according to (Newman et al., 1990). The RNA was recovered from aqueous phase by precipitation with the addition of 2.5 M LiCl final concentration overnight at −20°C. When needed, RNA preparations were treated with RQ1 DNase (Promega) according to manufacturer's instructions and RNA was precipitated with 1:10 volume of 3M sodium acetate and 2.5 volumes of absolute EtOH. Nucleic acids were quantified employing the 260/280 nm absorbance values on a NanoDrop spectrophotometer. Relative gene expression levels were measured in exponential phase cultures (~2–4 × 106 cells/mL) for all tested conditions. Cells were collected by centrifugation at 4,500 g for 4 min. The cDNAs were synthesized by reverse transcription reactions carried out with a High Capacity cDNA Synthesis kit (Applied Biosystems). The amplification cycles were conducted in a QuantStudio 5 Real‐Time PCR System (Thermo Fisher Scientifc) with PowerUp SYBR Green PCR Master Mix in 10 μl volume reactions through the use of 384‐well plates (reagents were purchased from Applied Biosystems, the MicroAmp Optical plates from Thermo Fisher Scientifc were sealed with Tape Pads 5, Qiagen). Melting curve analysis was performed right after the qPCR run in order to verify the amplification product. To ensure selective amplification, the primers were designed in the 3′‐UTR and blasted against the whole genome (Table S4). Expression level data were analyzed using the qBase+ software, Biogazelle. For normalization of expression levels, the β‐subunit‐like polypeptide (CBLP, Cre06.g278222) and ribosomal protein L13 (RPL13, Cre14.g630100) were adopted as reference transcripts. We verified with LinRegPCR software (v11.0) that amplification efficiency was at least 0.9.
2.7. Protein extraction for SDS–PAGE and immunoblot analyses
For total protein fraction, 5 mL of algal suspensions (corresponding to OD750 = 0.6–0.8) was centrifuged at 3,800 g for 3 min. Cell pellets were suspended in 2× extraction/loading buffer (0.1 M Tris‐HCl pH 6.8, 10% sodium dodecyl sulfate (SDS), 30% glycerol, 4% 2‐mercaptoethanol, 0.3% Coomassie Brilliant Blue G 250), and heated for 5 min at 95°C. Total hydrophobic (membrane) protein extractions were carried out according to (Remacle, Baurain, Cardol, & Matagne, 2001). Briefly, 8 × 107 cells were collected, suspended in MET buffer (280 mM mannitol, 0.1 mM EDTA, 10 mM Tris‐HCl pH 7, 0.1% BSA) supplemented with Protease inhibitor cocktails (P9599, Sigma‐Aldrich), and sonicated twice for 15 sec on ice. Serial centrifugations were used to remove soluble fraction and cell debris. The final pellet was suspended in MET buffer without BSA, mixed with equal volume of 2× loading buffer, and finally warmed at 42°C for 30 min in order to allow solubilization. Protein samples were quantified according to a Bradford assay (Bradford, 1976). To avoid residues, both extracts were centrifuged at 12,000 g for 5 min just before the gel loading. Total proteins and membrane preparations were resolved in 12% SDS–polyacrylamide gel electrophoresis (SDS–PAGE) run at 100 V/1.5 h and transferred onto polyvinylidene difluoride (PVDF) membranes (Amersham Hybond P 0.45, GE Healthcare) by overnight wet transfer at 4°C (~16 h) under constant voltage of 30 V. The tank bath was filled with precooled transfer buffer: 20% MeOH, 25 mM Tris, and 192 mM glycine. Western blocking reagent (Roche) was diluted in Tris‐buffered saline solution (TBS: 50 mM Tris‐HCl, pH 7.6, 150 mM NaCl) and used as 1× for blocking and 0.5× for antibody incubation. Washing steps were performed in TBS‐Tween‐20 0.1%. Primary GFP‐tag polyclonal antibody (1:5000, Invitrogen), followed by secondary antibody HRP‐conjugated anti‐Rabbit IgG (1:15000, Invitrogen), for both 1 h incubation. The peroxidase activity was detected by an enhanced chemiluminescence assay (Roche) in iBright Imaging System fl1000 (Thermo Fisher Scientific). All chemicals were purchased from Sigma‐Aldrich.
3. RESULTS
3.1. CrGFY1–5 genes code for paralogous membrane proteins
We first sought to investigate the genomic and amino acid structures of GFY isoforms 1–5 in order to gain more insight into their potential cellular role. When compared by multiple sequence alignment, these five isoforms exhibited similar amino acid sequence compositions (Figure 1a) with degrees of similarity ranging from 81% to 97% (Figure 1c). The central cores of these proteins are highly similar, diverging by only small variations in amino acid positions (Figure 1a). However, the N‐terminal regions (16–33 amino acids) of these five protein isoforms differ in length and amino acid composition (Figure 1a). The five protein sequences contain the typical signatures conserved in the GPR1/FUN34/YaaH superfamily (Pfam01184; PS01114; IPR000791) (Figure 1a, Table S1) (Qiu et al., 2018; Ribas et al., 2019). The secondary structure analysis indicates that CrGFY1–5 proteins are integral, multipass, membrane proteins composed of six transmembrane segments (Figure 1a) with location of both N‐ and C‐terminal regions on the same side of the membrane, which is predicted to be cytosolic [Phobius (Käll, Krogh, & Sonnhammer, 2004); TOPCONS (Tsirigos, Peters, Shu, Käll, & Elofsson, 2015); I‐TASSER (Yang et al., 2014)]. However, no other functional annotation of these proteins could be found in either InterProScan (Jones et al., 2014) or NCBI databases.
Figure 1.
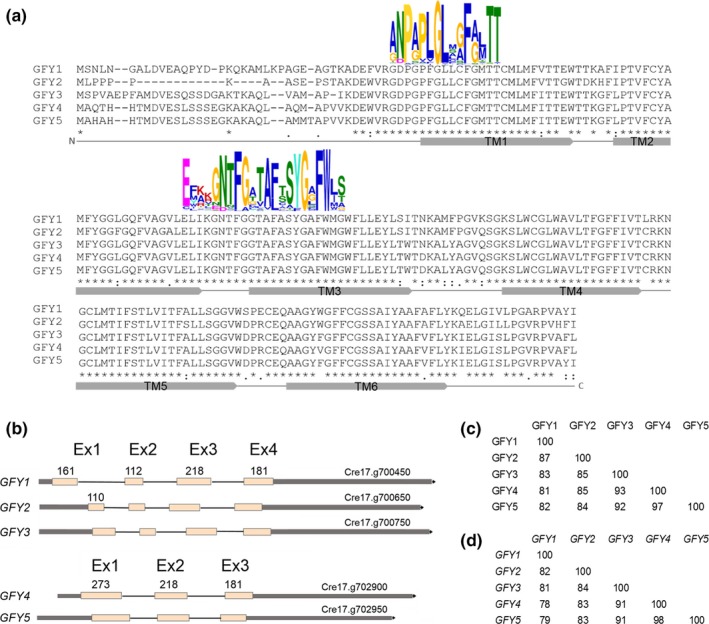
Analysis of CrGFY1–5 protein and gene sequences. (a) Multiple sequence alignment of CrGFY1–5 amino acid sequences. Colored letter signature logos depict the top two motifs from the 355 representative GFY protein sequences scanned by MEME Suite (Bailey, Johnson, Grant, & Noble, 2015). Gray bars below the alignment represent the schematic predicted transmembrane regions (TM) from 1 to 6. Alignment was generated using the PSI/TM‐Coffee multiple sequence analysis program. Gaps are represented by dashes; asterisks, colons, and dots indicate full, high, and low level of amino acid identity, respectively. (b) CrGFY1–5 gene structure diagrams, exons (Ex) with respective nucleotide lengths (numbers) are shown separated by introns (thin lines), 5′ and 3′ UTRs are indicated in gray outside of the terminal exons. Percent identity matrixes calculated from multiple alignment of amino acid sequences (c) or DNA gene coding sequences (d) are shown
Alignment of the CrGFY1–5 nucleotide sequences with PSI/TM‐Coffee (http://tcoffee.crg.cat/apps/tcoffee/do:tmcoffee) also revealed a very high percentage of similarity inside the coding sequences, between 78 and 98% (Figure 1d). However, the 5′‐ and 3′‐UTRs, as well as intron position and lengths, are not conserved across these five genes (Figure 1b). Based on Phytozome database (http://phytozome.jgi.doe.gov), all the five genes are located on chromosome 17, however, they are sorted into two distinct loci. One group is composed of the isoforms GFY1, 2, and 3, and the other group is composed of isoforms 4 and 5. Within the first group, the four exons of each GFY gene exhibit equal lengths to their respective counterparts, with the exception of first exon of GFY2 which is 110 bp compared to 161 bp for the other isoforms. The latter group exhibits only three exons, the first may be derived from a fusion of exons 1 and 2 (as in GFY1–3) (Figure 1b).
3.2. CrGFY1–5 gene transcripts have different expression patterns under various cultivation conditions
Relative transcript abundances for CrGFY1–5 were measured by RT‐qPCR experiments in cells grown to mid exponential phase in four different conditions: minimal medium in the light, acetate medium in the light, acetate medium in the dark, acetate medium in the light, followed by 6 h of anaerobic incubation in minimal medium. Data in Figure 2a are presented as the fold‐change of relative transcript abundances normalized to those obtained in cells grown in minimal medium without acetate. The results indicate that different expression patterns can be observed depending on the cultivation condition (Figure 2a). The expression of GFY1 and GFY2 genes are either reduced or unchanged in the presence of acetate in the light, respectively, and are both downregulated in the dark. Upon anaerobiosis, the transcript of GFY1 maintains a comparable expression level as in minimal medium, and GFY2 exhibits a small but significant upregulation. Conversely, a pronounced upregulation of GFY3–5 transcripts was observed in the light as well as in the dark when acetate was present compared to autotrophic conditions. This induction ranges from 1.8‐ to 8.5‐fold, similar to those previously reported for acetate addition to starved cultures (Goodenough et al., 2014). In anaerobically adapted cells, CrGFY3–5 expression levels were subject to strong downregulation.
Figure 2.
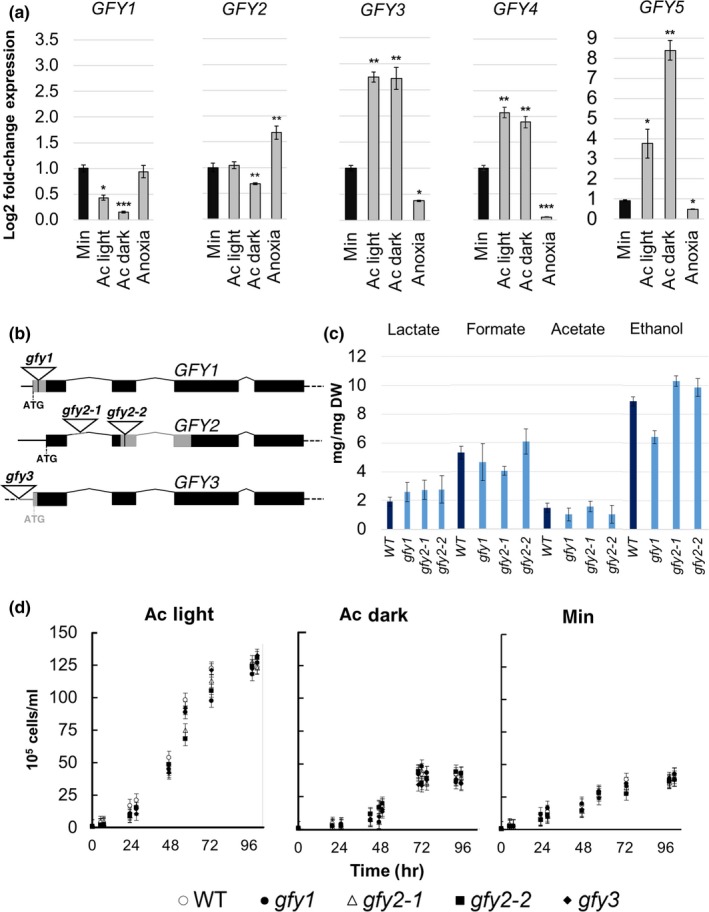
Transcript accumulation of CrGFY1–5 associated with different physiological conditions. (a) Data are presented as log2 fold‐changes of relative transcripts upon acetate addition or induction of anaerobiosis compared to values obtained from cells grown in minimal medium (min – absence of acetate). Transcripts were measured from cells grown in autotrophy (min), photoheterotrophy with light and acetate (ac light), complete heterotrophy with acetate (ac dark) grown cells, or 6 h after transfer of cultures grown in photoheterotrophy to minimal medium under anaerobic conditions (anoxia). Note the different scales for the GFY5 graph. Log2 values were calculated with 2−ΔΔCT method by using (CBLP, Cre06.g278222) and (RPL13, Cre14.g630100) transcripts as references for data normalization. Error bars represent standard deviation (SD) of three replicates. Asterisks denote statistical difference compared to min values (p < 0.05: *; <0.01: ** and <0.001: *** by two‐tailed Student's t test). (b) Genomic structure of CrGFY1, 2, and 3 gene disruptions in CLiP library insertion mutants. The location of the AphVIII cassette insertion sites were verified by sequencing and are depicted here with triangles. Gray bars indicate deletions in the genes caused by AphVIII cassette insertion. Mutant names are written in lowercase, gene names in uppercase. (c) HPLC measurements of fermentation products in the supernatant of cultures after 6 h in anaerobic conditions. WT strain (dark blue) compared to gfy1, gfy2‐1, gfy2‐2 mutant strains (light blue) normalized for cell dry weight (DW). Error bars indicated the SD of independent experiments (n = 4). (d) Growth curves of WT and gfy1, gfy2‐1, gfy2‐2, gfy3 CLiP library insertion mutants. Each series represents the average of three biological replicates, error bars represent SD
Coexpression analysis of CrGFY1–5 using either Phytozome or ALCOdb (Aoki, Okamura, Ohta, Kinoshita, & Obayashi, 2016) also confirmed distinct group behaviors: GFY1 and GFY2 were found to be coexpressed with genes related to fermentation, that is, pyruvate formate lyase (PFL1, Cre01.g044800), acetate kinase (ACK1, Cre09.g396700), phosphate acetyltransferase (PAT2, Cre09.g396650), and iron‐hydrogenase (HYDA2, Cre09.g396600), or involved in anaerobic response, such as hybrid cluster proteins (HCP1–4, Cre09.g391450; Cre09.g393543; Cre09.g393506; Cre09.g391650) (Olson & Carter, 2016) (Table S2). In contrast, GFY3–5 were found to be coexpressed with genes participating in acetate metabolism, in particular the glyoxylate cycle; peroxisomal acetyl‐CoA ligase/synthase 3 (ACS3, Cre07.g353450) and isocitrate lyase (ICL1, Cre06.g282800). The expression of GFY4/5 also correlates with the peroxisomal enzymes citrate synthase (CIS2, Cre03.g149100), malate synthase (MAS1, Cre03.g144807), the chloroplastic counterpart acetyl‐CoA synthase 2 (ACS2, Cre01.g055408), and one of the subunits of succinate dehydrogenase (SDH1, Cre14.g619133). Additionally, GFY3–5 were coexpressed with phosphoenolpyruvate carboxykinase (PCK1, Cre02.g141400) drawing a link to gluconeogenesis. Genes related to amino acid metabolism have high correlation indexes with GFY4/5, such as aspartate aminotransferase (AST3, Cre02.g097900), glyoxylate reductase 2 (GLYR, Cre06.g278148), alanine‐glyoxylate transaminase (AGT1, Cre06.g294650), and 3‐hydroxyisobutyryl‐CoA hydrolase (HIBYL‐CoA, Cre06.g278215). Finally, a limited number of genes concerning the endogenous reserve metabolism were identified for fatty acids (acyl‐CoA oxidase [ACO1, Cre16.g689050]; enoyl‐CoA hydratase [ECH1, Cre10.g463150]) and gene coding for alpha‐amylase (AMA1, Cre08.g385500), implying a link to starch metabolism (Table S2). The difference of expression pattern between GFY1–2 and GFY3–5 has already been reported previously (Goodenough et al., 2014). An expanded and detailed coexpression gene list can be consulted by using the accession numbers in the corresponding databases.
In order to analyze the function of the GFY proteins, individual mutants with knockout of GFY1, 2, or 3 genes were obtained from the CLiP library ( https://www.chlamylibrary.org/). The AphVIII cassette insertion site was mapped in the genomic sequence as illustrated in Figure 2b. Each strain exhibited no transcription of the desired corresponding GFY gene while the others were still comparably expressed (Table S3). No discernible phenotype in anaerobic fermentation products or aerobic growth rates could be detected for any of the strains analyzed under different cultivation conditions (Figure 2c,d). We analyzed fermentation products from the CLiP mutants in anaerobiosis because CrGFY1–2 genes were expressed more than other isoforms under these conditions (Figure 2a).
3.3. In silico protein modeling suggests CrGFY structural similarity to a bacterial acetate‐succinate channels
To further understand the roles CrGFY1–5 play in the green‐algal cell, we used several in silico prediction algorithms based on homology modeling to infer the structures of these proteins. For the five proteins, a pore‐forming structure was the resulting lowest free‐energy conformation calculated from I‐TASSER (Yang et al., 2014), SWISS‐MODEL (Waterhouse et al., 2018), Phyre2 (Kelley, Mezulis, Yates, Wass, & Sternberg, 2015), and RaptorX servers (Källberg et al., 2012). The predicted structures contain six alpha‐helical transmembrane (TM) segments forming an inner pore along with a soluble N‐terminal region. The servers suggest that the target‐template models share main structural features with the succinate‐acetate channel monomer of Citrobacter koseri (SatP_Ck, UniprotID: A8ALU5) (Qiu et al., 2018) and E. coli (SatP_Ec (also known as YaaH): P0AC98) (Sá‐Pessoa et al., 2013; Sun et al., 2018). These multipass membrane proteins also belong to GFY protein superfamily and their structures were recently resolved by X‐ray crystallography (PDB ID SatP_Ck: 5ys3; SatP_Ec: 5ZUG). However, only SatP_Ck was crystallized in complex with acetate ions along the channel pore. Both proteins exhibit hexameric oligomerization in their crystal structure, where the resultant central cavity formed by monomer arrangement does not function as an anion pathway. A multiple sequence alignment of CrGFY1–5 with SatP_Ck shows that regions which align between the bacterial and algal proteins are transmembrane associated regions and their adjacent amino acid positions (Figure 3a). Numerous residues that have been determined to form interactions with acetate ions along the channel pore of SatP_Ck crystal structure were found conserved in CrGFY1–5 (green, Figure 3a). The SatP_Ec protein differs from SatP_Ck by only 15 residues. The sequence similarity with CrGFY1–5 is conserved to ~32–33% (Figure 3b) and the positions forming ligand interactions are maintained. The global structural alignments of SatP_Ck with CrGFY1–5 proteins calculated by the I‐TASSER server present a high template modeling score (TM‐score 0.799–0.885, RMDS ~0.5–0.7 Å; normalized Z‐score up to 10, sequence coverage > 0.81). Iterative homology modeling via the SWISS‐MODEL server indicates that although mediocre QMEAN4 values were obtained, the transmembrane containing channel core presents high local quality (~0.8) and exhibits a highly similar Z‐score solvation potential of the template SatP_Ck (Figure 3c). Iterative modeling displays that the central CrGFY transmembrane regions maintain the structural conformation of the conserved residues found in the SatP_Ck PDB template within the expected range for ligand interactions with acetate ions (Figure 3d). Due to the almost identical transmembrane sequences of CrGFY1–5 (89–99% similarity if N‐terminal regions are excluded), all presented server results are comparable for the five protein sequences. The N‐terminal regions of CrGFY3–5 contained one or two alpha‐helices or beta‐sheets based on the server utilized; however, these structures were characterized from very low confidence matches to structures in the respective databases, giving no clues about their function.
Figure 3.
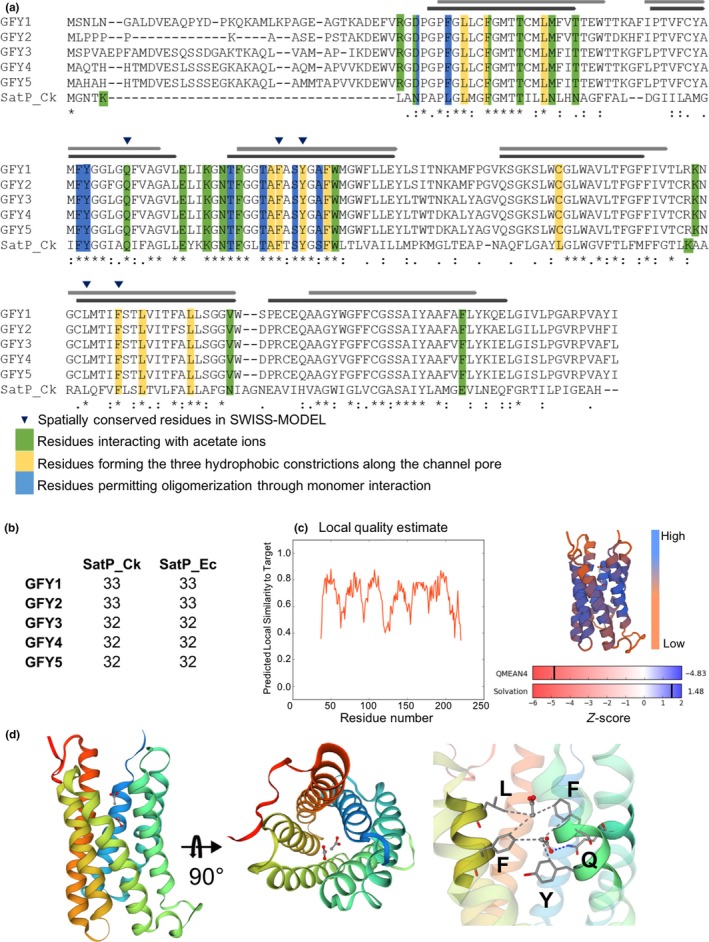
Conserved key amino acid positions of SatP_Ck and CrGFY1–5 isoforms and graphical representation of CrGFY3 protein modeling. (a). Multiple sequence alignment of CrGFY1–5 with SatP_Ck protein sequences. The residues involved in the acetate ion pathway along the pore are highlighted accordingly to their color for specific function as determined in SatP_Ck crystal structure analysis (Qiu et al., 2018). Light and dark gray bars represent the comparison of predicted CrGFY3 transmembrane regions (TM, light gray) with those experimentally determined for SatP_Ck (dark gray). Blue triangles depict residues conserving the relative coordinates of the original template structure and positioned within the expected range for ligand interactions. (b) Percentage identity matrix of CrGFY1–5 protein sequences separately aligned with bacterial proteins SatP_Ck and SatP_Ec. (c) Protein modeling quality estimation. Left: local quality plot showing high score in correspondence of TM core. Top right: ribbon diagram of protein model colored by QMEAN value depicting regions with low or high model quality. Bottom right: Match degree between CrGFY3 and template (SatP_Ck), expressed as Z‐score for QMEAN4 and solvation potential value. (d) Homology modeling of CrGFY3. Left and central panels: ribbon diagram of tertiary structure calculated using SatP_Ck structure as template, front and top view, respectively. The color gradient indicates the amino acid sequence from N‐ (blue) toward C‐terminus (red). Acetate ions and amino acid sidechains are shown in ball‐and‐stick and licorice representation, respectively. Gray and blue dashed lines represent the hydrophobic interactions and hydrogen bond, respectively. Right panel: magnification of the channel pore showing amino acid position from structure overlapping and those residues involved in acetate ion interactions marked with blue triangles in (a)
3.4. CrGFY1–5 reporter fusions localize to microbody membranes
Targeting signal prediction analysis performed with ChloroP (Emanuelsson, Nielsen, & Von Heijne, 1999) or TargetP (Emanuelsson, Brunak, von Heijne, & Nielsen, 2007), did not indicate targeting domains of CrGFY1–5 to either algal chloroplast or mitochondria. The values for endoplasmic reticulum (ER) secretory and non‐classical secretion pathways were found to be under the threshold of reliability using both PredAlgo (Tardif et al., 2012) and SecretomeP (Bendtsen, Jensen, Blom, Von Heijne, & Brunak, 2004) suggesting no ER targeting and consequent outer membrane localization.
In order to elucidate the subcellular protein location of CrGFY1–5, we individually amplified the genomic sequences of each CrGFY isoform from ATG to the last amino acid before the stop codon by PCR. Amplified products were cloned into the pOpt_mVenus_Paro vector (Lauersen et al., 2015) in order to create N‐ and C‐terminal fusions of the CrGFY proteins to the mVenus yellow fluorescent reporter protein (hereafter, YFP) (Figure 4A). Wide‐field fluorescence microscopy images were taken of single C. reinhardtii cells expressing these fusion proteins (Figure 4B). Fluorescence signals of fusion constructs were observed as small spots spread throughout the cell for all CrGFY variants expressed as either at N‐terminal (Figure 4B, d+i, d+ii, d+iii, d+iv, d+v) or C‐terminal fusions (d+vi, d+vii, d+viii, d+ix, d+x). YFP signals matched patterns of a malate synthase 1 (MAS1) fluorescent‐PTS1 control construct (Lauersen, Willamme, et al., 2016), which was previously characterized to be localized to the algal peroxisomes (Figure 4B, b). Transformations were carried out using a recipient strain expressing CFP (Lauersen et al., 2015) (mCerulean3) targeted to the ER by the cCA secretion signal and containing a C‐terminal reticulum endoplasmic‐retention signal KDEL* (Figure 4B, d). The YFP fluorescence patterns derived from GFY fusions clearly differentiated from ER‐localized CFP signal, further suggesting the GFY proteins were targeted to peroxisomal microbodies and not the ER. CrGFY‐linked fluorescence signals were observed on the inner side of the chloroplast but were distinctly different from the soluble CFP of the ER. The ER‐localized CFP protein was only detectable in soluble protein fractions (Figure 4C, left) analyzed by western blot and immunodetection while CrGFY chimeric YFP fusion products were found enriched in total membrane preparations (Figure 4C, right).
Figure 4.
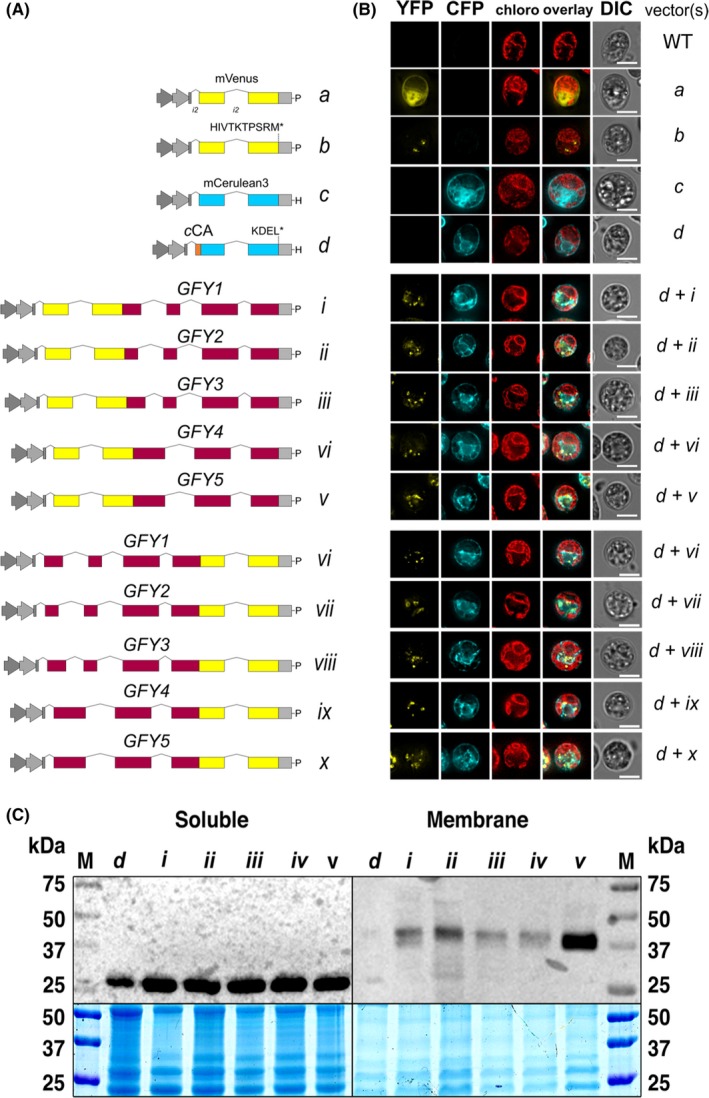
In vivo CrGFY1–5 protein localization and immunoblotting analyses. (A) Schematic overview of vectors used for CrGFY‐fluorescent reporter expression in this study: (a) cytosolic YFP and (b) peroxisomal YFP containing the PTS1 corresponding to the MAS1_PTS1 malate synthase C‐terminal peptide of C. reinhardtii (HIVTKTPSRM*) (Lauersen, Willamme, et al., 2016); (c) cytosolic CFP and (d) CFP carrying 20 amino acid secretion signal from CrCAH1 enzyme (cCA) and the KDEL* peptide at its C‐terminus for ER retention. Two gray arrows represent the hybrid promoter HSP70‐RBCS2; a gray box depicts the RBCS2 3′UTR; selection markers conferring resistance to hygromycin B (H) or paromomycin (P) are indicated; RBCS2 introns 1 and 2 of the pOpt vectors are depicted from left to right by thin bridge lines in vectors a–d. Vectors for expression of YFP fusion proteins cloned at N‐terminal (i–v) or C‐terminal position (vi‐x) of CrGFY1–5 proteins, native introns are indicated by thin bridge lines. (B) Fluorescence microscopy images of individual selected clones, the corresponding vector(s) used to transform strains are indicated to the left of each image. WT – parental strain UVM4. Channels and false colors are as follows: mVenus (YFP, yellow) and mCerulean3 (CFP, cyan), chlorophyll autofluorescence (chloro, red), the merged previous three channels (overlay), and the differential interference contrast (DIC). Scale bars represent 5 μm. (C) Left: immunoblotting analysis with anti‐GFP antibody detects ~28 kDa signals in the soluble extracts corresponding to CFP expressed and retained in the ER (30 μg protein per lane). Right: ~45 kDa signals were detected in the membrane fractions representing the YFP‐GFY fusion proteins (60 μg protein per lane). Individual lines used in microscopy were used in this analysis indicated by the lane labels corresponding to the vector constructs in A. M ‐ protein ladder marker
In order to verify the peroxisomal localization of the CrGFY proteins, we performed another round of transformations, using a citrate synthase 2 (CIS2) fluorescent‐PST2 recipient strain. CIS2 is known to localize in peroxisomes due to its role in the glyoxylate cycle (Lauersen, Willamme, et al., 2016). Like in Figure 4, fluorescent signals of fusion constructs were observed as small spots spread throughout the cell for all CrGFY variants (Figure 5). However, none of them colocalized with the glyoxylate cycle‐containing peroxisomes, regardless of CrGFY isoform considered. This last result suggests that CrGFY proteins are localized in small spherical microbodies, which are surprisingly distinct from the peroxisomes.
Figure 5.
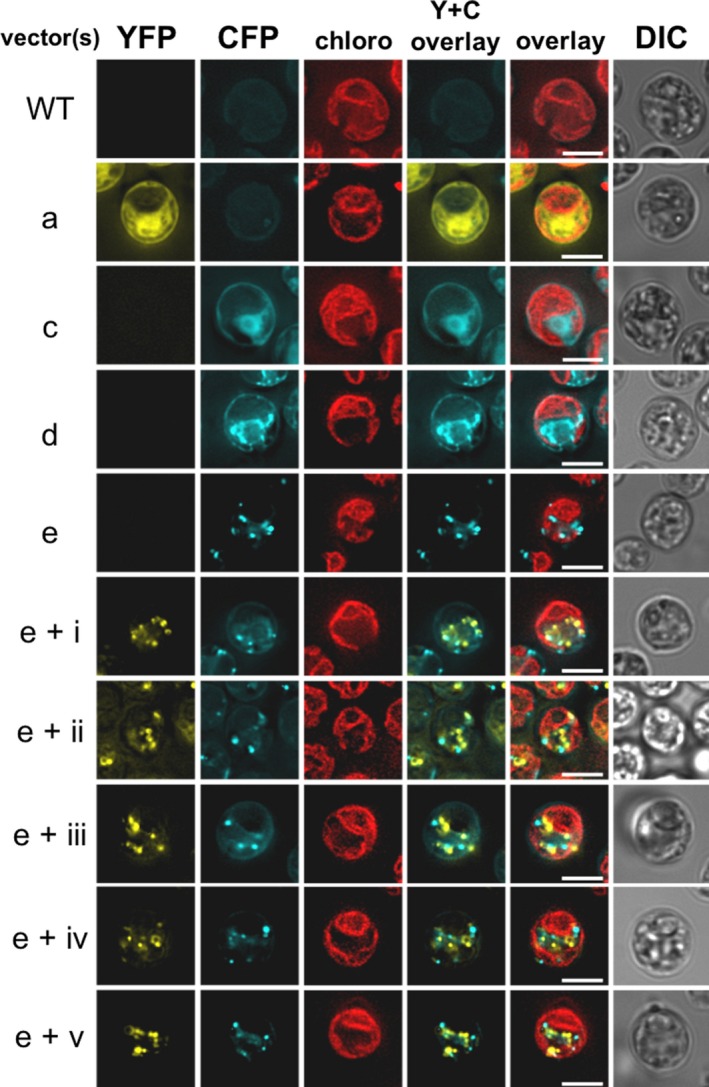
Localization of CrGFY proteins compared to peroxisomal glyoxylate cycle marker CIS2. Vector labels and fluorescent reporter channels as presented in Figure 4. New vector ‘e’ was generated by addition of the CIS2 N‐terminal PTS2 targeting peptide (Lauersen, Willamme, et al., 2016) into vector ‘c’ containing the cyan fluorescent reporter mCerulean3 (CFP). CIS2‐CFP is known to localize in peroxisomes due to its role in the glyoxylate cycle, note the differences in localization patterns of cyan fluorescence between peroxisomes (e) and ER (d). A strain containing CIS2‐CFP was used as a parent for coexpression of CrGFY1–5 proteins tagged with YFP for colocalization analysis. In all instances, YFP signals are observed separate from CIS2‐CFP signals of the glyoxylate cycle‐containing peroxisomes. Scale bar represents 5 μm
4. DISCUSSION
In this study, we analyzed the five GFY isoforms found in C. reinhardtii (CrGFY1–5) and provide evidence for their putative function as channel proteins. CrGFY1–5 isoforms exhibit the hallmarks of GFY protein superfamily and share critical structural features with the bacterial acetate‐succinate channels SatP_Ck (Qiu et al., 2018) and Sat_Ec (Sun et al., 2018). We provide experimental evidence that CrGFY1–5 proteins are localized in microbody membranes and have different expression patterns caused by cultivation conditions although they likely share functional redundancies. Detailed in silico structural analyses performed here contributed to the elucidation of their cellular role and suggest that CrGFY1–5 participate in unidirectional metabolite (likely acetate) trafficking across membranes.
The prokaryotic origin of the algal GFY proteins has been previously predicted to have occurred by a horizontal bacterial gene transfer event, followed by rapid gene evolution in algal lineages (Goodenough et al., 2014). It is probable that the similarity of CrGFY1–5 gene structures and proximity in the algal genome is due to gene duplication events, as previously suggested for the GFY homologs in S. cerevisiae (Ribas et al., 2019). The algal GFY isoforms share only ~30% sequence similarity with their bacterial counterparts SatP_Ck and SatP_Ec; however, homology modeling demonstrates strong conservation of their tertiary structures, especially in the transmembrane domain regions (Figure 3b–d). As acetate metabolism is a key carbon source for green algae such as C. reinhardtii, it is likely that a strong evolutionary pressure exists to maintain structural conformations which enable transport of this molecule across membranes. The structural conservation of CrGFY proteins with SatP_Ck and SatP_Ec is within the threshold of similarity previously reported for conservation of function (Gilson, Marshall‐Christensen, Choi, & Shakhnovich, 2017). Indeed, key residues specifically involved in acetate interactions are conserved between the algal and bacterial channel proteins, suggesting functional similarity (Figure 3a,d). Similar structural conservation has been reported for Gpr1 and Ady2 which both maintain acetate permease activity in two phylogenetically distant yeast species S. cerevisiae and Y. lipolytica (Ribas et al., 2019).
The similarity of core CrGFY1–5 sequences, conserved acetate interaction residues (Figure 1a), and localization to membranes of small spherical microbodies (Figures 4B,C, 5) suggest analogous functions for all five GFY isoforms. It remains unclear why C. reinhardtii has so many GFY genes, as each conceivably could permit unidirectional acetate import into these microbodies. However, multiple GFY genes are present in several organisms, for instance Y. lipolytica exhibits six GPR1 protein homologs (Gentsch & Barth, 2005).
The analysis of gene expression indicates that these isoforms can be separated into two distinct behavioral groups with different expression patterns that do not coincide with their genomic proximity (Figure 2a,b). These expression patterns may indicate involvement of different GFY isoforms with different metabolic pathways/states of the algal cell. CrGFY3–5 were found to expressed more prominently when acetate was present while the expression of CrGFY1–2 was unchanged or even slightly upregulated during anaerobiosis (anoxia/fermentation) (Figure 2a). Coexpression databases indicate that CrGFY3–5 genes are coexpressed with genes related to the glyoxylate cycle, strongly suggesting their involvement in acetate transport dedicated to this cycle, although the transporters do not colocalize with glyoxylate cycle‐containing peroxisomes. CrGFY3–5 were also found coexpressed with genes related to amino acid metabolism. The interrelatedness of acetate metabolism and amino acid biosynthesis is not yet determined in C. reinhardtii; however, Arabidopsis thaliana is known to contain several enzymes of amino acid metabolism in its peroxisomes (Hildebrandt, Nunes Nesi, Araújo, & Braun, 2015). In contrast to CrGFY3–5, CrGFY1–2 exhibited reduced or unchanged expression upon acetate addition, and CrGFY2 exhibited upregulation after 6 h of anaerobiosis (Figure 2a). Anoxic/hypoxic conditions involve the use of alternative acceptors for oxygen along with the fermentative pathways that convert pyruvate, mainly derived from starch breakdown, to different reduced and secreted end products (Yang, Catalanotti, Wittkopp, Posewitz, & Grossman, 2015). This process is usually considered unconnected to glyoxylate cycle. However, a study recently revealed a strong increase in ICL1 transcript and protein along with additional ICL1/MAS1 enzymatic activities under fermentative conditions which were comparable to levels in aerobic conditions (Subramanian et al., 2014). It was proposed that starch and residual acetate, but not fatty acids, may feed glyoxylate cycle under these conditions with the purpose of synthesizing amino acid precursors (Subramanian et al., 2014). CrGFY1–2 isoforms may be active in channeling of acetate in these conditions, or in the organellar metabolic crosstalk which occurs under anoxia.
All the information extracted from the coexpression databases argue for a connection between the microbodies and the glyoxylate cycle‐containing peroxisomes. However, we cannot exclude that these microbodies could also function in interorganellar trafficking between other compartments such as chloroplasts. Indeed, we noticed a close relationship between these microbodies and the chloroplast where their location seemed always to be within the interior cup of the chloroplast where it may share connections to the ER (Figures 4 and 5). Different kinds of microbodies have been described in C. reinhardtii. Microbodies accumulating exogenous fatty acids appear in the cytoplasm after feeding fluorescently labeled fatty acids (Kato, Dong, Bailey, Lum, & Ingram, 2013); electron‐dense vacuoles similar to polyphosphate bodies (acidocalcisomes) have been purified and shown to possess vacuolar proton ATPase (V‐H+‐ ATPase) and H+ ‐PPase activities (Ruiz, Marchesini, Seufferheld, & Docampo, 2001). But to our knowledge, this is the first time that putative acetate transporters have been found to be localized in microbodies. The microbodies detected here thus represent either one type of existing microbody or another kind of these organelles whose role and content are unknown at this time.
In addition to the differential gene regulation, it may be possible that regulation of CrGFY1–5 isoforms also occurs at protein level, mediated by variability in their N‐terminal regions (Figure 1a). CrGFY3–5 were found to have several phosphorylated residues at the N‐terminal regions during cultivation with acetate (Wang et al., 2014) and in dark anaerobic adapted cells compared to photoautotrophic conditions (Bergner et al., 2015). Similar regulation was observed in eukaryotic yeast GFY proteins (Ato2, Ato3, Ady2, S. cerevisiae (Reinders et al., 2007), and GPR1, Y. lipolytica)which contain serine residues phosphorylated in a carbon source‐dependent manner (Gentsch & Barth, 2005). Indeed, many channel families are targets of intracellular signaling pathways, including the regulation by phosphorylation (Davis et al., 2001).
In order to determine whether any of the CrGFY isoforms participated in specific cellular metabolic behaviors, we investigated knockout mutants in GFY1–3 genes. No insertional mutants for GFY4 or 5 were available from the CLiP library, and it was not possible to generate double knockouts by crossing of the available strains due to the proximity of the genes within the genome. Growth analysis of gfy1–3 and fermentation products in gfy1/2 knockout strains did not differ compared to their parental strain (Figure 2c,d). It is possible that the lack of apparent phenotype in these strains indicates a functional redundancy of the other isoforms of this family and that gene expression variations discussed above are irrelevant to cellular function. Future studies using targeted genome editing techniques to generate multiple knockouts in these genes may be able to determine whether specific isoforms have specific cellular roles, however, these investigations are beyond the scope of this report.
The CrGFY1–5 proteins exhibit the distinct hallmarks of GFY superfamily members, such as two key amino acid signatures and six alpha‐helix TM segments (Figure 1a). These features were found in the majority of 355 representative GFY protein sequences (Goodenough et al., 2014) analyzed by prediction servers (Table S1). In both the GFY conserved amino acid signatures, mutations were demonstrated to drastically reduce the flux of acetate across the channel in the Ady2 protein of S. cerevisiae (Ribas et al., 2019), and in the two bacterial channels SatP_Ck (Qiu et al., 2018) and SatP_Ec (Ribas et al., 2019; Sun et al., 2018). Because of the pivotal role involving attraction of substrates to the channel vestibule and stabilization of acetate ions along the inner pore (Qiu et al., 2018) (Figure 3a,d), the correspondence in all the five GFY proteins for these subset of residues would suggest that CrGFY1–5 have very similar substrate selectivity and transport mechanisms. In addition, these algal proteins exhibit conservation of the residues found to be responsible for the hexameric oligomerization observed in SatP_Ck and SatP_Ec proteins (Qiu et al., 2018; Sun et al., 2018), suggesting that similar secondary folding could be expected as well as the oligomeric organization (Figure 3a). Here, in vivo chimeric protein localization to microbody membranes of all five CrGFY isoforms and their predicted structural similarity to bacterial GFY proteins argue in favor of a channel allowing a passive yet selective transport of acetate (and potentially other substrates), across the membrane. Our modeling prediction indicates that GFY proteins have both N‐ and C‐terminal regions facing the cytosol which would commit the GFY channels to import of acetate, based on similarities to SatP_Ck (Qiu et al., 2018). Analyses of SatP_Ec channel conductance suggest a bidirectional transport across the membrane (Sun et al., 2018), which cannot be ruled‐out for the algal GFY isoforms. Indeed, the direction of metabolite transport mediated by CrGFY isoforms as well as the selectivity for acetate will need to be elucidated in future experiments.
The capacity for acetate metabolism has undoubtedly played a role in the evolution of C. reinhardtii as well as other eukaryotic microbes. For this green alga, acetate is the only exogenous carbon source able to be used for growth other than carbon dioxide. However, the mechanisms and subcellular compartmentalization of this metabolism are only now receiving increased research interest. The mechanisms by which acetate uptake occurs for the algal cell remain unclear. Here, we provided evidence that a family of five GFY proteins with structural homology to bacterial acetate channels is found in microbody membranes of C. reinhardtii. It is likely that these protein channels facilitate selective metabolite transport into these organelles, although their function is unknown. Although these proteins exhibit high structural similarity with one another, it remains unclear if individual isoforms participate in distinct metabolic states, organellar cross talk, or cellular pathways. Future research will be needed to determine individual transport properties, potential functional redundancies, and directionality in metabolite transport mechanisms of this protein family. This work will encourage increased investigation of metabolite transport mechanisms in algal cells.
CONFLICT OF INTEREST
The authors declare no conflict of interest associated with the work described in this manuscript.
AUTHOR CONTRIBUTION
LD performed the research, analyzed the data, and wrote the manuscript; WH performed the research; KL performed the research, analyzed the data, and wrote the manuscript; CR designed the research, analyzed the data, and wrote the manuscript.
Supporting information
ACKNOWLEDGMENTS
LD is a recipient of a fellowship of the Fund for research training in industry and agriculture (FRIA). CR acknowledges FRS‐FNRS (CDR J.0265.17). We thank Dr. M. Hanikenne and A. Corato (University of Liege) for help with RT‐qPCR experiments and HPLC analyses, respectively, and M. Radoux (University of Liege) for expert technical assistance. This work has been supported by the technology platform and infrastructure at the Center for Biotechnology (CeBiTec) of Bielefeld University (KL).
Durante L, Hübner W, Lauersen KJ, Remacle C. Characterization of the GPR1/FUN34/YaaH protein family in the green microalga Chlamydomonas suggests their role as intracellular membrane acetate channels. Plant Direct. 2019;00:1–14. 10.1002/pld3.148
The authors responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors are: L. Durante (Lorenzo.Durante@uliege.be) and C. Remacle (c.remacle@uliege.be).
References
- Aoki, Y. , Okamura, Y. , Ohta, H. , Kinoshita, K. , & Obayashi, T. (2016). ALCOdb: gene coexpression database for microalgae. Plant and Cell Physiology, 57, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augstein, A. , Barth, K. , Gentsch, M. , Kohlwein, S. D. , & Barth, G. (2003). Characterization, localization and functional analysis of Gpr1p, a protein affecting sensitivity to acetic acid in the yeast Yarrowia lipolytica. Microbiology, 149, 589–600. [DOI] [PubMed] [Google Scholar]
- Bailey, T. L. , Johnson, J. , Grant, C. E. , & Noble, W. S. (2015). The MEME suite. Nucleic Acids Research, 43, W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen, J. D. , Jensen, L. J. , Blom, N. , Von Heijne, G. , & Brunak, S. (2004). Feature‐based prediction of non‐classical and leaderless protein secretion. Protein Engineering, Design & Selection, 17, 349–356. [DOI] [PubMed] [Google Scholar]
- Bergner, S. V. , Scholz, M. , Trompelt, K. , Barth, J , Gäbelein, P , Steinbeck, J , … Hippler, M (2015). STATE TRANSITION7‐dependent phosphorylation is modulated by changing environmental conditions, and its absence triggers remodeling of photosynthetic protein complexes. Plant Physiology, 168, 615–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Analytical Biochemistry, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Davis, M. J. , Wu, X. , Nurkiewicz, T. R. , Kawasaki, J. , Gui, P. , Hill, M. A. , & Wilson, E. (2001). Regulation of ion channels by protein tyrosine phosphorylation. American Journal of Physiology, Heart and Circulatory Physiology, 281, H1835–H1862. [DOI] [PubMed] [Google Scholar]
- Elbourne, L. D. H. , Tetu, S. G. , Hassan, K. A. , & Paulsen, I. T. (2017). TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Research, 45, D320–D324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson, O. , Brunak, S. , von Heijne, G. , & Nielsen, H. (2007). Locating proteins in the cell using TargetP, SignalP and related tools. Nature Protocols, 2, 953–971. [DOI] [PubMed] [Google Scholar]
- Emanuelsson, O. , Nielsen, H. , & Von Heijne, G. (1999). ChloroP, a neural network‐based method for predicting chloroplast transit peptides and their cleavage sites. Protein Science, 8, 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentsch, M. , & Barth, G. (2005). Carbon source dependent phosphorylation of the Gpr1 protein in the yeast Yarrowia lipolytica. FEMS Yeast Research, 5, 909–917. [DOI] [PubMed] [Google Scholar]
- Gilson, A. I. , Marshall‐Christensen, A. , Choi, J. M. , & Shakhnovich, E. I. (2017). The role of evolutionary selection in the dynamics of protein structure evolution. Biophysical Journal, 112, 1350–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough, U. , Blaby, I. , Casero, D. , Gallaher, SD , Goodson, C , Johnson, S , … Wulan, T (2014). The path to triacylglyceride obesity in the sta6 strain of Chlamydomonas reinhardtii . Eukaryotic Cell, 13, 591–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, E. H. (1989). The Chlamydomonas sourcebook. Elsevier Inc.. [Google Scholar]
- Hildebrandt, T. M. , Nunes Nesi, A. , Araújo, W. L. , & Braun, H. P. (2015). Amino acid catabolism in plants. Molecular Plant, 8, 1563–1579. [DOI] [PubMed] [Google Scholar]
- Johnson, X. , & Alric, J. (2013). Central carbon metabolism and electron transport in chlamydomonas reinhardtii: Metabolic constraints for carbon partitioning between oil and starch. Eukaryotic Cell, 12, 776–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H. Y. , Fraser, M. , Li, W , McAnulla, C. , … Hunter, S. (2014). InterProScan 5: Genome‐scale protein function classification. Bioinformatics, 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll, L. , Krogh, A. , & Sonnhammer, E. L. L. (2004). A combined transmembrane topology and signal peptide prediction method. Journal of Molecular Biology, 338, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Källberg, M. , Wang, H. , Wang, S. , Peng, J. , Wang, Z. , Lu, H. , & Xu, J. (2012). Template‐based protein structure modeling using the RaptorX web server. Nature Protocols, 7, 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, N. , Dong, T. , Bailey, M. , Lum, T. , & Ingram, D. (2013). Triacylglycerol mobilization is suppressed by brefeldin A in Chlamydomonas reinhardtii . Plant and Cell Physiology, 54, 1585–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, L. , Mezulis, S. , Yates, C. , Wass, M. , & Sternberg, M. (2015). The Phyre2 web portal for protein modelling, prediction, and analysis. Nature Protocols, 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg, H.L. , & Krebs, H.A. (1957). Synthesis of cell constituents from C2‐units by a modified tricarboxylic acid cycle. Nature, 179, 988–991. [DOI] [PubMed] [Google Scholar]
- Kornberg, H. L. , & Madsen, N. B. (1957). Synthesis of C4‐dicarboxylic acids from acetate by a “glyoxylate bypass” of the tricarboxylic acid cycle. Biochimica et Biophysica Acta, 24, 651–653. [DOI] [PubMed] [Google Scholar]
- Kunze, M. , & Hartig, A. (2013). Permeability of the peroxisomal membrane: Lessons from the glyoxylate cycle. Frontiers in Physiology, 4, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze, M. , Pracharoenwattana, I. , Smith, S. M. , & Hartig, A. (2006). A central role for the peroxisomal membrane in glyoxylate cycle function. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1763, 1441–1452. [DOI] [PubMed] [Google Scholar]
- Lauersen, K. J. , Baier, T. , Wichmann, J. , Wördenweber, R. , Mussgnug, J. H. , Hübner, W. , … Kruse, O. (2016). Efficient phototrophic production of a high‐value sesquiterpenoid from the eukaryotic microalga Chlamydomonas reinhardtii . Metabolic Engineering, 38, 331–343. [DOI] [PubMed] [Google Scholar]
- Lauersen, K.J. , Kruse, O. , & Mussgnug, J.H. (2015). Targeted expression of nuclear transgenes in Chlamydomonas reinhardtii with a versatile, modular vector toolkit. Applied Microbiology and Biotechnology, 99, 3491–3503. [DOI] [PubMed] [Google Scholar]
- Lauersen, K. J. , Willamme, R. , Coosemans, N. , Joris, M. , Kruse, O. , & Remacle, C. (2016). Peroxisomal microbodies are at the crossroads of acetate assimilation in the green microalga Chlamydomonas reinhardtii . Algal Research, 16, 266–274. [Google Scholar]
- Li, X. , Zhang, R. , Patena, W. , Gang, SS , Blum, SR , Ivanova, N , … Jonikas, MC (2016). An Indexed, mapped mutant library enables reverse genetics studies of biological processes in Chlamydomonas reinhardtii . Plant Cell, 28, 367–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert, J. , Karcher, D. , & Bock, R. (2009). Generation of Chlamydomonas strains that efficiently express nuclear transgenes. Plant Journal, 57, 1140–1150. [DOI] [PubMed] [Google Scholar]
- Newman, S. M. , Boynton, J. E. , Gillham, N. W. , Randolph‐Anderson, B. L. , Johnson, A. M. , & Harris, E. H. (1990). Transformation of chloroplast ribosomal RNA genes in Chlamydomonas: Molecular and genetic characterization of integration events. Genetics, 126, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, A. C. , & Carter, C. J. (2016). The involvement of hybrid cluster protein 4, HCP4, in anaerobic metabolism in Chlamydomonas reinhardtii . PLoS ONE, 11, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva, S. , Devaux, F. , Barbosa, S. , Jacq, C. , & Casal, M. (2004). Ady2p is essential for the acetate permease activity in the yeast Saccharomyces cerevisiae . Yeast, 21, 201–210. [DOI] [PubMed] [Google Scholar]
- Plancke, C. , Vigeolas, H. , Höhner, R. , Roberty, S. , Emonds‐Alt, B. , Larosa, V. , … Remacle, C. (2014). Lack of isocitrate lyase in Chlamydomonas leads to changes in carbon metabolism and in the response to oxidative stress under mixotrophic growth. Plant Journal, 77, 404–417. [DOI] [PubMed] [Google Scholar]
- Qiu, B. , Xia, B. , Zhou, Q. , Lu, Y. , He, M. , Hasegawa, K. , … Liao, J. (2018). Succinate‐acetate permease from Citrobacter koseri is an anion channel that unidirectionally translocates acetate. Cell Research, 28, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders, J. , Wagner, K. , Zahedi, R. P. , Stojanovski, D. , Eyrich, B. , van der Laan, M. , … Meisinger, C. (2007). Profiling phosphoproteins of yeast mitochondria reveals a role of phosphorylation in assembly of the ATP synthase. Molecular & Cellular Proteomics: MCP, 6, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Remacle, C. , Baurain, D. , Cardol, P. , & Matagne, F. (2001). Mutants of Chlamydomonas reinhardtii deficient in mitochondrial complex I: Characterization of two mutations affecting the nd1 coding sequence. Genetics, 158, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas, D. , Soares‐Silva, I. , Vieira, D. , Sousa‐Silva, M , Sá‐Pessoa, J , Azevedo‐Silva, J , … Casal, M (2019). The acetate uptake transporter family motif “NPAPLGL(M/S)” is essential for substrate uptake. Fungal Genetics and Biology, 122, 1–10. [DOI] [PubMed] [Google Scholar]
- Robellet, X. , Flipphi, M. , Pégot, S. , MacCabe, A. P. , & Vélot, C. (2008). AcpA, a member of the GPR1/FUN34/YaaH membrane protein family, is essential for acetate permease activity in the hyphal fungus Aspergillus nidulans . Biochemical Journal, 412, 485–493. [DOI] [PubMed] [Google Scholar]
- Rochaix, J. D. (2002). Chlamydomonas, a model system for studying the assembly and dynamics of photosynthetic complexes. FEBS Letters, 529, 34–38. [DOI] [PubMed] [Google Scholar]
- Ruiz, F. A. , Marchesini, N. , Seufferheld, M. , Docampo, R. , & Govindjee (2001). The polyphosphate bodies of chlamydomonas reinhardtii possess a proton‐pumping pyrophosphatase and are similar to acidocalcisomes. Journal of Biological Chemistry, 276, 46196–46203. [DOI] [PubMed] [Google Scholar]
- Sá‐Pessoa, J. , Paiva, S. , Ribas, D. , Silva, I. J. , Viegas, S. C. , Arraiano, C. M. , & Casal, M. (2013). SATP (YaaH), a succinate–acetate transporter protein in Escherichia coli . Biochemical Journal, 454, 585–595. [DOI] [PubMed] [Google Scholar]
- Shimogawara, K. , Fujiwara, S. , Grossman, A. , & Usuda, H. (1998). High‐efficiency transformation of Chlamydomonas reinhardtii by electroporation. Genetics, 148, 1821–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizova, I. , Fuhrmann, M. , & Hegemann, P. (2001). A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii . Gene, 277, 221–229. [DOI] [PubMed] [Google Scholar]
- Subramanian, V. , Dubini, A. , Astling, D. P. , Laurens, L. M. L. , Old, W. M. , Grossman, A. R. , … Seibert, M. (2014). Profiling chlamydomonas metabolism under dark, anoxic H2‐producing conditions using a combined proteomic, transcriptomic, and metabolomic approach. Journal of Proteome Research, 13, 5431–5451. [DOI] [PubMed] [Google Scholar]
- Sun, P. , Li, J. , Zhang, X. , Xiao, Q. , Zhao, C. , Song, M. , … Deng, D. (2018). Crystal structure of the bacterial acetate transporter SatP reveals that it forms a hexameric channel. Journal of Biological Chemistry, 293, 19492–19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif, M. , Atteia, A. , Specht, M. , Cogne, G , Rolland, N , Brugière, S , … Cournac, L (2012). Predalgo: A new subcellular localization prediction tool dedicated to green algae. Molecular Biology and Evolution, 29, 3625–3639. [DOI] [PubMed] [Google Scholar]
- Tsirigos, K. D. , Peters, C. , Shu, N. , Käll, L. , & Elofsson, A. (2015). The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Research, 43, W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschoppe, K. , Augstein, A. , Bauer, R. , Kohlwein, S. D. , & Barth, G. (1999). Trans‐dominant mutations in the GPR1 gene cause high sensitivity to acetic acid and ethanol in the yeast Yarrowia lipolytica. Yeast, 15, 1645–1656. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Gau, B. , Slade, W. O. , Juergens, M. , Li, P. , & Hicks, L. M. (2014). The global phosphoproteome of Chlamydomonas reinhardtii reveals complex organellar phosphorylation in the Flagella and Thylakoid membrane. Molecular & Cellular Proteomics: MCP, 13, 2337–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, A. , Bertoni, M. , Bienert, S. , Studer, G. , Tauriello, G. , Gumienny, R. , … Schwede, T. (2018). SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W. , Catalanotti, C. , Wittkopp, T.M. , Posewitz, M.C. , & Grossman, A.R. (2015). Algae after dark: Mechanisms to cope with anoxic/hypoxic conditions. Plant Journal, 82, 481–503. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Yan, R. , Roy, A. , Xu, D. , Poisson, J. , & Zhang, Y. (2014). The I‐TASSER Suite: Protein structure and function prediction. Nature Methods, 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials