

Abstract
Background
The increasing frequency and complexity of cancer genomic profiling represents a challenge for the oncology community. Results from next-generation sequencing–based clinical tests require expert review to determine their clinical relevance and to ensure patients are stratified appropriately to established therapies or clinical trials.
Methods
The Sarah Cannon Research Institute UK/UCL Genomics Review Board (GRB) was established in 2014 and represents a multidisciplinary team with expertise in molecular oncology, clinical trials, clinical cancer genetics and molecular pathology. Prospective data from this board were collated.
Results
To date, 895 patients have been reviewed by the GRB, of whom 180 (20%) were referred for clinical trial screening and 62 (7%) received trial therapy. For a further 106, a clinical trial recommendation was given.
Conclusions
Numerous challenges are faced in implementing a GRB, including the identification of potential germline variants, the interpretation of variants of uncertain significance and consideration of the technical limitations of pathology material when interpreting results. These challenges are likely to be encountered with increasing frequency in routine practice. This GRB experience provides a model for the multidisciplinary review of molecular profiling data and for the linking of molecular analysis to clinical trial networks.
Keywords: molecular oncology, molecular tumour board, genomic medicine, clinical genetics
Significance of their study.
What is already known about this subject?
Molecular profiling of routine cancer specimens is both increasingly common and increasingly complex, requiring considerable expertise to distil the most relevant findings and make appropriate recommendations.
What does this study add?
A multidisciplinary genomics review board (GRB) of clinicians is able to make appropriate recommendations regarding the stratification of patients for established therapy, referral to clinical trials, clinical genetics review and advise on further testing.
How might this impact on clinical practice?
The experience of the Sarah Cannon Research Institute GRB provides a framework for the delivery of this service and highlights the challenges frequently encountered when reviewing molecular profiling results from patients with a range of tumour types.
Introduction
The field of cancer genomics has progressed rapidly in recent years with a growing use of molecular profiling, partly driven by an increasing accessibility to commercial and academic providers and the evolution from single-gene assays to massively parallel DNA sequencing, also known as next-generation sequencing (NGS).1–3 The implementation of NGS use for routine clinical samples is a significant step towards the personalisation of cancer medicine and is due to considerable reduction in time and cost of DNA sequencing, allowing for a growing implementation of genomics in cancer diagnosis and prediction of response to therapy, via the identification of molecular alterations within tumour cells which are associated with targeted cancer treatments such as protein kinase inhibitors and monoclonal antibodies.4 5 The growing use of NGS poses a challenge on how to properly interpret the vast output of data, merge it with existing knowledge databases and translate it into valid therapeutic decisions.6 7 Clinically, there is a need for increasing genomic literacy—one study of 160 physicians in a tertiary care National Cancer Institute–designated comprehensive cancer centre reported 22% of physicians having low confidence in genomic knowledge.8 As adoption rates of incorporating NGS technology into clinical practice continue to rise,9 there is a need for expert analysis of NGS results to assess the use of these data in the context of an individual patient.
The implementation of genomics-driven cancer medicine, including the technical infrastructure required and challenges faced, have been well described, confirming broad implementation of NGS testing in a clinical setting is feasible.10–12 For example, at Sarah Cannon Research Institute (SCRI) US, 936 patients with advanced cancers underwent NGS to guide clinical trial selection—of the 103 patients who were enrolled on clinical trials—50 of these patients were enrolled onto clinical trials matched to mutational status.13 NGS technologies typically generate far larger volumes of data than is usual in clinical tests and these data require expert interpretation if these data are to be used appropriately in the clinical setting.
Although well established in the USA, molecular tumour boards (MTBs) are not extensively established in UK practice. Following the completion of the 100K Genomes Project and subsequent establishment of The NHS National Genomic Test Directory, molecular testing of adult solid tumours through genomic medicine hubs using NGS is likely to require increasing MTB input in the management of routine cases.
A Genomics Review Board (GRB) was established at SCRI in August 2014 UK by assembling experts in oncology, clinical genetics and molecular pathology to scrutinise molecular profiling results, setting a standard for quality interpretation. The GRB currently consists of GRB physicians, a molecular oncologist, a clinical geneticist, a molecular pathologist and a GRB coordinator (see table 1). The SCRI UK GRB interprets molecular profiling results for clinicians, thoroughly assesses the validity of the results alongside patient medical information, including a review, where relevant, of the variant allele frequency (VAF) of detected variants in relation to tumour content in the pathology sample. It also identifies potential patients for clinical trials and makes recommendations where appropriate for clinical genetics review.
Table 1.
Members of the Genomics Review Board (GRB) and responsibilities
| Referring Physician | Responsible for identifying patients for review by the GRB, completing the GRB Patient Referral Form and providing supportive information relevant to the assessment of the patient |
| GRB Physician | Responsible for assessing clinical information provided on patients referred by the Referring Physician and making recommendations as to the suitability of the aforementioned patients for referral to SCRI/other UK trial sites for clinical trial eligibility assessment |
| GRB Molecular Oncologist | Responsible for assessing molecular information provided on patients referred by the Referring Physician and making recommendations as to the suitability of the aforementioned patients for referral to SCRI/other UK trial sites for clinical trial eligibility assessment |
| GRB Co-ordinator or Delegate | Responsible for liaising with the Referring Physician to ensure all information provided on referred patients is complete and correct, coordination of the GRB meeting, completion of the GRB Feedback Form, entry of data in the GRB Database |
| GRB Clinical Geneticist | Responsible for advising appropriate onward genetic counselling and testing based on molecular profiling results and clinical information |
| GRB Molecular Pathologist | Responsible for reviewing pathology reports and for liasing between the GRB and the SCMD molecular pathology laboratory |
SCMD, Sarah Cannon Molecular Diagnostics; SCRI, Sarah Cannon Research Institute.
This report represent the prospective analysis of cases reviewed by a solid tumour GRB over its first 45 months of activity. The breakdown of cases reviewed, profiling outcomes for those tested in the local molecular pathology laboratory, recommendations given and clinical trial recruitment are described. In light of this GRB experience, the challenges faced in bridging molecular profiling findings with making recommendations for patient therapy are discussed.
Methods
All cases reviewed by the GRB from 11 August 2014 to the cut-off date of 7 May 2018 were prospectively assessed and included in the cohort. Data were collated regarding tumour type, the molecular profiling test reviewed, recommendations given by the GRB and clinical trial recruitment where available. Additional data regarding driver mutations and samples effected by formalin artefact were collated for all cases where multigene panel testing had been performed in the local molecular diagnostic laboratory associated to the GRB.
GRB process
Molecular profiling reports were submitted to the GRB coordinator, either directly from treating physicians or as part of the genetic testing pathway from the healthcare provider. Concise patient clinical summaries and family histories where appropriate were collated for GRB review, and were distributed to the GRB membership alongside NGS reports weekly for review the following working day by teleconference. The GRB molecular oncologist examined the results in the context of the patient’s clinical and family history, specimen tumour content, detected variants and the VAF. These were also reviewed by the GRB clinical geneticist to determine whether the patient requires clinical genetics review in light of the detected variants and the wider clinical context. The variants were evaluated, with reference to the published literature and conference abstracts, Catalogue of Somatic Mutations in Cancer (COSMIC) database and other single-nucleotide polymorphism (SNP) databases such as Exome Aggregation Consortium (ExAC) database, International Agency for Research on Cancer (IARC), the gastrointestinal malignancy variant database InSiGHT, the archive of clinically relevant variants ClinVar and the cancer predisposition gene Variant Database (CAVADA). Reviews of recent conference proceedings and the use of algorithms including Polyphen and Sorts Intolerant From Tolerant (SIFT) algorithms were used to predict whether a particular mutation may be deleterious. This information was then collated and discussed at the GRB, with results fed back to the referring physician, typically the same day, including identification of any actionable mutation targetable by a drug as per the level of evidence for target prioritisation,2 and whether the patient may have be suitable for a clinical trial (see table 2). Where a recommendation was made for clinical genetics referral, this was also fed back through the GRB co-ordinator.
Table 2.
Level evidence scale for target prioritisation—adapted from Andre et al 2
| Level of evidence | A | B | C | Clinical implications |
| I: Molecular alteration validated in several robust early phase trials or at least one phase III randomised trial | Alteration validated in the disease under consideration, targeted therapies have shown to be ineffective in patients who are lacking the genomic alteration | No evidence that the therapy does not work in the absence of the molecular alteration | Level I molecular alteration, but not in the disease under consideration | A/B: Patients must be treated with the targeted therapy C: Patients must enter clinical trials testing the targeted therapy |
| II: Molecular alteration suggested in single and underpowered phase I/II trials | Alteration validated in the disease under consideration, targeted therapies have shown to be ineffective in patients who are lacking the genomic alteration | No evidence that the therapy does not work in the absence of the molecular alteration | Level I molecular alteration, but not in the disease under consideration | Patients must enter clinical trials testing the targeted therapy |
| III : Target suggested by preclinical studies | Preclinical studies include human samples, cell lines and animal models | Preclinical studies that lack either cell lines or animal models | NA | Inclusion in clinical trials is optional |
| IV: Target predicted but lack of clinical or preclinical data | Genomic alteration is a known cancer-related gene | Genomic alteration is not known as cancer-related gene | NA | Inclusion in clinical trials is optional |
NA, not available.
Somatic variant analysis
Multigene panel testing provided at the molecular diagnostic facility aligned to the GRB, Sarah Cannon Molecular Diagnostics (SCMD), was performed using the CE-IVD marked Oncomine solid tumour NGS panel (Life Technologies, Carlsbad, California, USA) run on the IonTorrent Personal Genome Machine (PGM). All specimens were received as formalin-fixed, paraffin-embedded tissue blocks, unstained slides or tissue curls. All specimens received as blocks and slides underwent preanalytical pathological assessment to estimate tumour cell fraction and to ensure there was sufficient material to process for DNA extraction. Samples over 5% tumour cell fraction were accepted for analysis. Following DNA extraction, the target regions were amplified, barcoded and then sequenced on the PGM. Data were analysed using a hybrid bioinformatics pipeline incorporating both Torrent Suite VariantCaller (V.5.0.4.0) and in-house developed software. This pipeline ultimately generated draft clinical reports which assigned an analytical status to >5000 hotspot variants. Hotspot variants were defined as unique coding variants in the COSMIC database (V.79) which mapped to the gene panel. Prior to manual review of the draft clinical reports, all variants above 2.5% VAF which met required quality thresholds were classified as detected. Variants below 1% VAF were classified as not detected, and variants between 1% and 2.5% VAF or those not meeting the necessary quality thresholds (regardless of VAF) were classed as equivocal variants. Prior to report authorisation and despatch, all reports underwent manual review by two independent assessors, during which the status of any variant could be reassigned if appropriate. Manual review also included the flagging of cases where the background level transition variants was indicated that certain observed variants may represent formalin fixation artefacts. Clinically appropriate comments were included in reports in light of the tumour type, detected variants, incidental finding and any potentially relevant variants whose status remained equivocal. Turnaround time for this assay is typically between 7 and 8 working days.14
In view of the heterogeneous group of patients receiving different targeted drugs and without any standard comparator for the respective disease, we have not formally assessed outcome.
Results
A total of 895 patients were prospectively reviewed as part of the GRB process over the 45 months studied. Non-small cell lung cancer and colorectal cancer were the most common tumour types submitted and in combination these accounted for over half of the patients reviewed. An extensive breakdown of the tumour types reviewed by the GRB is given in table 3.
Table 3.
Tumour type breakdown of cases reviewed by the Genomics Review Board
| Tumour type | Cases | % of total |
| Lung | 276 | 30.8 |
| Colorectal | 198 | 22.1 |
| Breast | 81 | 9.1 |
| Melanoma | 48 | 5.4 |
| Ovarian | 32 | 3.6 |
| Carcinoma of unknown primary | 31 | 3.5 |
| Not specified in clinical details | 27 | 3.0 |
| Cholangiocarcinoma | 27 | 3.0 |
| Pancreatic | 21 | 2.3 |
| Endometrial | 18 | 2.0 |
| Cervical | 13 | 1.5 |
| Bladder | 12 | 1.3 |
| Oesophageal | 11 | 1.2 |
| Gastric | 9 | 1.0 |
| Gastro-oesophageal junction | 7 | 0.8 |
| Anal | 6 | 0.7 |
| Renal | 6 | 0.7 |
| Gallbladder | 5 | 0.6 |
| Glioma | 5 | 0.6 |
| Oral | 5 | 0.6 |
| Prostate | 4 | 0.4 |
| Uterine | 4 | 0.4 |
| Hepatocellular | 3 | 0.3 |
| Leiomyosarcoma | 3 | 0.3 |
| Parotid | 3 | 0.3 |
| Salivary gland | 3 | 0.3 |
| Thymus | 3 | 0.3 |
| Ureter | 3 | 0.3 |
| Vulval | 3 | 0.3 |
| Appendiceal | 2 | 0.2 |
| Fallopian | 2 | 0.2 |
| Mesothelioma | 2 | 0.2 |
| Retroperitoneal sarcoma | 2 | 0.2 |
| Urachal carcinoma | 2 | 0.2 |
| Adrenal | 1 | 0.1 |
| Ampullary | 1 | 0.1 |
| Chondrosarcoma | 1 | 0.1 |
| Duodenal | 1 | 0.1 |
| Fibromyxosarcoma | 1 | 0.1 |
| Lacrimal gland | 1 | 0.1 |
| Myxofibrosarcoma | 1 | 0.1 |
| Nasopharynx | 1 | 0.1 |
| Optic nerve sheath | 1 | 0.1 |
| Osteoblastic chondrosarcoma | 1 | 0.1 |
| Osteosarcoma | 1 | 0.1 |
| Penile | 1 | 0.1 |
| Peripheral nerve sheath | 1 | 0.1 |
| Pneumocytoma | 1 | 0.1 |
| Kaposi sarcoma | 1 | 0.1 |
| Thyroid | 1 | 0.1 |
| Tracheal | 1 | 0.1 |
| Vaginal | 1 | 0.1 |
Of the 895 patients reviewed, 91 patients had multiple molecular profiling results reviewed, with a maximum of 8, and the majority of these 91 patients were discussed at more than one GRB meeting. Seventy-six of the patients discussed had a recommendation made regarding an established therapy, based on genomic profiling results (8.5%). A total of 180 patients (20.1%) were referred to specific clinical trials at either SCRI (166) or UCLH (14) and a further 10 patients were referred to other trial sites. Of the 180, 58 were screened for trial at SCRI with 32 subsequently enrolled onto and treated on a trial at SCRI. Five were screened at UCLH, with three enrolled and treated as part of a trial. A further 19 patients were treated on a trial at SCRI at a later time point, of whom 9 were enrolled onto the trial on the basis of their molecular profiling results. This gives a total of 73 patients enrolled on trials after GRB review.
Eight patients were not eligible for trials but were granted compassionate access to trial therapy at SCRI following GRB review. In total, therefore, 62 patients (7%) discussed at GRB received a trial therapy following GRB review.
For a further 106 patients, a general trial recommendations was included in the GRB report submitted to their clinician, but without a specific trial being identified. Due to the practicalities of the GRB operation and the lack of robust mechanisms for follow-up of many of the patients reviewed, it was not possible to determine how many of these recommendations led to trial referral and recruitment.
Clinical genetics referral was advised in 117 patients (13.1%) and further somatic molecular testing was advised in 94 patients (10.5%). The full breakdown of patients is given in figure 1.
Figure 1.

Consort diagram of the stratification of patients reviewed by the Genomics Review Board (GRB) for standard therapy, to trials for targeted therapies and general recommendations returned to the treating clinician. In total, 62 patients received trial therapy (8 under compassionate access programmes). 19 of these patients were enrolled to trial directly following on from the GRB as a result of variants detected from molecular profiling. SCRI, Sarah Cannon Research Institute.
Of all results reviewed by the SCRI GRB, the majority of reports were issued by SCMD, most of which used a diagnostic multigene panel (71%) and a smaller fraction single gene tests (4%). Of the gene panels, 38 of these represented a combined DNA and RNA somatic mutation and fusion panel. No positive fusion events were detected in this cohort. Other reports reviewed by the GRB were issued from Guardant ctDNA (9%), Foundation Medicine (8%), Caris (3%) and the remainder issued from a range of other providers (5%).
Of the cases reported at SCMD, the proportion of cases showing driver mutations in the most commonly affected genes are shown in figure 2.
Figure 2.
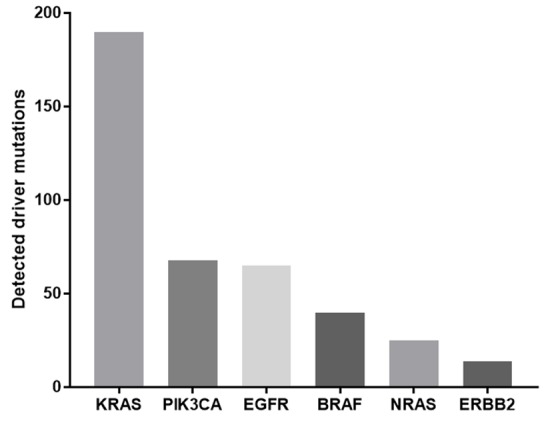
Number of reported cases with driver mutations in commonly affected cancer genes reported from Sarah Cannon Molecular Diagnostics multigene panel results.
Breakdown of variants in MET and KRAS have been further detailed to exemplify the challenges in GRB analysis of these results, specifically the problems of variants of unknown significance in common drivers, and likely germline variants which are reported as COSMIC-annotated variants in assays where germline DNA is not tested for comparison.
The vast majority of driver mutations reported in KRAS occurred within codons 12 and 13, with other recognised drivers being located in codons 59, 61, 117 and 146. In the GRB review of SCMD reports, four KRAS mutations occurring outside of these codons were reported and were determined to be of uncertain clinical significance (see figure 3).
Figure 3.

Total number of KRAS variants identified and discussed by the Genomics Review Board from all Sarah Cannon Molecular Diagnostics reports. All recognised driver mutations in grey, variants of unknown clinical significance in black. KRAS variants identified as likely formalin fixation artefacts excluded.
Several COSMIC-annotated variants from the reference genome in MET have been reviewed by the GRB following detection on molecular profiling, many of which are commonly reported at a VAF of around 50%, independent of tumour fraction suggesting that they represent germline SNPs, as represented in figure 4. Due to the potential for SNPs to be misinterpreted as functional mutations, careful matching by the GRB with genome reference databases such as ExAC is critical.15
Figure 4.

MET variants identified in Sarah Cannon Molecular Diagnostics multigene panel testing with variant allele frequencies (VAFs). Several of these variants are consistently reported close to 50% VAF, which suggests these are likely single-nucleotide polymorphisms rather than tumour-specific mutations.
Of the 723 profiling results undergoing GRB review which were tested using a multigene panel at SCMD, 88 (12.2%) had evidence of formalin fixation artefacts as was noted in the molecular pathology report. Overfixed samples were characterised by multiple transition variants (C>T, G>A) detected with a VAF above the limit of detection for the assay and not in keeping with the sample tumour fraction. Table 4 shows an example of detected variants from a sample with evidence of formalin fixation artefact. There was marked variation in the proportion of cases from differing institutions showing this signature, ranging from 0% to 26.9% in those centres submitting ≥10 cases, suggesting variation based on tissue processing protocols in different laboratories (see figure 5).
Table 4.
Example of detected variants (COSMIC-annotated coding variants at >2.5% VAF) from a single sample with estimated tumour cell fraction of between 50% and 75%
| Detected variant | Variant allele frequency |
| AKT1 p.(Pro51Leu), c.152 C>T, COSM4468165 | 4% |
| BRAF p.(Gly466Glu), c.1397 G>A, COSM453 | 3% |
| BRAF p.(Asp587Asn), c.1759 G>A, COSM21608 | 3% |
| CTNNB1 p.(Ala21Val), c.62 C>T, COSM1422998 | 3% |
| CTNNB1 p.(Trp25*), c.74 G>A, COSM3593969 | 3% |
| DDR2 p.(Gly584Arg), c.1750 G>A, COSM5446941 | 4% |
| EGFR p.(Glu872Lys), c.2614 G>A, COSM133589 | 3% |
| SMAD4 p.(Ser171Leu), c.512 C>T, COSM5610594 | 3% |
| TP53 p.(His168Arg), c.503 A>G, COSM43545 | 34% |
| TP53 p.(Glu285Lys), c.853 G>A, COSM10722 | 4% |
9 of 10 detected variants are low frequency (<5% VAF) transition variants (C>T or G>A), indicating likely formalin fixation artefact in the sample, with one apparent genuine tumour-specific variant: TP53 p.(His168Arg).
VAF, variant allele frequency.
Figure 5.

Proportion of cases demonstrating formalin-fixed, paraffin-embedded artefact from the six main referring institutions referring samples to Sarah Cannon Molecular Diagnostics which were then discussed at the Genomics Review Board (range, 0%–26.9%).
Discussion
A number of challenges exist in correctly translating molecular profiling results for clinical implementation, making expert GRB review a vital process in patient stratification.
Identification of potential germline variants and variants of unknown clinical significance
Most somatic variant analysis assays do not include parallel germline testing alongside tumour samples as part of the analysis, and this is not common practice currently in the UK, although there are some exceptions.16 17 The MSK-IMPACT study reported ‘presumed pathological germline variants’ in 15.7% of patients with advanced cancer.18 Germline testing should ideally be performed following genetic counselling as results can have wide-ranging implications for patients and their families. Traditionally, germline testing is typically limited to those for whom the tumour in question is high risk for a specific genetic predisposition syndrome, such as in the Lynch screening pathway, or those with a relevant family history. Suspicious germline variants which may be clinically relevant are however frequently detected by somatic variant analysis, presenting with variant allele frequencies between 40% and 60%, reflecting germline heterozygosity. These may be germline variants seen frequently in the population, with no implications for cancer risk (SNPs), rarer variants with an unknown effect on risk or those which have been described as being relevant to cancer susceptibility.
This consideration highlights the importance of reporting tumour fraction and VAF in somatic variant analysis reports, as included in the SCMD multigene assays, or ideally analysing germline DNA in parallel. The GRB references online databases to determine whether a given mutation is likely to represent a somatic alteration frequently identified in cancer specimens or a germline mutation.
In a study conducted by Meric-Bernstam et al which consisted of 1000 patients undergoing somatic mutation screening using a 202-gene panel, pathogenic germline variants (PGVs) were identified in 43 patients of which 23 were previously unrecognised and the remaining 20 variants were known to be pathogenic. The study also showed that 99% of patients indicated interest in being notified of these findings, and a protocol of determining whether the PGV results are significant and to be returned to the patient and the family, with genetic counselling, was established.19 20
It is crucial to consider the level of risk a particular PGV is likely to carry.20 For example, some PGVs have a well-described association with an increased risk of cancer, while others are less well understood in their effect on cancer predisposition and consideration of an experienced GRB clinical geneticist is crucial for appropriate recommendations to be made.
Other variants which pose a challenge are variants of uncertain clinical significance (VUCSs). These are variants which are reported in cancer-associated genes, which appear to be genuine (ie, non-artefactual), but are not located in codons with well-described functional relevance. These variants may or may not confer a oncogenic effect on the relevant gene.21–23 Recently published guidelines will provide an important resource in enabling newly established GRBs to classify reported variants appropriately.24 25
Established driver KRAS mutations are predictors of resistance to anti-EGFR therapy in both lung and colorectal cancer. Where the KRAS mutation is of uncertain clinical significance with no functional evidence in the literature of pathogenicity, the GRB cannot positively warrant refraining from anti-EGFR therapy options. VUCSs can also be found in cancer predisposition genes and this requires a decision to be made as to whether a VUCS should be followed up with confirmatory testing of the germline.
As molecular profiling becomes even more prevalent and population-level tumour genomic profiling data are available, the role of many variants whose clinical significance is currently unknown is likely to be elucidated.
Tumour heterogeneity
Tumour heterogeneity presents another challenge in determining whether a given mutation is a relevant driver in oncogenesis or is a subclonal event, arising de novo in an expanding tumour, reflecting the branched clonal evolution of the tumour.3 26 As an example, in EGFR-driven lung cancer, subclonal T790M mutation occurs as a resistance mechanism to EGFR tyrosine kinase inhibitors following treatment, representing a subclonal mutation with crucial clinical relevance. Subclonal mutations may show lower variant allele frequencies than clonal mutations, although any difference may depend on the size of the subclone, local DNA copy number aberrations, and how this relates to the resection or biopsy specimen which has been sampled in the sequencing assay. Since the content of the tumour can change rapidly with time, it is helpful to compare molecular profiling of samples collected recently with any previous results to identify the emergence of subclonal mutations. Indeed, comparison of primary and metastatic tumour samples profiled at different time points can also help confirm that they are clonally related and do not represent dual primary tumours. The GRB determines the likelihood that a particular mutation might be subclonal and the relevance of this to selection of treatment. Significant changes can be seen between specimens taken from the same tumour at different time points and from different biopsy sites.
Fixation artefacts
Surgical pathology specimens are routinely fixed in formalin, processed through alcohol and embedded in paraffin to aid histopathological processes and to preserve the tissue indefinitely. The majority of tumour molecular profiling is currently performed on specimens which have been processed in this way. Formalin fixation has a range of effects on DNA and RNA including deamination of nucleotides causing artefactual transition variants (cytosine to thymine and guanine to adenine). Specimens which have been fixed in formalin for longer show much greater numbers of transition type variants27 and in somatic variant analysis fixation artefacts can be falsely interpreted as genuine somatic mutations. Overfixed specimens usually demonstrate multiple background mutations with VAFs <10% on multigene molecular profiling. The GRB identifies cases with this signature and interprets molecular profiling results with these considerations in mind such that GRB reports reflect this issue.
Identifying actionable mutations and feedback of results
If an oncogenic somatic mutation has been identified, the GRB is required to determine whether this can be effectively targeted by a therapeutic agent. Some activating mutations are established predictors of standard of care targeted therapies, while other mutations identified from cancer genomic profiling can potentially stratify patients for clinical trials and the GRB has the ability to link these patients with relevant clinical trials, with rates of trial referrals and trial recruitment detailed above. Although follow-up in this cohort is not entirely comprehensive, specifically the breadth of referrals making it impossible to know how many general trial recommendations led to recruitment and the lack of robust outcome data for those patients reviewed, the size of the cohort and the detailed local trial recruitment and clinical genetic referral data do highlight the potential for stratifying patients uniformly and effectively using molecular profiling data.
Where a driver mutation is identified which is not a marker of response to a standard of care treatment, the availability of a suitable clinical trial and the logistics of linking patients with the centres offering these trials present further challenges for GRBs and co-ordinators of patients with cancer. However, with an increasing use of GRBs in clinical institutions, supported by the UK NHS Genomic Medicine Service recommendation for Genomic Tumour Advisory Boards to be established across NHS Trusts as an integral part of the cancer pathway, it should be possible to expand the communication between molecular profiling laboratories, clinics and clinical trial centres, allowing patients greater access to suitable clinical trials targeting their specific mutations. In addition, in UK practice, this may help to identify patients suitable for therapy outside of either through compassionate access programmes or the NHS Cancer Drug Fund.
At present, molecular profiling is not well integrated into routine pathology in the UK and as such comprehensive review of patients typically requires the manual collation of reports from across unconnected laboratories. The GRB co-ordinator is therefore crucial in ensuring that the clinically relevant data are available. Ideally, pathology reporting systems would allow for combined reporting, able to ensure that all relevant information for patients can be delivered as fully integrated pathology reports.
The data extracted from this large cohort do question the need for applying broad NGS panels as a routine test and the limited number of actionable alterations, even within the clinical trials environment, suggests that using more focused NGS panels for first-line molecular profiling is more appropriate.
Molecular profiling reports vary widely between providers and reports from some providers can include large quantities of data which may not be clearly presented, making these complex and difficult for clinicians to interpret. The GRB serves to help clinicians interpret these results, discerning between therapeutically relevant or potentially relevant mutations and those which are not relevant to therapy, as well as making any relevant clinical genetics recommendations. As the GRB network expands and becomes more interconnected with a growing number of referrers using it on a regular basis, GRBs will need the facility to communicate in order to share practices and experience. Ultimately, the GRB makes molecular profiling results clear and accessible to the treating physician and patient, increasing the implementation and use of NGS in clinical practice.
Conclusion
The SCRI UK/UCL GRB provides an example of a framework which can be implemented for proper use of molecular profiling in clinical practice and in referral to clinical trials by generating a system for communication between physicians, molecular laboratories and study sites. The interpretation of NGS molecular profiling and generation of appropriate clinical recommendations requires a multidisciplinary team with expertise in the analysis of sequencing data, clinical cancer genetics and clinical trials. Patients as consumers are increasingly aware of the availability of such testing platforms, and their perceptions and expectations need to be adequately managed.9
The challenge of distinguishing between somatic and germline mutations in tumour samples may eventually lead to the parallel sequencing of tumour samples and matched germline samples as routine in molecular diagnostics in collaboration with clinical genetics to follow-up results from these analyses. Eventual prospective clinical validation will be needed for cost–benefit analysis of employing parallel testing as standard in clinical practice however, and at present the standard of testing the tumour sample alone needs to be interpreted appropriately with clinical genetics expertise on GRBs.
As the SCRI UK GRB network expands, the communication channels between clinics and study sites will increase awareness of study availability, giving patients greater access to appropriate clinical trials.
SCMD is now introducing a combined mutation and gene fusion panel and has an additional 50-gene panel assay, which will provide further data on causal and potentially actionable mutations and oncogenic fusion events. With a growing availability of trials and widened molecular profiling, we foresee a greater number of patients coming through the GRB to be enrolled in clinical trials and an increasing need for GRB review to optimise cancer patient stratification.
Footnotes
DAM and MK contributed equally.
Contributors: DAM, HW, MF, AK, CS and H-TA contributed to Genomic Review Board discussions. HW, TC, MV, MM and NR-M reviewed clinical cases. DAM collated molecular diagnostic data. MK collated clinical data. KB extracted molecular diagnostic data. DAM, MK, GM and H-TA wrote the manuscript. HW, PB, MF, AK, DH, CS and H-TA reviewed the manuscript. H-TA conceived the project and had oversight throughout.
Funding: The clinical work evaluated was funded as part of the healthcare pathway for these patients. No specific funding was sought to evaluate and collate these data.
Competing interests: PB has participated in ThermoFisher’s European Clinical Oncology Advisory Board meetings, and has delivered sponsored, but not for profit, presentations at the invitation of ThermoFisher UK on a number of occasions. CS reports grant support from Cancer Research UK, UCLH Biomedical Research Council, Rosetrees Trust and AstraZeneca. Personal fees from Boehringer Ingelheim, Novartis, Eli Lilly, Roche Ventana, GlaxoSmithKline, Pfizer, Genentech and Celgene. Stock options in GRAIL, APOGEN Biotechnologies and EPIC Bioscience and has stock options and is cofounder of Achilles Therapeutics.
Patient consent for publication: Not required.
Ethics approval: Ethical approval was not sought for this project. The project represents prospective service evaluation of a genomic review board in clinical practice and as such it was not deemed appropriate to seek ethics approval for this work.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: The data used were collated clinical report data which are not publicly available.
References
- 1. Lewin J, Siu LL. Cancer genomics: the challenge of drug accessibility. Curr Opin Oncol 2015;27:250–7. 10.1097/CCO.0000000000000185 [DOI] [PubMed] [Google Scholar]
- 2. Andre F, Mardis E, Salm M, et al. . Prioritizing targets for precision cancer medicine. Ann Oncol 2014;25:2295–303. 10.1093/annonc/mdu478 [DOI] [PubMed] [Google Scholar]
- 3. Tran B, Dancey JE, Kamel-Reid S, et al. . Cancer genomics: technology, discovery, and translation. JCO 2012;30:647–60. 10.1200/JCO.2011.39.2316 [DOI] [PubMed] [Google Scholar]
- 4. Ciardiello F, Arnold D, Casali PG, et al. . Delivering precision medicine in oncology today and in future—the promise and challenges of personalised cancer medicine: a position paper by the European Society for Medical Oncology (ESMO). Ann Oncol 2014;25:1673–8. 10.1093/annonc/mdu217 [DOI] [PubMed] [Google Scholar]
- 5. Garraway LA, Verweij J, Ballman KV. Precision oncology: an overview. J Clin Oncol 2013;31:1803–5. 10.1200/JCO.2013.49.4799 [DOI] [PubMed] [Google Scholar]
- 6. Mardis ER. The translation of cancer genomics: time for a revolution in clinical cancer care. Genome Med 2014;6 10.1186/gm539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tyner JW. Functional genomics for personalized cancer therapy. Sci Transl Med 2014;6 10.1126/scitranslmed.3009586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gray SW, Hicks-Courant K, Cronin A, et al. . Physicians' attitudes about multiplex tumor genomic testing. JCO 2014;32:1317–23. 10.1200/JCO.2013.52.4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hall MJ, Forman AD, Montgomery SV, et al. . Understanding patient and provider perceptions and expectations of genomic medicine. J Surg Oncol 2015;111:9–17. 10.1002/jso.23712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meric-Bernstam F, Brusco L, Shaw K, et al. . Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol 2015;33:2753–62. 10.1200/JCO.2014.60.4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meric-Bernstam F, Farhangfar C, Mendelsohn J, et al. . Building a personalized medicine infrastructure at a major cancer center. JCO 2013;31:1849–57. 10.1200/JCO.2012.45.3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meric-Bernstam F, Johnson A, Holla V, et al. . A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst 2015;107 10.1093/jnci/djv098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bauer TM, Spigel DR, Ma Z, Arkenau H-T, et al. . Next-generation sequencing (NGS) in 936 patients (PTS) with advanced cancers to prospectively guide clinical trial selection: the SARAH Cannon Research Institute (SCRI) experience. Journal of Clinical Oncology 2014;32(15_suppl). 10.1200/jco.2014.32.15_suppl.2534 [DOI] [Google Scholar]
- 14. Moore DA, Balbi K, Ingham A, et al. . Analysis of a large cohort of non-small cell lung cancers submitted for somatic variant analysis demonstrates that targeted next-generation sequencing is fit for purpose as a molecular diagnostic assay in routine practice. J Clin Pathol 2018;71:1001–6. 10.1136/jclinpath-2018-205319 [DOI] [PubMed] [Google Scholar]
- 15. Jung H, Bleazard T, Lee J, et al. . Systematic investigation of cancer-associated somatic point mutations in SNP databases. Nat Biotechnol 2013;31:787–9. 10.1038/nbt.2681 [DOI] [PubMed] [Google Scholar]
- 16. Zehir A, Benayed R, Shah RH, et al. . Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–13. 10.1038/nm.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Middleton G, Crack LR, Popat S, et al. . The National Lung matrix trial: translating the biology of stratification in advanced non-small-cell lung cancer. Ann Oncol 2015;26:mdv394–9. 10.1093/annonc/mdv394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schrader KA, Cheng DT, Joseph V, et al. . Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol 2016;2:104–11. 10.1001/jamaoncol.2015.5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mak G, Moschetta M, Arkenau HT. Reporting incidental germline variants in the context of day-to-day somatic genomic profiling. Ann Oncol 2016. [DOI] [PubMed] [Google Scholar]
- 20. Meric-Bernstam F, Brusco L, Daniels M, et al. . Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann Oncol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Domchek SM, Bradbury A, Garber JE, et al. . Multiplex genetic testing for cancer susceptibility: out on the high wire without a net? J Clin Oncol 2013;31:1267–70. 10.1200/JCO.2012.46.9403 [DOI] [PubMed] [Google Scholar]
- 22. Van Allen EM, Wagle N, Levy MA. Clinical analysis and interpretation of cancer genome data. JCO 2013;31:1825–33. 10.1200/JCO.2013.48.7215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dienstmann R, Rodon J, Barretina J, et al. . Genomic medicine frontier in human solid tumors: prospects and challenges. J Clin Oncol 2013;31:1874–84. 10.1200/JCO.2012.45.2268 [DOI] [PubMed] [Google Scholar]
- 24. Mateo J, Chakravarty D, Dienstmann R, et al. . A framework to RANK genomic alterations as targets for cancer precision medicine: the ESMO scale for clinical Actionability of molecular targets (ESCAT). Ann Oncol 2018;29:1895–902. 10.1093/annonc/mdy263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li MM, Datto M, Duncavage EJ, et al. . Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19:4–23. 10.1016/j.jmoldx.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gray SW, Park ER, Najita J, et al. . Oncologists' and cancer patients' views on whole-exome sequencing and incidental findings: results from the CanSeq study. Genet Med 2016;18:1011–9. 10.1038/gim.2015.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2ddcafb6-2da9-4b89-86b7-236dec9909c3.2ddcafb6-2da9-4b89-86b7-236dec9909c3. Wong SQ, Li J, Tan AY-C, et al. . Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics 2014;7 10.1186/1755-8794-7-23 [DOI] [PMC free article] [PubMed] [Google Scholar]