

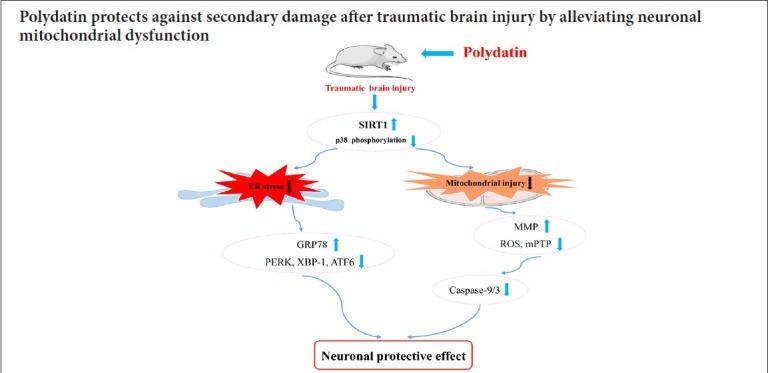
Keywords: nerve regeneration, traumatic brain injury, polydatin, mitochondria, endoplasmic reticulum stress, SIRT1, reactive oxygen species, p38, mitochondrial membrane potential, mitochondrial permeability transition pore, lateral fluid percussion, neural regeneration
Abstract
Polydatin is thought to protect mitochondria in different cell types in various diseases. Mitochondrial dysfunction is a major contributing factor in secondary brain injury resulting from traumatic brain injury. To investigate the protective effect of polydatin after traumatic brain injury, a rat brain injury model of lateral fluid percussion was established to mimic traumatic brain injury insults. Rat models were intraperitoneally injected with polydatin (30 mg/kg) or the SIRT1 activator SRT1720 (20 mg/kg, as a positive control to polydatin). At 6 hours post-traumatic brain injury insults, western blot assay was used to detect the expression of SIRT1, endoplasmic reticulum stress related proteins and p38 phosphorylation in cerebral cortex on the injured side. Flow cytometry was used to analyze neuronal mitochondrial superoxide, mitochondrial membrane potential and mitochondrial permeability transition pore opened. Ultrastructural damage in neuronal mitochondria was measured by transmission electron microscopy. Our results showed that after treatment with polydatin, release of reactive oxygen species in neuronal mitochondria was markedly reduced; swelling of mitochondria was alleviated; mitochondrial membrane potential was maintained; mitochondrial permeability transition pore opened. Also endoplasmic reticulum stress related proteins were inhibited, including the activation of p-PERK, spliced XBP-1 and cleaved ATF6. SIRT1 expression and activity were increased; p38 phosphorylation and cleaved caspase-9/3 activation were inhibited. Neurological scores of treated rats were increased and the mortality was reduced compared with the rats only subjected to traumatic brain injury. These results indicated that polydatin protectrd rats from the consequences of traumatic brain injury and exerted a protective effect on neuronal mitochondria. The mechanisms may be linked to increased SIRT1 expression and activity, which inhibits the p38 phosphorylation-mediated mitochondrial apoptotic pathway. This study was approved by the Animal Care and Use Committee of the Southern Medical University, China (approval number: L2016113) on January 1, 2016.
Chinese Library Classification No. R453; R364
Introduction
Traumatic brain injury (TBI) is the leading cause of death and severe disability in trauma patients. Moreover, TBI is becoming a serious public health problem and TBI events are gradually increasing worldwide (Menon et al., 2010; Lyeth, 2016; Li et al., 2019a). In Europe, TBI resulted in approximately 2.1 million hospital discharges and 82,000 deaths in 2012 (Majdan et al., 2016). In the United States, approximately 2.5 million people each year sustain a TBI, resulting in 282,000 hospitalizations and 56,000 deaths (Taylor et al., 2017). In China, overall TBI-related mortality has remained at a relatively high level, ranging from 12.99 to 17.06 cases per 100,000 people between 2006 and 2013 (Cheng et al., 2017). Moreover, TBI survivors typically face prolonged disruption to normal personal, family and work functions (Lyeth, 2016). Patients with severe TBI experience disabilities, which often require extensive rehabilitation (Wagner and Kumar, 2018; Bown et al., 2019). Because the pathophysiological mechanisms of TBI are complex and unclear, effective pharmacotherapies for TBI are lacking. Thus, there is increasing need to elucidate the mechanisms underpinning TBI pathophysiology and to develop treatments and interventions for TBI patients.
TBI events include both primary and secondary injuries (Hiebert et al., 2015). Mechanical displacement of brain tissue at the injury moment causes the primary insult. Therefore, the primary injury is almost impossible to treat (Gajavelli et al., 2015). TBI-caused secondary brain injury has become the main focus of much research. There are many potential intervention targets to explore and select for the treatment of TBI because it occurs gradually, developing after hours or even days. Secondary injury involves several cellular processes including reactive oxygen species generation, mitochondrial dysfunction, endoplasmic reticulum stress, vascular abnormalities, neuroinflammation, apoptosis, and excitotoxicity (Cao et al., 2016; Sun et al., 2016; Yang et al., 2017; Tan et al., 2018; Corbett et al., 2019; Lee et al., 2019). Mitochondria illustrate the negative effect of TBI secondary effects (Gajavelli et al., 2015; Wang et al., 2015; Yang et al., 2017; Fraunberger et al., 2019). TBI causes neuronal mitochondrial dysfunction, as indicated by the accumulation of reactive oxygen species in the mitochondria and decreased mitochondrial membrane potential (MMP), resulting in mitochondrial permeability transition pore (mPTP) opening (Yang et al., 2017; Tan et al., 2018). It has been shown that silent information regulator family protein 1 (SIRT1) activation and p38 mitogen activated protein kinase (MAPK) pathway inhibition can exert a neuroprotective effect by preventing secondary injury resulting from TBI (Zhao et al., 2012; Yang et al., 2017). Endoplasmic reticulum stress occurs before the mitochondrial damage after secondary brain injury post-TBI (Sun et al., 2017; Tan et al., 2018). Thus, intervention in these mechanisms could provide a new therapeutic strategy for TBI.
Polydatin (3,4′,5-trihydroxystibene-3-monoglucoside) is a monocrystalline drug that can be isolated from a traditional Chinese medicine herb, Polygonum cuspidatum (Wang et al., 2013; Li et al., 2015b; Liao et al., 2018). Polydatin protects the mitochondria of neuronal cells, hepatocytes, myocardial and arterial smooth muscle cells after severe ischemia/reperfusion injury (Wang et al., 2012, 2013; Li et al., 2015b; Ling et al., 2016). Polydatin has been identified as a potential therapy in sepsis-induced multiple organ dysfunction syndrome to alleviate mitochondria damage in the liver, kidney, lung and intestine (Li et al., 2014, 2015b; Zeng et al., 2015a, b). Notably, its effect is superior to resveratrol, which is another well-known mitochondrial protector (Wang et al., 2012, 2013; Li et al., 2015b; Ni et al., 2017). Like resveratrol, polydatin is also a SIRT1 activator and can reinstate SIRT1 activity in the intestine after sepsis, in pulmonary arteriolar smooth muscle cells during hypoxia/reoxygenation, as well as in the kidney and hepatocytes after hemorrhagic injury (Li et al., 2015b, 2017; Zeng et al., 2015b). However, the effect of polydatin on neuronal mitochondria following TBI-induced secondary brain injury has not been investigated.
This present study aimed to explore the protective effect of polydatin on TBI-induced secondary brain injury, and to clarify the relationship of polydatin with SIRT1 and the p38 MAPK pathway.
Materials and Methods
Animals
A total of 148 specific-pathogen-free male Sprague-Dawley rats aged 12 weeks and weighing 220–250 g were housed individually under controlled environmental conditions with a 12-hour light/dark cycle and unrestricted access to pellet food and water. The animals were purchased from the Experimental Animal Center of the Southern Medical University in China (certification number: SYXK (Yue) 2016-0167). All surgical interventions were performed under anesthesia with a mixture of 13.3% urethane and 0.5% chloralose (0.65 mL/100 g body weight, intraperitoneally). All efforts were made to reduce the number of animals used and to minimize animal discomfort. This study was approved by the Animal Care and Use Committee of the Southern Medical University, China (approval number: L2016113) on January 1, 2016. The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No 85-23, revised 1985).
Lateral fluid percussion brain injury
Surgical procedures were performed as previously described (Chen et al., 2013; Yang et al., 2017; Tan et al., 2018). Briefly, anesthetized rats were placed in a stereotaxic frame. After incision of the scalp, the temporal muscles were reflected and a 4.8 mm craniotomy was drilled (2.5 mm lateral to the sagittal sinus and centered between bregma and lambda). A hollow female Luer-Lok fitting was placed directly over the dura and rigidly fixed using dental cement. Prior to the induction of trauma, the female Luer-Lok was connected to the fluid percussion injury device via a transducer (Biomedical Engineering Facility, Medical College of Virginia, USA). For the infliction of TBI, a metal pendulum was released from a pre-selected height, leading to a rapid injection of normal saline into the closed cranial cavity. The pulse of increased intracranial pressure of 21–23 ms duration was controlled and recorded by an oscilloscope (Agilent 54622D, MEGAZoom, Germany). The severity of injury inflicted on the animal was altered by adjusting the amount of pressure generated by the pendulum. For the present experiment, a severe injury level was induced (3.5 ± 0.2 atm) (Chen et al., 2013; Tan et al., 2018). Sham animals underwent identical preparatory procedures, including craniotomy, but were not injured.
Experimental groups and drug administration
The first experiment analyzed ROS production, mitochondrial injury-related indicators, ER stress and apoptosis-related proteins. The rats were randomized into the sham group, the TBI group, the TBI + SRT1720 group and the TBI+ polydatin group. SRT1720 (SIRT1 agonist, 20 mg/kg; Merck Millipore, Boston, MA, USA) was intraperitoneally injected immediately following TBI (Khader et al., 2017). Polydatin (30 mg/kg; Haiwang, Shenzhen, China) was intraperitoneally injected immediately following TBI (n = 18/group) (Wang et al., 2013). Each rat injured-side cortex was isolated at 6 hours post-TBI. The second experiment analyzed neurological score, including a sham-treated control group, as well as groups sacrificed at 6, 12, 24, 72, and 168 hours post-TBI (n = 6/group). The third experiment analyzed survival. The rats were randomized into the sham group, the TBI group, the TBI + SRT1720 group and the TBI + polydatin group (n = 10/group).
Isolation of rat cortical neurons
Neurons are sensitive to hypoxic ischemia and require constant immersion in Dulbecco’s Modified Eagle’s minimal essential medium (DMEM) during neuronal isolation (Brewer and Torricelli, 2007; Ray et al., 2009). To rapidly isolate cortical neurons from the injured side of the rat brain, we followed a previously described method with some minor modifications. After the TBI procedures, the injured-side cortex was cut into fragments, and cells were dissociated by incubation for 30 minutes at 37°C with 2 mg/mL papain in DMEM. The immune adherence method was used to achieve a pure cell population. Cell suspensions were poured into anti-neural cell adhesion molecule-coated petri dishes (Millipore, Boston, MA, USA) and placed on a shaker for 1 hour, after which the adhered cells were collected. Trypan blue was used to identify and exclude nonviable cells.
Mitochondrial reactive oxygen species measurement
Neurons were isolated, as described above, from the cortex on the injured side at 6 hours post-TBI. Mitochondrial reactive oxygen species (O2 –.) was detected using the fluorescent MitoSOX probe (Molecular Probes, Carlsbad, CA, USA). Neurons were incubated with 4 µM MitoSOX (red fluorescence) for 30 minutes at 37°C in the dark. The fluorescence intensities of reactive oxygen species (O2–.) probes were analyzed by flow cytometry (BD FACSVerse™; BD, Franklin, NJ, USA). Data were analyzed by BD FAC Suite software (BD FACSVerse™).
Malondialdehyde (MDA) determination
The injured-side cortices from 6 rats were isolated at 6 hours post-TBI. The cortex tissue was placed into a 1.5 mL centrifuge tube. 250 μL of RIPA buffer was added with protease inhibitors. Homogenate was then centrifuged at 11,000 × g for 10 minutes at 4°C. MDA content was determined using a thiobarbituric acid assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The optical density at 532 nm was measured using a spectrophotometer. 1,1,3,3-Tetramethoxypropane was used as an external standard. MDA level is expressed as nanomoles per milligram of protein.
Superoxide dismutase activity (SOD) determination
The injured-side cortices from rats were isolated at 6 hours post-TBI. The cortex tissue was placed into a 1.5 mL centrifuge tube. 250 μL of RIPA buffer was added with protease inhibitors. Homogenate was then centrifuged at 11,000 × g for 10 minutes at 4°C. SOD activity of the cortex tissue was determined using Xanthine Oxidase Assay Kits (Nanjing Jiancheng Bioengineering Institute) and expressed as units per milligram of protein.
SIRT1 deacetylase activity detection
SIRT1 deacetylase activity of the isolated injured-side cortical tissue was performed as previously described using the Cyclex SIRT1/Sir2 Deacetylase Fluorometric Assay Kit (Catalog number CY-1151; Cyclex, Nagano, Japan) (Li et al., 2015b). Briefly, the cortex tissue (100 mg) was homogenized in 500 μL of immunoprecipitation buffer (T-PER Tissue Protein Extraction Reagent 78510; Pierce, Rockford, IL, USA). For immunoprecipitation assay of SIRT1, primary antibody against SIRT1 (10 μg) was incubated overnight with precleared lysates (800 μg) at 4°C before the addition of 20 μL of protein agarose A/G beads (Protein A/G PLUS agarose: sc-2003; Boston, MA, USA). The reactions were rotated for 2 hours at 4°C. The beads were extensively washed and centrifuged followed by a deacetylation assay. The final reaction mixture (50 μL) contained 50 mM Tris-HCl (pH 8.8), 0.5 mM dithiothreitol, 0.25 mAU/mL lysyl endopeptidase, 1 μM trichostatin A, 200 μM NAD+ and 10 μL of tissue extraction. The mixtures were mixed well and incubated for 30 minutes at room temperature. The fluorescence intensity (excitation wavelength 340 nm, emission wavelength 460 nm) was measured using an automatic microplate reader (SpectraMax M5, Shanghai, China).
Neurological score evaluation
Neurological deficiency was assessed by a blinded investigator 6, 12, 24, 72 and 168 hours post-TBI as previously reported (Yang et al., 2017). Behavioral tests included the ability to maintain position on an inclined plane (vertical, right horizontal, and left horizontal positions), left and right forelimb flexion after suspension by the tail, and degree of resistance to right and left lateral pulsion. The seven individual scores, each graded from 0 (severely impaired) to 5 (normal function) according to Faden et al. (1989), were added to yield a composite neurological score ranging from 0 to 35.
MMP detection
Neurons were isolated from the injured-side cortex at 6 hours post-TBI, as described above. The MMP was measured using the fluorescent JC-1 probe (Invitrogen, Carlsbad, CA, USA). In mitochondria with normal membrane potentials, JC-1 forms aggregates that fluoresce red, whereas in damaged, depolarized mitochondria, JC-1 forms monomers that fluoresce green. Isolated neurons were incubated in DMEM containing 5 µM JC-1 for 15 minutes at 37°C. Relative fluorescence was subsequently measured by flow cytometry. Data were analyzed using BD FAC Suite software (Franklin, NJ, USA).
mPTP determination
Neurons were isolated from the injured-side cortex of each rat at 6 hours post-TBI. The mPTP opening was measured by incubating the isolated neurons at room temperature for 15 minutes in the dark in DMEM containing 1 µM calcein-AM (Invitrogen) and 2 mM CoCl2. Cells were analyzed by flow cytometry (BD FACSVerse™; BD Biosciences, San Jose, CA, USA) to quantify green fluorescence. Resulting mPTP values were determined by BD FACSuite software (BD Biosciences; Franklin Lakes, NJ, USA).
Morphological observation
The rat injured-side cortex was isolated at 6 hours post-TBI. The morphological changes of neuronal mitochondria were observed by transmission electron microscopy (Philips CM10; Philips, Eindhoven, The Netherlands). The injured-side cortex tissues were fixed with 2.5% glutaraldehyde and stained with cacodylate-buffered osmium tetroxide (OsO4). Sections were prepared and examined under an electron microscope (Philips CM10; Philips).
The mitochondria of the injured lateral cerebral cortex were observed under an electron microscope and neuronal mitochondria were semi-quantitatively analyzed according to a previously published method (Flameng et al., 1980). Five fields of view were randomly selected at the same magnification, and approximately 20 mitochondria were randomly selected from each field of view. A total of 80 mitochondria were analyzed per animal. According to the degree of mitochondrial damage, scores of 0–4 grades were made for each mitochondrion (the higher the level, the heavier the damage). The average score of each group of mitochondria was calculated and compared. The details of Flameng’s scale (Flameng et al., 1980) are listed in Table 1.
Table 1.
Flameng’s scale to evaluate mitochondrial damage
| Grade | Morphological change |
|---|---|
| Level 0 (0 points) | Normal structure |
| Level I (1 point) | The structure is basically normal, but the matrix particles are lost (slight swelling, matrix density is reduced, cristae separation) |
| Level II (2 points) | Mitochondrial swelling (reduced matrix density, cristae separation); matrix is transparent, cristae is not broken |
| Level III (3 points) | Mitochondrial cristae rupture, matrix coagulation (severe swelling) |
| Level IV (4 points) | Mitochondrial cristae rupture, the integrity of the inner and outer membranes disappears, and becomes vacuolated (severe swelling, rupture of the inner and outer membranes) |
Western blot assay
Protein was extracted from the injured side cortical tissue of all treatment groups using a Total Protein Extraction Kit or a Mitochondria Fractionation Kit (Beyotime Institute of Biotechnology, Haimen, China) following the manufacturer’s protocols. The protein concentrations of the extracts were determined using an Enhanced BCA Protein Assay Kit (Beyotime Institute of Biotechnology). Western blot assay was performed as described previously using rabbit monoclonal anti-SIRT1, rabbit polyclonal anti-p-p38, rabbit polyclonal-anti p38, rabbit polyclonal anti-cleaved caspase-9 and caspase-3 (apoptosis related proteins), rabbit monoclonal anti-phospho-PERK, rabbit monoclonal anti-PERK, rabbit monoclonal anti-spliced XBP-1, mouse monoclonal anti-cleaved ATF6, rabbit monoclonal anti-GRP78 (endoplasmic reticulum stress related proteins) (1:1000; all Cell Signaling Technology, Danvers, MA, USA), and mouse monoclonal anti-GAPDH antibodies (1:1000; Abcam, Cambridge, UK). A horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibody was used as the secondary antibody (1:2000; Zhongshan Golden Bridge Biotechnology, Beijing, China). The signal was visualized using enhanced chemiluminescence (Pierce, Rockford, IL, USA). Band optical density values were determined using Gel-Pro Analyzer Software (version 4.0, Media Cybernetics, Silver Spring, MD, USA). Quantities of optical densities were normalized using glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Statistical analysis
Data are expressed as the mean ± SD. All data were analyzed for statistical significance using SPSS 20.0 software (IBM, Armonk, NY, USA). All data were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. The Kaplan-Meier method was used to calculate the survival rate of each group and the survival curve was drawn. Results were compared using the log-rank test. A P value < 0.05 was considered statistically significant.
Results
Polydatin upregulates SIRT1 expression and activity post-TBI
As shown in Figure 1A–C, polydatin and SRT1720 increased SIRT1 expression and activity to similar levels in injured cortices after TBI (P < 0.05).
Figure 1.

Polydatin or the SIRT1 activator SRT1720 upregulates SIRT1 expression and activity post-TBI.
TBI was induced by lateral fluid percussion in rats, followed by administration of PD (30 mg/kg, intraperitoneally) or SRT1720 (20 mg/kg, intraperitoneally). The rat injured-side cortex was isolated at 6 hours post-TBI. (A) SIRT1 levels were assessed by western blot assay. (B) Relative band densities of SIRT1. The densities of the protein bands were analyzed and normalized to GAPDH. (C) Relative fluorescence intensity of SIRT1. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. SIRT1: Silent information regulator family protein 1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; PD: polydatin; TBI: traumatic brain injury.
Polydatin suppresses mitochondrial reactive oxygen species accumulation and oxidative stress response post-TBI
To explore whether polydatin suppresses the oxidative stress response by upregulating SIRT1, we treated rats with SRT1702 and polydatin post-TBI. Compared with the sham group, neuronal mitochondrial reactive oxygen species levels were lower in the TBI + polydatin and TBI + SRT1720 groups (P < 0.05; Figure 2A and B). Furthermore, oxidative stress response and homeostasis were restored in the TBI + polydatin and TBI + SRT1720 groups. Treatment with polydatin and SRT1720 inhibited TBI-induced MDA expression and increased SOD levels in injured cortices post-TBI (P < 0.05; Figure 2C and D).
Figure 2.

Polydatin suppresses mitochondrial reactive oxygen species accumulation and oxidative stress response post-TBI.
(A) Neurons were isolated from the rat injured-side cortex, and were labeled with 4 µM MitoSOX for detection of mitochondrial superoxide. Mitochondrial reactive oxygen species (O2 –.) was detected using the fluorescent MitoSOX probe. Flow cytometry was used to analyze TBI-induced mitochondrial superoxide. (B) Quantification of mitochondrial superoxide post-TBI. (C) Quantification of MDA levels in the rat injured-side cortex detected by enzyme linked immunosorbent assay. (D) Quantification of SOD levels in the rat injured-side cortex detected by enzyme linked immunosorbent assay. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. PD: Polydatin; TBI: traumatic brain injury; MDA: malondialdehyde; SOD: superoxide dismutase; SRT1720: SIRT1 activator.
Polydatin attenuates neuronal mitochondrial damage post-TBI
To detect whether polydatin has a protective effect on neuronal mitochondria, the mitochondrial morphology change in the SRT1720 or polydatin treated rats was detected by transmission electron microscope (Figure 3A). In the sham group, neuronal mitochondria showed intact mitochondrial membranes and cristae. In contrast, in the TBI group, mitochondria were swollen and irregularly shaped. The mitochondrial cristae appeared disordered and poorly demarcated. However, both polydatin and SRT1720 treatment showed markedly fewer alterations to neuronal mitochondria. The results were confirmed after the structural damage of neuronal mitochondria was quantified using Flameng’s score (P < 0.05; Figure 3B).
Figure 3.

Polydatin attenuates neuron mitochondrial damage post-TBI.
(A) Ultrastructure of neuronal mitochondria observed by transmission electron microscopy. Scale bar: 500 nm. (a) Normal neuronal mitochondria showed an intact mitochondrial membranes and cristae in the sham group (yellow arrows). (b). In the TBI group, mitochondria were swollen, irregularly shaped; the mitochondrial cristae appeared disordered and poorly demarcated (red arrows). (c and d) In the TBI + SRT1720 (SIRT1 activator) and TBI + PD groups, PD or SRT1720 treatment markedly improved neuron mitochondrial alterations (blue arrows), respectively. (B) Change of neuronal mitochondria structural damage by Flameng’s score. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. PD: Polydatin; TBI: traumatic brain injury.
Polydatin inhibits MMP collapse and mPTP opening post-TBI
We next investigated whether polydatin could prevent TBI-induced neuronal MMP loss through upregulating SIRT1. Because JC-1 fluorescence change from red to green represents mitochondrial depolarization (low MMP) level, the neurons with low MMP could be detected. The percentage of low MMP was 9.7% in the sham group, and increased to 42.7% in the TBI group (P < 0.05; Figure 4Aa, b and B). In contrast, either polydatin or SRT1720 treatment decreased the TBI effect to restore the neuronal MMP (P < 0.05; Figure 4Ac, d and B). Calcein-AM was loaded into cells that were simultaneously incubated with 1 mM CoCl2, which quenches calcein fluorescence in the cytosol but not in mitochondria, because Co2+ ions have difficultly penetrating the inner membrane of mitochondria. However, if mPTP opening occurs Co2+ can enter the mitochondria, resulting in quenching of the matrix calcein fluorescence. Flow cytometry results showed that the green calcein fluorescence intensity of mitochondria in calcein-AM + Co2+ exposed cells decreased in the TBI group but remained higher after treatment with polydatin and SRT1720 (P < 0.05; Figure 5 Ac, d and B).
Figure 4.

Polydatin inhibits the collapse of mitochondrial membrane potential post-TBI.
(A) Neurons were isolated from the injured-side cortices of rats, and were labeled with 5 µM JC-1 to detect mitochondrial depolarization by flow cytometry (low mitochondrial membrane potential), upper right: normal cells (JC-1 aggregates), low right: injured cells (JC-1 monomer), upper and low left: undetected by JC-1. (B) Percentage of mitochondrial membranes with low potential visualized with JC-1 fluorescence. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. PD: Polydatin; TBI: traumatic brain injury; JC-1: tetrachloro-tetraethylbenzimidazolocarbocyanine iodide; SRT1720: SIRT1 activator.
Figure 5.

Polydatin inhibits mitochondrial permeability transition pore opening post-TBI.
(A) Neurons were isolated from the injured-side cortices of rats, and the opening of mitochondrial permeability transition pore was detected by calcein-AM and CoCl2 staining with green fluorescence, and tested by flow cytometry. (B) Green fluorescence intensity of mitochondria. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. PD: Polydatin; TBI: traumatic brain injury; SRT1720: SIRT1 activator.
Polydatin inhibits p38 MAPK signaling and mitochondrial apoptotic pathway
Since SIRT is an upstream factor involved in p38 phosphorylation initiation of neuronal mitochondria damage and apoptosis (Zhao et al., 2012; Yang et al., 2017), we asked whether polydatin could inhibit p38 phosphorylation by upregulating SIRT1. Both polydatin and SRT1720 treatments markedly suppressed TBI-induced p38 phosphorylation (Figure 6A and B). Cleaved caspase-9 and -3 expression was dramatically reduced in the TBI + SRT1720 and TBI+ polydatin groups, indicating inhibition of the mitochondrial apoptotic pathway (Figure 6A, C and D).
Figure 6.

Polydatin inhibits p38 MAPK signaling and the mitochondrial apoptotic pathway post-TBI.
(A) Western blot assay of p-p38, p38, cleaved caspase-9 and -3. (B) Relative band densities of p-p38 protein: The densities of the protein bands were analyzed and normalized to p38. (C, D) Relative band densities of cleaved caspase-9 and -3 proteins, respectively: The densities of the protein bands were analyzed and normalized to GAPDH. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. PD: Polydatin; TBI: traumatic brain injury; p38 MAPK: p38 mitogen activated protein kinase; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; SRT1720: SIRT1 activator.
Polydatin inhibits the endoplasmic reticulum stress response post-TBI
In addition to mitochondrial damage, TBI induces endoplasmic reticulum stress, then triggers the unfolded protein response cascade, which leads to secondary damage post-TBI. Therefore, we tested whether polydatin alleviated the endoplasmic reticulum stress response in injured cortices post-TBI. Endoplasmic reticulum stress related unfolded protein activation was markedly reduced in the TBI + SRT1720 and TBI + polydatin groups, including suppressed PERK phosphorylation, decreased spliced XBP-1 and cleaved ATF6 expression, and increased GRP78 expression (P < 0.05; Figure 7).
Figure 7.

Polydatin inhibits the endoplasmic reticulum stress response post-TBI.
(A) Western blot assay of p-PERK, PERK, spliced XBP-1, cleaved ATF6, and GRP78. (B) Relative band densities of p-PERK protein: The densities of the protein bands were analyzed and normalized to PERK. (C–E) Relative band densities of spliced XBP-1, spliced cleaved ATF6 and GRP78 proteins, respectively: The densities of the protein bands were analyzed and normalized to GAPDH. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. PD: Polydatin; TBI: traumatic brain injury; p-PERK: phosphorylated protein kinase R-like endoplasmic reticulum kinase; PERK: protein kinase R-like endoplasmic reticulum kinase; XBP-1: X box-binding protein-1; ATF6: activating transcription factor 6; GRP78: glucose-regulated protein 78; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; SRT1720: SIRT1 activator.
Polydatin increases neurological scores and survival duration in TBI rats
To investigate whether polydatin promoted the recovery of neurological score in TBI rats, we monitored neurological function at different indicated times post-TBI (6, 12, 24, 72 and 168 hours). Treatment with polydatin and SRT1720 both restored neurological score within 168 hours post-TBI (P < 0.05; Figure 8A). Moreover, although a log-rank test suggested that there was no significant overall difference among these groups, polydatin and SRT1720 treatment both reduced rat mortality from 50% to 40% post-TBI (log-rank test, χ2 = 6.281, P = 0.099 > 0.05; HR: 1.657, 95%CI: 0.943–2.911; Figure 8B).
Figure 8.

Polydatin improves neurological score and increases survival duration of rats post-TBI.
(A) Neurological scores were evaluated at different times (6, 12, 24, 72 and 168 hours post-TBI; n = 6; one-way analysis of variance followed by Tukey’s multiple-comparisons test). (B) Kaplan-Meier program for survival duration of rats in each group. All data are expressed as the mean ± SD (n = 10; log-rank test). *P < 0.05, vs. sham group. PD: Polydatin; TBI: traumatic brain injury; SRT1720: SIRT1 activator.
Discussion
TBI is a major socioeconomic problem and a common cause of death and disability worldwide (Menon et al., 2010; Lyeth, 2016). Due to the untreatable nature of the TBI primary injury, the subsequent secondary brain injury has become the main focus of most research to explore potential intervention targets for treatment of TBI (Chiu and Hinson, 2017). We recently found that SIRT1 upregulation has a protective effect on neuronal mitochondria, preventing the secondary damage after TBI (Yang et al., 2017). In cerebral ischemia/reperfusion injury, evidence suggests that polydatin can exert its neuroprotective effect because it crosses the blood-brain barrier (Cheng et al., 2006; Su and Hsieh, 2011; Ruan et al., 2019). Furthermore, polydatin is a SIRT1 activator and can reinstate SIRT1 activity as demonstrated in the kidney and hepatocytes after hemorrhagic injury (Li et al., 2015b; Zeng et al., 2016). Here, whether polydatin could upregulate neuronal SIRT1 expression and reinstate SIRT1 activity post-TBI was tested by treating rats with a SIRT1 activator (SRT1720) or polydatin after inducing TBI. The results showed that polydatin noticeably promotes SIRT1 expression and activity, indicating that polydatin has a neuroprotective role in preventing secondary damage via upregulation of SIRT1 expression and activity post-TBI.
Mitochondria are critical to maintaining neuronal homeostasis through a myriad of intricate processes (Gajavelli et al., 2015). The main cause of secondary deleterious cascades in TBI is neuronal damage, which is closely linked to mitochondrial function (Boeck et al., 2013; Wang et al., 2013, 2015). Reactive oxygen species can directly cause mitochondrial damage and the opening of mPTP (Song et al., 2011; Li et al., 2015b). Furthermore, TBI causes neuronal mitochondrial dysfunction, as a result of reactive oxygen species accumulation in mitochondria, decreased MMP and mPTP opening (Yang et al., 2017; Khatri et al., 2018; Tan et al., 2018). Various studies have demonstrated that polydatin is a mitochondrial protector that maintains mitochondrial function in the kidney, small intestine, liver, neurons, ventricular myocytes and smooth muscle cells after hemorrhagic injury or sepsis (Wang et al., 2012, 2015; Li et al., 2015b; Zeng et al., 2015a, b, 2016). In this study, polydatin showed clear mitochondrial protective effects, by preventing mitochondrial reactive oxygen species accumulation, alleviating ultrastructural damage of neuronal mitochondria, restoring MMP and inhibiting mPTP opening. Moreover, the mitochondrial protective effect of polydatin might be associated with increased expression and activity of SIRT1 post-TBI.
MAPK cascades, for instance the p38 extracellular signal-regulated kinase cascade or c-Jun N-terminal kinase cascades are involved in most brain injuries (Nozaki et al., 2001; Zhang et al., 2015). Several studies have suggested that activated p38 MAPK phosphorylation exerts an essential effect on stress-induced neuronal death; however, both in vivo and in vitro experiments have shown that inhibition of p38 MAPK phosphorylation reduces ischemic brain injury and leads to neuroprotection (Takeda and Ichijo, 2002; Li et al., 2015a). More importantly, TBI induces a significant increase in p38 phosphorylation, whereas a p38 MAPK inhibitor reduces this effect and inhibits activation of the mitochondrial apoptotic pathway (Yang et al., 2017). In addition, the SIRT1 inhibitor activates the p38 phosphorylation-mediated mitochondrial apoptotic pathway after TBI (Yang et al., 2017). Consistent with this, we found that treatment of TBI rats with polydatin or the SIRT1 activator SRT1720 reduced TBI-induced p38 phosphorylation and cleavage of caspase-9 and -3. Therefore, we speculate that polydatin inhibits p38 phosphorylation, which results in mitochondrial apoptosis, through SIRT1 upregulation, thereby providing a protective effect on neuronal mitochondria and blocking secondary brain injury post-TBI. Nonetheless, the underlying mechanism requires further investigation.
Endoplasmic reticulum stress is clearly linked to the fates of neurons after brain injury (Logsdon et al., 2014; Gulyaeva, 2015; Go et al., 2017). Pathological and behavioral deficits resulting from secondary injury cascades after TBI might contribute to endoplasmic reticulum stress, and reducing such stress in the hippocampus could improve learning and memory post-TBI (Dash et al., 2015). Inhibition of endoplasmic reticulum stress response could down-regulate proinflammatory cytokine levels and exert neuroprotection post-TBI (Gao et al., 2018). Consistent with this, we found that the major endoplasmic reticulum stress activated unfolded protein response signal transduction pathway initiators PERK-eIF2α-ATF4, IRE1XBP-1, and ATF6 are activated post-TBI, which further damage neuronal mitochondria (Tan et al., 2018). We found that treatment of TBI-rats with polydatin or the SIRT1 activator SRT1720 significantly inhibited the expression of TBI-induced endoplasmic reticulum stress-related proteins. Specifically, p-PERK phosphorylation, spliced XBP-1, and cleaved ATF6 expression were all reduced.
In conclusion, polydatin can protect neuronal mitochondria by suppressing endoplasmic reticulum stress, inhibiting the accumulation of reactive oxygen species in mitochondria, alleviating ultrastructural damage of neuronal mitochondria, restoring MMP and inhibiting mPTP opening. The underlying mechanisms seem to be associated with increased SIRT1 expression and activity, which inhibits the p38 phosphorylation-mediated mitochondrial apoptotic pathway and blocks secondary brain injury post-TBI. Furthermore, polydatin restored neurological scores and reduced the mortality of TBI rats. Therefore, polydatin may be a good choice for mitochondrial protection and treatment of TBI, which will hopefully be confirmed in future clinical trials.
Acknowledgments:
We thank Song Zhou from Zhujiang Hospital, Southern Medical University, China for helping us to establish TBI rat models.
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81501690 (to ZTG); the Scientific Research Staring Foundation for Talent Introduction for Southern Medical University (to MM). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: The study was approved by the Animal Care and Use Committee of the Southern Medical University, China (approval number: L2016113) on January 1, 2016.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This study was supported by the National Natural Science Foundation of China, No. 81501690 (to ZTG); the Scientific Research Staring Foundation for Talent Introduction for Southern Medical University (to MM).
C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Dawes EA, de Souza M, Qiu Y, Song LP; T-Editor: Jia Y
References
- 1.Boeck CR, Carbonera LS, Milioli ME, Constantino LC, Garcez ML, Rezin GT, Scaini G, Streck EL. Mitochondrial respiratory chain and creatine kinase activities following trauma brain injury in brain of mice preconditioned with N-methyl-D-aspartate. Mol Cell Biochem. 2013;384:129–137. doi: 10.1007/s11010-013-1790-8. [DOI] [PubMed] [Google Scholar]
- 2.Bown D, Belli A, Qureshi K, Davies D, Toman E, Upthegrove R. Post-traumatic stress disorder and self-reported outcomes after traumatic brain injury in victims of assault. PLoS One. 2019;14:e0211684. doi: 10.1371/journal.pone.0211684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- 4.Cao Y, Gao Y, Xu S, Bao J, Lin Y, Luo X, Wang Y, Luo Q, Jiang J, Neale JH, Zhong C. Glutamate carboxypeptidase II gene knockout attenuates oxidative stress and cortical apoptosis after traumatic brain injury. BMC Neurosci. 2016;17:15. doi: 10.1186/s12868-016-0251-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen B, Mutschler M, Yuan Y, Neugebauer E, Huang Q, Maegele M. Superimposed traumatic brain injury modulates vasomotor responses in third-order vessels after hemorrhagic shock. Scand J Trauma Resusc Emerg Med. 2013;21:77. doi: 10.1186/1757-7241-21-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng P, Yin P, Ning P, Wang L, Cheng X, Liu Y, Schwebel DC, Liu J, Qi J, Hu G, Zhou M. Trends in traumatic brain injury mortality in China, 2006-2013: apopulation-based longitudinal study. PLoS Med. 2017;14:e1002332. doi: 10.1371/journal.pmed.1002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Y, Zhang HT, Sun L, Guo S, Ouyang S, Zhang Y, Xu J. Involvement of cell adhesion molecules in polydatin protection of brain tissues from ischemia-reperfusion injury. Brain Res. 2006;1110:193–200. doi: 10.1016/j.brainres.2006.06.065. [DOI] [PubMed] [Google Scholar]
- 8.Chiu AW, Hinson HE. Future directions for hypothermia following severe traumatic brian injury. Semin Respir Crit Care Med. 2017;38:768–774. doi: 10.1055/s-0037-1607989. [DOI] [PubMed] [Google Scholar]
- 9.Corbett JM, Ho KM, Honeybul S. Prognostic significance of abnormal hematological parameters in severe traumatic brain injury requiring decompressive craniectomy. J Neurosurg. 2019;8:1–7. doi: 10.3171/2018.10.JNS182293. [DOI] [PubMed] [Google Scholar]
- 10.Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, Tsvetkov AS, Moore AN. Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma. 2015;32:1608–1620. doi: 10.1089/neu.2014.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 12.Flameng W, Borgers M, Daenen W, Stalpaert G. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J Thorac Cardiovasc Surg. 1980;79:413–424. [PubMed] [Google Scholar]
- 13.Fraunberger EA, Shutt TE, Esser MJ. Sex-dependent and chronic alterations in behavior and mitochondrial function in a rat model of pediatric mild traumatic brain injury. Brain Inj. 2019;19:1–9. doi: 10.1080/02699052.2019.1565898. [DOI] [PubMed] [Google Scholar]
- 14.Gajavelli S, Sinha VK, Mazzeo AT, Spurlock MS, Lee SW, Ahmed AI, Yokobori S, Bullock RM. Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. J Bioenerg Biomembr. 2015;47:133–148. doi: 10.1007/s10863-014-9589-1. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Zhang MY, Wang T, Fan YY, Yu LS, Ye GH, Wang ZF, Gao C, Wang HC, Luo CL, Tao LY. IL-33/ST2L signaling provides neuroprotection through inhibiting autophagy, endoplasmic reticulum stress, and apoptosis in a mouse model of traumatic brain injury. Front Cell Neurosci. 2018;12:95. doi: 10.3389/fncel.2018.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Go BS, Kim J, Yang JH, Choe ES. Psychostimulant-induced endoplasmic reticulum stress and neurodegeneration. Mol Neurobiol. 2017;54:4041–4048. doi: 10.1007/s12035-016-9969-0. [DOI] [PubMed] [Google Scholar]
- 17.Gulyaeva NV. Brain ischemia, endoplasmic reticulum stress, and astroglial activation: new insights. J Neurochem. 2015;132:263–265. doi: 10.1111/jnc.13016. [DOI] [PubMed] [Google Scholar]
- 18.Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci. 2015;350:132–138. doi: 10.1097/MAJ.0000000000000506. [DOI] [PubMed] [Google Scholar]
- 19.Khader A, Yang WL, Hansen LW, Rajayer SR, Prince JM, Nicastro JM, Coppa GF, Wang P. SRT1720, a sirtuin 1 activator, attenuates organ injury and inflammation in sepsis. J Surg Res. 2017;219:288–295. doi: 10.1016/j.jss.2017.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK. Oxidative stress: major threat in traumatic brain Injury. CNS Neurol Disord Drug Targets. 2018;17:689–695. doi: 10.2174/1871527317666180627120501. [DOI] [PubMed] [Google Scholar]
- 21.Lee SW, de Rivero Vaccari JP, Truettner JS, Dietrich WD, Keane RW. The role of microglial inflammasome activation in pyroptotic cell death following penetrating traumatic brain injury. J Neuroinflammation. 2019;16:27. doi: 10.1186/s12974-019-1423-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li F, Lu L, Chen H, Wang P, Zhang H, Chen YC, Yin X. Neuroanatomical and functional alterations of insula in mild traumatic brain injury patients at the acute stage. Brain Imaging Behav. 2019a doi: 10.1007/s11682-019-00053-3. doi: 10.1007/s11682-019-00053-3. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Zhou S, Wu L, Liu K, Zhang Y, Ma G, Wang L. The role of p38MAPK signal pathway in the neuroprotective mechanism of limb postconditioning against rat cerebral ischemia/reperfusion injury. J Neurol Sci. 2015a;357:270–275. doi: 10.1016/j.jns.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Li P, Liu Y, Burns N, Zhao KS, Song R. SIRT1 is required for mitochondrial biogenesis reprogramming in hypoxic human pulmonary arteriolar smooth muscle cells. Int J Mol Med. 2017;39:1127–1136. doi: 10.3892/ijmm.2017.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, Wang X, Zhao M, Song R, Zhao KS. Polydatin protects hepatocytes against mitochondrial injury in acute severe hemorrhagic shock via SIRT1-SOD2 pathway. Expert Opin Ther Targets. 2015b;19:997–1010. doi: 10.1517/14728222.2015.1054806. [DOI] [PubMed] [Google Scholar]
- 26.Li Q, Wang P, Huang C, Chen B, Liu J, Zhao M, Zhao J. N-Acetyl serotonin protects neural progenitor cells against oxidative stress-induced apoptosis and improves neurogenesis in adult mouse hippocampus following traumatic brain injury. J Mol Neurosci. 2019b doi: 10.1007/s12031-019-01263-6. doi: 10.1007/s12031-019-01263-6. [DOI] [PubMed] [Google Scholar]
- 27.Li T, Liu Y, Li G, Wang X, Zeng Z, Cai S, Li F, Chen Z. Polydatin attenuates ipopolysaccharide-induced acute lung injury in rats. Int J Clin Pathol. 2014;7:8401–8410. [PMC free article] [PubMed] [Google Scholar]
- 28.Liao P, He Y, Yang F, Luo G, Zhuang J, Zhai Z, Zhuang L, Lin Z, Zheng J, Sun E. Polydatin effectively attenuates disease activity in lupus-prone mouse models by blocking ROS-mediated NET formation. Arthritis Res Ther. 2018;20:254. doi: 10.1186/s13075-018-1749-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ling Y, Chen G, Deng Y, Tang H, Ling L, Zhou X, Song X, Yang P, Liu Y, Li Z, Zhao C, Yang Y, Wang X, Kitakaze M, Liao Y, Chen A. Polydatin post-treatment alleviates myocardial ischaemia/reperfusion injury by promoting autophagic flux. Clin Sci (Lond) 2016;130:1641–1653. doi: 10.1042/CS20160082. [DOI] [PubMed] [Google Scholar]
- 30.Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, Matsumoto RR, Huber JD, Rosen CL. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyeth BG. Historical review of the fluid-percussion tbi model. Front Neurol. 2016;7:217. doi: 10.3389/fneur.2016.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majdan M, Plancikova D, Brazinova A, Rusnak M, Nieboer D, Feigin V, Maas A. Epidemiology of traumatic brain injuries in Europe: a cross-sectional analysis. Lancet Public Health. 2016;1:e76–e83. doi: 10.1016/S2468-2667(16)30017-2. [DOI] [PubMed] [Google Scholar]
- 33.Menon DK, Schwab K, Wright DW, Maas AI. Demographics Clinical Assessment Working Group of the I, Interagency Initiative toward Common Data Elements for Research on Traumatic Brain I, Psychological H. Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil. 2010;91:1637–1640. [Google Scholar]
- 34.Ni Z, Tao L, Xiaohui X, Zelin Z, Jiangang L, Zhao S, Weikang H, Hongchao X, Qiujing W, Xin L. Polydatin impairs mitochondria fitness and ameliorates podocyte injury by suppressing Drp1 expression. J Cell Physiol. 2017;232:2776–2787. doi: 10.1002/jcp.25943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nozaki K, Nishimura M, Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Mol Neurobiol. 2001;23:1–19. doi: 10.1385/MN:23:1:01. [DOI] [PubMed] [Google Scholar]
- 36.Ray B, Bailey JA, Sarkar S, Lahiri DK. Molecular and immunocytochemical characterization of primary neuronal cultures from adult rat brain: differential expression of neuronal and glial protein markers. J Neurosci Methods. 2009;184:294–302. doi: 10.1016/j.jneumeth.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruan W, Li J, Xu Y, Wang Y, Zhao F, Yang X, Jiang H, Zhang L, Saavedra JM, Shi L, Pang T. MALAT1 up-regulator polydatin protects brain microvascular integrity and ameliorates stroke through C/EBPbeta/MALAT1/CREB/PGC-1alpha/PPARgamma pathway. Cell Mol Neurobiol. 2019;2:265–286. doi: 10.1007/s10571-018-00646-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song R, Bian H, Wang X, Huang X, Zhao KS. Mitochondrial injury underlies hyporeactivity of arterial smooth muscle in severe shock. Am J Hypertens. 2011;24:45–51. doi: 10.1038/ajh.2010.184. [DOI] [PubMed] [Google Scholar]
- 39.Su SY, Hsieh CL. Anti-inflammatory effects of Chinese medicinal herbs on cerebral ischemia. Chin Med. 2011;6:26. doi: 10.1186/1749-8546-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun D, Chen X, Gu G, Wang J, Zhang J. Potential roles of mitochondria-associated ER membranes (MAMs) in traumatic brain injury. Cell Mol Neurobiol. 2017;37:1349–1357. doi: 10.1007/s10571-017-0484-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun GZ, Gao FF, Zhao ZM, Sun H, Xu W, Wu LW, He YC. Endoplasmic reticulum stress-induced apoptosis in the penumbra aggravates secondary damage in rats with traumatic brain injury. Neural Regen Res. 2016;11:1260–1266. doi: 10.4103/1673-5374.189190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeda K, Ichijo H. Neuronal p38 MAPK signalling: an emerging regulator of cell fate and function in the nervous system. Genes Cells. 2002;7:1099–1111. doi: 10.1046/j.1365-2443.2002.00591.x. [DOI] [PubMed] [Google Scholar]
- 43.Tan HP, Guo Q, Hua G, Chen JX, Liang JC. Inhibition of endoplasmic reticulum stress alleviates secondary injury after traumatic brain injury. Neural Regen Res. 2018;13:827–836. doi: 10.4103/1673-5374.232477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths-United States, 2007 and 2013. MMWR Surveill Summ. 2017;66:1–16. doi: 10.15585/mmwr.ss6609a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner AK, Kumar RG. TBI rehabilomics research: conceptualizing a humoral triad for designing effective rehabilitation interventions. Neuropharmacology. 2018;145:133–144. doi: 10.1016/j.neuropharm.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang WX, Visavadiya NP, Pandya JD, Nelson PT, Sullivan PG, Springer JE. Mitochondria-associated microRNAs in rat hippocampus following traumatic brain injury. Exp Neurol. 2015;265:84–93. doi: 10.1016/j.expneurol.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Song R, Bian HN, Brunk UT, Zhao M, Zhao KS. Polydatin, a natural polyphenol, protects arterial smooth muscle cells against mitochondrial dysfunction and lysosomal destabilization following hemorrhagic shock. Am Journal Physiol Regul Integr Comp Physiol. 2012;302:R805–R814. doi: 10.1152/ajpregu.00350.2011. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Song R, Chen Y, Zhao M, Zhao KS. Polydatin-a new mitochondria protector for acute severe hemorrhagic shock treatment. Expert Opin Investig Drugs. 2013;22:169–179. doi: 10.1517/13543784.2013.748033. [DOI] [PubMed] [Google Scholar]
- 49.Yang H, Gu ZT, Li L, Maegele M, Zhou BY, Li F, Zhao M, Zhao KS. SIRT1 plays a neuroprotective role in traumatic brain injury in rats via inhibiting the p38 MAPK pathway. Acta Pharmacol Sin. 2017;38:168–181. doi: 10.1038/aps.2016.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeng Z, Chen Z, Li T, Zhang J, Gao Y, Xu S, Cai S, Zhao KS. Polydatin: a new therapeutic agent against multiorgan dysfunction. J Surg Res. 2015a;198:192–199. doi: 10.1016/j.jss.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 51.Zeng Z, Chen Z, Xu S, Song R, Yang H, Zhao KS. Polydatin alleviates small intestine injury during hemorrhagic shock as a SIRT1 activator. Oxid Med Cell Longev. 2015b;2015:965961. doi: 10.1155/2015/965961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeng Z, Chen Z, Xu S, Zhang Q, Wang X, Gao Y, Zhao KS. Polydatin protecting kidneys against hemorrhagic shock-induced mitochondrial dysfunction via SIRT1 activation and p53 deacetylation. Oxid Med Cell Longev. 2016;2016:1737185. doi: 10.1155/2016/1737185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang XM, Zhang L, Wang G, Niu W, He Z, Ding L, Jia J. Suppression of mitochondrial fission in experimental cerebral ischemia: the potential neuroprotective target of p38 MAPK inhibition. Neurochem Int. 2015;90:1–8. doi: 10.1016/j.neuint.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y, Luo P, Guo Q, Li S, Zhang L, Zhao M, Xu H, Yang Y, Poon W, Fei Z. Interactions between SIRT1 and MAPK/ERK regulate neuronal apoptosis induced by traumatic brain injury in vitro and in vivo. Exp Neurol. 2012;237:489–498. doi: 10.1016/j.expneurol.2012.07.004. [DOI] [PubMed] [Google Scholar]