

Abstract
Neuroinflammation is initiated as a result of traumatic brain injury and can exacerbate evolving tissue pathology. Immune cells respond to acute signals from damaged cells, initiate neuroinflammation, and drive the pathological consequences over time. Importantly, the mechanism(s) of injury, the location of the immune cells within the brain, and the animal species all contribute to immune cell behavior following traumatic brain injury. Understanding the signals that initiate neuroinflammation and the context in which they appear may be critical for understanding immune cell contributions to pathology and regeneration. Within this paper, we review a number of factors that could affect immune cell behavior acutely following traumatic brain injury.
Keywords: traumatic brain injury, inflammation, neuroinflammation, microglia, macrophage, acute, diffuse brain injury, cytokines, adenosine 5‘-triphosphoate, glutamate, calcium
Traumatic Brain Injury Initiates Neuroinflammation
Traumatic brain injury (TBI) is a major health problem resulting in 2.8 million new incidences every year (Taylor et al., 2017). TBI is unique from other neurological afflictions in that it is caused by a discrete biomechanical event, termed the primary injury, and therefore affects individuals irrespective of age, sex, and/or baseline health status. Secondary injury, initiated by the primary mechanical insult, is driven by a number of positive-feedback cascades including neuroinflammation, cell death, excitotoxicity, blood-brain barrier (BBB) disruption, and mitochondrial dysfunction, all of which exacerbate neuronal dysfunction and tissue loss (Loane and Faden, 2010).
It has long been established that neuroinflammation is a major contributor to secondary injury. Indeed, it has been postulated that in many cases, TBI-induced neuroinflammation is more detrimental to pathological progression than the primary injury itself (Patterson and Holahan, 2012). Enhancing our understanding of signals that initiate and drive neuroinflammation is essential to understanding TBI-induced secondary injury. Simultaneously, a more comprehensive understanding of neuroinflammatory cascades over time may inform translational therapeutics that mitigate pathology. Here, we review the signals that can initiate immune cell reactivity in the brain in the acute timeframe following trauma. Literature searches were completed in PubMed using keywords including but not limited to neuroinflammation, microglia, cytokines, inflammasomes, damage-associated molecular patterns, ATP, oxidative damage, excitotoxicity, and monocyte-derived macrophages.
Neuronal Permeability Initiates Inflammation
In a landmark neuroimmunology paper, Davalos et al. (2005) observed that microglia adopt a more reactive morphology within minutes following a laser-generated lesion in the mouse cortex. However, it was unclear if rapid microglia reactivity was also sensitive to subtler pathologies that are more common in clinical presentations of diffuse TBI. In order to test this, we utilized a porcine model of closed-head, non-impact rotational acceleration TBI using a HYGE pneumatic actuator (HYGE Inc., Kittanning, PA, USA) (Cullen et al., 2016; Wofford et al., 2017). Importantly, this large animal model closely replicates the biomechanical parameters most relevant to closed-head TBI in humans, and has been shown to generate reproducible neurological and neuropathological deficits (Cullen et al., 2016). Prior to injury, we introduced a membrane impermeant dye into the parenchyma of the brain (Wofford et al., 2017). TBI-induced mechanical forces generated transient membrane perturbations in the neurons (Farkas et al., 2006), resulting in dye accumulation in cytoplasmic spaces of damaged cells before plasma membrane integrity was reestablished. Fifteen minutes after injury, residual dye was flushed out of the parenchyma, tissue was fixed, and immunohistochemistry was employed to assess neuropathology (Wofford et al., 2017). We observed that neurons were exclusively labeled with the cell-impermeant dye while astrocytes and microglia were not. These data suggest that neurons are preferentially vulnerable to mechanical stress-strain fields generated during closed-head diffuse TBI.
Furthermore, in this study microglia density increased and morphology became more reactive around permeabilized neurons acutely after injury (Figure 1). Specifically, microglia around permeabilized neurons exhibited shorter process lengths and larger cell body diameters but did not exhibit the rod-shape that has been recently described in some rodent models of diffuse TBI (Ziebell et al., 2014; Witcher et al., 2018). Of note, microglia in regions absent neuronal permeability within injured brains were not significantly different in density or morphology from microglia found in uninjured control animals (Figure 1). These data suggest that neuronal membrane perturbation – directly resulting from mechanical tissue deformation – could be a driver of acute immune activation in TBI. Additionally, animals experiencing multiple TBIs exhibited enhanced neuronal permeability and a qualitative increase in microglia reactivity suggesting that there could be interplay between baseline inflammatory states and ongoing pathophysiological cascades in driving the extent and consequences of neuronal injury (Figure 1).
Figure 1.
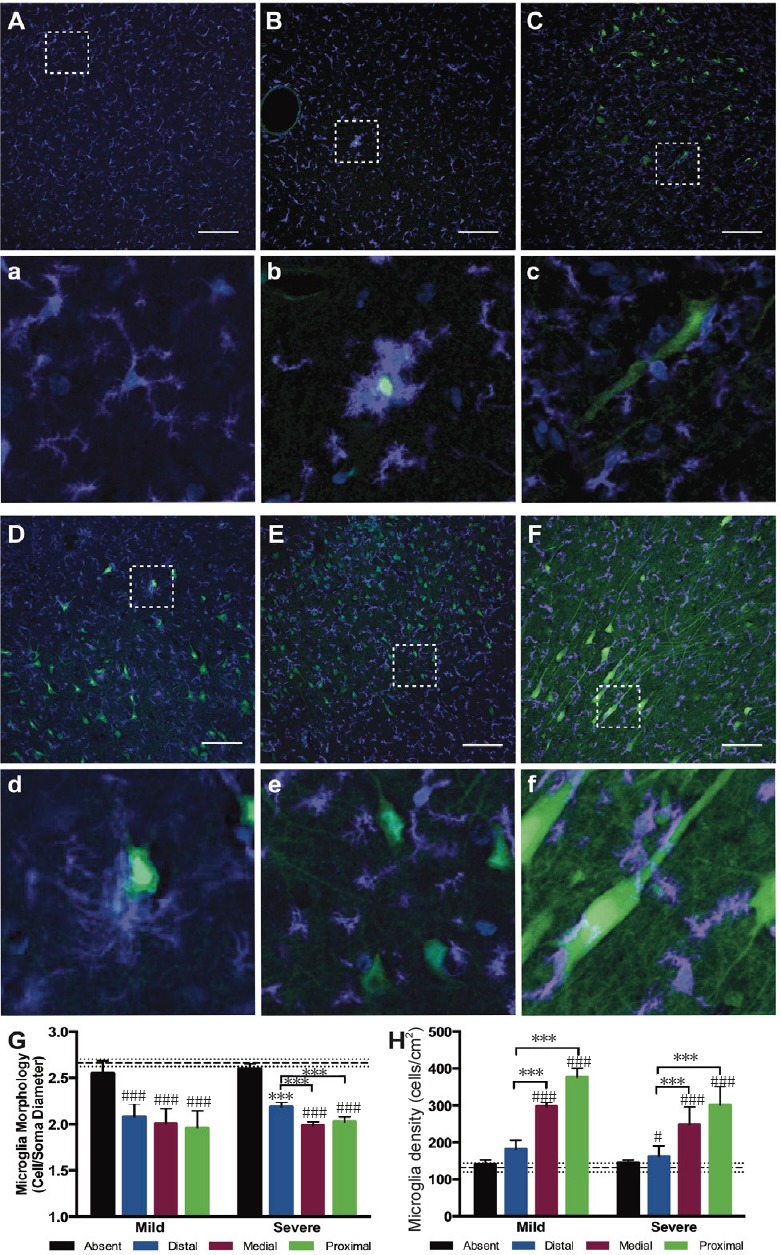
Immune cells exhibit a reactive morphology around permeabilized neurons following diffuse traumatic brain injury (TBI).
(A, a) Lucifer yellow (green), a cell impermeant dye, was taken up by permeabilized neurons following TBI. Microglia (ionized calcium binding adapter molecule 1 positive (Iba1+), purple) in the cortex of sham injured animals exhibit a scanning, ramified morphology (A) with enlarged call out box (a). (B–F, b–f) However, following TBI, microglia in the cortex (B, C, E, F) and hippocampus (D) localize around permeabilized neurons and adopt a more reactive morphology following severe (C, D, E) or mild (B, F) TBI with enlarged call out boxes (b–f). Repetitive moderate injuries separated by 15 minutes (E) and repetitive mild injuries separated by 7 days also generated reactive microglia (F). (G, H) Single cell quantification metrics indicated a significant change in immune cell morphology (G) and density (H) in regions proximal to permeabilized neurons. Scale bars: 100 µm. Images were adapted from Wofford et al. (2017).
Importantly, within this study we observed neuronal permeability in the cortex, sub-cortical white matter, and in the hippocampus. It remains to be seen if distribution of permeabilized neurons is ubiquitous across all brain regions. However, previous research suggests that diffuse axonal injury at acute timepoints following diffuse TBI in swine exhibits different patterns across brain regions (Lafrenaye et al., 2015). Furthermore, recent work suggests that microglia exhibit unique morphologies and variable densities across gray and white matter and across different brain regions (Lawson et al., 1990; De Biase and Bonci, 2018). Indeed, it has been postulated that microglia exhibit unique, region-specific functions even during baseline brain function (De Biase et al., 2017; De Biase and Bonci, 2018). Moreover, microglia reactivity has been shown to vary significantly across species even when species are subjected to the same injury paradigm (Gorse and Lafrenaye, 2018). Together, these findings suggest that microglia are a highly sensitive cell population and generalization about microglia functionality is challenging since these cells can change their reactivity and function based on brain region and species, as well as with mechanism of neuronal injury induction and injury severity.
While it is tempting to use density and morphology characteristics to generate conclusions about immune cell behavior, it is important to note that morphology and density of immune cells cannot directly predict microglial phenotype or behavior. Recent evidence suggests that immune cells in the body, including microglia, can exist across a spectrum of behavioral phenotypes. Microglia have diverse effector functions ranging from secreting neuroprotective factors (Colton, 2009), releasing neurotoxic factors (Donat et al., 2017), and phagocytosing cells and cell debris (Takahashi et al., 2005). Interestingly, a recent report found that microglia are capable of selectively sequestering pathological proteins from the intracellular space of neurons without affecting non-pathological proteins (Spiller et al., 2018). Notably, the behavioral phenotype of immune cells is driven by environmental stimuli. Better elucidating environmental cues that are released as a result of primary mechanical trauma and secondary sequelae could better predict immune cell behavior following TBI.
Here, we consider a number of acute cues that could be passively or actively released following TBI to initiate immune cell activation. Passive cues released from either damaged or necrotic cells may contribute to immune activation. As we demonstrated, rapid mechanical stress-strain fields in the brain – unique to TBI – can cause membrane perturbation and passive release of chemoattractant cues and immune activators that are traditionally sequestered in the intracellular space (Table 1). Additionally, unregulated necrosis can release immune activators into the parenchyma, facilitating immune cell activation. Separately, upregulated transcription and translation of neuroinflammatory drivers can lead to an active amplification of local immune cell reactivity (Figure 2).
Table 1.
Passive and active immunomodulatory cues are detectable following traumatic brain injury (TBI)
| Signal | Detection | Source |
|---|---|---|
| Examples of passively released cues | ||
| ATP | Detectable within 15 minutes | Davalos et al. (2005) |
| Glutamate | Peaks within 2 minutes | Katayama et al. (1990) |
| HMGB1 | Elevated from 3–24 hours | Okuma et al. (2012) |
| Potassium | Peaks within 2 minutes | Katayama et al. (1990) |
| Examples of actively released cues | ||
| TNF | Detectable within 17 minutes | Frugier et al. (2010) |
| IL-1β | Peaks at 8 hours | Taupin et al. (1993) |
| IL-6 | Peaks at 8 hours | Taupin et al. (1993) |
| MCP1 | Peaks 4–12 hours after TBI | Semple et al. (2010) |
| SP | Peaks at 30 minutes | Donkin et al. (2009) |
ATP: Adenosine 5’-triphosphoate; HMGB1: high mobility group box 1; TNF: tumor necrosis factor; IL: interleukin; MCP1: monocyte chemoattractant protein 1; SP: substance P.
Figure 2.

Schematic of passive and active drivers of neuroinflammation following mechanical trauma.
In the healthy central nervous system, cells rest at a steady state with crosstalk that balances extracellular signaling molecules, osmotic pressure, ionic concentrations, and vascular integrity. Following mechanoporation to neuronal membranes, a number of passive signals are leaked from the cell including DAMPs and glutamate which facilitate immune cell reactivity, intracellular calcium influx, ROS formation, and mitochondrial damage. After passive drivers initiate neuroinflammation, active drivers such as inflammasomes, proteases, ion transporters, caspases, exosomes, and infiltrating immune cells further amplify neuroinflammation and can even initiate a second wave of released passive signals. ATP: Adenosine 5′-triphosphoate; TNF: tumor necrosis factor; IL: interleukin; DAMPs: damage-associated molecular patterns; ROS: reactive oxygen species; EVs: extracellular vesicles.
Passive Drivers of Neuroinflammation
Transient membrane permeabilization can release damage-associated molecular patterns (DAMPs), ions, and cytokines. DAMPs are proteins, nucleic acids, or other molecules that are present in the cells prior to injury and then passively released by damaged cells following trauma, resulting in immune cell activation. Importantly, DAMPs are released from damaged or necrotic cells but not from apoptotic cells (Bianchi, 2006), suggesting that immune cell behavior towards damaged and necrotic cells is distinct from immune cell behavior towards apoptotic cells. DAMPs are capable of initiating a robust immune response acutely following trauma (Bianchi, 2006; Kumar and Loane, 2012; Braun et al., 2017). DAMPs such as high mobility group box 1 protein, heat shock proteins, S100 proteins, uric acid, and heparin sulfate can all serve as acute immune cell initiators following trauma (Laird et al., 2014; Simon et al., 2017). Degradation of the extracellular matrix protein hyaluronic acid via hyaluronidases and reactive oxygen species (ROS), which are elevated following TBI, produces the lower molecular weight hyaluronan, initiating a pro-inflammatory response in damaged tissue (Xing et al., 2014). For a comprehensive review of DAMPs following TBI see Braun et al. (2017).
Additionally, adenosine 5′-triphosphoate (ATP), a storage modulus of chemical energy, acts as an important DAMP when released from damaged neurons into the extracellular environment. ATP has been well established as an acute immune cell initiator. ATP can be passively released when neurons are transiently permeabilized or actively released by astrocytes following injury (Davalos et al., 2005). Active release of ATP from astrocytes may act to amplify extracellular signals and function as a chemoattractant signal for immune cells. Interestingly, removal of ATP chemoattractive gradients after laser injury prevents microglia from rapidly extending their processes toward the injury site, suggesting that ATP may precede other acute immune signals (Davalos et al., 2005). Indeed, purinergic receptor activation in microglia can induce calcium influx, an essential secondary messenger, that induces microglial proliferation, chemotaxis, and inflammatory protein synthesis (Honda et al., 2001; Fields and Burnstock, 2006). Importantly, microglial stimulation with low or with high concentrations of ATP can generate unique microglia responses (Hide et al., 2000; Shigemoto-Mogami et al., 2001). For example, simulating microglia with 30 µM of ATP results in rapid but transient plasminogen release (Inoue et al., 1998). In contrast, stimulating microglia with 1 mM of ATP, a concentration associated with microglia reactivity in vivo, induces tumor necrosis factor-alpha (TNF-α) release followed by a later release of interleukin (IL)-6 (Hide et al., 2000; Shigemoto-Mogami et al., 2001; Davalos et al., 2005).
In addition to DAMPs, mechanically damaged neurons release high concentrations of potassium and glutamate into the extracellular space (Katayama et al., 1990). Extracellular glutamate activates N-methyl-D-aspartate (NMDA) receptors on local neurons to facilitate an influx of calcium (Özsüer et al., 2005). Unregulated calcium influx can lead to excitotoxicity, dysregulated energy transduction, mitochondrial damage, and generation of free radicals and ROS in the cytoplasmic space by overwhelming cellular antioxidant responses (Xiong et al., 1997; Bains and Hall, 2012). Increases in intracellular ROS can react with polyunsaturated fatty acids, ultimately resulting in destabilized cellular and mitochondrial membranes (Bains and Hall, 2012). Destabilization of cellular and mitochondrial membranes can initiate mitochondrial dysregulation as well as passive leakage of intracellular molecules into the extracellular space and thus generate iterative waves of DAMPs and passive initiators of neuroinflammation (Xiong et al., 1997; Verweij et al., 2000). Likewise, reactions between ROS and other biological molecules including proteins, carbohydrates, and nucleic acids can permanently alter the structure, and thus the function, of these biologics. In murine models of TBI, ROS levels were elevated one minute after trauma and maintained elevated levels for up to 30 minutes after injury (Hall et al., 1993). Following close head TBI in rats, lipid peroxidation was elevated from 30 minutes (the earliest time considered) to 48 hours after injury (Özsüer et al., 2005). Indeed, administration of memantine fifteen minutes after injury (to prevent NMDA channel opening), reduced the extent of lipid peroxidation in rats (Özsüer et al., 2005). Together, these data suggest that dysregulated calcium gradients initiate a number of potent and detrimental cascades in and around mechanically permeabilized neurons.
As well as inducing deleterious intracellular signaling cascades, accumulation of extracellular glutamate can simultaneously function as an initiator of immune cell reactivity (Faden et al., 1989; Weber et al., 1999). During healthy conditions, astrocytes sequester glutamate from synapses and recycle it to neurons (Tanaka et al., 1997), however, following trauma, excessive glutamate is released from neurons and impaired astrocytic clearance of glutamate generates excessive accumulation of glutamate in the parenchyma (Yi and Hazell, 2006; Cantu et al., 2015). Glutamate receptors, which are expressed on the surface of microglia, monocytes, and macrophages, facilitate an acute, inflammatory response to excessive glutamate in the tissue parenchyma (Kumar and Loane, 2012). Taken together, these data suggest that membrane mechanoporation can passively initiate a number of intracellular and intercellular signaling cascades that all contribute to neuroinflammation and secondary injury following TBI.
Active Drivers of Neuroinflammation
In addition to passive cues, active upregulation of the synthesis and release of various molecules can also drive immune cell reactivity (Woodcock and Morganti-Kossmann, 2013; Simon et al., 2017). It is well established that TNF-α, IL-6, and IL-1β are major drivers of neuroinflammation while IL-10 acts as an anti-inflammatory molecule. TNF-α mRNA and proteins are rapidly generated, producing detectable levels within minutes and reaching peak concentrations within a few hours after TBI in pre-clinical animal models (Taupin et al., 1993; Fan et al., 1996), and is also detectable within minutes in clinical presentations (Frugier et al., 2010). Likewise, IL-1β and IL-6 mRNA and proteins are also detectable at early time points after injury in pre-clinical models (Taupin et al., 1993; Fan et al., 1995; Hans et al., 1999; Kinoshita et al., 2002; Patterson and Holahan, 2012) and exhibit a slightly delayed expression profile in clinical presentations (Frugier et al., 2010). IL-10 also exhibited sub-acute expression in pre-clinical rodent models of TBI (Kamm et al., 2006) and elevated levels at sub-acute timepoints in clinical cases of severe TBI (Hensler et al., 2000; Hayakata et al., 2004).
Proteases can be another source of active inflammatory cues because they can process enzymes and cytokines into their bioactive form. For example, matrix metalloproteinases (MMP) activation has been associated with neuroinflammation, vascular dysregulation, BBB breakdown, white matter damage, neuronal death, and brain edema (Lo et al., 2002). In a closed head weight-drop murine model of TBI, MMP-2 (also known as gelatinase A), MMP-9 (also known as gelatinase B), and tumor necrosis factor-α-converting enzyme (TACE, also known as ADAM-17) were found at detectable levels within 10 minutes of injury and peaked within 1 hour of injury in the cortex (Zhang et al., 2016). Interestingly, TBI generated with an open head injury model of TBI exhibited a delayed production of all these proteinases, suggesting that injury mechanism could play an important regulatory role for these enzymes and their downstream byproducts (Zhang et al., 2016). MMP-2 and MMP-9 are important signaling molecules in neuroinflammation because they can cleave IL-1β from its inactive precursor form into its potent form in a caspase-1-independent pathway (Schönbeck et al., 1998). MMP-9 can rapidly generate active IL-1β within minutes while MMP-2 generated IL-1β after 24 hours of incubation. Notably, MMP-9-generated IL-1β was active and stable for up to 72 hours (Schönbeck et al., 1998). Following a closed head model of TBI, TACE exhibits a very pronounced and acute amplification in the brain (Zhang et al., 2016). Importantly, TACE is responsible for, among other things, converting membrane-bound pro-TNF-α to shed its ectodomain and become the soluble, paracrine signaling molecule TNF-α (Le Gall et al., 2009). Soluble TNF-α has been associated with more pro-inflammatory activities following injury (Brambilla et al., 2011). TACE can be turned “on” and “off” through a number of physiological signaling pathways including thrombin, epidermal growth factor, lysophosphatidic acid, dibenzoyl-ATP, and TNF-α (Le Gall et al., 2010). In clinical cases of TBI, MMP-2 levels were elevated in patient plasma 72 hours after injury. MMP-9 levels were also elevated in the cerebral spinal fluid at both acute and sub-acute timepoints relative to control patients (Grossetete et al., 2009; Guilfoyle et al., 2015).
In addition to proteases, formation of inflammasomes in both microglia and neurons can amplify the production of inflammatory signaling molecules. Inflammasomes are intracellular complexes that assemble in response to DAMPs and stimulate the production of pro-inflammatory factors. The inflammasome protein composition is largely dictated by the inflammatory stimulus that the cell perceives. Inflammasome-generating stimuli can be DAMPs such as ROS, host ectopic dsDNA or signals from dysfunctional mitochondria that are expressing cardiolipin on their outer membrane (Zhou et al., 2010; Iyer et al., 2013). The inflammasome complexes nucleotide-binding, leucine-rich repeat pyrin domain containing protein 1 (NLRP1) and absent in melanoma 2 (AIM2) are found within neurons (de Rivero Vaccari et al., 2009; Adamczak et al., 2014; Simon et al., 2017). These inflammasomes can generate large amounts of caspase 1 which subsequently amplifies IL-1β levels (de Rivero Vaccari et al., 2009; Adamczak et al., 2014). Interfering with the formation of these inflammasome complexes can reduce inflammation and contusion volume in a rodent model of diffuse TBI (de Rivero Vaccari et al., 2009). AIM2 inflammasome activation can induce neuronal death by generating plasmalemmal pores, ultimately leading to neuronal pyroptosis (Adamczak et al., 2014). Loss of membrane integrity may induce cell death via osmotic swelling and simultaneously release pro-inflammatory components into the parenchyma. Interestingly, selectively blocking pannexin1 pores before inflammasomes formed reduced cellular pyroptosis, suggesting that pannexin1 may be an important channel in neuronal cell death via pyroptosis (Adamczak et al., 2014). Separately, the inflammasome complex nucleotide-binding, leucine-rich repeat pyrin domain containing protein 3 (NLRP3) is found within microglia, astrocytes, and neurons (Fann et al., 2013; Liu et al., 2013; Simon et al., 2017). Following a weight drop model of TBI, NLRP3 mRNA expression increased incrementally over the first week (Liu et al., 2013). Activation of NLRP3 was also correlated with pro-caspase expression and cleaved caspase 1 expression (Liu et al., 2013). Likewise, IL-18 concentrations followed the same trend as increasing NLRP3 while IL-1β trends peaked acutely at 6 hours and decreased thereafter (Liu et al., 2013). Modulation of inflammasome formation in murine pre-clinical models results in reduced microglial activation, leukocyte infiltration, pro-inflammatory cytokine production, brain edema, and lesion volume while simultaneously improving neurological outcomes (Irrera et al., 2017; Ismael et al., 2018; Xu et al., 2018). Following clinical cases of severe TBI in a pediatric population, NLRP1 was detectable in some injured patients at acute timepoints while NLRP3 was elevated in all injured patients across all acute and sub-acute timepoints (Wallisch et al., 2017). Preclusion of inflammasome formation could be a unique way to prevent amplification of pro-inflammatory cytokines and neuronal pyroptosis following TBI.
In addition to inflammatory stimuli, BBB dysregulation can also drive acute neuroinflammation following TBI. Indeed, BBB permeability often exhibits a bi-modal increase over time following TBI, exhibiting elevated levels of BBB permeability acutely (within several hours) after injury and again sub-acutely (within days) after injury (Başkaya et al., 1997; Saatman et al., 2006). Likewise, this bi-phasic trend of BBB permeability has been observed in porcine models of closed-head diffuse mild TBI, where permeability was elevated both at 6 hour and 72 hour timepoints (Johnson et al., 2018). During TBI, diffuse shear forces throughout the brain can lead to mechanical disruption of the vasculature and generate microhemorrhages. Microhemorrhages can permit passive immune cell accumulation in the brain following TBI, allowing the infiltration of circulating erythrocytes, leukocytes, and lymphocytes (Bigler and Maxwell, 2012).
BBB permeability within capillaries can be increased through non-mechanical mechanisms. For example, the Na+-K+-2Cl– cotransporter (NKCC1) uses ATP to transport sodium, potassium, and chloride into cells (Kahle et al., 2010). NKCC1 controls intracellular chloride concentrations in order to maintain intracellular solute concentrations and thus cellular volume at physiologically relevant levels (Kahle et al., 2010). Following TBI, NKCC1 mRNA and protein expression is upregulated from 2 to 24 hours after injury with maximal expression peaking at 8 hours (Lu et al., 2006, 2008). NKCC1 may play a role in driving secondary injury because inhibition of NKCC1 with bumetanide reduced neuronal damage, edema, and contusion volume following a rodent model of closed head TBI (Lu et al., 2006, 2008). Additionally, NKCC1 expression has been associated with BBB disruption. Inhibiting NKCC1 expression with bumetanide reduces the extent of BBB breakdown, edema, and behavioral deficits (Zhang et al., 2017). This effect most likely occurs because bumetanide prevents unregulated osmotic swelling, edema, and progressive secondary hemorrhages which amplify extravasation of blood components into the brain parenchyma (Kahle et al., 2010). Glutamate excitotoxicity and subsequent NMDA and AMPA stimulation can activate NKCC1 in neurons which increases intracellular sodium and chloride (Beck et al., 2003). Indeed, preventing NKCC1 activity in models of excitotoxicity can significantly ameliorate sodium and chloride accumulation in neurons and reduce the extent of cell death (Beck et al., 2003). Likewise, NKCC1 activity in astrocytes can be induced when the surrounding environment contains high concentrations of potassium – a common characteristic of the environment following TBI (Su et al., 2001). Elevated potassium concentration correlated with increased intracellular chloride concentration and cell swelling, all of which could be prevented by administering an NKCC1 inhibitor (Su et al., 2001). Together, these data suggest that mechanical, as well as chemical factors contribute to reduced efficacy of the BBB after trauma.
In addition to local soluble signals and regulated receptor expression, activated immune cells can release extracellular vesicles (EVs) into the circulation to amplify neuroinflammation. Exosomes and microvesicles are cell-derived EVs that facilitate paracrine signaling (Raposo and Stoorvogel, 2013). Importantly, EV composition, and thus their effect, is largely dictated by the state of the cell that is producing the exosomes or microvesicles. Following TBI, microglia in the brain generate EVs that are released into the circulation (Kumar et al., 2017). Interestingly, separate reports have characterized microglia-derived EVs to be inflammatory and anti-inflammatory following TBI (Kumar et al., 2017; Huang et al., 2018). Previous literature suggests that microglial-derived EVs can initiate neuroinflammation even in the absence of injury (Kumar et al., 2017), but they can also play a protective role by downregulating neuronal inflammation (Huang et al., 2018). More characterization is needed to identify EV type (exosome versus microvesicle), timing, and contributions of inflammatory and anti-inflammatory microglia-derived EVs on injury progression after TBI.
When BBB permeability increases again days after injury, this facilitates a wave of peripheral immune cell activation and infiltration. Many factors that are actively secreted by cells can modulate the integrity and permeability of the BBB. For example, monocyte chemoattractant protein 1 (MCP1, also known as CCL2) is expressed by neurons, astrocytes, microglia, and endothelial cells and acts as a chemokine gradient for infiltrating leukocytes and simultaneously works to decrease tight junctions in the BBB (Semple et al., 2010; Yao and Tsirka, 2014). Previous studies in mice and humans identified that MCP1 levels are elevated in the brain and cerebral spinal fluid following TBI and levels of MCP1 are directly proportional to monocyte infiltration (Semple et al., 2010). Likewise, tissue plasminogen activator is expressed on endothelial cells and can facilitate transient BBB permeability for peripheral monocyte-derived macrophages (Reijerkerk et al., 2008; Yao and Tsirka, 2014). Finally, substance P (SP) is a neuropeptide released by sensory neurons to promote plasma extravasation and capillary permeability, leading to vasogenic edema (Corrigan et al., 2016). Indeed, SP levels increase within minutes of TBI in both pre-clinical animal models and human cases and can induce leukocyte recruitment, augment inflammatory protein production, and stimulate the expression of adhesion molecules on endothelial cells (Corrigan et al., 2016).
Interestingly, literature is somewhat contradictory about the consequences of immune cell infiltration into the brain following trauma. Preventing monocyte infiltration into the injured brain following a controlled cortical impact injury by knocking out the essential chemokine receptor, CCR2, resulted in larger lesion volumes, enhanced neuronal loss, and elevated cell death within one week following TBI (Semple et al., 2010). However, the same knockout study also resulted in reduced astrogliosis and smaller lesion volumes at chronic timepoints (Semple et al., 2010). Similar knockout studies of CCR2 resulted in improved functional recovery and elevated neuronal density after TBI (Hsieh et al., 2014). Separately, monocyte ablation studies with clodronate liposomes enhanced functional recovery following controlled cortical impact TBI in mice (Makinde et al., 2017). Decreasing the extend of macrophage recruitment following a murine model of controlled cortical impact resulted in reduced NOX2, indicative of ROS production, and simultaneously improved functional outcome (Morganti et al., 2015). In diffuse murine models of TBI, preventing monocyte infiltration was associated with reduced lesion volume, reduced axonal pathology, and increased mislocalization of pathological proteins (Gyoneva et al., 2015).
To further complicate interpretation, it is important to note that microglia and infiltrating brain macrophages can exist across a spectrum of behavioral phenotypes. Typically, the different phenotypes of microglia and macrophages can be dichotomized into the pro-inflammatory (M1) phenotype and the anti-inflammatory (M2) phenotype (Mosser and Edwards, 2008; Xu et al., 2017). The pro-inflammatory phenotype, or classically-activated cells, generate signals that amplify inflammation while the anti-inflammatory phenotype, or alternatively-activated cells, generate anti-inflammatory signals that resolve inflammation. Importantly, the pro-inflammatory versus anti-inflammatory dichotomy grossly oversimplifies the scope of immune cell activation and behaviors. Indeed, immune cells can have a number of different behavioral phenotypes which is largely driven by environmental stimuli. For example, following TBI, both pro- and anti-inflammatory phenotypes have been observed within the same tissue and occasionally have been observed within the same immune cell (Wang et al., 2013; Kim et al., 2016; Kumar et al., 2016; Morganti et al., 2016). These data may suggest that oversimplification of immune cell behavior could facilitate erroneous conclusions about the contributions of these cells to pathology and tissue regeneration (Ransohoff, 2016). Historically, scientists theorized that pro-inflammatory immune cells were responsible for secondary injury pathology so attempts to pharmacologically control microglia or macrophage behaviors were limited to eliminating the pro-inflammatory response and promoting the anti-inflammatory response (Cao et al., 2012; Xu et al., 2017). However, more recently, scientists have begun to appreciate that both phenotypes could play a significant role in regeneration (Simon et al., 2017). Specifically, one common hypothesis is that transient pro-inflammatory phase followed by a prolonged anti-inflammatory response could guide resolution of inflammation, repair of tissue, and generate trophic support for compromised cells (Spiller and Koh, 2017).
Most data suggest that peripheral macrophages contribute to secondary injury pathology by homing to the site of injury and amplifying inflammation (Hsieh et al., 2014; Morganti et al., 2015). While these data cannot be overlooked, contradictory results in the field of neuroinflammation could also suggest that peripheral macrophages play an important role in tissue preservation and regeneration (Semple et al., 2010). It remains to be seen if these differences in immune cell contribution to pathology and regeneration varies across species, injury model, and timepoint, or if the variability in macrophage contribution is simply the result of incomplete characterization of the phenotype of these cells. Indeed, it is widely established that immune cell behavior is highly variable across species (Zelnickova et al., 2008; Fairbairn et al., 2011; Spiller et al., 2016). Identification of pre-clinical models that most closely recapitulate immune cell contributions in clinical presentations of TBI, such as our translational TBI model in swine, are essential to develop a fundamental understanding of neuroinflammatory cascades in humans as well as to develop clinically relevant therapeutic interventions.
Concluding Remarks and Future Perspectives
In conclusion, we observed immune cell reactivity around permeabilized neurons within minutes of closed-head diffuse TBI in swine (Wofford et al., 2017). Importantly, this animal model generates pathology mimetic of clinical TBI and could be utilized to better investigate drivers of acute and chronic neuroinflammation that are translationally relevant to clinical presentations (Cullen et al., 2016). In the future, we aim to elucidate the specific passive and active immune modulators that are received by immune cells and influence their phenotype. Within the context of these findings, we suspect that passive cues may primarily drive the immune cell reactivity that we observed within minutes of injury. Comprehending the chronology and extent of these signals across neuroanatomical regions following TBI may better inform immune cell characteristics such as cell phenotype, behavior, and fate. While therapeutic intervention within minutes is likely unrealistic for clinical cases, we suspect that elucidating the specific acute drivers of neuroinflammation will uncover phenotypes of the neuroinflammatory environment and present targets for later beneficial modulation.
Understanding how immune cell reactivity is initiated as well as the consequences of immune reactivity on neuronal health is essential for preserving the potentially beneficial effects of acute inflammation while minimizing the presumed negative consequences of prolonged inflammation. However, the behavior of immune cells, and thus the pathological implications, are highly variable. Within this paper, we reviewed recent evidence suggesting that the mechanism of injury, the injury severity, the location of the immune cells within the brain, and the animal species all contribute to immune cell behavior following TBI. As a result of these findings, we believe that a major challenge of the neuroinflammation field is extrapolation of data from pre-clinical model systems to generate information that has clinical applicability. We caution that interpretation of experimental results from model systems should appreciate that immune cell behavior often varies across species, brain region, and injury type and can exhibit both neurotoxic and neuroprotective functions.
Footnotes
Conflicts of interest: None declared.
Financial support: The work was supported by the Department of Veterans Affairs, USA (Merit Review I01-RX001097 & I01-BX003748).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: The work was supported by the Department of Veterans Affairs, USA (Merit Review I01-RX001097 & I01-BX003748).
C-Editors: Zhao M, Yu J; T-Editor: Jia Y
References
- 1.Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ, Nonner D, Bullock MR, Dahl GP, Dietrich WD, Keane RW. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab. 2014;34:621–629. doi: 10.1038/jcbfm.2013.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012;1822:675–684. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Başkaya MK, Rao a M, Doğan a, Donaldson D, Dempsey RJ. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett. 1997;226:33–36. doi: 10.1016/s0304-3940(97)00239-5. [DOI] [PubMed] [Google Scholar]
- 4.Beck J, Lenart B, Kintner DB, Sun D. Na-K-Cl cotransporter contributes to glutamate-mediated excitotoxicity. J Neurosci. 2003;23:5061–5068. doi: 10.1523/JNEUROSCI.23-12-05061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2006;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 6.Bigler ED, Maxwell WL. Neuropathology of mild traumatic brain injury: Relationship to neuroimaging findings. Brain Imaging Behav. 2012;6:108–136. doi: 10.1007/s11682-011-9145-0. [DOI] [PubMed] [Google Scholar]
- 7.Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, Bethea JR. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain. 2011;134:2736–2754. doi: 10.1093/brain/awr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun M, Vaibhav K, Saad NM, Fatima S, Vender JR, Baban B, Hoda MN, Dhandapani KM. White matter damage after traumatic brain injury: a role for damage associated molecular patterns. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2614–2626. doi: 10.1016/j.bbadis.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantu D, Walker K, Andresen L, Taylor-weiner A, Hampton D, Tesco G, Dulla CG. Traumatic brain injury increases cortical glutamate network activity by compromising gabaergic control. Cereb Cortex. 2015;25:2306–2320. doi: 10.1093/cercor/bhu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012;225:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corrigan F, Mander KA, Leonard AV, Vink R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation. 2016;13:264. doi: 10.1186/s12974-016-0738-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cullen DK, Harris JP, Browne KD, Wolf JA, Duda JE, Meaney DF, Margulies SS, Smith DH. A porcine model of traumatic brain injury via head rotational acceleration. Methods Mol Biol. 2016;1462:289–324. doi: 10.1007/978-1-4939-3816-2_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davalos D, Grutzendler J, Yang G, Kim J V, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 15.De Biase LM, Bonci A. Region-specific phenotypes of microglia: the role of local regulatory cues. Neuroscientist. 2018 doi: 10.1177/1073858418800996. doi: 10.1177/1073858418800996. [DOI] [PubMed] [Google Scholar]
- 16.De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, Zhang H, Liu Q-R, Shen H, Xi ZX, Goldman D, Bonci A. Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron. 2017;95:341–356. doi: 10.1016/j.neuron.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralisation of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb blood flow Metab. 2009;29:1251–1261. doi: 10.1038/jcbfm.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donat CK, Scott G, Gentleman SM, Sastre M. Microglial activation in traumatic brain injury. Front Aging Neurosci. 2017;9:208. doi: 10.3389/fnagi.2017.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 20.Fairbairn L, Kapetanovic R, Sester DP, Hume DA. The mononuclear phagocyte system of the pig as a model for understanding human innate immunity and disease. J Leukoc Biol. 2011;89:855–871. doi: 10.1189/jlb.1110607. [DOI] [PubMed] [Google Scholar]
- 21.Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, Mcintosh TK. Experimental brain injury induces expression of interleukin-lb mRNA in the rat brain. Mol Brain Res. 1995;30:125–130. doi: 10.1016/0169-328x(94)00287-o. [DOI] [PubMed] [Google Scholar]
- 22.Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, Mclntosh TK. Experimental brain injury induces differential expression of tumor necrosis factor-a mRNA in the CNS. Mol Brain Res. 1996;36:287–291. doi: 10.1016/0169-328x(95)00274-v. [DOI] [PubMed] [Google Scholar]
- 23.Fann DY, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, Bernreuther C, Glatzel M, Cheng YL, Thundyil J, Widiapradja A, Lok KZ, Foo SL, Wang YC, Li YI, Drummond GR, Basta M, Magnus T, Jo DG, Mattson MP, et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013;4:e790. doi: 10.1038/cddis.2013.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farkas O, Lifshitz J, Povlishock JT. Mechanoporation induced by diffuse traumatic brain injury: an irreversible or reversible response to injury? J Neurosci. 2006;26:3130–3140. doi: 10.1523/JNEUROSCI.5119-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fields RD, Burnstock G. Purinergic signalling in neuron–glia interactions. Nat Rev Neurosci. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma. 2010;27:497–507. doi: 10.1089/neu.2009.1120. [DOI] [PubMed] [Google Scholar]
- 27.Gorse KM, Lafrenaye AD. The importance of inter-species variation in traumatic brain injury-induced alterations of microglial-axonal interactions. Front Neurol. 2018;9:778. doi: 10.3389/fneur.2018.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grossetete M, Phelps J, Arko L, Yonas H, Rosenberg GA. Elevation of MMP-3 and MMP-9 in CSF and blood in patients with severe traumatic brain injury. Neurosurgery. 2009;65:702–708. doi: 10.1227/01.NEU.0000351768.11363.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guilfoyle MR, Carpenter KLH, Helmy A, Pickard JD, Menon DK, Hutchinson PJA. Matrix metalloproteinase expression in contusional traumatic brain injury: a paired microdialysis Study. J Neurotrauma. 2015;32:1553–1559. doi: 10.1089/neu.2014.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gyoneva S, Kim D, Katsumoto A, Kokiko-Cochran ON, Lamb BT, Ransohoff RM. Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. J Neuroinflammation. 2015;12:228. doi: 10.1186/s12974-015-0443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall ED, Andrus PK, Yonkers PA. Brain hydroxyl radical generation in acute experimental head injury. J Neurochem. 1993;60:588–594. doi: 10.1111/j.1471-4159.1993.tb03189.x. [DOI] [PubMed] [Google Scholar]
- 32.Hans VHJ, Kossmann T, Lenzlinger PM, Probstmeier R, Imhof H-G, Trentz O, Morganti-Kossmann MC. Experimental axonal injury triggers interleukin-6 mRNA, protein synthesis and release into cerebrospinal fluid. J Cereb Blood Flow Metab. 1999;19:184–194. doi: 10.1097/00004647-199902000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Hayakata T, Shiozaki T, Tasaki O, Ikegawa H, Inoue Y, Toshiyuki F, Hosotubo H, Kieko F, Yamashita T, Tanaka H, Shimazu T, Sugimoto H. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock. 2004;22:102–107. doi: 10.1097/01.shk.0000131193.80038.f1. [DOI] [PubMed] [Google Scholar]
- 34.Hensler T, Sauerland S, Riess P, Hess S, Helling HJ, Andermahr J, Bouillon B, Neugebauer EAM. The effect of additional brain injury on systemic interleukin (IL)-10 and IL-13 levels in trauma patients. Inflamm Res. 2000;49:524–528. doi: 10.1007/s000110050626. [DOI] [PubMed] [Google Scholar]
- 35.Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, Nakata Y. Extracellular ATP triggers tumor necrosis factor-a release from rat microglia. J Neurochem. 2000;75:965–972. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- 36.Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen ML, Charo I, Huang EJ, Liu J, Nakamura MC. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma. 2014;31:1677–1688. doi: 10.1089/neu.2013.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang S, Ge X, Yu J, Han Z, Yin Z, Li Y, Chen F, Wang H, Zhang J, Lei P. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. 2018;32:512–528. doi: 10.1096/fj.201700673R. [DOI] [PubMed] [Google Scholar]
- 39.Inoue K, Nakajima K, Morimoto T, Kikuchi Y, Koizumi S, Illes P, Kohsaka S. ATP stimulation of Ca2+-dependent plasminogen release from cultured microglia. Br J Pharmacol. 1998;123:1304–1310. doi: 10.1038/sj.bjp.0701732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irrera N, Pizzino G, Calò M, Pallio G, Mannino F, Famà F, Arcoraci V, Fodale V, David A, Francesca C, Minutoli L, Mazzon E, Bramanti P, Squadrito F, Altavilla D, Bitto A. Lack of the Nlrp3 inflammasome improves mice recovery following traumatic brain injury. Front Pharmacol. 2017;8:459. doi: 10.3389/fphar.2017.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ismael S, Nasoohi S, Ishrat T. MCC950 the selective NLRP3 inflammasome inhibitor protects mice against traumatic brain injury. J Neurotrauma. 2018;1303:1294–1303. doi: 10.1089/neu.2017.5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson VE, Weber MT, Xiao R, Cullen DK, Meaney DF, Stewart W, Smith DH. Mechanical disruption of the blood–brain barrier following experimental concussion. Acta Neuropathol. 2018;135:711–726. doi: 10.1007/s00401-018-1824-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kahle KT, Gerzanich V, Simard JM. Molecular mechanisms of microvascular failure in CNS injury - synergistic roles of NKCC1 and SUR1/TRPM4. J Neurosurg. 2010;113:622–629. doi: 10.3171/2009.11.JNS081052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamm K, VanderKolk W, Lawrence C, Jonker M, Davis AT. The effect of traumatic brain injury upon the concentration and expression of interleukin-1β and interleukin-10 in the rat. J Trauma. 2006;60:152–157. doi: 10.1097/01.ta.0000196345.81169.a1. [DOI] [PubMed] [Google Scholar]
- 46.Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889–900. doi: 10.3171/jns.1990.73.6.0889. [DOI] [PubMed] [Google Scholar]
- 47.Kim CC, Nakamura MC, Hsieh CL. Brain trauma elicits non-canonical macrophage activation states. J Neuroinflammation. 2016;13:117. doi: 10.1186/s12974-016-0581-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinoshita K, Chatzipanteli K, Vitarbo E, Truettner JS, Alonso OF, Dietrich WD. Interleukin-1beta messanger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: importance of injury severity and brain temperature. Neurosurgery. 2002;51:195–203. doi: 10.1097/00006123-200207000-00027. [DOI] [PubMed] [Google Scholar]
- 49.Kumar A, Alvarez-Croda D-M, Stoica BA, Faden AI, Loane DJ. Microglial/macrophage polarization dynamics following traumatic brain injury. J Neurotrauma. 2016;33:1732–1750. doi: 10.1089/neu.2015.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 51.Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N, Kumar A, Thom SR, Faden AI. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation. 2017;14:47. doi: 10.1186/s12974-017-0819-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lafrenaye AD, Todani M, Walker SA, Povlishock JT. Microglia processes associate with diffusely injured axons following mild traumatic brain injury in the micro pig. J Neuroinflammation. 2015;12:186–201. doi: 10.1186/s12974-015-0405-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, Youssef P, Yanasak N, Vender JR, Dhandapani KM. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of Toll-like receptor 4. Glia. 2014;62:26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lawson LJJ, Perry VHH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 55.Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor a, L-selectin, and tumor necrosis factor a. Mol Biol Cell. 2009;20:1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Gall SM, Maretzky T, Issuree PDA, Niu X-D, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci. 2010;123:3913–3922. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, Zhou ML, Zhu L, Hang CH. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res. 2013;38:2072–2083. doi: 10.1007/s11064-013-1115-z. [DOI] [PubMed] [Google Scholar]
- 58.Lo EH, Wang X, Louise Cuzner M. Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res. 2002;69:1–9. doi: 10.1002/jnr.10270. [DOI] [PubMed] [Google Scholar]
- 59.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu KT, Cheng NC, Wu CY, Yang YL. NKCC1-mediated traumatic brain injury-induced brain edema and neuron death via Raf/MEK/MAPK cascade. Crit Care Med. 2008;36:917–922. doi: 10.1097/CCM.0B013E31816590C4. [DOI] [PubMed] [Google Scholar]
- 61.Lu KT, Wu CY, Cheng NC, Wo YYP, Yang JT, Yen HH, Yang YL. Inhibition of the Na+-K+-2Cl–-cotransporter in choroid plexus attenuates traumatic brain injury-induced brain edema and neuronal damage. Eur J Pharmacol. 2006;548:99–105. doi: 10.1016/j.ejphar.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 62.Makinde HM, Cuda CM, Just TB, Perlman HR, Schwulst SJ. Nonclassical monocytes mediate secondary injury, neurocognitive outcome, and neutrophil infiltration after traumatic brain injury. J Immunol. 2017;199:3583–3591. doi: 10.4049/jimmunol.1700896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. 2015;35:748–760. doi: 10.1523/JNEUROSCI.2405-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morganti JM, Riparip LK, Rosi S. Call off the dog(ma): M1/M2 polarization is concurrent following traumatic brain injury. PLoS One. 2016;11:e0148001. doi: 10.1371/journal.pone.0148001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Özsüer H, Görgülü A, Kiriş T, Çobanoğlu S. The effects of memantine on lipid peroxidation following closed-head trauma in rats. Neurosurg Rev. 2005;28:143–147. doi: 10.1007/s10143-004-0374-1. [DOI] [PubMed] [Google Scholar]
- 67.Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci. 2012;6:58. doi: 10.3389/fncel.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- 69.Raposo G, Stoorvogel W. Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol. 2013;2000:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reijerkerk A, Kooij G, van der Pol SMA, Leyen T, van het Hof B, Couraud P-O, Vivien D, Dijkstra CD, de Vries HE. Tissue-type plasminogen activator is a regulator of monocyte diapedesis through the brain endothelial barrier. J Immunol. 2008;181:3567–3574. doi: 10.4049/jimmunol.181.5.3567. [DOI] [PubMed] [Google Scholar]
- 71.Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- 72.Schönbeck U, Mach F, Libby P. Generation of biologically active IL-1β by matrix metalloproteinases: a novel Caspase-1-independent pathway of IL-1β processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- 73.Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): Evidence from severe TBI patients and CCL2–/– mice. J Cereb Blood Flow Metab. 2010;30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shigemoto-Mogami Y, Koizumi S, Tsuda M, Ohsawa K, Kohsaka S, Inoue K. Mechanisms underlying extracellular ATP-evoked interleukin-6 release in mouse microglial cell line, MG-5. J Neurochem. 2001;78:1339–1349. doi: 10.1046/j.1471-4159.2001.00514.x. [DOI] [PubMed] [Google Scholar]
- 75.Simon DW, McGeachy MJ, Bayır H, Clark RSB, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:171–191. doi: 10.1038/nrneurol.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, Roberts CJ, Miller KR, Ransohoff RM, Trojanowski JQ, Lee VMY. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci. 2018;21:329–340. doi: 10.1038/s41593-018-0083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spiller KL, Koh TJ. Macrophage-based therapeutic strategies in regenerative medicine. Adv Drug Deliv Rev. 2017;122:74–83. doi: 10.1016/j.addr.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spiller KL, Wrona EA, Romero-Torres S, Pallotta I, Graney PL, Witherel CE, Panicker LM, Feldman RA, Urbanska AM, Santambrogio L, Vunjak-Novakovic G, Freytes DO. Differential gene expression in human, murine, and cell line-derived macrophages upon polarization. Exp Cell Res. 2016;347:1–13. doi: 10.1016/j.yexcr.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 79.Su G, Kintner DB, Sun D. Contribution of Na+-K+-Cl– cotransporter to high-[K+]o- induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol. 2001;282:C1136–1146. doi: 10.1152/ajpcell.00478.2001. [DOI] [PubMed] [Google Scholar]
- 80.Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 82.Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in I IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. J Neuroimmunol. 1993;42:177–186. doi: 10.1016/0165-5728(93)90008-m. [DOI] [PubMed] [Google Scholar]
- 83.Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury–related emergency department visits, hospitalizations, and deaths — United States, 2007 and 2013. MMWR Surveill Summ. 2017;66:1–16. doi: 10.15585/mmwr.ss6609a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verweij BH, Muizelaar JP, Vinas FC, Peterson PL, Xiong Y, Lee CP. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg. 2000;93:815–820. doi: 10.3171/jns.2000.93.5.0815. [DOI] [PubMed] [Google Scholar]
- 85.Wallisch JS, Simon DW, Bayır H, Bell MJ, Kochanek PM, Clark RSB. Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit Care. 2017;27:44–50. doi: 10.1007/s12028-017-0378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK, Gao Y, Chen J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1864–1874. doi: 10.1038/jcbfm.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber JT, Rzigalinski BA, Willoughby KA, Moore SF, Ellis EF. Alterations in calcium-mediated signal transduction after traumatic injury of cortical neurons. Cell Calcium. 1999;26:289–299. doi: 10.1054/ceca.1999.0082. [DOI] [PubMed] [Google Scholar]
- 88.Witcher KG, Bray CE, Dziabis JE, Mckim DB, Benner BN, Rowe RK, Kokiko-cochran ON, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP. Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia. 2018;66:2719–2736. doi: 10.1002/glia.23523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wofford KL, et al. Rapid neuroinflammatory response localized to injured neurons after diffuse traumatic brain injury in Swine. Exp Neurol. 2017;290:85–94. doi: 10.1016/j.expneurol.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xing G, Ren M, Verma A. Divergent temporal expression of hyaluronan metabolizing enzymes and receptors with craniotomy vs. controlled-cortical impact injury in rat brain: A pilot study. Front Neurol. 2014;5:173. doi: 10.3389/fneur.2014.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP. Mitochondrial dysfunction and calcium perturbation by traumatic brain injury. J Neurotrauma. 1997;14:23–34. doi: 10.1089/neu.1997.14.23. [DOI] [PubMed] [Google Scholar]
- 93.Xu H, Wang Z, Li J, Wu H, Peng Y, Fan L, Chen J, Gu C, Yan F, Wang L, Chen G. The polarization states of microglia in TBI: A new paradigm for pharmacological intervention. Neural Plast. 2017;2017:5405104. doi: 10.1155/2017/5405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu X, Yin D, Ren H, Gao W, Li F, Sun D, Wu Y, Zhou S, Lyu L, Yang M, Xiong J, Han L, Jiang R, Zhang J. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis. 2018;117:15–27. doi: 10.1016/j.nbd.2018.05.016. [DOI] [PubMed] [Google Scholar]
- 95.Yao Y, Tsirka SE. Monocyte chemoattractant protein-1 and the blood-brain barrier. Cell Mol Life Sci. 2014;71:683–697. doi: 10.1007/s00018-013-1459-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yi J, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 97.Zelnickova P, Matiasovic J, Pavlova B, Kudlackova H, Kovaru F, Faldyna M. Quantitative nitric oxide production by rat, bovine and porcine macrophages. Nitric Oxide. 2008;19:36–41. doi: 10.1016/j.niox.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 98.Zhang J, Pu H, Zhang H, Wei Z, Jiang X, Xu M, Zhang L, Zhang W, Liu J, Meng H, Stetler RA, Sun D, Chen J, Gao Y. Inhibition of Na+-K+-2Cl– cotransporter attenuates blood-brain-barrier disruption in a mouse model of traumatic brain injury. Neurochem Int. 2017;111:23–31. doi: 10.1016/j.neuint.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 99.Zhang S, Kojic L, Tsang M, Grewal P, Liu J, Namjoshi D, Wellington CL, Tetzlaff W, Cynader MS, Jia W. Distinct roles for metalloproteinases during traumatic brain injury. Neurochem Int. 2016;96:46–55. doi: 10.1016/j.neuint.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 100.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2010;469:221–226. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 101.Ziebell JM, Adelson PD, Lifshitz J. Microglia: dismantling and rebuilding circuits after acute neurological injury. Metab Brain Dis. 2014:393–400. doi: 10.1007/s11011-014-9539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]