

Abstract
Neuroinflammation plays a fundamental role on the pathophysiology of acute and chronic neural disorders. Microglia activation is a major event following central nervous system inflammation displaying different phenotypes with beneficial and detrimental actions (a Janus face). The reason for this apparent duality is unknown. We have previously shown that following experimental middle cerebral artery occlusion in the rat brain, microglia seem to support and impair adult neurogenesis in the same ischemic striatum. Based on these results, we raised the hypothesis that in the same pathologic environment, gradients of different ligands distributed over different anatomical niches might contribute to both detrimental and beneficial microglial phenotypes. These ligands (“danger signals”) are released by dying cells and bind to microglial receptors in their membranes. Activation of different microglial receptors induces downstream biochemical pathways culminating in a spectrum of microglial phenotypes like M1 and M2 and others. In this paper, we first review the immune functions of microglia and the role of toll-like receptors on the fight against infections. We then briefly revise the dual role of microglia after neural disorders. We then propose a novel hypothesis to explain the Janus face of microglia during the pathophysiology of central nervous system diseases: the “friendly fire hypothesis”. According to this idea “danger signals” or danger associated molecular patterns released by stressed, damaged and/or dying cells during stroke, trauma and other diseases might activate microglial pattern-recognition receptors (i.e., toll like receptors) or other unidentified receptors normally activated by pathogens. This could activate the same genetic and biochemical machinery used by microglia to fight against pathogens even in the absence of infection. According to this notion, microglia may cause bystander neuronal damage with a kind of blind “friendly fire”, fighting against a non-existing infection during non-infectious disorders, like stroke and trauma. The “friendly fire hypothesis” is a novel proposal to explain why microglia may be detrimental and beneficial after acute and chronic neural disorders and may direct future investigations for developing of neuroprotective agents.
Keywords: stroke, trauma, Alzheimer's disease, neuroinflammation, glial cells, phenotypes, degeneration, neuroprotection
Microglia are resident macrophages derived from primitive myeloid progenitors from the yolk sac and the main components of the central nervous system (CNS) innate immune system (Mariani and Kielian, 2009). These glial cells play important roles on both developing and adult CNS (Vainchtein et al., 2018). They engulf synapses during CNS development interacting with astrocytes, which release interleukin 33 - a fundamental mechanism for controlling synapse maturation and neural circuit development (Vainchtein et al., 2018). These resident macrophages regulate neurogenesis in embryos and shape CNS ontogeny, for example in the retina, cortex, cerebellum and striatum, mainly by phagocytosis of excessive progenitor cells (Low and Ginhoux, 2018). Experimental studies suggest that the appropriate distribution of specific neuronal types and their connections depend on the interaction between microglia and neurons (Squarzoni et al., 2014). In the mature adult brain, individual microglial cells occupy their own spatial territory continuously scanning CNS parenchyma through stochastic movements of their branches looking for minor tissue damage (Nimmerjahn et al., 2005).
As part of the CNS innate immune system, microglia are the first line of defense against pathogens (Mariani and Kielian, 2009). These glial cells were evolutivelly programmed to fight against pathogens and protect the CNS against infection. This is one of the most prominent beneficial actions of microglia (Mariani and Kielian, 2009). The CNS is protected by the blood-brain barrier, but several types of virus, bacteria, fungi, and protozoa can cause CNS damage and illness (Mariani and Kielian, 2009). During infection, pathogens replicate and release molecules that can be recognized by innate immune cells. These molecules are known as pathogen-associated molecular patterns and they are identified by pattern-recognition receptors (PRRs) expressed on microglial membrane as well as in other immune cells (Mariani and Kielian, 2009). PRRs include toll-like receptors (TLRs), receptors for advanced glycation endproducts, nucleotide binding oligomerisation domain-like receptors, c-type lectin receptors, RIG-I-like receptors, and intra-cytosolic DNA sensors. The interaction between PRRs and pathogen-associated molecular patterns activates adaptor molecules like myeloid differentiation primary response 88, which in turn interacts with several other receptors to activate protein kinases for inducing nuclear factor kappa B translocation to the nucleus culminating in activation of several pro-inflammatory genes (Mariani and Kielian, 2009). This induces synthesis and release of pro-inflammatory cytokines (for example, tumor necrosis factor-α, interleukin-1β), chemokines and reactive oxygen/nitrogen species by microglia in order to eliminate pathogens (Mariani and Kielian, 2009).
Microglia also play a dual role following acute and chronic neural disorders (Gomes-Leal, 2012). Inhibition of microglia activation with minocycline reduces neuroinflammation and infarct area in rats following middle cerebral artery occlusion (Yrjänheikki et al., 1999). Overactivated microglia are also detrimental to neurons in several other experimental conditions (Gomes-Leal, 2012). Paradoxically, microglia are also neuroprotective in the same stroke model. Selective genetic ablation of proliferating microglia exacerbates ischemic damage following middle cerebral artery occlusion in rats (Lalancette-Hébert et al., 2007). Similar results were reported following oxygen-glucose deprivation. Oxygen-glucose deprivation induces neuronal damage in hippocampal organotypic culture, but the presence of BV2 microglia reduces neuronal loss (Neumann et al., 2006). In addition, microglia are also neuroprotective in the oxygen-glucose deprivation ischemic model by performing direct engulfment of neutrophils (Neumann et al., 2008). This Janus-faced actions of microglia/macrophages are also reported for chronic neurodegenerative diseases (Heppner et al., 2015).
There is no explanation for microglial Janus face, but we believe this can be related to the immune functions of microglia. We first reported differential patterns of microglial activation and their implication for adult neurogenesis in different anatomical niches in the ischemic striatum following middle cerebral artery occlusion in rats (Gomes-Leal, 2012). In the dorsal striatum, overactivated microglia seem to impair neuroblast migration, but more ramified microglia seem to support migration of immature neurons in the ventral striatum (Figure 1). We proposed that gradients of different ligands may be present in the same pathological environment inducing different microglial phenotypes contributing to repair or secondary damage to neurons (Gomes-Leal, 2012). This is in agreement with the notion that microglia display beneficial (M2) and detrimental (M1) phenotypes, although they can be dynamically changed depending on the pathological environment (Gomes-Leal, 2012).
Figure 1.
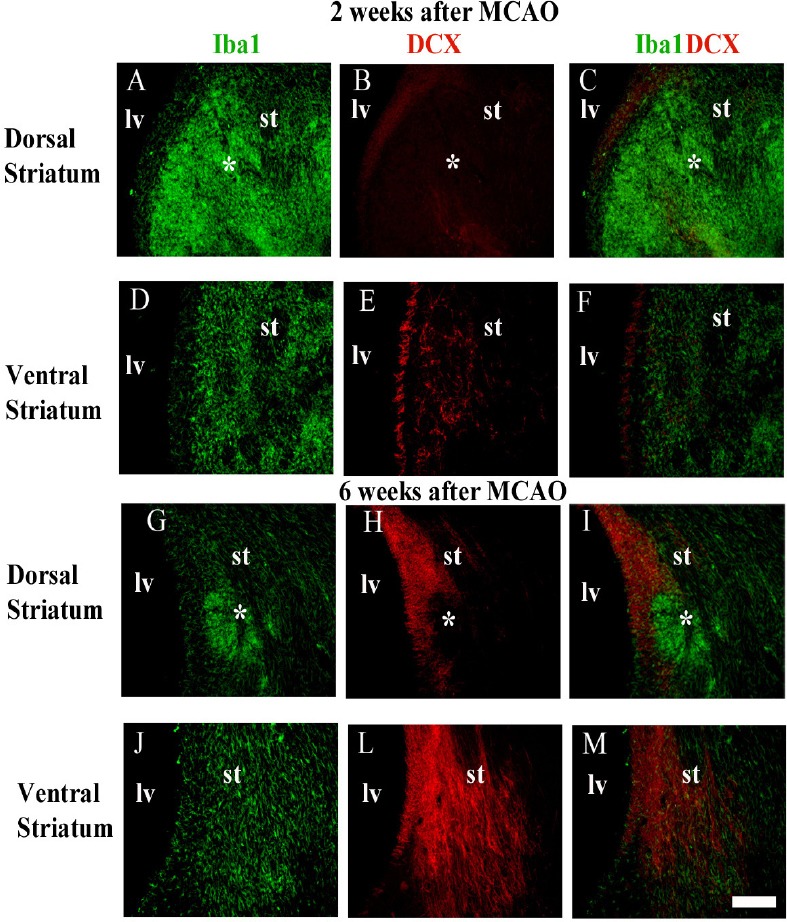
Spatial correlation between activated microglia and migrating neuroblasts in the striatum after middle cerebral artery occlusion (MCAO).
(A–C, G–I) Microglia were labeled by anti-Iba1 (green) and migrating neuroblasts by anti-doublecortin (DCX) (red) double immunofluorescence. Aggregations (*clustering) of overactivated microglia/macrophages are present in the dorsal striatum at 2 (A–C) and 6 weeks (G–I) after MCAO. (D–F, J–M) Neuroblasts were less frequent in the ischemic striatal regions containing overactivated microglia/macrophages clustering (dorsal striatum), but intermingled with moderately activated microglia in striatal regions outside microglia/macrophages clustering (ventral striatum). lv: Lateral ventricle; st: striatum. Scale bar: 100 μm. is adapted from Gomes-Leal (2012).
Matzinger (1994) proposed the “danger hypothesis”. According to this idea, the adaptive immune system evolved not only to respond to pathogens but to “danger signals” released by damaged, stress or dying cells. In our opinion, this notion gives an important clue to explain the microglial Janus face after CNS disorders. Here, we propose a novel hypothesis to explain the dual role of microglia after acute neural disorders: the “friendly fire hypothesis”. According to this idea, “danger signals” or danger associated molecular patterns (DAMPs) released by stressed, damaged and/or dying cells during stroke and trauma might activate microglial PRRs (i.e., toll like receptors) or other unidentified receptors normally activated by pathogens, even in the absence of infection. This could activate the same genetic and biochemical machinery used by microglia to fight against pathogens even in the absence of infection. According to this notion, microglia might cause bystander neuronal damage with a kind of blind friendly fire, fighting against a non-existing infection during non-infectious disorders (Gomes-Leal, 2012).
Several “danger signals” have been described to be released by different cell types after stroke, including adenosine triphosphate (ATP), heat shock proteins, high-mobility group box 1 protein, hyaluronic acid, lipids, peroxiredoxins, S100A8, S100A9, myeloid-related protein 8, myeloid-related protein 14, and cold-inducible RNA binding protein (Gelderblom et al., 2015). These DAMPs bind to different receptors on immune cells activating inflammasome cascade culminating in the expression of several pro-inflammatory genes resulting in synthesis and release of interleukin-1β, interleukin-17, tumor necrosis factor-α, perforin, granzyme, reactive oxygen species and several other pro-inflammatory and damaging molecules (Gelderblom et al., 2015). Microglia express a multitude of different receptors on the membrane with an immense variety of functions (Mariani and Kielian, 2009; Gelderblom et al., 2015; Hug et al., 2018). These include P2Y family, toll-like, fractalkine, tumor necrosis factor-α, complement, Fractalkine receptor 1, immunoglobulin, triggering receptor expressed on myeloid cells 2, low density lipoprotein receptor-related protein 1- and low density lipoprotein protein-1 receptors. The expression of different receptors and the outcome following their activation depend on the functional state and pathological condition. Downstream intracellular biochemical reactions will translate the activation of a specific receptor in the microglia membrane into beneficial or detrimental effects, which depends on the nature of the ligand (Gomes-Leal, 2012). ATP binds to different purinergic receptors with beneficial and detrimental actions after stroke (Gelderblom et al., 2015). Nevertheless, activation of P2×7R receptor, which is important to eliminate pathogens in tuberculosis and toxoplasmosis, is also activated by ATP in the event of stroke leading to neuronal damage (Gelderblom et al., 2015). In addition, knockout mice for P2×7R receptor (P2×7R−/−) present smaller ischemic damage and blockage of P2×7R receptor induces neuroprotection (Domercq et al., 2010).
The toll-like family of PRRs are the main receptors activated by different DAMPS following stroke normally with deleterious consequences (Gelderblom et al., 2015). TLR-2 mediates CNS injury in focal cerebral ischemia (Lehnardt et al., 2007) and activation of TLR-4 by a specific type of heat shock protein may be an endogenous molecular pathway common to many forms of neuronal injury (Lehnardt et al., 2008). It is likely that non-selective activation of different PRRs in macrophages by non-infectious stimuli is a general phenomenon present in all tissues. This hypothesis is strongly supported by the fact that activation of TLRs causes bystander damage to non-neural tissues, including gut and kidney (Hug et al., 2018; Panchapakesan and Pollock, 2018). Most of the papers suggests a detrimental role for TLRs activation after stroke and other neural disorders (Lehnardt et al., 2007, 2008; Gelderblom et al., 2015). Nevertheless, the role of TLRs on the pathophysiology of neural disorders is a complex issue, considering that recent evidences suggest that activation of some types of TLRs in microglia membrane may also contribute to neuroprotection and plasticity (Tajalli-Nezhad et al., 2019).
Although we have discussed the “friendly fire hypothesis” in the context of acute neural disorders, this idea can also be applied to the pathophysiology of chronic neurodegenerative diseases. Protein aggregates including neuromelanin, α-synuclein, fibrillar amyloid-βfibrils, prion proteins, viral RNA play a detrimental role on chronic neurodegenerative diseases by inducing a detrimental phenotype in microglia (Heppner et al., 2015). In experimental models of Alzheimer’s disease, microglia can be beneficial by removing plaques of amyloid-β, but this protein can activate receptors on microglia membrane rendering them dystrophic with low phagocytic capacity or highly pro-inflammatory and neurotoxic (Heppner et al., 2015). It is likely that danger signals released by damaged, stressed or dying cells together with protein aggregates like α-synuclein and fibrillary amyloid-β also cause neuronal damage by activating PRRs or other receptors in the membrane of microglia (Gomes-Leal, 2012; Heppner et al., 2015).
As a first step to test the “friendly fire hypothesis,” we started to investigate the injury outcome and microglial phenotypes in ischemic rats with and without meningoencephalitis induced by Staphylococcus aureus. We have data from an ongoing investigation showing that the presence of bacterial infection exacerbates ischemic damage (Figure 2).
Figure 2.
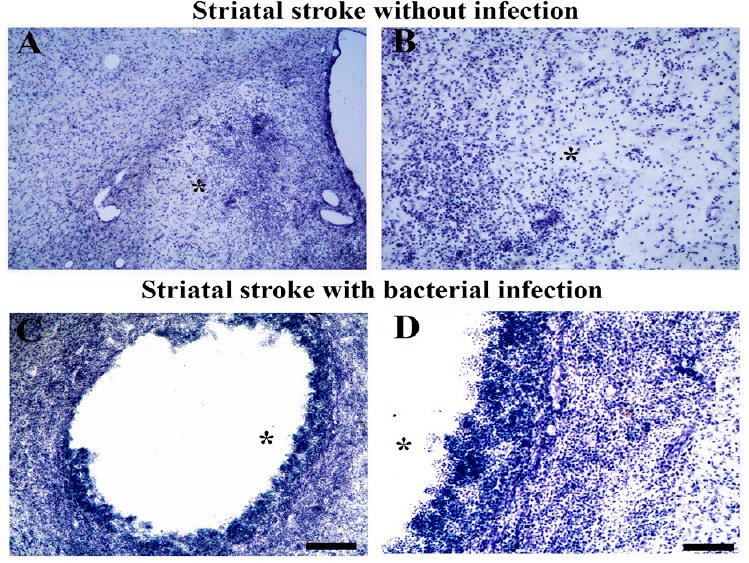
Bacterial infection exacerbates striatal ischemic damage.
(A–D) Striatal ischemic damage induced by microinjections of endothelin-1 without (A, B) and with bacterial infection (C, D). Experimental bacterial infection exacerbates ischemic damage causing extensive cystic cavities in the rat striatum (C, D). B and D are higher power of A and C. Asterisks point to the center of ischemic damage. lv: Lateral ventricle. Scale bars: 100 μm in A, B; 25 μm in C, D.
Here, we discussed a novel idea to explain the dual role of microglia after acute neural disorders. The core of this idea is that PRRs, normally used to sense, fight and eliminate pathogens during infections, are activated during sterile inflammation by DAMPs released by damaged cells. This may be a general phenomenon present in all tissues in which PRRs in the membranes of resident macrophages are activated by non-infectious stimuli leading to bystander tissue damage. In the CNS, identification of the danger signals and the putative receptors that they activate in deleterious microglia can lead to development of new neuroprotective agents.
The following searches were performed in the US National Library of Medline (PubMed) (Table 1).
Table 1.
Search strategy
| Key words | Search time | Search results | Best matches |
|---|---|---|---|
| Microglia and review | 1962–2019 | 4666 | 3 |
| Microglia and CNS diseases and phenotypes | 1986–2019 | 1153 | 3 |
| Microglia and CNS diseases and detrimental | 1997–2019 | 339 | 3 |
| Microglia and CNS diseases and beneficial | 1994–2019 | 742 | 3 |
| Microglia and infection | 1931–2019 | 2377 | 3 |
| Microglia and infection and toll like receptors | 2001–2019 | 134 | 3 |
| Microglia and danger signals | 2007–2019 | 52 | 3 |
| Danger signals and stroke | 1968–2019 | 26 | 3 |
| Membrane receptors in microglia | 1983–2019 | 959 | 3 |
CNS: Central nervous system.
Additional file: Open peer review report 1 (80.3KB, pdf) .
Acknowledgments:
The author is grateful to Ijair R. dos Santos and Leonardo M. T. Sousa (Federal University of Pará-Brazil) for performing some of the experiments mentioned as preliminary results in this paper.
Footnotes
Conflicts of interest: All authors disclose no conflicts of interest.
Financial support: The work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ) -Brazil, Banco da Amazônia and Organização não governamental (ONG) Iluminando A Vida.
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Javier Francisco-Morcillo, Universidad de Extremadura, Spain.
Funding: The work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ)-Brazil, Banco da Amazônia and Organização não governamental (ONG) Iluminando A Vida.
P-Reviewer: Francisco-Morcillo J; C-Editors: Zhao M, Yu J; T-Editor: Jia Y
References
- 1.Domercq M, Perez-Samartin A, Aparicio D, Alberdi E, Pampliega O, Matute C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2010;58:730–740. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- 2.Gelderblom M, Sobey CG, Kleinschnitz C, Magnus T. Danger signals in stroke. Ageing Res Rev. 2015;24:77–82. doi: 10.1016/j.arr.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Gomes-Leal W. Microglial physiopathology: how to explain the dual role of microglia after acute neural disorders? Brain Behav. 2012;2:345–356. doi: 10.1002/brb3.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- 5.Hug H, Mohajeri MH, La Fata G. Toll-like receptors: regulators of the immune response in the human gut. Nutrients. 2018;10:E203. doi: 10.3390/nu10020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehnardt S, Schott E, Trimbuch T, Laubisch D, Krueger C, Wulczyn G, Nitsch R, Weber JR. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J Neurosci. 2008;28:2320–2331. doi: 10.1523/JNEUROSCI.4760-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 9.Low D, Ginhoux F. Recent advances in the understanding of microglial development and homeostasis. Cell Immunol. 2018;330:68–78. doi: 10.1016/j.cellimm.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Mariani MM, Kielian T. Microglia in infectious diseases of the central nervous system. J Neuroimmune Pharmacol. 2009;4:448–461. doi: 10.1007/s11481-009-9170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 12.Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. 2006;20:714–716. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- 13.Neumann J, Sauerzweig S, Ronicke R, Gunzer F, Dinkel K, Ullrich O, Gunzer M, Reymann KG. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci. 2008;28:5965–5975. doi: 10.1523/JNEUROSCI.0060-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 15.Panchapakesan U, Pollock C. The role of toll-like receptors in diabetic kidney disease. Curr Opin Nephrol Hypertens. 2018;27:30–34. doi: 10.1097/MNH.0000000000000377. [DOI] [PubMed] [Google Scholar]
- 16.Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, Bessis A, Ginhoux F, Garel S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8:1271–1279. doi: 10.1016/j.celrep.2014.07.042. [DOI] [PubMed] [Google Scholar]
- 17.Tajalli-Nezhad S, Karimian M, Beyer C, Atlasi MA, Azami Tameh A. The regulatory role of Toll-like receptors after ischemic stroke: neurosteroids as TLR modulators with the focus on TLR2/4. Cell Mol Life Sci. 2019;76:523–537. doi: 10.1007/s00018-018-2953-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, Akil O, Joshita S, Barres BA, Paz JT, Molofsky AB, Molofsky AV. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269–1273. doi: 10.1126/science.aal3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yrjänheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.