

Our common home, planet Earth, is inhabited by more than 7 billion human beings. The average life expectancy currently estimated for men is ~69 years, with strong differences among countries. Currently, neurodegenerative diseases are currently the leading cause of death and disability worldwide. In clinical use and neuroscience research, a solution for an effective diagnosis is in progress, but a prophylactic treatment to counteract this long-lasting plague for humanity is still unidentified.
At the end of the 20th century, it is known that brain cells de novo synthesize erythropoietin (EPO) and that the cytokine has neuroprotective effects against brain damage caused by permanent focal ischemia in mice (Morishita et al., 1997). Since then, neuroprotective effects of EPO in preclinical studies have been widely reported for a variety of neurodegenerative diseases, including stroke, Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis. Although each of these different neuropathologies has a different etiology, they all share excitotoxicity as one of the main mechanisms that leads to neuronal cell loss (Dong et al., 2009). Excitotoxicity is defined as cell death due to excessive stimulation of neurons by excitatory amino acids neurotransmitters, particularly glutamate.
EPO is a glycoprotein cytokine, initially known for its essential role in the regulation of erythropoiesis. EPO is a single polypeptide chain of 165 amino acids with a molecular mass of 30 kDa, depending on the carbohydrate content. The overall composition of mature EPO is ~60% protein and 40% carbohydrate, containing four glycosylated chains including three N-linked and one O-linked acidic oligosaccharide side chains that are important for the biological activity of EPO. In the last 30 years, recombinant human EPO (rh-EPO) and its analogues have improved the quality of life of more than a million patients with anemia associated with chronic renal failure. In addition, many studies have shown that EPO also possesses various other non-hematopoietic biological functions, such as neuroprotection, neurogenesis and angiogenesis.
The fact that EPO was widely used in clinical practice makes rh-EPO a highly attractive candidate drug for neuroprotection/neurodegeneration. After some encouraging initial results in clinical trials to human patients with ischemic stroke, a German multicenter EPO trial fails to detect beneficial effects. This was partly due to the erythropoietic effect of EPO, with a high percentage of patients showing edema and cerebral hemorrhage (Ehrenreich et al., 2009). The use of rh-EPO in the treatment of neurological diseases requires higher doses and prolonged application, in a way that likely produces an increase of hematocrit and blood viscosity, finally leading to serious cardiovascular events such as infarct or stroke. To address this issue, an approach may be the use of non-hematopoietic EPO derivatives that retain neuroprotective activity against neuronal injury (Leist et al., 2004) but appear devoid of erythropoietic activity. Moreover, given the pleiotropic actions of EPO in the mammalian brain, any dysbalance due to alterations in either its synthesis or its regulation could in turn produce a progressive damage in the brain region involved, leading with time to different clinical manifestations (Broxmeyer, 2013). In this sense, it is important to develop non-erythropoietic variant of EPO, ideally with minor chemical modifications, and Neuro-EPO appears as a promising derivative that meets these requirements.
Neuro-EPO is a recombinant human glycoprotein produced in Chinese hamster ovary cells supplied by the Center of Molecular Immunology (CIM, Havana, Cuba). It is characterized by its low sialic acid content and consequently Neuro-EPO is devoid of erythropoietic activity, while it exhibits high neuroprotective properties (Rodríguez-Cruz et al., 2010). Neuro-EPO is rapidly degraded in the liver and must be preferentially applied by intranasal route (García-Rodriguez and Sosa-Teste, 2009). After intranasal administration, the molecule reaches the brain rapidly and does not stimulate erythropoiesis after acute treatments. It has been shown that Neuro-EPO exerts neuroprotective effects such as improving viability and cognitive functions in animal models of stroke (Rodríguez-Cruz et al., 2010) or in non-transgenic and transgenic rodent models of Alzheimer’s disease (AD) (Maurice et al., 2013; Rodríguez-Cruz et al., 2017). In fact, Neuro-EPO, injected intranasally was first compared with rh-EPO, injected intraperitoneally, in mice injected intracerebroventricularly with aggregated amyloid-β(Aβ)25–35 peptide (Maurice et al., 2013). Both rh-EPO and Neuro-EPO led to a significant prevention of learning deficits induced by Aβ25–35 and prevented the induction of lipid peroxidation in the hippocampus, showing an antioxidant activity. rh-EPO or Neuro-EPO prevented the increase induced by Aβ25–35 at the Bax level, the production of tumor necrosis factor-α and interleukin 1β and the decrease in Akt activation. A significant prevention of cell loss induced by Aβ25–35 in hippocampal CA1 was also observed. These observations confirmed that EPO is neuroprotective in a non-transgenic mouse model of AD, and identified Neuro-EPO as a promising new therapeutic agent in AD with efficacy, ease and safety (Maurice et al., 2013).
More recently, the protective effect of Neuro-EPO was analyzed in APPSwe mice, a transgenic mouse model of AD reference (Rodríguez-Cruz et al., 2017). The mice were intransally treated with Neuro-EPO (125 or 250 μg/kg), between 12 and 14 months of age. Motor responses, general activity and memory responses were analyzed during and after treatment. Deficiencies in spontaneous alternation, learning of place in the water maze, and recognition of new objects observed in APPSwe mice were alleviated by the low dose of Neuro-EPO. Oxidative stress, neuroinflammation, trophic factor levels and a synaptic marker were analyzed in the hippocampus or cortex of animals. Increases in lipid peroxidation or in the contents of glial fibrillary acidic protein and Iba-1, respectively markers of astroglial and microglial reactions, in APPSwe mice were significantly reduced after Neuro-EPO. The activation of intrinsic and extrinsic apoptotic pathways was analyzed. Increases in the Bax/B-cell lymphoma-2 (Bcl-2), tumor necrosis factor-α ratio or Fas ligand levels observed in APPSwe mice were reduced with Neuro-EPO. Finally, analyzes of Aβ1–42 levels in the cortex and hippocampus of APPSwe mice showed a marked reduction in Aβ deposits and soluble and insoluble forms of Aβ1–42. Therefore, this study confirmed the neuroprotective activity of this Neuro-EPO formulation delivered by intranasal route without erythropoietic side effects, in a transgenic mouse model of AD reference, the APPSwe mouse (Rodríguez-Cruz et al., 2017).
To achieve a clinical use of Neuro-EPO as a neuroprotector, it is critical to understand the cellular mechanisms that mediate neuronal injury and are susceptible to be modulated by Neuro-EPO. The brain has cellular mechanisms helping to maintain its functionality against deleterious factors that provoke neurological damages. It is known that in response to brain injury, endogenous survival mechanisms are activated, such as antioxidant response, anti-inflammatory cytokines, antiapoptotic systems, and growth factors. All of these components of the endogenous response tend to counteract the cascades promoting neuronal death, including oxidative stress, inflammation, and apoptosis. In this scheme, given the encouraging results provided by previous studies with Neuro-EPO in animal models of stroke and AD (Rodríguez-Cruz et al., 2010, 2017; Maurice et al., 2013), it was necessary to understand the direct effect of Neuro-EPO on neuronal damage, and we considered first an excitotoxicity model.
Excitototoxicity has been linked to oxidative stress and mitochondrial dysfunction (Lin and Beal, 2006). Excitotoxicity leads to a number of deleterious consequences, including impairment of cellular calcium homeostasis, generation of free radicals (oxidative stress), mitochondrial dysfunction, and activation of several transcription factors. Although each of these mechanisms may individually cause neuronal death, they act synergistically. Oxidative stress causes mitochondrial dysfunction and increased production of free radicals, which in turn increases mitochondrial dysfunction, a vicious circle that leads to greater oxidative stress and intensifies the neuronal damage.
Experimental in vitro models are often used to study morphological and biochemical changes related to degeneration processes, because they allow an easier and precise control of the extracellular environment compared to in vivo models. However, as it happens with all studies carried out with cell cultures, it has a series of limitations that prevent the extrapolation of the results to in vivo conditions. We chose primary cortical neurons culture with very low presence of glial cells. This condition has the advantage that it allows to associate the results to a direct effect of Neuro-EPO on neurons, but it has the disadvantage that the interaction between neurons and glial cells is lacking.
We analyzed the effect of Neuro-EPO in an in vitro excitotoxic model using a primary culture of cortical neurons exposed to glutamate (Garzón et al., 2018a, b). The neurotoxicity induced by exposure to high doses of glutamate (100 μM) was attenuated by treatment with Neuro-EPO (100 ng/mL). Neuro-EPO reduced neuronal mortality caused by glutamate. Untreated cortical neurons retained normal morphology characterized by many large cells with thick and abundant cellular extensions (dendrites) that make contact with the neighboring cells (Figure 1IA). Exposure to glutamate showed an evident morphological deterioration. Neurons exhibited cell body shrinkage, formation of cell surface “blebs”, and a dramatic loss of dendritic processes in the cell culture were found. Small cells, retracted with thin processes that do not make contact with neighboring cells could be observed (Figure 1IB). Treatment with Neuro-EPO showed a tendency to retain the morphology seen in the health neurons, large cells had thick processes and preserved contact with neighboring cells (Figure 1IC). Immunocytochemistry images showed that glutamate causes cell mortality (Figure 1IIB) that could be partially avoided when the culture medium was supplemented with Neuro-EPO (Figure 1IIC). We reported that Neuro-EPO has a neuroprotective effect against oxidative stress associated with increases of antioxidant activity without significant changes in the oxidant activity (Garzón et al., 2018a). Like EPO, Neuro-EPO acts as powerful antioxidant keeping the cellular antioxidant activity, which allows neurons to regulate their intracellular redox condition.
Figure 1.
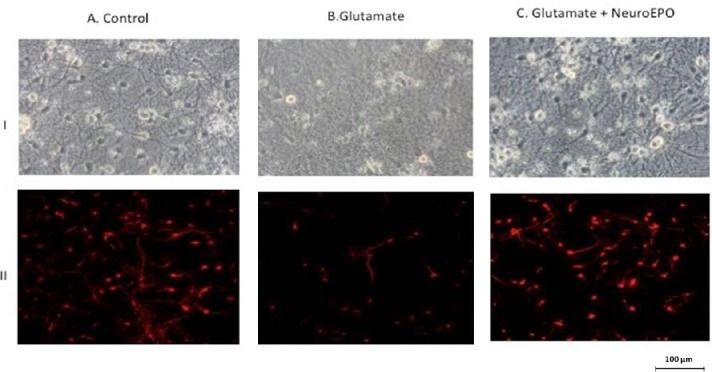
Morphological changes and characterization of neuron culture after exposure to glutamate: neuroprotective effect to Neuro-EPO.
Primary cortical cells obtained from the cerebral hemisphere of embryos of Wistar rats after 17 days of gestation. I. Phase-contrast photomicrographs of cortical neurons after 9 days of culture. (A) Control neurons show the presence of large cell with thick and thin processes that make contact with neighboring cells. (B) Cortical neurons exposed to glutamate show small cells with thin processes fragmented that not make contact with neighboring cells, and with cell body shrunk. (C) Cortical neurons treated with Neuro-EPO after exposure to glutamate show a tendency to retain the morphology seen in neurons control: large cells had thick processes and preserve contact to neighboring neurons. II. Immunostanding using MAP-2 (red), and GFAP (green), specific by neurons and glia cells reveals: (A) Control, the presence of neurons and that only very few small number of glia were present in the culture (not found in these images). (B) Glutamate exposure induces death of cortical neurons while that (C) neurons treated with Neuro-EPO attenuate the loss of viability. MAP-2: Microtubule-associated protein 2; GFAP: glial fibrillary acidic protein; EPO: erythropoietin.
Activation of intrinsic apoptotic pathways was analyzed. The decreased Bcl-2/Bax ratio, increased cytochrome c release, and increased expression and activity of caspase-3 observed in cells treated with glutamate, were restored by Neuro-EPO. Neuro-EPO therefore protected cortical neurons from glutamate-induced apoptosis via upregulation of Bcl-2 and inhibited glutamate-induced activation of caspase-3 (Garzón et al., 2018b). Although Neuro-EPO showed a reduction in the expression of the main markers of the mitochondrial apoptotic pathway, these effects could be explained as a result of increased expression of the anti-apoptotic Bcl-2 protein. In this sense, it is noteworthy that increase in the expression of Bcl-2 induced by Neuro-EPO in glutamate-treated neurons is also observed in untreated neurons. It is well known that Bcl-2 plays a pivotal role in maintaining the integrity of the outer membrane structure of the mitochondria counteracting the activation of pro-apoptotic proteins by regulating the permeabilization of mitochondrial outer membrane and consequently the release of cytochrome c into cytoplasm and the mitochondrial intrinsic apoptotic pathway. The integrity of the mitochondria implies the maintenance of its redox state and in this situation the mitochondria would cease to be the main source of reactive oxygen species, necessary for the appearance of a state of oxidative stress. This would justify the hypothesis that the neuroprotective effect of Neuro-EPO against situations of excitotoxicity would be through the induction of increased expression of Bcl-2. By preserving the integrity of mitochondria, Bcl-2 cancels the main source of reactive oxygen species induced by glutamate. This neuroprotective effect of Neuro-EPO confirmed those reported in AD animals (Maurice et al., 2013; Rodríguez-Cruz et al., 2017) or after focal ischemia (García-Rodriguez and Sosa-Teste, 2009; Rodríguez-Cruz et al., 2010). However, the extrapolation of the results obtained in cultures of neurons to chronic neurodegenerative pathologies must take into account the existence of multiple and complex pathways responsible for delayed death in each pathological process.
It is important to note that the neuroprotective effects of Neuro-EPO against excitotoxic mechanisms occur once excitotoxicity has been induced. We characterized the time-course effect of Neuro-EPO, immediately added after inducing excitotoxicity, and its effect is limited to 24 hours. Without a doubt and considering potential therapeutic application, it would be interesting to know the therapeutic window effect of Neuro-EPO.
While we do not have an effective methodology to find a way to activate the endogenous production of EPO by the brain, the neuroprotective effects of Neuro-EPO make it a very attractive biosimilar as a candidate for its use in neuroprotective therapies.
Additional file: Open peer review report 1 (96.6KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Gabriele Siciliano, Universita degli Studi di Pisa, Italy.
P-Reviewer: Siciliano G; C-Editor: Zhao M, Sun Y; T-Editor: Liu XL
References
- 1.Broxmeyer HE. Erythropoietin: multiple targets, actions, and modifying influences for biological and clinical consideration. J Exp Med. 2013;210:205–208. doi: 10.1084/jem.20122760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30:379–387. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, Schellinger PD, Bohn M, Becker H, Wegrzyn M, Jähnig P, Herrmann M, Knauth M, Bähr M, Heide W, Wagner A, Schwab S, Reichmann H, Schwendemann G, Dengler R, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–656. doi: 10.1161/STROKEAHA.109.564872. [DOI] [PubMed] [Google Scholar]
- 4.García-Rodriguez JC, Sosa-Teste I. The nasal route as a potential pathway for delivery of erythropoietin in the treatment of acute ischemic stroke in humans. ScientificWorldJournal. 2009;9:970–981. doi: 10.1100/tsw.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garzón F, Rodríguez-Cruz Y, García-Rodriguez JC, Rama R. Neuroprotective effects of neuroEPO using an in vitro model of stroke. Behav Sci (Basel) 2018a;8 doi: 10.3390/bs8020026. pii: E26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garzón F, Coimbra D, Parcerisas A, Rodriguez Y, García JC, Soriano E, Rama R. NeuroEPO preserves neurons from glutamate-induced excitotoxicity. J Alzheimers Dis. 2018b;65:1469–1483. doi: 10.3233/JAD-180668. [DOI] [PubMed] [Google Scholar]
- 7.Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, Savino C, Bianchi M, Nielsen J, Gerwien J, Kallunki P, Larsen AK, Helboe L, Christensen S, Pedersen LO, Nielsen M, Torup L, Sager T, Sfacteria A, Erbayraktar S, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305:239–242. doi: 10.1126/science.1098313. [DOI] [PubMed] [Google Scholar]
- 8.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 9.Maurice T, Mustafa MH, Desrumaux C, Keller E, Naert G, de la C García-Barceló M, Rodríguez Cruz Y, Garcia Rodríguez JC. Intranasal formulation of erythropoietin (EPO) showed potent protective activity against amyloid toxicity in the Aβ25-35 non-transgenic mouse model of Alzheimer’s disease. J Psychopharmacol. 2013;27:1044–1057. doi: 10.1177/0269881113494939. [DOI] [PubMed] [Google Scholar]
- 10.Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience. 1997;76:105–116. doi: 10.1016/s0306-4522(96)00306-5. [DOI] [PubMed] [Google Scholar]
- 11.Rodríguez-Cruz Y, Mengana Támos Y, Muñoz Cernuda A, Subirós Martines N, González-Quevedo A, Sosa Testé I, García Rodríguez JC. Treatment with nasal neuro-EPO improves the neurological, cognitive, and histological state in a gerbil model of focal ischemia. ScientificWorldJournal. 2010;10:2288–2300. doi: 10.1100/tsw.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodríguez-Cruz Y, Strehaiano M, Rodríguez Obaya T, García Rodriguez JC, Maurice T. An intranasal formulation of erythropoietin (Neuro-EPO) prevents memory deficits and amyloid toxicity in the APPSwe transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2017;55:231–248. doi: 10.3233/JAD-160500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.