

Summary
Osteogenesis imperfecta (OI) is a genetic disorder characterized by bone fragility and blue sclerae, which are mainly caused by a mutation of the COL1A1 or COL1A2 genes that encode type I procollagen. Mutations in the splice site of type I collagen genes are one of the mutations that cause OI and usually lead to a mild or moderate OI phenotype. A heterozygous A to G point mutation in intron 9 at the -2 position of the splice receptor site of COL1A1 was identified in a family with type I or IV OI. Three affected individuals in four generations of one family all presented with several clinical symptoms. They all had pectus carinatum, flat feet, gray-blue sclerae, and normal stature, teeth, hearing, and vision. Forearm fractures, small joint dislocations, and muscle weakness were all present in the patient's father and grandmother, who presented with a moderate type IV phenotype. The 10-year-old proband with type I OI had suffered a fracture twice, but had no history of joint dislocation or skin hyperextensibility. Charting the family helped to identify clinical symptoms in patients with mutations at the N-terminal of type I collagen genes.
Keywords: Osteogenesis imperfecta, splice receptor-site mutation, COL1A1, N-terminal of type I collagen
1. Introduction
Osteogenesis imperfecta (OI), also called brittle bone disease, is a genetic connective tissue disorder with a broad phenotypic variation and genetic heterozygosity. Autosomal dominant mutations in COL1A1 and COL1A2 are the main cause of classic osteogenesis imperfecta, which is characterized by bone fragility,
blue sclerae, defects in the teeth, and deficits in hearing and vision (1-3). Epidemiological data on OI varies greatly, with an average prevalence of 1 in 15,000 to 20,000 births worldwide (4). The proportion of OI and its subtypes differs markedly among certain races or ethnicities, the ratio of OI type I to OI type III is seven to one in white Australians (5). In North America, the number of black patients with type III OI is six times that of black patients with type I OI (6). A total of 3,548 patients with OI have been registered (http://oi.gene.le.ac.uk). Epidemiological data on Chinese patients with OI have not been available until now.
A splice site mutation occurs in the processing of pre-mRNA into mature mRNA and can lead to a substitution, insertion, deletion, or frameshift. Mutations in either splice-donor or splice-acceptor sequences may lead to retention of intronic DNA or exon skipping, hence the production of abnormal proteins (7). Over 300 splice site mutations have been documented in the OI mutation database; most affect the COL1A1 gene and most are substitutions (8,9).
The type I collagen gene is also the gene responsible for other genetic bone diseases including Caffey disease (10), arthrochalasia Ehlers-Danlos syndrome (aEDS) (11-13), cardiac valvular EDS (cvEDS), and OI/EDS disease (14,15). Given the clinical and genetic heterozygosity of these diseases, their diagnosis and treatment are always challenging, and this is particularly true for rare diseases. Hence, detailed clinical characteristics of rare diseases are extremely important. A substitution of c.697-2A>G in the COL1A1 gene has been reported three times, though information on clinical phenotypes is limited. The current report describes a splice site mutation of c.697- 2A>G in COL1A1 that was identified for the first time in Chinese patients with OI.
2. Case Report
The proband was a 10-year-old girl of normal height and weight at birth. She was 50 cm tall and weighed 3.3 kg at delivery. She could not walk until she was 1 and half years old. Delayed closure of the fontanelle was noted, and pectus carinatum was mild. She suffered a forearm fracture when she was 11 months old and a lower leg fracture at the age of 4, as shown on X-rays (Figure 1). Teeth, hearing, and vision were all normal in the proband and her father and grandmother, both of whom had OI. Flat feet, gray-blue sclerae, and pes planus were present in all three family members. The proband's affected father and grandmother also had a relatively normal height and weight, dislocations of the ankle and elbow, limb muscle weakness, and upper limb fractures. Joint laxity was not evident in the proband. The proband had a low bone mass, with a Z score of -2 in the lumbar spine according to dual energy X-ray absorptiometry (DXA).
Figure 1.
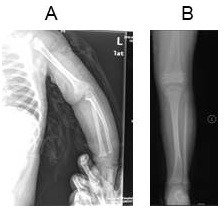
X-rays from the proband. (A) 11 months of age; (B) age 4.
A heterozygous mutation of c.697-2A>G in intron 9 was identified in the proband and her affected family members (Figure 2) according to molecular analysis (16).
Figure 2.
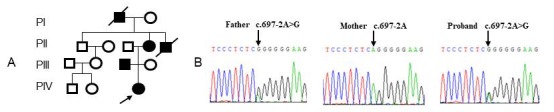
Molecular analysis of a family with OI. (A) Pedigree of the proband's family; (B) Electropherograms showing the partial sequence of COL1A1 in the proband and her parents.
3. Discussion
This report is the first to describe an A to G point mutation in intron 9 at the -2 position of the splice receptor site of the COL1A1 gene in a Chinese patient with OI and her family. There are no Chinese patients with this mutation according to a literature and database search (http://oi.gene.le.ac.uk) (8,9). The current patient and her family members had fractures of the upper limb, frequent small joint dislocations, blue sclerae, flat feet, and normal teeth, sight, and hearing. Since an RNA analysis was not performed, so errors in COL1A1 transcription were not analyzed.
A total of 12 individuals with OI and four different mutations at position of 697 have been reported; these mutations include 11 splice site mutations and 1 missense mutation. A splice site mutation of c.697- 2A>G was reported 3 times in patients with OI type I or IV; one of the three patients with type I OI was 35 years old and presented with blue sclerae, normal hearing, and bone fractures (17). The mutations -1G>T and -1G>C at the same site have also been identified in patients with mild or moderate OI who lacked clinical symptoms (http://oi.gene.le.ac.uk) (8,9,17). A mutation of G > C lead to glycine to arginine substitution, which was identified in one 68-year-old female patient with type IV OI; the patient did not have blue sclerae or a history of childhood fractures and extravertebral fractures, but she had suffered a vertebral fracture once (18). The splice site of c.697-2delA has been identified in one 14-year-old Korean female patient with type I OI (19).
A study has reported that mutations in the 5' splice donor site and 3' splice acceptor site consensus sequences are related to diseases (7). RNA mis-splicing underlies one of the main types of mutations in patients with OI and type I collagen genes. Type I collagen consists of 2 type I collagen α1 chains and 1 α2 chain coil in a triple helix structure, with 3 repeating amino acids: Gly-X-Y. COL1A1 and COL1A2 contain approximately 52 intronic sequences that are particularly susceptible to RNA splicing mutations (20). A mutation of c.697-2A>G introduces a cryptic splice acceptor site in intron 9 of the COL1A1 gene and may lead to retention of the intron in the abnormally spliced mRNA.
Mutations near the N-telopeptide, which links the N-propeptide to the triple helical domain of collagen, are often associated with aEDS or OI/EDS. Cases of OI/EDS are rarely reported (15,21-27). The molecular mechanism for OI/EDS is closely related to defective procollagen and cross-linking due to interference with N-propeptide processing (26,28). Patients with OI/EDS have generalized joint hyperlaxity and skin hyperextensibility, early progressive scoliosis, and OI symptoms such as blue sclerae and fractures.
Clinical symptoms of OI and OI/EDS overlap. The relationship between clinical characteristics of those diseases and mutations in the N-terminal region of type I collagen will be more apparent once more data on their clinical phenotypes are available.
Acknowledgements
The study was supported by a grant from the Shandong Key Research and Development Plan and the Natural Science Foundation (2016ZDJS07A10, 2016GSF201222 and 2015ZRC03171). The authors wish to thank the patient and her family members who participated in this study.
References
- 1. Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007; 28:209-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu Y, Ren X, Wang Y, Bardai G, Sturm M, Dai Y, Riess O, Zhang Y, Li H, Li T, Zhai N, Zhang J, Rauch F, Han J. Novel WNT1 mutations in children with osteogenesis imperfecta: Clinical and functional characterization. Bone. 2018; 114:144-149. [DOI] [PubMed] [Google Scholar]
- 3. Lu Y, Ren X, Wang Y, Han J. Molecular mechanisms of osteogenesis imperfecta. Progress in Biochemistry and Biophysics. 2015; 42:511-518. [Google Scholar]
- 4. Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011; 7:540-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979; 16:101-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin E, Shapiro JR. Osteogenesis imperfecta: Epidemiology and pathophysiology. Curr Osteoporos Rep. 2007; 5:91-97. [DOI] [PubMed] [Google Scholar]
- 7. Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016; 17:19-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dalgleish R. The human type I collagen mutation database. Nucleic Acids Res. 1997; 25:181-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dalgleish R. The human collagen mutation database 1998. Nucleic Acids Res. 1998; 26:253-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gensure RC, Makitie O, Barclay C, Chan C, Depalma SR, Bastepe M, Abuzahra H, Couper R, Mundlos S, Sillence D, Ala Kokko L, Seidman JG, Cole WG, Juppner H. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest. 2005; 115:1250-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cole WG, Chan D, Chambers GW, Walker ID, Bateman JF. Deletion of 24 amino acids from the pro-α 1(I) chain of type I procollagen in a patient with the Ehlers-Danlos syndrome type VII. J Biol Chem. 1986; 261:5496-5503. [PubMed] [Google Scholar]
- 12. Weil D, D'Alessio M, Ramirez F, de Wet W, Cole WG, Chan D, Bateman JF. A base substitution in the exon of a collagen gene causes alternative splicing and generates a structurally abnormal polypeptide in a patient with Ehlers- Danlos syndrome type VII. EMBO J. 1989; 8:1705-1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eyre DR, Shapiro FD, Aldridge JF. A heterozygous collagen defect in a variant of the Ehlers-Danlos syndrome type VII. Evidence for a deleted amino-telopeptide domain in the pro-α 2(I) chain. J Biol Chem. 1985; 260:11322-11329. [PubMed] [Google Scholar]
- 14. Schwarze U, Hata R, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE, Byers PH. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet. 2004; 74:917-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu Y, Wang Y, Rauch F, Li H, Zhang Y, Zhai N, Zhang J, Ren X, Han J. Osteogenesis imperfecta type III/Ehlers- Danlos overlap syndrome in a Chinese man. Intractable Rare Dis Res. 2018; 7:37-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu Y, Ren X, Wang Y, Li T, Li F, Wang S, Xu C, Wu G, Li H, Li G, Zhao F, Wang Z, Mo X, Han J. Mutational and structural characteristics of four novel heterozygous C-propeptide mutations in the proα1(I) collagen gene in Chinese osteogenesis imperfecta patients. Clin Endocrinol (Oxf). 2014; 80:524-531. [DOI] [PubMed] [Google Scholar]
- 17. Hartikka H, Kuurila K, Korkko J, Kaitila I, Grenman R, Pynnonen S, Hyland JC, Ala-Kokko L. Lack of correlation between the type of COL1A1 or COL1A2 mutation and hearing loss in osteogenesis imperfecta patients. Hum Mutat. 2004; 24:147-154. [DOI] [PubMed] [Google Scholar]
- 18. Rolvien T, Sturznickel J, Schmidt FN, Butscheidt S, Schmidt T, Busse B, Mundlos S, Schinke T, Kornak U, Amling M, Oheim R. Comparison of bone microarchitecture between adult osteogenesis imperfecta and early-onset osteoporosis. Calcif Tissue Int. 2018; 103:512-521. [DOI] [PubMed] [Google Scholar]
- 19. Lee KS, Song HR, Cho TJ, Kim HJ, Lee TM, Jin HS, Park HY, Kang S, Jung SC, Koo SK. Mutational spectrum of type I collagen genes in Korean patients with osteogenesis imperfecta. Hum Mutat. 2006; 27:599. [DOI] [PubMed] [Google Scholar]
- 20. Bateman JF, Chan D, Moeller I, Hannagan M, Cole WG. A 5' splice site mutation affecting the pre-mRNA splicing of two upstream exons in the collagen COL1A1 gene. Exon 8 skipping and altered definition of exon 7 generates truncated pro α1(I) chains with a non-collagenous insertion destabilizing the triple helix. Biochem J. 1994; 302 (Pt 3):729-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nicholls AC, Oliver J, Renouf DV, McPheat J, Palan A, Pope FM. Ehlers-Danlos syndrome type VII: A single base change that causes exon skipping in the type I collagen α 2(I) chain. Hum Genet. 1991; 87:193-198. [DOI] [PubMed] [Google Scholar]
- 22. Vasan NS, Kuivaniemi H, Vogel BE, Minor RR, Wootton JA, Tromp G, Weksberg R, Prockop DJ. A mutation in the pro α 2(I) gene (COL1A2) for type I procollagen in Ehlers-Danlos syndrome type VII: Evidence suggesting that skipping of exon 6 in RNA splicing may be a common cause of the phenotype. Am J Hum Genet. 1991; 48:305-317. [PMC free article] [PubMed] [Google Scholar]
- 23. Chiodo AA, Hockey A, Cole WG. A base substitution at the splice acceptor site of intron 5 of the COL1A2 gene activates a cryptic splice site within exon 6 and generates abnormal type I procollagen in a patient with Ehlers- Danlos syndrome type VII. J Biol Chem. 1992; 267:6361-6369. [PubMed] [Google Scholar]
- 24. Feshchenko S, Brinckmann J, Lehmann HW, Koch HG, Muller PK, Kugler S. Identification of a new heterozygous point mutation in the COL1A2 gene leading to skipping of exon 9 in a patient with joint laxity, hyperextensibility of skin and blue sclerae. Mutations in brief no. 166. Online. Hum Mutat. 1998; 12:138. [DOI] [PubMed] [Google Scholar]
- 25. Nicholls AC, Valler D, Wallis S, Pope FM. Homozygosity for a splice site mutation of the COL1A2 gene yields a non-functional proα2(I) chain and an EDS/ OI clinical phenotype. J Med Genet. 2001; 38:132-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cabral WA, Makareeva E, Colige A, Letocha AD, Ty JM, Yeowell HN, Pals G, Leikin S, Marini JC. Mutations near amino end of α1(I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing. J Biol Chem. 2005; 280:19259-19269. [DOI] [PubMed] [Google Scholar]
- 27. Shi X, Lu Y, Wang Y, Zhang YA, Teng Y, Han W, Han Z, Li T, Chen M, Liu J, Fang F, Dou C, Ren X, Han J. Heterozygous mutation of c.3521C>T in COL1A1 may cause mild osteogenesis imperfecta/Ehlers-Danlos syndrome in a Chinese family. Intractable Rare Dis Res. 2015; 4:49-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Makareeva E, Cabral WA, Marini JC, Leikin S. Molecular mechanism of α 1(I)-osteogenesis imperfecta/ Ehlers-Danlos syndrome: Unfolding of an N-anchor domain at the N-terminal end of the type I collagen triple helix. J Biol Chem. 2006; 281:6463-6470. [DOI] [PubMed] [Google Scholar]