

Abstract
The clinical presentations of papillomavirus (PV) infections come in many different flavors. While most PVs are part of a healthy skin microbiota and are not associated to physical lesions, other PVs cause benign lesions, and only a handful of PVs are associated to malignant transformations linked to the specific activities of the E5, E6, and E7 oncogenes. The functions and origin of E5 remain to be elucidated. These E5 open reading frames (ORFs) are present in the genomes of a few polyphyletic PV lineages, located between the early and the late viral gene cassettes. We have computationally assessed whether these E5 ORFs have a common origin and whether they display the properties of a genuine gene. Our results suggest that during the evolution of Papillomaviridae, at least four events lead to the presence of a long noncoding DNA stretch between the E2 and the L2 genes. In three of these events, the novel regions evolved coding capacity, becoming the extant E5 ORFs. We then focused on the evolution of the E5 genes in AlphaPVs infecting primates. The sharp match between the type of E5 protein encoded in AlphaPVs and the infection phenotype (cutaneous warts, genital warts, or anogenital cancers) supports the role of E5 in the differential oncogenic potential of these PVs. In our analyses, the best-supported scenario is that the five types of extant E5 proteins within the AlphaPV genomes may not have a common ancestor. However, the chemical similarities between E5s regarding amino acid composition prevent us from confidently rejecting the model of a common origin. Our evolutionary interpretation is that an originally noncoding region entered the genome of the ancestral AlphaPVs. This genetic novelty allowed to explore novel transcription potential, triggering an adaptive radiation that yielded three main viral lineages encoding for different E5 proteins, displaying distinct infection phenotypes. Overall, our results provide an evolutionary scenario for the de novo emergence of viral genes and illustrate the impact of such genotypic novelty in the phenotypic diversity of the viral infections.
Keywords: oncogenes, virus evolution, papillomavirus, genome evolution, infections and cancer, de novo genes
Introduction
Papillomaviruses (PVs) constitute a numerous family of small, nonencapsulated viruses infecting virtually all mammals, and possibly all amniotes and bony fishes. According to the International Committee on Taxonomy of Viruses (ICTV: https://talk.ictvonline.org/taxonomy/, last accessed: May 9 2019), the Papillomaviridae family currently consists of 53 genera, which can be organized into a few crown groups according to their phylogenetic relationships (Gottschling et al. 2011). The PV genome consists of a double stranded circular DNA genome, roughly organized into three parts: an early region coding for six open reading frames (ORFs: E1, E2, E4, E5, E6, and E7) involved in multiple functions including viral replication and cell transformation; a late region coding for structural proteins (L1 and L2); and a noncoding regulatory region (URR) that contains the cis-elements necessary for replication and transcription of the viral genome. The major oncoproteins encoded by PVs are E6 and E7, which have been extensively studied (Münger et al. 1992; Moody and Laimins 2010; Tomaić 2016). However, there is also a minor oncoprotein termed E5, whose functions and origin remain to be fully elucidated (DiMaio and Petti 2013).
The E5 ORFs are located in the intergenic region between the E2 and the L2 genes. This inter-E2–L2 region is highly variable between PV genomes. In most PV lineages the early and late gene cassettes are located in direct apposition. In a few, nonmonophyletic PV lineages, this region accommodates both coding and noncoding genomic segments, which may have gained access to the PV genomes through recombination events with hitherto nonidentified donors (Bravo and Felez-Sanchez 2015). PVs within the Alpha- (infecting primates) and DeltaPV (infecting cetartiodactyles) genera encode different E5 proteins in the inter-E2–L2 region (Bravo and Alonso 2004). Additionally members of the Lambda-MuPV (infecting, i.a., bats, humans, rabbits, rodents, canids, hyaenids, and felids) and Beta-XiPV (infecting, i.a., primates, rodents, cetartiodactyles, canids, and felids) crown groups present in the inter-E2–L2 region large noncoding stretches of unknown significance and/or function (García-Pérez et al. 2014).
The largest wealth of scientific literature about PVs deals with AlphaPVs. These are a clinically important group of PVs that infect primates, and are associated to largely different clinical manifestations: nononcogenic PVs causing anogenital warts; oncogenic and nononcogenic PVs causing mucosal lesions; and nononcogenic PVs causing cutaneous warts. The E5 proteins in AlphaPVs can be classified into four different groups according to their hydrophobic profiles and phylogeny (Bravo and Alonso 2004). The presence of a given E5 type sharply correlates with the clinical presentation of the corresponding PV infection: viruses that contain E5α (e.g., HPV16) are associated with malignant mucosal lesions such as cervical cancer; viruses coding for E5β (e.g., HPV2) are associated with benign cutaneous lesions, commonly warts on fingers and face; and viruses that contain two putative E5 proteins, termed E5γ and E5δ (e.g., HPV6) are associated with benign mucosal lesions such as anogenital warts (Bravo and Alonso 2004). Two additional putative E5 proteins, E5ϵ and E5ζ (PaVE; https://pave.niaid.nih.gov, last accessed: May 9 2019), have been identified in AlphaPVs infecting Cercopithecinae (macaques and baboons). Contrary to the other E5 proteins, the E5ϵ and E5ζ are not associated with a specific clinical presentation, although our knowledge about the epidemiology of the infections in primates other than humans is still very limited. It has been suggested that the integration of an E5 proto-oncogene in the ancestor of (AlphaPVs) supplied the viruses with genotypic novelty, which triggered an adaptive radiation through exploration of phenotypic space, and eventually generated the extant three clades of PVs (Willemsen and Bravo 2019).
The only feature that all E5 proteins have in common is their highly hydrophobic nature and their location in the inter-E2–L2 region of the PV genome. It remains unclear whether all E5 proteins are evolutionary related, and moreover, the E5 ORFs do not seem to have close relatives (Puustusmaa et al. 2017). The E5 proteins of HPV16 and of bovinepapillomavirus1 (BPV1) are the only E5s for which the biology is partially known. Despite the absence of sequence similarity and the differences in immediate interaction partners, their cellular roles during infection are comparable. HPV16 E5 is a membrane protein that localizes in the Golgi apparatus and in the early endosomes. It has been associated with different oncogenic mechanisms related to the induction of cell replication through manipulation of the epidermal growth factor receptor response (Pim et al. 1992; Conrad et al. 1993; Straight et al. 1993), as well as to immune evasion by modifying the membrane chemistry (Bravo et al. 2005; Suprynowicz et al. 2008) and decreasing the presentation of viral epitopes (Ashrafi et al. 2005). BPV1 E5 is a very short protein (barely 40 amino acids, half the size of HPV16 E5) that also localizes in the membranes. It displays a strong transforming activity, largely by activating the platelet-derived growth factor receptor (Petti et al. 1997; DiMaio and Mattoon 2001), and it downregulates as well the presentation of viral epitopes in the context of the MHC-I molecules (Ashrafi et al. 2002).
In this study, we have explored the evolutionary history of the E5 ORFs found within the inter-E2–L2 region in PVs. First, we identified the PV clades that contain a long intergenic region between E2 and L2, and therewith putative E5 ORFs. Then, we assessed whether the E5 ORFs in the identified clades originated from a single common ancestor. Next, we verified whether the evolutionary history of the inter-E2–L2 region and of the E5 ORFs therein encoded is similar to that of the other PVs genes, by comparing their sequences and phylogenies. Finally, we examined whether the different E5 ORFs exhibited the characteristics of a bona fide gene to exclude the conjecture that these are simply spurious ORFs.
Materials and Methods
DNA and Protein Sequences
We collected 354 full length PV genomes from the PaVE (https://pave.niaid.nih.gov/, last accessed: May 9 2019) and GenBank (https://www.ncbi.nlm.nih.gov/genbank/, last accessed: May 9 2019) databases (supplementary table S1, Supplementary Material online). The corresponding E5 sequences were retrieved from these genomes as well as the intergenic region between the E2 and L2 genes (hereafter named inter-E2–L2). Based on the size of the inter-E2–L2 region in which E5s are present, we selected those with a minimum length of 250 nucleotides (fig. 1 and supplementary fig. S1, Supplementary Material online). For comparison in the tree figures, we extended our analysis and also indicated inter-E2–L2 regions with a minimum length of 125 nucleotides. The genomes containing and inter-E2–L2 region where E5 was not annotated, were scanned for possible unannotated E5-like ORFs with the NCBI ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/, last accessed: May 9 2019). The URR, E6, E7, E1, E2, L2, and L1 were also extracted from the collected genomes and analyzed in parallel to the E5 sequences. We excluded the E4 ORFs from our analyses as most of its coding sequence overlaps the E2 gene in a different reading frame and it is supposed to be under different evolutionary pressures (Hughes and Hughes 2005; Félez-Sánchez et al. 2015). Genes were aligned individually at the amino acid level using MAFFT v.7.271 (Katoh and Standley 2013), corrected manually, and backtranslated to nucleotides using PAL2NAL v.14 (Suyama et al. 2006). Before concatenating the E1, E2, L2, and L1 genes, the alignments were filtered using Gblocks v.0.91b (Castresana 2000). The URR and the inter-E2–L2 region (noncoding regions) were aligned at the nucleotide level.
Fig. 1.

—Best-known maximum likelihood phylogenetic tree of the concatenated E1E2L2L1 amino acid sequences of 343 PVs and identification of clades containing an intergenic E2–L2 region. Clade color codes highlight the four PV crown groups: red, Alpha-OmikronPVs; green, Beta-XiPVs; yellow, Lambda-MuPVs; blue, Delta-ZetaPVs; gray, a yet unclassified crown group consisting of PVs infecting birds and turtles; and white, PVs without well-supported phylogenetic relationships. Outer labels, Mucosal Lesions, Genital Warts, and Cutaneous Warts, indicate the most common tropism for the AlphaPVs. Values on branches correspond to ML bootstrap support values. Asterisks indicate a maximal support of 100, and values under 50 are not shown. Branches in light-gray correspond to PV genomes containing an inter-E2–L2 region with a minimum size of 250 nt; branches in dark-gray correspond to PV genomes with an inter-E2–L2 region with a minimum size of 125 nt. The basal nodes of the four clades containing a relatively long intergenic region between the E2 and the L2 ORFs are labeled with a star. The basal node of the lineages containing an E5 coding sequence is indicated with an arrow, and the corresponding terminal taxa are labeled with a color-coded dot indicating the E5 type. Purple dots indicate: E5, orange dots: E5α, light green dots: E5γδ, dark green dots: E5δ, blue dots: E5β, and black dots are lineages containing unclassified E5 types.
Phylogenetic Analyses
The phylogenetic relationships were inferred using the core PV genome of the E1, E2, L2, and L1 genes (Willemsen and Bravo 2019). The previously identified recombinant PVs isolated from Cetaceans (PphPV1-2, TtPV1-7, DdPV1, PsPV1) (Rector et al. 2008; Gottschling et al. 2011; Robles-Sikisaka et al. 2012) were removed before alignment, leaving us with a data set of 343 PVs. The concatenated E1–E2–L2–L1 alignment was used to construct Maximum Likelihood (ML) trees with RAxML v.8.2.9 (Stamatakis 2014) under the GTR + Γ4 model for the nucleotide alignment (supplementary fig. S1, Supplementary Material online), using 12 partitions (three for each gene corresponding to each codon position), or under the LG+I + Γ model for the amino acid alignment (fig. 1) using four partitions (one for each gene), and using 1,000 bootstrap replicates.
To measure the distances between the URR, E6, E7, E1, E2, E5, inter-E2–L2, L2, and L1 trees, we reduced the data set to 69 PVs so that all terminal taxa were present in all trees. We reconstructed a phylogenetic tree for each gene separately, for the concatenated E6E7E1E2L2L1 genes, as well as for the URR and the inter-E2–L2 region. ML trees were constructed at the nucleotide level using RAxML v.8.2.9 under the GTR + Γ4 model. For tree construction of the concatenated alignment, we introduced six partitions (one per gene). To measure the topological distances between trees, we calculated the weighted Robinson–Foulds (RF) distance (Robinson and Foulds 1981) and the K-tree score (Soria-Carrasco et al. 2007). The weighted RF distance is a topological distance measure that considers the edge weights of the tree, and the K-tree score is the minimum branch length distance one can get from one tree to another after scaling one of them. Because of this initial scaling step the K-tree score is not a metric, that is, the distance from A to B is not equal to the distance from B to A. Therefore, we obtained a symmetrical distance matrix for the weighted RF distance and an asymmetrical distance matrix for the K-tree score. Using these matrices, a correspondence analysis was performed to identify similarities between the topologies of the trees reconstructed for each PV ORF, the inter-E2–L2 region, and the URR.
Testing for Common Ancestry Using Bali-Phy
In order to evaluate the common ancestry of the E5 ORFs, we used the BAli-Phy algorithm (Suchard and Redelings 2006). Under this Bayesian framework, the input data are the unaligned sequences, as the alignment itself is one of the parameters of the model to be treated as an unknown random variable (Redelings and Suchard 2005). We ran our analysis under the null hypothesis of common ancestry of the intergenic regions. We used the marginal likelihood calculated as the harmonic mean of the sample likelihood to estimate the Bayes Factor between the null hypothesis Common Ancestry (CA) and the alternative hypothesis Independent Origin (IO) (de Oliveira Martins and Posada 2014). Therefore, we have ΔBF = log[Prob(CA)] − log[Prob(IO)], such that positive values support CA and negative values support IO. The likelihood for the CA model was obtained running the software for all the E5 sequences together. For the IO scenarios, we ran one analysis for each group independently. We started with the different PV clades that contain an E5 ORF in the inter-E2–L2 region, located within the Alpha-Omikron (red) and Delta-Zeta (blue) crown groups (fig. 1 and supplementary fig. S1, Supplementary Material online). In the cases where two putative E5 ORFs were located in the same inter-E2–L2 fragment (for instance for E5γ and E5δ, and for E5ϵ and E5ζ) sequences were concatenated. Then we ran the analyses on the E5 ORFs within AlphaPVs stratifying by the different E5 types that are associated to three distinct clinical presentations; mucosal lesions, cutaneous warts, and genital warts. The values for the independent groups of E5, E5, E5β, E5γδ, E5δ, and E5, and the sum of combinations of these, rendered the likelihood for the IO models. For instance, (α1 − α2 − ) + (γδ − δ) + β denotes a hypothesis of three independent ancestries, one tree for the E5 types associated to mucosal lesions (E5α1, E5α2, and E5 together), another separate tree for the E5 types associated to genital warts (E5γδ and E5δ together), and another tree for the E5 type associated to cutaneous warts (E5β). The likelihood of this example was obtained running BAli-Phy three times: one run for E5α1, E5α2, and E5, one for E5γδ and E5δ, and one for E5β. The sum of these three analyses corresponded to the likelihood of the model. We only considered the IO scenarios that were biologically plausible based on the phylogeny of PVs (fig. 1 and supplementary fig. S1, Supplementary Material online). The same procedure was applied to the E5 sequences belonging to both the Alpha-Omikron and Delta-Zeta crown groups. This analysis was performed at the amino acid level using the LG substitution model. For each model, three independent MCMC chains were run for at least 100,000 iterations. The three runs were combined and checked for convergence.
Random Permutations to Test for Common Ancestry
To support the results of the BAli-Phy analyses, we performed a random permutation test as described in de Oliveira Martins and Posada (2016). In this test the sequences for one of the groups are randomly shuffled and the statistics are recalculated after realignment with MUSCLE (Edgar 2004), providing information about how much the results using the original data depart from those with phylogenetic structure partially removed. The statistics used in this test are ML tree length and Log Likelihood calculated with PhyMLv3.0 (Guindon et al. 2010). As for the BAli-Phy test, these analyses were performed at the amino acid level using the LG substitution model. We obtained a distribution by reshuffling one of the groups (e.g., the E5 sequences) 100 times, each time realigning against the other groups from the data set, and comparing the resulting phylogeny with those if we separate again the groups. For each iteration, the alignment is always optimized and the statistics are calculated. To make the statistics comparable, the same alignment is used for both the IO and CA hypotheses. We compare the distribution for the CA and IO hypotheses with a Kruskal–Wallis rank sum test and a multiple comparison test after Kruskal–Wallis. The results were confirmed by performing Wilcoxon rank sum tests with continuity correction. Lower ML tree length and superior Log likelihood values are expected to support the best model.
Generation of Random ORFs
In order to assess whether the E5 sequences were larger than expected by chance, we estimated first the median A/T/G/C composition of the inter-E2–L2 regions of AlphaPVs (A: 0.22; T: 0.41; G: 0.20; C: 0.17). Using in-house perl scripts, we created a set of 10,000 random DNA sequences with this median nucleotide composition and with a median length of 400 nt. Then, we computed the length of all putative ORFs that may have appeared in this set of randomly generated DNA sequences. The lengths of the actual and of the simulated sequences were compared with a one-way ANOVA followed by a post hoc Tukey’s HSD test (supplementary table S2, Supplementary Material online).
dN/dS Values
To determine whether the E5 ORFs are protein-coding sequences, we computed the dN/dS values for all E5 ORFs as well as for all other PV ORFs (E1, E2, E6, E7, L1, L2). The dN/dS values were computed with SELECTON (http://selecton.tau.ac.il/, last accessed: May 9 2019; Doron-Faigenboim et al. 2005; using the MEC model; Doron-Faigenboim and Pupko 2007). The likelihood of MEC model was tested against the M8a model (Yang et al. 2000), which does not allow for positive selection. As these models are not nested, AIC scores were compared. For all the sequence sets, the MEC model was preferred over the M8a model (supplementary table S3, Supplementary Material online).
Pairwise Distances
To evaluate the diversity of the AlphaPV genes, we calculated the pairwise distances between aligned sequences within each group of the E5 ORFs, the other PV ORFs (E1, E2, E6, E7, L1, L2), the URR, and randomly generated intergenic CDSs. These were generated by extracting the noncoding region of the E2–L2 fragments of the AlphaPVs. Then, for each noncoding region, we extracted a random subregion with the same length as the E5 ORF of this PV. These random intergenic regions were truncated at the 5′ to get a sequence length multiple of 3. All internal stop codons were replaced by N’s. Pairwise distances between aligned DNA sequences were calculated using the TN93 model. Besides analyzing the raw distances, the distances were also normalized with respect to the corresponding one obtained for L1.
Codon Usage Preferences
We calculated the codon usage preferences (CUPrefs) for the E5 AlphaPV ORFs. The relative frequencies for each of the 18 families of synonymous codons were calculated using COUSIN v.1.0 (https://cousin.ird.fr, last accessed: May 9 2019). We only considered the frequencies of the 59 codons with redundancy (i.e., excluding Met, Trp, and stop codons). We performed the same analysis for all other ORFs in the same genomes (E1, E2, E6, E7, L1, and L2) as well as for the randomly generated intergenic CDSs. A matrix was created in which the rows corresponded to the ORFs on one PV genome and the columns to the 59 relative frequency values, such that each row had the codon usage information for a specific ORF. We performed a Kruskal nonmetric multidimensional scaling (MDS) analysis using Euclidean distances as well as a principal component analysis (PCA) in order to assess similarities in CUPrefs of the E5 ORFs with respect to the other AlphaPV ORFs, as described in (Félez-Sánchez et al. 2015). In parallel, we performed a hierarchical clustering analysis using Ward’s method and standardizing the variable’s by using Z-scores. The optimal number of clusters was determined according to the majority rule of the Hubert index and the D index.
GRAVY Index
For all E5 proteins, the grand average hydropathy (GRAVY) (Kyte and Doolittle 1982) was calculated by adding the hydropathy value for each residue and dividing this value by the length of the protein sequence.
Statistics and Graphics
Statistical analyses and graphics were done using R (R Core team 2017), with the aid of the packages “ape,” “ade4,” “dplyr,” “factoextra,” “ggplot2,” “ggpubr,” “lawstat,” “MASS,” “NbClust,” “pgirmess,” “phangorn,” and “RColorBrewer.” The final display of the graphics was designed using Inkscape v.0.92 (https://inkscape.org/en/, last accessed: May 9 2019).
Results
Do the E5 ORFs Present in the Genomes of PVs Belonging to Different Crown Groups Have a Common Ancestor?
We collected 354 full length PV genomes from the PaVE and GenBank databases (supplementary table S1, Supplementary Material online). After removing 11 recombinant sequences, we constructed a maximum likelihood phylogenetic tree of the concatenated E1E2L2L1 sequences at the nucleotide and amino acid levels. Out of the 354 PV genomes, we identified 339 with an intergenic region (of at least one nucleotide) between the E2 and L2 genes. Of these, 83 contain an E5 ORF in the inter-E2–L2 region (fig. 1 and supplementary fig. S1, Supplementary Material online). The E5 ORFs have a median size of 144 nucleotides (min: 126, max: 306). Based on the size of inter-E2–L2 region in which E5s are present (min: 289, median: 517, max: 938), we identified four PV clades containing an intergenic region selecting for a minimum size of 250 (min: 262, median: 512, max: 1,579). We further lowered this threshold to 125 nucleotides to identify possible unannotated E5-like ORFs in the inter-E2–L2 region. The identified clades are indicated in figure 1 and supplementary figure S1, Supplementary Material online, and are located in the four PV crown groups: Alpha-Omikron (colored red), Delta-Zeta (colored blue), Lambda-Mu (colored yellow), and Beta-Xi (colored green). Additionally, three recombinant bottlenose dolphin PVs (TtPV1-3) belonging to the UpsilonPV genus, also present an inter-E2–L2 region. Only the clades identified in the Alpha-Omikron and Delta-Zeta crown groups, have an E5 ORF present within the inter-E2–L2 region. The two other clades that locate within the Lambda-Mu and Beta-Xi crown groups also contain this relatively long intergenic region. Although, for these clades the inter-E2–L2 region does not contain any apparent ORFs. Interestingly, an ORF named E5 is present in the Lambda-Mu clade in two rabbit PV genomes (SfPV1 and OcPV1), where no intergenic noncoding region is present and where this E5 largely overlaps with both the E2 and L2 genes in the case of SfPV1 and with L2 in the case of OcPV1. There are other cases, like HPV16, where E5 partially overlaps with the E2 gene. Nonetheless this overlap is small (four nucleotides) compared with the almost complete overlap of E5 with L2 in the rabbit PV genomes. All things being equal, the E5 ORFs in the rabbit PV genomes seem unique in a way that no inter-E2–L2 region is present at all.
In order to determine whether the E5 ORFs in the different PV crown groups share a single common ancestor, we tested for common ancestry using BAli-Phy as described in de Oliveira Martins and Posada (2014). We named the clades according to their colored crown groups, therefore, we have the red clade (including 69 E5 sequences), the blue clade (12 E5 sequences), and the yellow clade (two E5 sequences). For the common ancestry test, trees were inferred for all groups combined as well as separately (see Materials and Methods). Therefore, we could not include the yellow clade in this test, as this clade contains only two sequences and consequently no trees can be inferred. We performed the analysis on the full data set (excluding the two yellow clade sequences) containing 81 sequences as well as on a reduced data set containing 24 sequences; 12 representative E5 sequences from the red clade; and the 12 E5 sequences from the blue clade. We made the choice between the alternative hypotheses Common Ancestry (CA) and Independent Origin (IO) by computing the marginal likelihoods using the stabilized harmonic mean estimator. We ran our analysis under the null hypothesis of CA of the E5 ORF. Therefore, we have ΔBF = log[Prob(CA)] − log[Prob(IO)], such that positive values support CA and negative values support IO. Other statistics that we analyzed were the alignment length and the Bayesian tree length, calculated as the sum of the branch lengths. For both the alignment length and tree length, lower values support the best model.
The results of the BAli-Phy analyses are contradictory between the different statistics tested. On the one hand, based on the marginal likelihood the best supported model is CA for the E5 ORFs in both the Alpha-Omikron and Delta-Zeta PV crown groups (table 1). Nonetheless, the difference in likelihood (ΔBF) between the CA and IO hypotheses is very small for both the full and reduced data sets. On the other hand, the alignment length and tree length statistics for the BAli-Phy analyses support the IO hypothesis. Such conflicts are indeed not novel. It has been shown that CA tests that use alignments as primary data provide misleading conclusions, spuriously favoring the CA hypothesis, as in the case of alignments without any phylogenetic structure (Koonin and Wolf 2010), or built using unrelated families of protein coding sequences (Yonezawa and Hasegawa 2012). Because all these approaches started from a fixed alignment there could be an initial bias toward CA (Yonezawa and Hasegawa 2010; Theobald 2011; de Oliveira Martins and Posada 2014). The BAli-Phy approach used here partly reduces this bias, as it starts from unaligned sequences and estimates simultaneously the alignment and the phylogeny.
Table 1.
Results from the BAli-Phy Analyses for the Hypothesis Testing on the Origin of the E5 ORFs in the Alpha-Omikron (red in fig. 1 ) and Delta-Zeta (blue in fig. 1 ) PV Crown Groups
| Model | Full Data Set |
Reduced Data Set |
||||||
|---|---|---|---|---|---|---|---|---|
| P(dataM) | ΔBF | Ali Length | Tree Length | P(dataM) | ΔBF | Ali Length | Tree Length | |
| H0: (red–blue) | −7,129.167 | 0 | 402 | 26.946 | −2,608.666 | 0 | 344 | 10.893 |
| H1: red + blue | −7,139.029 | 9.862 | 373 | 24.301 | −2,617.114 | 8.448 | 335 | 10.706 |
Note.—For each hypothesis tested, common ancestry (H0) and independent origin (H1), we show the marginal likelihood (P(data|M)) value, the ΔBF, the alignment (ali) length, and tree length. H0 is the best supported model according to the marginal likelihood, while H1 is the best supported model according to the alignment and tree lengths.
Given the inconclusive results, we performed a random permutation test as described in de Oliveira Martins and Posada (2016). In this test, the columns of the alignment for one of the groups are randomly shuffled and statistics are recalculated after realignment. Contrary to the BAli-Phy test, all trees are produced within a maximum likelihood (ML) framework (see Materials and Methods). We performed this test on both the full and the reduced data sets, using 100 iterations. For each iteration, we recovered the ML tree length and Log likelihood, and estimated the empirical distribution of these value. If the E5 ORFs have an IO, we expect lower ML tree length and superior Log likelihood values for this hypothesis (H1). The results of the permutation test show that for the full data set no significant differences were found between the ML tree length and Log likelihood distributions of CA and IO (supplementary fig. S2A and C, Supplementary Material online). We did obtain significant differences for the reduced data set, where the IO hypothesis is favored for the ML tree length, while the CA hypothesis is slightly favored for the Log likelihood (supplementary fig. S2B and D, Supplementary Material online.
The initial idea of the authors that developed the alignment-based permutation test (de Oliveira Martins and Posada 2016) was to resort only to simple summary statistics such as the ML tree length, rather than to rely on Log likelihood values. If we only regard the ML tree length values, the best supported model for the red and for the blue clades is IO. Nevertheless, we cannot ignore the Log likelihood values of the permutation test (supplementary fig. S2, Supplementary Material online), nor the results of the alignment-independent BAli-Phy test (table 1), and therefore, we cannot make a conclusive choice between the alternative hypotheses CA and IO. Finally, when considering the final trees produced by BAli-Phy supplementary figure S3, Supplementary Material online, we observe that the branch lengths leading to each group are long compared with the other branches, compatible rather with IO being the preferred model. These same trees further suggest that the E5 ORFs within the AlphaPVs (red clade) also originate from different ancestors, which may have introduced a bias in other alignment-driven CA tests, as discussed below.
Do the E5 ORFs Present in the Genomes of the AlphaPVs Clade Have a Common Ancestor?
In the AlphaPVs clade within the Alpha-Omikron crown group (red), the six types of E5 proteins are present in five different clades (fig. 1 and supplementary fig. S1, Supplementary Material online): E5α exists in two different clades of PVs associated to mucosal lesions, hereafter named E5α1 and E5α2, consisting of eight and nine sequences, respectively. E5β is present in all PVs associated to cutaneuous warts, consisting of 28 sequences. E5δ exists in all PVs associated to anogenital warts. Of these, only four PV genomes contain E5δ alone. The other seven PV genomes contain two E5 types; E5γ and E5δ, hereafter named E5γδ. Finally, E5 is present in 12 nonhuman AlphaPV genomes from viruses that infect Cercopithecinae and that are associated to mucosal lesions.
The BAli-Phy trees obtained in the CA test above, suggest that the E5 ORFs encoded in the AlphaPVs may have an IO (supplementary fig. S3, Supplementary Material online). These trees, that are based on the E5 amino acid sequences, show a clear separation of the protein clades depending on the clinical presentation of the corresponding viral infections: mucosal lesions (E5α1, E5α2, and E5), genital warts (E5γ, and E5γδ), and cutaneous warts (E5β). One exception is HPV54 –associated to genital warts, but whose E5 of unclassified type– clusters with the E5α2, characteristic of viruses associated to mucosal lesions. To address whether the E5 ORFs present in the genomes of the AlphaPVs have a CA, we applied the same protocol described earlier. We considered different plausible IO scenarios based on the E5 types and the phylogeny of the AlphaPVs (fig. 1 and supplementary fig. S1, Supplementary Material online). The BAli-Phy analysis showed that the CA hypothesis was the best-supported model for all statistics, while the hypothesis of each clade having an IO (H6) had the lowest support (table 2). The second best-supported IO model H1—where E5β has an IO—has a small difference in Log likelihood with the CA model (H0). As in the results described earlier, the results from the random permutation tests strongly disagree with the results of the BAli-Phy approach. The results of the random permutation test suggest that based on ML tree length the IO H6 is the best supported model, while based on Log likelihood the CA model (H0) and IO models H1 and H2 are equally probable (supplementary fig. S4, Supplementary Material online). Although the CA tests performed here give inconclusive results, the IO H1 model is also supported by the trees produced, where long branches separate E5β and the other E5 types (supplementary fig. S3, Supplementary Material online). In this H1 scenario E5, E5, γδ, E5δ, and E5 (encoded in PVs with mucosal and anogenital tropism) have a CA, while E5β (encoded in PVs with cutaneous tropism) has an IO. We therefore propose that at least E5, E5, E5γδ, E5δ, and E5 have a single ancestor, and originated from the same recombination donor and/or gained access to the ancestral genome through a single integration event. Further tests are needed to conclude whether E5β originated from the same ancestor as the other E5 types or whether it has an independent origin.
Table 2.
Results from the BAli-Phy Analyses for the Hypothesis Testing on the Origin of the E5 ORFs within the AlphaPV Clade (red in fig. 1 )
| Model | Full Data Set |
Reduced Data Set |
||||||
|---|---|---|---|---|---|---|---|---|
| P(dataM) | ΔBF | Ali Length | Tree Length | P(dataM) | ΔBF | Ali Length | Tree Length | |
| H0: (α1–α2–β–γδ–δ–) | −6,400.049 | 0 | 305 | 1.059 | −3,288.708 | 0 | 216 | 1.207 |
| H1: (α1–α2–γδ–δ–) + β | −6,415.579 | 15.530 | 328 | 2.238 | −3,300.208 | 11.500 | 275 | 2.533 |
| H2: (α1–α2–γδ–δ) + β + | −6,460.830 | 60.781 | 370 | 3.288 | −3,336.950 | 48.242 | 344 | 3.581 |
| H3: (α1–α2–) + (γδ–δ) + β | −6,460.851 | 60.802 | 401 | 3.329 | −3,333.322 | 44.614 | 384 | 3.689 |
| H4: (α1–α2–) + β + γδ + δ | −6,515.861 | 115.812 | 444 | 4.306 | −3,388.247 | 99.539 | 431 | 4.641 |
| H5: (α1–α2) + (γδ–δ)+ β + | −6,491.504 | 91.455 | 438 | 4.431 | −3,362.185 | 73.477 | 414 | 4.767 |
| H6: α1 + α2 + γδ + δ + β + | −6,609.832 | 209.783 | 551 | 6.489 | −3,472.122 | 183.414 | 535 | 6.879 |
Note.—For each hypothesis tested, common ancestry (H0) and independent origins (H1–H6), we show the marginal likelihood (P(data | M)) value, the ΔBF, the alignment (ali) length, and tree length. H0 is the best supported model according to all statistics tested here.
In AlphaPVs, the Evolutionary History of the Inter-E2–L2 Region Is Different from That of E5
In order to look deeper into the evolutionary history of the inter-E2–L2 region within AlphaPVs, we performed phylogenetic analyses and compared the tree topology for the inter-E2–L2 fragment sequences and for the E5 ORFs with the topologies obtained for each of the other PV ORFs (E6, E7, E1, E2, L2, and L1), the concatenated E6E7E1E2L2L1 ORFs, as well as for the noncoding URR. We calculated the weighted RF distances and K-tree scores between paired trees and we performed a correspondence analysis on the distance matrix in order to identify similarities among the topologies of the PV gene trees (fig. 2). For both distance measures, the first axis captured a large fraction of the variance (>50%) and splitted the E5 ORF from all other PV genes. For the weighted RF distance the second axis explained 15.7% of the overall variance and splitted the topologies of the early genes E6, E7, E1, and E2, from those of the late genes L2 and L1, and the URR. Interestingly, in this second axis the inter-E2–L2 clustered together with the late genes, while the E5 genes clustered together with the early genes. For the K-tree score the second axis explains 12% of the variance and separates the early genes E1 and E2 from the late genes. For both measures of tree distance, the concatenated E6E7E1E2L2L1 maps in between the main core genes in the PV genomes, namely E1, E2, L2, and L1. Altogether, these results suggest that E5 genes have a different evolutionary history from that of the PV core genome, from that of the other PV oncogenes, but, more intriguingly, different from that of the inter-E2–L2 region where the E5 genes reside.
Fig. 2.

—Correspondence analysis of the weighted Robinson–Foulds (RF) tree distance (A) and the K-tree score (B) comparing maximum likelihood trees constructed for each of the PV ORFs, the concatenated E6E7E1E2L2L1 ORFs, the inter-E2–L2 region, and the URR. The weighted RF distance matrix is symmetrical and only one series of results are generated (blue symbols in panel A). For the K-tree score the distance matrix is not symmetrical and two series of results are thus generated when exchanging rows and columns, corresponding to the blue and red symbols in panel B). Values in parenthesis represent the percentage of total variance explained by the corresponding axis.
The E5 ORFs in AlphaPVs Display the Characteristics of a Bona Fide Gene
It is often discussed whether the E5 ORFs in AlphaPVs are actual coding sequences. We have thus performed a number of analyses in order to assess whether the different E5 ORFs exhibit the characteristics of a bona fide gene. To determine whether the E5 ORFs are larger than expected by chance, we constructed first 1,000 random DNA sequences with the same median nucleotide composition as the inter-E2–L2 region of AlphaPVs, we identified all putative ORFs appearing by chance in these randomly generated DNA sequences and we computed their nucleotide length. Figure 3 shows the cumulative frequency of the length of the E5 and random ORFs. A one-way ANOVA followed by a post hoc Tukey’s HDS test, with gene as a factor (supplementary table S2, Supplementary Material online) showed that ORFs in randomly generated sequences are shorter than any of the E5 ORFs (Tukey’s HSD: P < 0.0001).
Fig. 3.
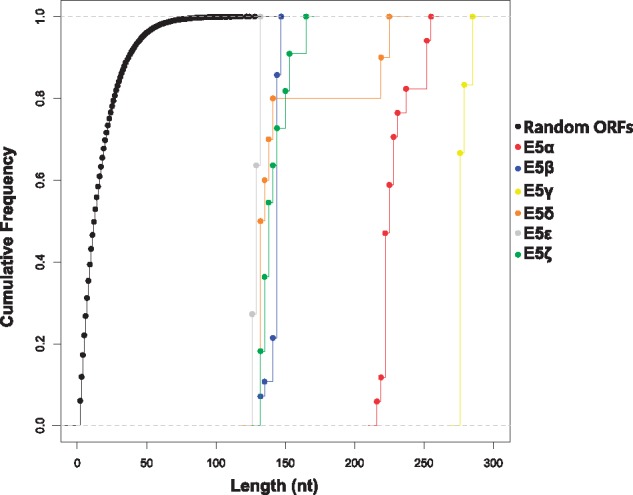
—Cumulative frequency of the nucleotide length for each group of the E5 genes and random ORFs. The random ORFs correspond to identified putative ORFs on a set of randomly generated DNA sequences with a median nucleotide composition and a median length of the inter-E2–L2 region of the AlphaPV genomes. The different types of E5 are color-coded as indicated in the legend.
Besides length, evidence of selective pressure is another signature of bona fide genes. We calculated the dN/dS values per codon position for all E5 sequences (fig. 4). Our results showed that the E5 ORFs display a per position dN/dS distribution that is significantly <1 (Wilcoxon–Mann–Whitney one-sided test: V = 1,416.5, P < 2.2e-16), with median values ranging from 0.09 to 0.40. All other PV genes presented median dN/dS values lower than the E5 sequences (Wilcoxon–Mann–Whitney two-sided test: W = 608,800, P < 2.2e-16). Although these results indicate that the vast majority of codons in all PV genes are under purifying selection, the analysis did reveal sites under positive selection in most E5 ORFs as well as in all other PV ORFs, except in E5β, E5ϵ, and L1 (supplementary table S3, Supplementary Material online).
Fig. 4.

—Box and whisker plots of the dN/dS values calculated per position for all genes encoded by AlphaPVs. For each distribution the median is labeled with a bar, the box encompasses the first and third quartile, and the whiskers cover the 95% confidence interval.
We next calculated the pairwise distances between terminal taxa for all ORFs and the URR in AlphaPVs, as well as for a set of randomly generated intergenic CDSs (fig. 5). These random intergenic CDSs were generated by extracting the inter-E2–L2 regions of the AlphaPV genomes, and subsequently extracting random subregions with the same length as the E5 ORF in the corresponding PV genome (see Materials and Methods). Pairwise differences were highest among the random intergenic CDSs region, as expected (fig. 5). The E5 ORFs displayed larger pairwise differences than any other PV gene (Wilcoxon–Mann–Whitney two-sided test: W = 24,943,000, P < 2.2e-16), but they were significantly lower than those for the random sequences (Wilcoxon–Mann–Whitney two-sided test: W = 251,230, P < 2.2e-16). The E5α, E5β, E5δ, and E5ζ show larger pairwise distances compared with the URR, while the E5γ and E5ϵ show lower rates (supplementary table S4, Supplementary Material online). When pairwise differences were normalized with respect to the corresponding L1 distance, the earlier described results are the same (supplementary fig. S5 and table S4, Supplementary Material online).
Fig. 5.

—Box and whisker plots of the pairwise distances between terminal taxa for the corresponding trees inferred for all genes encoded by AlphaPVs, the URR, and randomly generated intergenic CDSs. These random CDSs were generated by using the inter-E2–L2 region of the corresponding AlphaPV genome. For each distribution the median is labeled with a bar, the box encompasses the first and third quartile, and the whiskers cover the 95% confidence interval.
In PVs, CUPrefs are different from those of their hosts, and viral genes with similar expression patterns display similar CUPrefs (Félez-Sánchez et al. 2015). To analyze the CUPrefs of the E5 genes in the context of those of other PV genes, we calculated the relative frequencies of the 59 codons in synonymous families and performed a nonmetric multidimensional scaling (MDS) analysis as well as a PCA on the 59D codon usage vectors. Since the AlphaPVs clade contains both human and nonhuman PVs and in order to prevent a host-based bias, we performed the analysis on all AlphaPVs (fig. 6 and supplementary figs. S6–S8, Supplementary Material online) and on only the human AlphaPVs, separately (supplementary figs. S6–S8, Supplementary Material online). In the MDS plot (fig. 6A) the PV core genes E1 and E2, as well as L2 and L1 cluster together in the center, and thus show to have similar CUPrefs. From the PV oncogenes, E6 shows to have CUPrefs that are close to that of the early genes, while E5 and E7 have more disperse CUPrefs. In the PCA plot (fig. 6B) it is shown that the first and mainly the second axis separate the CUPrefs of the early (E6, E7, E1, and E2) and late (L2 and L1) genes, where certain synonymous codons are preferred by the early genes while others are preferred by the late genes (supplementary fig. S7, Supplementary Material online). The CUPrefs of E5 are in between, but more closely related to the late genes. Interestingly, in both the MDS and PCA, E5s cluster with the randomly generated intergenic CDSs. Since these random CDSs were generated using the composition characteristics of the genomic region in which the E5 ORFs reside, we interpret that such similarity in CUPrefs highlights the importance of nucleotide composition as the main factor governing CUPrefs (Pouyet et al. 2017).
Fig. 6.

—Nonmetric multidimensional scaling analysis (A) and principal component analysis (B) of the codon usage preferences for all AlphaPV ORFs. Additionally, the ORFs were independently clustered by a hierarchical clustering algorithm based also on their codon usage preferences. The results from the best hierarchical clustering (two clusters) have been plotted onto both plots, with a color code as described in the legend. Grossly, cluster 1 consists of the E6, E1, E2, L2, and L1 genes, whereas cluster 2 consists of the E7 and E5 genes, and the randomly generated intergenic CDSs. The values for the total stress of the data explained by the MDS as well as for the percentage of the total variance explained by the each of the first two principal components are given in each plot.
In parallel, a hierarchical clustering analysis based on the 59D codon usage vectors was performed. When considering all AlphaPVs the optimal number of clusters was two (supplementary fig. S8A, Supplementary Material online): one cluster containing the core PV genes (E1, E2, L2, and L1), almost all E6 genes (97.1%), some E7 genes (23.2%), and a few E5 genes (5.8%); and a second cluster containing most E7 genes (76.8%), almost all E5 genes (94.2%) and all randomly generated intergenic CDSs. When considering only human AlphaPVs the optimal number of clusters was either two or seven. When using two clusters, the results were cleaner compared with the nonhuman and human AlphaPVs together (supplementary fig. S8B, Supplementary Material online): the first cluster contained all E6, E1, E2, L2, and L1 genes, some E7 (17.9%) and one of the E5 genes; while the second cluster contained most E7 and E5 genes, and all random sequences (supplementary table S5, Supplementary Material online). When using seven clusters most of the clustering can be explained by the different ORFs present, as virtually one cluster per ORF was retrieved (supplementary fig. S8C and table S5, Supplementary Material online). Interesting is that the first cluster, consisting of only E1 genes (50% of total), contains mainly ORFs that come from PV lineages associated to mucosal lesions and genital warts (supplementary table S6, Supplementary Material online), while only one ORF in this cluster comes from a PV lineage associated to cutaneous warts. The other half of the E1 genes are present in the second cluster together with all E2 genes. Here, PVs associated to all three phenotypes are present. In addition, cluster six, consisting of the late L2 and L1 genes, is essentially associated to cutaneous warts, while cluster seven contains mainly the late genes from PV lineages associated to mucosal lesions and genital warts.
As the best-studied E5 proteins are transmembrane proteins, we hypothesized that a bona fide E5 protein should be more hydrophobic than expected by chance. We calculated the GRAVY index for the E5 proteins as well as for the ORFs encoded in the randomly generated intergenic CDSs (fig. 7). We found that E5α, E5β, E5γ, E5δ, and E5ϵ are more hydrophobic than the random intergenic CDSs (Wilcoxon–Mann–Whitney test, P < 0.0001). The E5ζ is the only E5 protein that did not tested significantly more hydrophobic than the random intergenic CDSs (Wilcoxon–Mann–Whitney, P = 0.125).
Fig. 7.
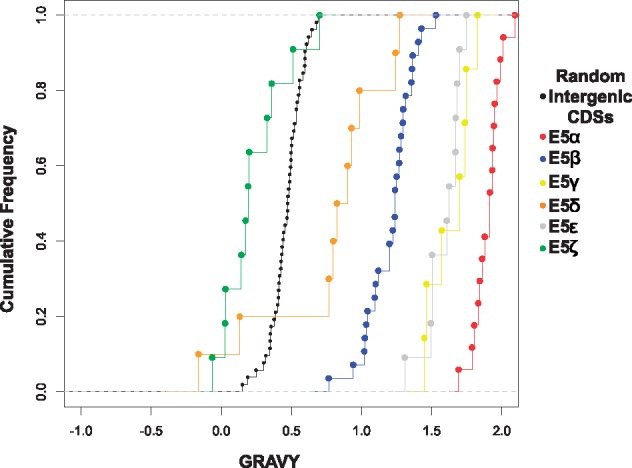
—Cumulative frequency of the GRAVY index for the E5 ORFs and the randomly generated intergenic CDSs. The different types of E5 are color-coded as indicated in the legend.
Discussion
Reconstructing how PV genes have originated and evolved is crucial for explaining the genetic basis of the origin and evolution of phenotypic diversity found in PVs, if we eventually aim to understand why certain PVs are oncogenic while their close relatives cause anodyne infections (Willemsen and Bravo 2019). In this work our main aim was to study the origin of the E5 oncogenes in AlphaPVs. This viral genus hosts ∼50 viral genotypes with a relative narrow host distribution (they seem to be restricted to primates), but with very diverse phenotypic presentations of the infections: many of them are associated to asymptomatic infections of the skin, but also of the oral, nasal, or anogenital mucosas; some of them cause productive infections that result in common skin warts, or in genital warts; and a number of them cause chronic infections that may result in anogenital or oropharyngeal cancers (Doorbar et al. 2012; Forman et al. 2012). All AlphaPVs present a region between the E2 and L2 genes, potentially encoding in all cases for conserved ORFs. With few exceptions (Cartin and Alonso 2003), actual gene expression and protein function for E5 oncogenes have only been characterized for the more oncogenic HPVs, which carry E5 proteins of type E5α (Bravo and Alonso 2004). These E5α behave as oncoproteins, promoting cell division and allowing the infected cells to avoid immune recognition (Ashrafi et al. 2005; Bravo et al. 2005; Suprynowicz et al. 2008).
Since all the E5 ORFs in AlphaPVs map between the E2 and L2 genes, we extended our analysis to the evolution of this intergenic region in the Alpha-Omikron crown group. Finally, since a number of nonmonophyletic PVs also contain a sometimes long noncoding region between the E2 and L2 genes in their genomes that may also encode for genes named E5, we expanded our analyses to the full set of PV sequences containing a long noncoding region at this genomic location. PVs displaying an intergenic region between E2 and L2 are not monophyletic, and belong instead to four polyphyletic clades in the PV tree (fig. 1 and supplementary fig. S1, Supplementary Material online). It can be argued that the ancestral PV genomes could have already presented an inter-E2–L2 region, which may have undergone several loss events. Such repeated losses have been invoked as a mechanism to explain the repeated absence of early genes (E6 and E7) in certain PVs (Van Doorslaer and McBride 2016). Alternatively, the different inter-E2–L2 regions present in extant PV genomes could derive from one or from several genetic events in which an ancestral sequence could have gained access to one ancestral PV genome.
We can formulate two main nonexclusive mechanisms to explain the origin of the four extant groups of inter-E2–L2 regions in the PVs genomes: random nucleotide addition and recombination. Random nucleotide addition is a plausible mechanism, based on the way the PV genome replicates. The replication of the PV genome occurs bidirectionally during the nonproductive stages of the infection, yielding episomes (Flores and Lambert 1997). During the PV bidirectional replication, the replication forks start at the URR and converge opposite to the origin of replication, which happens to lay between the E2 and L2 genes. At this point, concerted DNA breaks are required for decatenation, which eventually generates two separate circular dsDNA molecules. The end joining of these DNA breaks is error prone. Indeed, the DNA close to the break site can be used as a template for de novo synthesis before the DNA ends are joined, resulting in the nontemplated introduction of a stretch of additional nucleotides (Roerink et al. 2014), which may lead to the emergence of an ancestral inter-E2–L2 region in one or in several instances during the evolutionary history of PVs.
Recombination can also be invoked as a mechanism that could result in the integration of novel DNA sequences into the PV genome. In parallel to the host keratinocyte differentiation, replication of the viral genome switches from bidirectional to unidirectional (Flores and Lambert 1997; McBride 2017), generating large linear molecules of concatenated viral genomes (Dasgupta et al. 1992). Unidirectional replication relies on homologous recombination, as this mechanisms is required for resolving, excising, and recircularizing the concatenated genomes into individual plasmid genomes (Gillespie et al. 2012; Sakakibara et al. 2013; Mehta and Laimins 2018). Additionally, productive replication concurs with a virus-mediated impairment of the cellular DNA damage repair mechanisms (Chappell et al. 2016; Wallace et al. 2017), thus rendering the overall viral replication process error-prone by increasing the probability of integrating exogenous DNA during recircularization. Phylogenetic evidence for the existence and fixation of such recombination events is provided by the incongruence in the reconstruction of the evolutionary history for different regions of the PV genome. In all cases, such inconsistencies appear when comparing the phylogenetic inference for the early and for the late genes of the genome, respectively, upstream and downstream the recombination-prone genomic region. Evidence for recombination has been described at several nodes in the PV tree. The first example occurs at the root of AlphaPVs, with the species containing oncogenic PVs being monophyletic according to the early genes (involved in oncogenesis and genome replication), and paraphyletic according to the late genes (involved in capsid formation) (Bravo and Alonso 2004; Narechania et al. 2005). The second example is provided by certain PVs infecting cetaceans, which display the early genes related to those in other cetacean PVs in the Alpha-Omikron crown group (in red in fig. 1) and the late genes related to those in bovine PVs in the Beta-Xi crown group (in green in fig. 1) (Rector et al. 2008; Gottschling et al. 2011; Robles-Sikisaka et al. 2012). Finally, the most cogent examples of recombination between distant viral sequences are two viruses isolated from bandicoots and displaying the early genes related to Polyomaviruses and the late genes related to PVs (Woolford et al. 2007; Bennett et al. 2008).
The inter-E2–L2 sequences may occasionally be very long and span >1 kb, a considerable size for an average genome length of ∼8 kb. Additionally, for many viral genomes, the sequences in the inter-E2–L2 region do not resemble other sequences in the databases, and do not seem to contain any functional elements, neither ORFs nor transcription factor binding sites or conserved regulatory regions (Rector et al. 2007; Schulz et al. 2009; García-Pérez et al. 2014). Despite the lack of obvious function and of their length, these sequences seem to belong bona fide in the viral genome in which they are found, as they are fixed and conserved in viral lineages (Rector et al. 2007). Although the two hypothesis referred above to explain the origin of the inter-E2–L2 regions (random nucleotide addition and recombination) are plausible, we interpret that the presence of long and conserved sequences in certain monophyletic clades (labeled with a star in fig. 1 and supplementary fig. S1, Supplementary Material online) suggests that the respective insertions of each of these long sequences in the ancestral genomes occurred during single episodes, pointing thus toward a recombination event.
The putative ORFs that emerged in the inter-E2–L2 region are often named E5. Notwithstanding, our results suggest the E5 proteins encoded in the different clades may not be monophyletic. Specifically, this would imply that the E5 ORFs in AlphaPVs (e.g., HPV16 E5) are not evolutionarily related to the E5 ORFs in DeltaPVs (e.g., BPV1 E5). This is an important change in perspective, because these two proteins are often referred to and their cellular activities compared as if they were orthologs (Ashby et al. 2001; Venuti et al. 2011). Yet, the E5 sequences are short and display similar amino acid composition because of their transmembrane nature, and these two facts combined reduce the power of the algorithms used to pinpoint common ancestry between genes. Further tests are needed to resolve the riddle on the origin of E5s: either in silico by improving the CA test or experimentally by evolving a predicted ancestor(s) of E5 or by performing de novo gene evolution on the inter-E2–L2 region.
When restricting our analysis to the E5 ORFs within the AlphaPVs, we found support for monophyly (table 2), indicating that a single event on the backbone of the ancestral AlphaPV genome could have led to its emergence. Nevertheless, the alternative hypothesis of E5β having an independent origin was not significantly worse. This hypothesis is supported by the different tropism of lineages within AlphaPVs: those containing an E5β display an essentially cutaneous tropism, while all other lineages encoding for E5α, E5γ, E5δ, E5ϵ, and E5ζ, display a mucosal tropism. Indeed, there is no evident sequence similarity between the E5 proteins, inasmuch as the evolutionary divergence between E5β and the other E5 ORFs rises to 80% (Bravo and Alonso 2004). Phylogenetic reconstruction based on the E5 ORFs showed a star-like pattern with the main branches emerging close to a putative central point (Bravo and Alonso 2004). These features could be related to the multiple ancestries of the different E5 ORFs.
It remains unclear how the different E5 genes emerged in the AlphaPV genomes. Our interpretation based on the evidence here provided is as follows. Under the hypothesis of recombination, within AlphaPVs, a noncoding sequence was integrated in a single event between the early and the late genes in the genome of an ancestral PV lineage, which infected the ancestors of Old World monkeys and apes. Mutations in this originally noncoding region gave birth to the different E5 ORFs. Such de novo birth of new protein-coding sequences from noncoding genomic regions is not unfamiliar and has been reported in for example Drosophila (Levine et al. 2006; Zhou et al. 2008; Zhao et al. 2014), yeast (Cai et al. 2008; Carvunis et al. 2012), and mammals (Toll-Riera et al. 2008). Experimentally, it has been shown that random, E5-like short peptide sequences can indeed insert in the cellular membranes and display a biological activity (Chacón et al. 2014). Using genetic selection, these small artificial transmembrane amino acid sequences that do not occur in nature were able to bind and activate the platelet derived growth factor (PDGF) β receptor (just like BPV1 E5 does), resulting in cell transformation and tumorogenicity (Chacón et al. 2014). Therefore, we consider de novo birth of the E5 genes in the inter-E2–L2 region a plausible hypothesis. The randomly appeared E5 genes, short and enriched in hydrophobic amino acids, could thus have provided with a rudimentary function by binding to membrane receptors or by modifying membrane environment. Such activities may have led to an increase in viral fitness and could have been selected and enhanced, resulting in the different E5 genes lineages observed today.
The location within the inter-E2–L2 region and the hydrophobic nature of the protein have up to date been the criteria to classify the E5 ORFs as putative genes. This is probably the reason for which we found all E5 ORFs, with the only exception of E5ζ, more hydrophobic than expected by chance (fig. 7). However, for most of these ORFs, we do not have evidence of their expression in vivo. Moreover, the possible independent origins of E5, raise the concern of whether all E5 ORFs are actually coding sequences. In this study, we have used several approaches in order to distinguish true E5 genes from spurious ORFs that are not functional. As orthologs of the E5 genes are not found in other viruses or in their hosts, we have studied the E5 ORFs in the context of orphan genes. In agreement with studies of orphan genes in other species (Toll-Riera et al. 2008; Wolf et al. 2009; Carvunis et al. 2012), the E5 genes are shorter than the other PV genes. It has previously been proposed that there is a direct relationship between the length of a gene and its age (Albà and Castresana 2005; Toll-Riera et al. 2008; Palmieri et al. 2014). However, a bona fide gene should be longer than expected by chance (Schlötterer 2015), and this is what we actually find for the different E5 ORFs (fig. 3).
For a new functional protein to evolve from randomly occurring ORFs, it needs to be produced in significant amounts. These proteins are expected to evolve under neutral selection, as these are unlikely to be functional at first. By combining ribosome profiling RNA sequencing with proteomics and SNP information Ruiz-Orera et al. (2018) found evidence to support this hypothesis. By analyzing mouse tissue they found hundreds of small proteins that evolve under no purifying selection. Regarding the E5 ORFs, we obtained dN/dS ratios <1 (fig. 4), indicating negative or purifying selection, reinforcing the idea that extant E5s may be functionally relevant. Gene CUPrefs have a strong effect on ORF translation, where a favorable codon composition may facilitate the translation of certain ORFs, while other ORFs with a less favorable codon composition may remain untranslated (Ruiz-Orera et al. 2018). We have thus evaluated whether CUPrefs in E5 resemble those in other AlphaPV genes. The E5 genes exhibited CUPrefs similar to those of the E7 genes (fig. 6), and both proteins are implicated in the transforming potential of oncogenic HPVs. This is in line with previous work reporting that genes expressed at similar stages during viral infection have similar CUPrefs (Félez-Sánchez et al. 2015). Provocative is that the CUPrefs of E5 are also similar to the randomly generated intergenic CDSs. However, as these random sequences are generated using the inter-E2–L2 region of AlphaPVs, we would expect this clustering as we hypothesize that E5 evolved de novo in this region. The observation that the E5 ORFs are under purifying selection and the clustering of the CPUrefs of E5 together with the E7 oncogene, reinforces the proposal of an oncogenic role of the different E5 proteins in the life cycle of oncogenic human AlphaPVs.
In summary, our results strongly suggest that E5 in AlphaPVs are bona fide genes and not merely spurious translations. This is supported by previous studies that already assigned different properties to E5, such as the alteration of membrane composition and dynamics (Bravo et al. 2005; Suprynowicz et al. 2008) and the downregulation of surface MHC class I molecules (Cartin and Alonso 2003; Campo et al. 2010) for immune evasion. However, many questions about E5 remain to be elucidated. Further experimental studies should be performed to provide evidence of the expression of the different E5 ORFs in vivo and to elucidate whether E5 originated through recombination, random nucleotide addition, or another unknown mechanism.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to the genotoul bioinformatics platform Toulouse Midi-Pyrenees (Bioinfo Genotoul) for providing computing and storage resources. The authors acknowledge the IRD itrop HPC (South Green Platform) at IRD Montpellier for providing HPC resources that have contributed to the research results reported within this article. We also thank the Peer Community In Evolutionary Biology for reviewing and recommending the preprint version of this article. This work was supported by the European Research Council Consolidator Grant CODOVIREVOL (contract number 647916) to I.G.B. and by the European Union Horizon 2020 Marie Sklodowska-Curie research and innovation program grant ONCOGENEVOL (contract number 750180) to A.W.
Data deposition: This project has been deposited at Zenodo under the accession https://doi.org/10.5281/zenodo.2647437.
Literature Cited
- Albà MM, Castresana J.. 2005. Inverse relationship between evolutionary rate and age of mammalian genes. Mol Biol Evol. 22(3):598–606. [DOI] [PubMed] [Google Scholar]
- Ashby AD, Meagher L, Campo MS, Finbow ME.. 2001. E5 transforming proteins of papillomaviruses do not disturb the activity of the vacuolar H+-ATPase. J Gen Virol. 82(10):2353–2362. [DOI] [PubMed] [Google Scholar]
- Ashrafi GH, et al. 2002. Down-regulation of MHC class I by bovine papillomavirus E5 oncoproteins. Oncogene 21(2):248–259. [DOI] [PubMed] [Google Scholar]
- Ashrafi GH, Haghshenas MR, Marchetti B, O’Brien PM, Campo MS.. 2005. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int J Cancer. 113(2):276–283. [DOI] [PubMed] [Google Scholar]
- Bennett MD, et al. 2008. Genomic characterization of a novel virus found in papillomatous lesions from a southern brown bandicoot (Isoodon obesulus) in Western Australia. Virology 376(1):173–182. [DOI] [PubMed] [Google Scholar]
- Bravo IG, Alonso A.. 2004. Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. J Virol. 78(24):13613–13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo IG, Crusius K, Alonso A.. 2005. The E5 protein of the human papillomavirus type 16 modulates composition and dynamics of membrane lipids in keratinocytes. Arch Virol. 150(2):231–246. [DOI] [PubMed] [Google Scholar]
- Bravo IG, Felez-Sanchez M.. 2015. Papillomaviruses: viral evolution, cancer and evolutionary medicine. Evol Med Public Health. 2015(1):32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Zhao R, Jiang H, Wang W.. 2008. De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics 179(1):487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo M, et al. 2010. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 407(1):137–142. [DOI] [PubMed] [Google Scholar]
- Cartin W, Alonso A.. 2003. The human papillomavirus HPV2a E5 protein localizes to the Golgi apparatus and modulates signal transduction. Virology 314(2):572–579. [DOI] [PubMed] [Google Scholar]
- Carvunis AR, et al. 2012. Proto-genes and de novo gene birth. Nature 487(7407):370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. [DOI] [PubMed] [Google Scholar]
- Chacón KM, et al. 2014. De novo selection of oncogenes. Proc Natl Acad Sci U S A. 111(1):E6–E14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell WH, et al. 2016. Homologous recombination repair factors Rad51 and BRCA1 are necessary for productive replication of human papillomavirus 31. J Virol. 90(5):2639–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Bubb VJ, Schlegel R.. 1993. The human papillomavirus type 6 and 16 E5 proteins are membrane-associated proteins which associate with the 16-kiloDalton pore-forming protein. J Virol. 67(10):6170–6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Zabielski J, Simonsson M, Burnett S.. 1992. Rolling-circle replication of a high-copy BPV-1 plasmid. J Mol Biol. 228(1):1–6. [DOI] [PubMed] [Google Scholar]
- de Oliveira Martins L, Posada D.. 2014. Testing for universal common ancestry. Syst Biol. 63(5):838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Martins L, Posada D.. 2016. Infinitely long branches and an informal test of common ancestry. Biol Dir. 11(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio D, Mattoon D.. 2001. Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene 20(54):7866–7873. [DOI] [PubMed] [Google Scholar]
- DiMaio D, Petti LM.. 2013. The E5 proteins. Virology 445(1–2):99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J, et al. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30(5):F55–70. [DOI] [PubMed] [Google Scholar]
- Doron-Faigenboim A, Pupko T.. 2007. A combined empirical and mechanistic codon model. Mol Biol Evol. 24(2):388–397. [DOI] [PubMed] [Google Scholar]
- Doron-Faigenboim A, Stern A, Mayrose I, Bacharach E, Pupko T.. 2005. Selection: a server for detecting evolutionary forces at a single amino-acid site. Bioinformatics 21(9):2101–2103. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félez-Sánchez M, et al. 2015. Cancer, warts, or asymptomatic infections: clinical presentation matches codon usage preferences in human papillomaviruses. Genome Biol Evol. 7(8):2117–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Lambert PF.. 1997. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J Virol. 71(10):7167–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman D, et al. 2012. Global burden of human papillomavirus and related diseases. Vaccine 30:F12–F23. [DOI] [PubMed] [Google Scholar]
- García-Pérez R, et al. 2014. Novel papillomaviruses in free-ranging Iberian bats: no virus host co-evolution, no strict host specificity, and hints for recombination. Genome Biol Evol. 6(1):94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie KA, Mehta KP, Laimins LA, Moody CA.. 2012. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol. 86(17):9520–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling M, Bravo IG, et al. 2011. Modular organizations of novel cetacean papillomaviruses. Mol Phylogenet Evol. 59(1):34–42. [DOI] [PubMed] [Google Scholar]
- Gottschling M, Göker M, et al. 2011. Quantifying the phylodynamic forces driving papillomavirus evolution. Mol Biol Evol. 28(7):2101–2113. [DOI] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Hughes M.. 2005. Patterns of nucleotide difference in overlapping and non-overlapping reading frames of papillomavirus genomes. Virus Res. 113(2):81–88. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT Multiple Sequence Alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI.. 2010. The common ancestry of life. Biol Dir. 5:64.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF.. 1982. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 157(1):105–132. [DOI] [PubMed] [Google Scholar]
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ.. 2006. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A. 103(26):9935–9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA. 2017. Mechanisms and strategies of papillomavirus replication. Biol Chem. 398(8):919–927. [DOI] [PubMed] [Google Scholar]
- Mehta K, Laimins L.. 2018. Human papillomaviruses preferentially recruit DNA repair factors to viral genomes for rapid repair and amplification. mBio 9(1):e00064–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody CA, Laimins LA.. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 10(8):550–560. [DOI] [PubMed] [Google Scholar]
- Münger K, Scheffner M, Huibregtse JM, Howley PM.. 1992. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 12:197–217. [PubMed] [Google Scholar]
- Narechania A, Chen Z, DeSalle R, Burk RD.. 2005. Phylogenetic incongruence among oncogenic genital alpha human papillomaviruses. J Virol. 79(24):15503–15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri N, Kosiol C, Schlötterer C.. 2014. The life cycle of Drosophila orphan genes. eLife 3:e01311.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petti LM, Reddy V, Smith SO, DiMaio D.. 1997. Identification of amino acids in the transmembrane and juxtamembrane domains of the platelet-derived growth factor receptor required for productive interaction with the bovine papillomavirus E5 protein. J Virol. 71(10):7318–7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pim D, Collins M, Banks L.. 1992. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 7(1):27–32. [PubMed] [Google Scholar]
- Pouyet F, Mouchiroud D, Duret L, Sémon M.. 2017. Recombination, meiotic expression and human codon usage. eLife 6: e27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puustusmaa M, Kirsip H, Gaston K, Abroi A.. 2017. The enigmatic origin of papillomavirus protein domains. Viruses 9(9):240.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
- Rector A, et al. 2007. Ancient papillomavirus-host co-speciation in Felidae. Genome Biol. 8(4):R57.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector A, et al. 2008. Genomic characterization of novel dolphin papillomaviruses provides indications for recombination within the Papillomaviridae. Virology 378(1):151–161. [DOI] [PubMed] [Google Scholar]
- Redelings BD, Suchard MA.. 2005. Joint Bayesian estimation of alignment and phylogeny. Syst Biol. 54(3):401–418. [DOI] [PubMed] [Google Scholar]
- Robinson D, Foulds L.. 1981. Comparison of phylogenetic trees. Math Biosci. 53(1–2):131–147. [Google Scholar]
- Robles-Sikisaka R, et al. 2012. Evidence of recombination and positive selection in cetacean papillomaviruses. Virology 427(2):189–197. [DOI] [PubMed] [Google Scholar]
- Roerink SF, Schendel R, Tijsterman M.. 2014. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 24(6):954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Orera J, Verdaguer-Grau P, Villanueva-Cañas JL, Messeguer X, Albà MM.. 2018. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nat Ecol Evol. 2(5):890–896. [DOI] [PubMed] [Google Scholar]
- Sakakibara N, Chen D, McBride AA.. 2013. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 9(7):e1003321.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C. 2015. Genes from scratch–the evolutionary fate of de novo genes. Trends Genet. 31(4):215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz E, et al. 2009. Genomic characterization of the first insectivoran papillomavirus reveals an unusually long, second non-coding region and indicates a close relationship to Betapapillomavirus. J Gen Virol. 90(3):626–633. [DOI] [PubMed] [Google Scholar]
- Soria-Carrasco V, Talavera G, Igea J, Castresana J.. 2007. The K tree score: quantification of differences in the relative branch length and topology of phylogenetic trees. Bioinformatics 23(21):2954–2956. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight SW, Hinkle PM, Jewers RJ, McCance DJ.. 1993. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol. 67(8):4521–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard MA, Redelings BD.. 2006. BAli-Phy: simultaneous Bayesian inference of alignment and phylogeny. Bioinformatics 22(16):2047–2048. [DOI] [PubMed] [Google Scholar]
- Suprynowicz FA, et al. 2008. HPV-16 E5 oncoprotein upregulates lipid raft components caveolin-1 and ganglioside GM1 at the plasma membrane of cervical cells. Oncogene 27(8):1071–1078. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P.. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34(Web Server):W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theobald DL. 2011. On universal common ancestry, sequence similarity, and phylogenetic structure: the sins of P-values and the virtues of Bayesian evidence. Biol Dir. 6(1):60.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll-Riera M, et al. 2008. Origin of primate orphan genes: a comparative genomics approach. Mol Biol Evol. 26(3):603–612. [DOI] [PubMed] [Google Scholar]
- Tomaić V. 2016. Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers 8(10):95.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Doorslaer K, McBride AA.. 2016. Molecular archeological evidence in support of the repeated loss of a papillomavirus gene. Sci Rep. 6(1):33028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venuti A, et al. 2011. Papillomavirus E5: the smallest oncoprotein with many functions. Mol Cancer. 10(1):140.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace NA, et al. 2017. High-risk alphapapillomavirus oncogenes impair the homologous recombination pathway. J Virol. 91(20):e01084–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen A, Bravo IG.. 2019. Origin and evolution of papillomavirus (onco)genes and genomes. Philos Trans R Soc B. 374(1773):20180303.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Novichkov PS, Karev GP, Koonin EV, Lipman DJ.. 2009. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc Natl Acad Sci U S A. 106(18):7273–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford L, et al. 2007. A novel virus detected in papillomas and carcinomas of the endangered Western barred bandicoot (Perameles bougainville) exhibits genomic features of both the Papillomaviridae and Polyomaviridae. J Virol. 81(24):13280–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen A.. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155(1):431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonezawa T, Hasegawa M.. 2010. Was the universal common ancestry proved? Nature 468(7326):E9–E9. [DOI] [PubMed] [Google Scholar]
- Yonezawa T, Hasegawa M.. 2012. Some problems in proving the existence of the universal common ancestor of life on Earth. ScientificWorldJournal 2012:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Saelao P, Jones CD, Begun DJ.. 2014. Origin and spread of de novo genes in Drosophila melanogaster populations. Science 343(6172):769–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, et al. 2008. On the origin of new genes in Drosophila. Genome Res. 18(9):1446–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.