

Abstract
Microcephalic osteodysplastic primordial dwarfism syndrome II (MOPDII) is microcephalic primordial dwarfism and is a very rare form of disproportionate short stature. This disorder shares common features with other forms of microcephalic primordial dwarfism, including severe prenatal and postnatal growth retardation with marked microcephaly. However, it includes characteristic skeletal dysplasia, abnormal dentition and increased risk for cerebrovascular diseases. Recent reports added more features, including café-au-lait lesions, cutis marmorata, astigmatism, Moyamoya disease, insulin resistance, obesity, abnormal skin pigmentation and acanthosis nigricans around the neck. Clearly, the more MOPDII reports that are produced, the more information will be added to the spectrum of MOPDII features that can improve our understanding of this disorder. In this paper, we reported a new case of MOPDII with more severe clinical features, earlier onset of common features, in addition to a homozygous novel variant in the PCNT gene.
Keywords: paediatrics, genetics, neonatal health, congenital disorders, developmental paediatrocs
Background
Short stature, also known as dwarfism or restricted growth, is defined as height of ≤2 SD below the mean for age and gender or height below the third centile in the same ethnic group.1 There are a variety of short stature types with very variable phenotypes. Short stature that is associated with other physical and/or developmental abnormalities can be divided into proportionate and disproportionate groups.2 In proportionate dwarfism, normal body proportions are maintained, while the overall height is reduced. The most common causes of proportionate short stature are hereditary, in addition to constitutional delay of growth and puberty,3 4 which are normal variants of growth. Abnormal upper-to-lower body segment ratios for age cause disproportionate short stature, mostly seen in cases of skeletal dysplasia such as achondroplasia, the most common type of disproportionate dwarfism.5 In addition, there are other less common forms of this type of short stature, including primordial dwarfism in forms of Seckel’s syndrome and Majewski microcephalic osteodysplastic primordial dwarfism (MOPD) syndromes I/III and II.
MOPDII (MIM# 210720) is a rare autosomal-recessive form of bird-headed dwarfism that was initially described in 1982 by Majewski et al.6 It is characterised by severe intrauterine growth retardation (IUGR), postnatal growth failure, disproportionate short stature, marked microcephaly, skeletal dysplasia and characteristic facial features.7 Other features of MOPDII include high-pitched nasal voice, café-au-lait spots, cutis marmorata, Moyamoya disease, elevated platelet count, astigmatism, insulin resistance, obesity, abnormal skin pigmentation, acanthosis nigricans around the neck, mild mental retardation, cerebral aneurysms and other vascular abnormalities.8–13 Recognising these features with the support of radiographic examinations aids the diagnosis of MOPDII. However, identifying bi-allelic loss-of-function variants in the pericentrin (PCNT) gene might be necessary to confirm the diagnosis and to differentiate MOPDII from other syndromes with overlapping features such as Seckel syndrome (MIM# 210600). The PCNT protein is involved in cell cycle and spindle organisation,14 and the absence of this protein results in disorganised mitotic spindles and mis-segregation of chromosomes.15
To date, over 150 individuals have been diagnosed with MOPDII.16 In Saudi Arabia, at least two MOPDII cases were previously reported.17 18 Here, we report a third case of MOPDII with typical and atypical features. Genetic analysis identified a novel homozygous pathogenic variant in the PCNT gene in addition to a novel homozygous variant of unknown significance in the GLRB gene.
Case presentation
We report a 17-month-old boy with MOPDII born to consanguineous parents who have two unaffected children. The mother had a history of unexplained intrauterine fetal demise at 8 months’ gestation. Our patient was a product of an uneventful 40+2 week pregnancy. Prenatal ultrasound showed anhydramnios, cardiomegaly, fetal distress and severe intrauterine growth retardation. He was delivered limp by emergent caesarean section with a birth weight of 1.31 kg, below the fifth growth percentile. APGAR scores were 1/1 min, 1/5 min and 2/10 min. He required prolonged resuscitation in the form of full cardiopulmonary resuscitation, intubation, positive pressure ventilation and three doses of epinephrine through an umbilical venous catheter. In the neonatal intensive care unit (NICU), he was started on high-frequency oscillatory ventilation (HFOV), dopamine and inhaled nitric oxide. Laboratory investigations showed that he was anaemic with low platelet and leucocytosis. Blood gas analysis from umbilical artery catheter revealed severe metabolic acidosis with a pH of 7.13, CO2 of 30 and H2CO3 of 10. He had a clinical and laboratory picture of cholestatic hepatitis that resolved spontaneously with a few weeks of birth.
Echocardiography revealed persistent pulmonary hypertension, an ostium secundum atrial septal defect, a large PDA with a bidirectional shunt and a D-shaped ventricular septum. Head ultrasound revealed a left-sided grade-4 intraventricular haemorrhage with a grade-1 germinal matrix haemorrhage on the right side. CT of the head revealed extensive hydrocephalus that was most likely an outcome of a post-haemorrhagic event, minimal brain tissue in the supratentorial area and a hypoplastic midbrain/cerebellum. MRI of the brain showed large dilatation of the lateral ventricles with loss of normal brain tissue in the form of cystic encephalomalacia (figures 1 and 2). Physical examination showed that his limbs were hypertonic and fixed in flexion with growth parameters below the fifth percentiles. He had moderate response to light, fair response to sound and poor response to tactile stimulation. Eye examination revealed a right eye retinal mass with vitreous haemorrhage and left eye vision loss due to retinal detachment. In addition to severe mental retardation, he had many dysmorphic features, including narrow face, fine hair, hazy corneas, beaked nose, deep-set ears, retrognathia, microdontia, widely spaced nipples, micropenis and abnormal skin hypopigmentation on the posterior trunk (figures 3–7). Based on this, our patient was highly suspected to have a form of primordial dwarfism. The molecular genetic investigation was indicated to confirm the diagnosis.
Figure 1.

Hypoplastic cerebellum/vermis with left orbital hypoplasia and haemorrhage.
Figure 2.
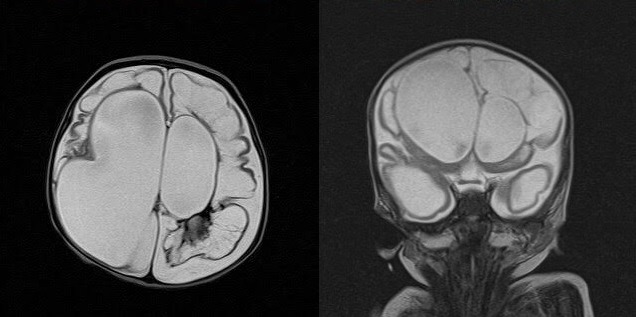
An extensive cystic encephalomalacic changes with normal basal ganglia. Remnant brain tissue can be seen mainly in the frontal and temporal regions.
Figure 3.

Generalised hypertonia in the flexion position with fixed rotation towards the right side.
Figure 4.
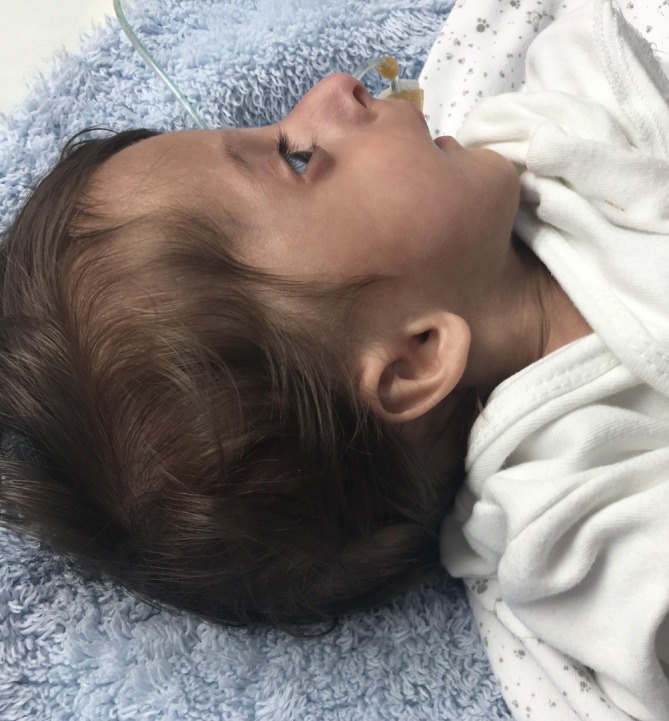
Severe microcephaly, fine hair and facial dysmorphic features in forms of beaked nose, retrognathia and low-set ears.
Figure 5.
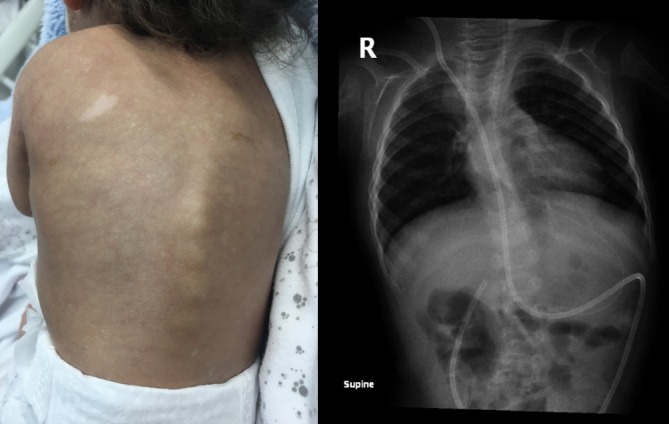
Thoracic scoliosis with spots of skin hypopigmentation and hyperpigmentation.
Figure 6.
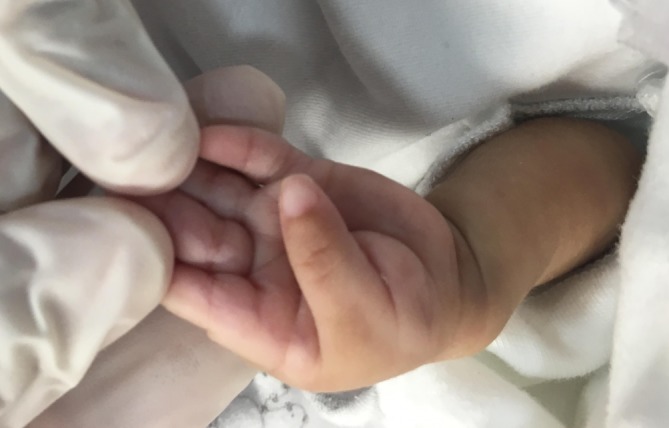
Inward flexed thumb with single fused interphalangeal crease in the fifth finger.
Figure 7.

Microdontia.
Investigations
Imaging is as mentioned in the text above.
Genetic analysis is as follows:
Karyotyping and chromosomal array were performed for the patient and both were negative. Whole-exome sequencing was then performed for the proband and his parents. Briefly, genomic DNA was prepared from leucocytes using the QIAamp DNA Blood Mini QIAcube Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Exome sequencing was performed using the SureSelect Human All Exon kit (Agilent, Santa Clara, California, USA) for enrichment and Illumina HiSeq4000 instrument for the actual sequencing. Variants were annotated using Annovar (http://annovar.openbioinformatics.org) and in-house ad hoc bioinformatics tools. Specific attention was paid to short stature-associated genes (summarised by Dauber et al 19 and Seaver and Irons2) and intellectual disability-associated genes (summarised by Pekeles et al 20). Identified variants were classified as per the standards and guidelines recommended by the American College of Medical Genetics and Genomics (ACMGG) and described according to the Human Variation Society (HGVS) nomenclature system. Neither pathogenic de novo variants nor de novo variants related to the patient’s phenotype were detected. However, the analysis identified a homozygous pathogenic variant in the PCNT gene (NM_006031.5: c.3760C>T, p(.Gln1254X); Chr21(GRCh37):g.47809266C>T) (figure 8A). This nonsense variant creates a premature stop codon that is expected to result in a non-functional truncated protein. Another homozygous variant of unknown significance was identified in the GLRB gene (NM_000824.4: c.1492T>A, p(*498fs); Chr4(GRCh37):g.158091878T>A) (figure 8B). This variant is expected to cause loss of stop codon and the addition of one amino acid. The parents were heterozygous for both variants while two healthy siblings were confirmed by Sanger sequencing to be heterozygous for one or both variants. To the best of our knowledge, both variants have never been reported previously in the literature. Based on the ACMGG guidance for the interpretation of sequence variants and the phenotype–genotype correlation, the variant identified in the PCNT gene was the disease-causing variant in our patient.
Figure 8.

Family pedigree and Sanger chromatogram. (A) A pedigree of the family included in this study. (B) Sanger sequencing traces showing the homozygosity of the affected index patient for both variants (in red rectangles) identified in the PCNT and GLRB genes.
Differential diagnosis
Seckel syndrome and Silver-Russell dwarfism.
Treatment
To date, there is no curative intervention for MOPD. The current treatment for this spectrum of disorders is outlined by treating the collateral developmental and dietary challenges in addition to anticipating any latent expected complications.
Outcome and follow-up
The child is tolerating food through a gastric feeding tube. His family is very supportive and are regularly following with our clinical paediatric department while he is showing moderate improvement in his general health condition.
Discussion
In 1982, Majewski et al characterised what we now call MOPD types I, II and III.6 21 Over time, it was recognised that MOPDI and MOPDIII were variants of the same disorder.22 Today, many other distinct forms of microcephalic primordial dwarfism are recognised either clinically or through molecular diagnosis. MOPDII is probably the most common type of microcephalic primordial dwarfism. Life expectancy is generally reduced to the 20–30 s range, with an exceptional case who lived to 39 years of age.16
Hall et al outlined the diagnostic criteria of MOPDII based on 58 affected individuals.8 The diagnosis of our patient was confirmed by identifying a homozygous variant in the PCNT gene. In addition to this disease-causing variant, our patient was found to carry a homozygous variant of unknown clinical significance in the GLRB gene. Compound heterozygous or homozygous pathogenic variants in this gene are associated with hyperekplexia type 2, an autosomal-recessive disorder (OMIM# 614619). Hereditary hyperekplexia is characterised by generalised stiffness immediately after birth that normalises during the first years of life, excessive startle reflex to unexpected (particularly auditory) stimuli and a short period of generalised stiffness following the startle response during which voluntary movements are impossible. These features were not present in our patient; therefore, the identified variant in the GLRB gene had no clear clinical significance, at least in the case of our patient.
MOPDII (OMIM# 210720) shares common features with other forms of microcephalic primordial dwarfism such as severe prenatal and postnatal growth retardation with marked microcephaly. However, MOPDII is known for its characteristic skeletal dysplasia, abnormal dentition, insulin resistance and increased risk for cerebrovascular disease.8 11 23 Individuals with MOPDII start to develop scoliosis around the age of 9 years at the time of the pubertal growth spurt.8 In our case, it started before the age of 10 months (figure 5), which is atypical for MOPDII. In addition, although intellectual development is typically normal or mildly affected in MOPDII, our patient had a severe intellectual disability. The reason for these atypical features is unknown. However, it is possible that different variants in the PCNT gene might result in more prominent or earlier-onset features compared with other variants in the same gene (phenotypic heterogeneity). More MOPDII reports are needed to obtain a better understanding of this disorder with the impact of its different PCNT variants and to reach more clear explanations that can explain such findings.
Learning points.
We report a novel homozygous pathogenic variant (c.3760C>T) in the PCNT gene that is associated with microcephalic osteodysplastic primordial dwarfism syndrome II (MOPDII) and, possibly, severe neurological manifestations.
Children with MOPD II might present early with severe neuroradiological manifestations which require early screening and annual follow-up.
With the proper management of general health and feeding issues, these individuals can live longer and have better life quality.
Acknowledgments
The co-authors would like to thank Dr Ahmed Karsou, the consultant of diagnostic radiology at King Abdulaziz Medical City-Jeddah/KSA, for his generous input and support into the radiological imaging figures of our study.
Footnotes
Contributors: All authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing or revision of the manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Parental/guardian consent obtained.
References
- 1. Mahoney CP. Evaluating the child with short stature. Pediatr Clin North Am 1987;34:825–49. 10.1016/S0031-3955(16)36289-7 [DOI] [PubMed] [Google Scholar]
- 2. Seaver LH, Irons M. American College of Medical Genetics (ACMG) Professional Practice and Guidelines Committee. ACMG practice guideline: genetic evaluation of short stature. Genet Med 2009;11:465–70. 10.1097/GIM.0b013e3181a7e8f8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sultan M, Afzal M, Qureshi SM, et al. Etiology of short stature in children. J Coll Physicians Surg Pak 2008;18:493–7. doi:08.2008/JCPSP.493497 [PubMed] [Google Scholar]
- 4. Garganta MD, Bremer AA. Clinical dilemmas in evaluating the short child. Pediatr Ann 2014;43:321–7. 10.3928/00904481-20140723-11 [DOI] [PubMed] [Google Scholar]
- 5. Bouali H, Latrech H. Achondroplasia: Current Options and Future Perspective. Pediatr Endocrinol Rev 2015;12:388–95. [PubMed] [Google Scholar]
- 6. Majewski F, Ranke M, Schinzel A. Studies of microcephalic primordial dwarfism II: the osteodysplastic type II of primordial dwarfism. Am J Med Genet 1982;12:23–35. 10.1002/ajmg.1320120104 [DOI] [PubMed] [Google Scholar]
- 7. Majewski F, Goecke TO. Microcephalic osteodysplastic primordial dwarfism type II: report of three cases and review. Am J Med Genet 1998;80:25–31. [DOI] [PubMed] [Google Scholar]
- 8. Hall JG, Flora C, Scott CI, et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. Am J Med Genet A 2004;130A:55–72. 10.1002/ajmg.a.30203 [DOI] [PubMed] [Google Scholar]
- 9. Unal S, Alanay Y, Cetin M, et al. Striking hematological abnormalities in patients with microcephalic osteodysplastic primordial dwarfism type II (MOPD II): a potential role of pericentrin in hematopoiesis. Pediatr Blood Cancer 2014;61:302–5. 10.1002/pbc.24783 [DOI] [PubMed] [Google Scholar]
- 10. Brancati F, Castori M, Mingarelli R, et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II) complicated by stroke: clinical report and review of cerebral vascular anomalies. Am J Med Genet A 2005;139:212–5. 10.1002/ajmg.a.31009 [DOI] [PubMed] [Google Scholar]
- 11. Huang-Doran I, Bicknell LS, Finucane FM, et al. Genetic defects in human pericentrin are associated with severe insulin resistance and diabetes. Diabetes 2011;60:925–35. 10.2337/db10-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kantaputra P, Tanpaiboon P, Porntaveetus T, et al. The smallest teeth in the world are caused by mutations in the PCNT gene. Am J Med Genet A 2011;155A:1398–403. 10.1002/ajmg.a.33984 [DOI] [PubMed] [Google Scholar]
- 13. Kannu P, Kelly P, Aftimos S. Microcephalic osteodysplastic primordial dwarfism type II: a child with café au lait lesions, cutis marmorata, and moyamoya disease. Am J Med Genet A 2004;128A:98–100. 10.1002/ajmg.a.30086 [DOI] [PubMed] [Google Scholar]
- 14. Doxsey SJ, Stein P, Evans L, et al. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell 1994;76:639–50. 10.1016/0092-8674(94)90504-5 [DOI] [PubMed] [Google Scholar]
- 15. Rauch A, Thiel CT, Schindler D, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 2008;319:816–9. 10.1126/science.1151174 [DOI] [PubMed] [Google Scholar]
- 16. Bober MB, Jackson AP. Microcephalic Osteodysplastic Primordial Dwarfism, Type II: a Clinical Review. Curr Osteoporos Rep 2017;15:61–9. 10.1007/s11914-017-0348-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alfares A, Alfadhel M, Wani T, et al. A multicenter clinical exome study in unselected cohorts from a consanguineous population of Saudi Arabia demonstrated a high diagnostic yield. Mol Genet Metab 2017;121:91–5. 10.1016/j.ymgme.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 18. Shaheen R, Faqeih E, Ansari S, et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res 2014;24:291–9. 10.1101/gr.160572.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab 2014;99:3080–92. 10.1210/jc.2014-1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pekeles H, Accogli A, Boudrahem-Addour N, et al. Diagnostic yield of intellectual disability gene panels. Pediatr Neurol 2019;92:30645–3. 10.1016/j.pediatrneurol.2018.11.005 [DOI] [PubMed] [Google Scholar]
- 21. Majewski F, Stoeckenius M, Kemperdick H. Studies of microcephalic primordial dwarfism III: an intrauterine dwarf with platyspondyly and anomalies of pelvis and clavicles–osteodysplastic primordial dwarfism type III. Am J Med Genet 1982;12:37–42. 10.1002/ajmg.1320120105 [DOI] [PubMed] [Google Scholar]
- 22. Meinecke P, Schaefer E, Wiedemann H-R. Microcephalic osteodysplastic primordial dwarfism: Further evidence for identity of the so-called types I and III. Am J Med Genet 1991;39:232–6. 10.1002/ajmg.1320390228 [DOI] [PubMed] [Google Scholar]
- 23. Bober MB, Khan N, Kaplan J, et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II): expanding the vascular phenotype. Am J Med Genet A 2010;152A:960–5. 10.1002/ajmg.a.33252 [DOI] [PubMed] [Google Scholar]