

Abstract
DNA ligases are a highly conserved group of nucleic acid enzymes that play an essential role in DNA repair, replication, and recombination. This review focuses on functional interaction between DNA polymerases and DNA ligases in the repair of single- and double-strand DNA breaks, and discusses the notion that the substrate channeling during DNA polymerase-mediated nucleotide insertion coupled to DNA ligation could be a mechanism to minimize the release of potentially mutagenic repair intermediates. Evidence suggesting that DNA ligases are essential for cell viability includes the fact that defects or insufficiency in DNA ligase are casually linked to genome instability. In the future, it may be possible to develop small molecule inhibitors of mammalian DNA ligases and/or their functional protein partners that potentiate the effects of chemotherapeutic compounds and improve cancer treatment outcomes.
Keywords: Genome stability, DNA repair, DNA polymerase, DNA ligase, ligation failure, oxidative stress, oxidized nucleotide pool, cancer
Introduction
DNA ligases are ubiquitous cellular proteins that belong to the large family of nucleotidyl transferases (NTases). These enzymes catalyze phosphodiester bond formation to seal the nick consisting of 5′-phosphate (P) and 3′-hydroxyl (OH) termini on ends of the broken DNA strand [1,2]. There are three DNA ligase genes, Lig I, III, and IV that encode mammalian DNA ligases I, III, and IV, respectively [3]. Lig III encodes two polypeptides, DNA ligase IIIα and β [4]. All three mammalian DNA ligases as well as prototypical bacterial DNA ligases contain a highly conserved C-terminal catalytic core consisting of the oligonucleotide binding domain (OBD) and the adenylation (AdD) or nucleotidyl transferase (NTase) domain [5] (Figure 1). The AdD domain contains the adenosine monophosphate (AMP) binding pocket and the active site lysine residue that becomes adenylated, while the OBD includes only catalytic residues [6,7]. Mammalian DNA ligases also harbor the DNA binding domain (DBD). This domain stimulates catalytic activity, most likely through its ability to stabilize DNA conformation in which 5′-P and 3′-OH DNA termini at the nick are well positioned to interact with the active site lysine in the DNA ligase catalytic core [8–10]. In mammalian DNA ligases, the DBD stabilizes the DNA, and interacts with both the minor groove of the nicked DNA as well as the OBD to make a ring-shaped protein structure that encircles the DNA during nick sealing (Figure 2), as shown in the crystal structure of the catalytic core of human DNA ligase I in complex with adenylated DNA [11–13]. This conformational flexibility allows DNA ligase to open and close around the DNA, thereby optimizing the position of the nick in the enzyme active site. In addition to the highly conserved catalytic core, mammalian DNA ligases contain amino (N)- and carboxyl (C)- terminal non-catalytic residues (Figure 1), that promote protein-protein interactions and subcellular localization [1,5,10]
Figure 1. Structural domain organization of nuclear human DNA ligases.
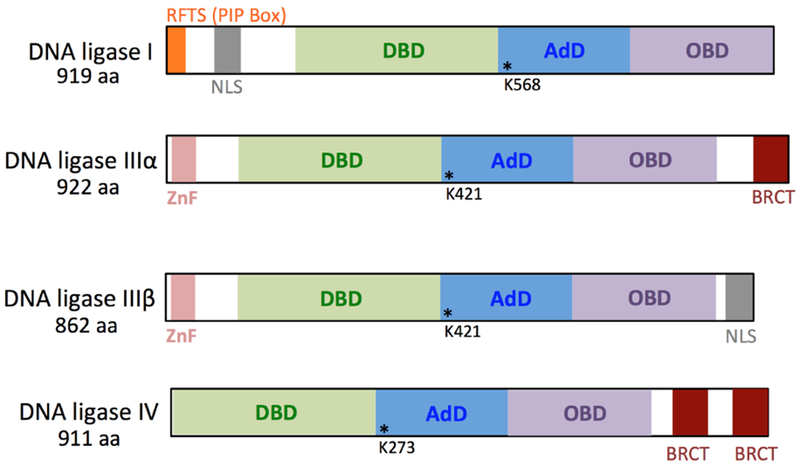
DNA ligases contain the N-terminal DNA binding domain (DBD, green) and the C-terminal catalytic core consisting of the adenylation domain (AdD, blue) and the oligonucleotide binding domain (OBD, purple). DNA ligase III isoforms a and β include an N-terminal zinc finger (ZnF, pink). DNA ligases IIIα and IV have a breast and ovarian cancer susceptibility protein-1 C-terminal domain (BRCT, red). While DNA ligases I and IIIβ have a nuclear localization signal (NLS, gray), the N-terminal region of DNA ligase I also contains a replication factory targeting sequence (RFTS, orange) also known as a PCNA-interacting peptide (PIP) box. The active site lysine residues that become adenylated during the first step of ligation reaction in the AdD domain that contains an AMP binding pocket are shown as asterisks.
Figure 2. Three chemical sequential steps of DNA ligation reaction.
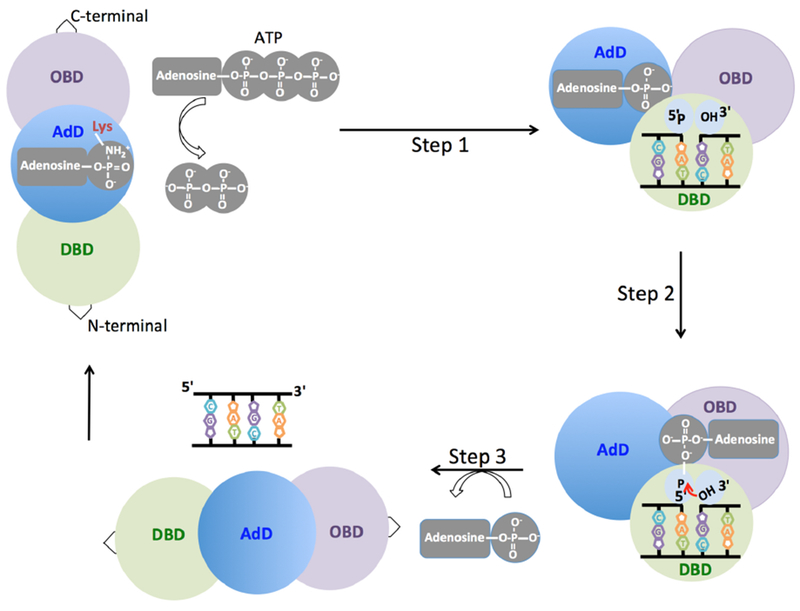
In the first step, the active site lysine residue in the adenylation domain (AdD, blue) of the DNA ligase interacts with adenosine triphosphate (ATP) and the enzyme becomes adenylated. In the second step, the adenylated ligase binds to and encircles the nicked DNA through the DNA binding (DBD, green) and the oligonucleotide binding (OBD, purple) domains. During this step, the adenosine monophosphate (AMP) is then transferred to the 5′-P end of the nicked DNA. In the third step, the non-adenylated DNA ligase utilizes the 3′-hydroxyl (3′-OH) terminus of the nick as a nucleophile to attack the 5′-adenylated DNA. This results in formation of the phosphodiester bond coupled to release of AMP.
DNA ligase structure and reaction mechanism
All three mammalian DNA ligases utilize adenosine triphosphate (ATP) as a high-energy cofactor during catalysis [5,10,14,15]. The universally conserved pathway of DNA ligation reaction involves three chemical sequential steps (Figure 2). In the first step, enzyme adenylation and formation of DNA ligase-adenylate intermediate occur. This step requires that the active site lysine residue in the AdD attacks and forms a covalent bond with the α-phosphate of ATP [16–18]. During the second step, DNA ligase binds to the nicked DNA, and the AMP is transferred from the adenylated DNA ligase to the 5′-P end of the nicked DNA, through the OBD, generating a 5′-adenylated (5′-AMP) DNA intermediate [19]. In the third and final step, DNA ligase catalyzes phosphodiester bond formation between adjacent 3′-OH and 5′-P DNA termini. In this reaction, the 3′-OH terminus of the nick attacks the 5′-P terminus downstream of the nick, and formation of the phosphodiester bond is coupled to release of AMP [14,20]. Pre-steady-state kinetic studies revealed that all three steps of DNA ligation require at least one divalent metal ion. Release of high-energy pyrophosphate coupled to adenylation of the active site lysine during the first step of ligation makes the overall reaction irreversible [19,21]. While the three chemical steps occur at similar rates, and the efficiency of ligation is high at saturating concentrations of ATP and Mg2+[22,23]. Also, DNA ligases show different propensity for the joining of DNA ends in response to the salt concentration in the reaction mixture during the ligation of single- and double-strand breaks [24].
Fidelity of DNA ligases and ligation failure
The joining of the 5′- and 3′-ends in the phosphodiester backbone of nicked DNA is critical for maintaining genome stability [25]. Single- and double-strand breaks in DNA can be generated as a consequence of normal DNA transactions during cellular mechanisms [26]. These strand breaks can also occur as a consequence of DNA lesion removal by DNA repair pathway or be caused directly by a DNA damage-inducing agent, such as oxidative stress-inducing chemicals or ionizing radiation [27,28]. DNA ligases may fail in the presence of damaged or modified DNA ends, resulting in the formation of a 5′-adenylated DNA intermediate [29,30]. This abortive ligation product with the 5′-AMP group is itself very serious damage at the nick and could become a persistent DNA strand break if left unrepaired or left until DNA end processing proteins come to repair them [31,32]. As persistent DNA breaks are expected to be toxic, to block transcription and to be converted into double-strand breaks upon DNA replication, the formation and repair of the ligation failure intermediates are expected to be critical in cellular viability and genome stability.
The multidomain architecture of DNA ligases enables them to sense the nick, which directs the enzyme towards appropriate DNA substrates for efficient ligation [1,5,6,10]. All three mammalian DNA ligases have different ligation fidelity depending on the structural differences in their three-domain core structures. For example, the zinc finger domain of DNA ligase IIIα is not required for catalytic activity but gives the enzyme a broader substrate range in the joining of blunt-ended duplex DNA than that of DNA ligase I, which is also significantly less efficient in the ligation of RNA-containing DNA substrates [24,33,34]. Biochemical studies have shown that heavy metals can also inhibit DNA ligases. For example, all three steps of the DNA ligation reaction are significantly inhibited by zinc (Zn) and cadmium (Cd), while the antitumor agent 9-βĐ-arabinofuranosyl-2-fluoroade (F-ara-A), known as an analog of adenosine and deoxyadenosine, specifically inhibits the formation of the DNA ligase-AMP complex during the first step of the ligation reaction [35,36]. Moreover, it has been reported that human DNA ligase I can also be inhibited by several DNA binding drugs, such as distamycin, ethidium bromide, and actinomycin [37].
DNA ligation at the end of single- and double-strand break repair
In mammalian cells, the base excision repair (BER) pathway is the major DNA repair mechanism in the cellular defense against single-strand breaks generated by reactive oxygen species, alkylating agents, and ionizing radiation [38–41]. There are two distinct BER subpathways: short-patch BER (SP-BER) and long-patch BER (LP-BER) [42,43]. These BER subpathways are differentiated by their repair patch sizes and the enzymes involved (Figure 3). In SP-BER, the repair pathway is initiated by a DNA glycosylase that removes the damaged base (i.e., 8-oxo-7,8-dihydroguanine or 8-oxoG) through hydrolysis of the N-glycosidic bond linking the base to the sugar of the phosphodiester backbone [44]. This hydrolysis results in an abasic or apurinic/apyrimidinic (AP) site in double-stranded DNA. In the widely accepted model of SP-BER, AP endonuclease 1 (APE1) cleaves the phosphodiester backbone at the AP site, leaving 3′-OH and 5′-deoxyribose phosphate (dRP) groups at the termini in a single-nucleotide gap [45]. DNA polymerase (pol) β removes the 5′-dRP group and fills the gap by inserting a single nucleotide [46,47]. However, in the presence of a modified dRP moiety or a reduced AP site, LP-BER becomes the major BER pathway [48]. This subpathway involves strand displacement DNA synthesis by proliferating cell nuclear antigen (PCNA) and pol δ/pol ε, followed by the removal of flap-like structures of 2–10 nucleotides by Flap Endonuclease 1 (FEN1). The studies also suggest that pol β plays roles in the nucleotide insertion and extension steps during LP-BER [49]. Finally, DNA ligases finalize both BER subpathways at the end. DNA ligase IIIα/X-ray repair cross-complementing protein 1 (XRCC1) complex and DNA ligase I join 3′-OH and 5′-P groups to seal the nicked DNA intermediate to complete the SP-BER and LP-BER subpathways, respectively (Figure 3). On the other hand, as shown in cell extracts immunodepleted of either DNA ligase I or IIIα, DNA ligase III can complement DNA ligase I during LP-BER, and DNA ligase I is able to efficiently substitute for DNA ligase IIIα/XRCC1 complex during SP-BER [50].
Figure 3. Substrate channeling of DNA repair intermediates during single-strand break repair.
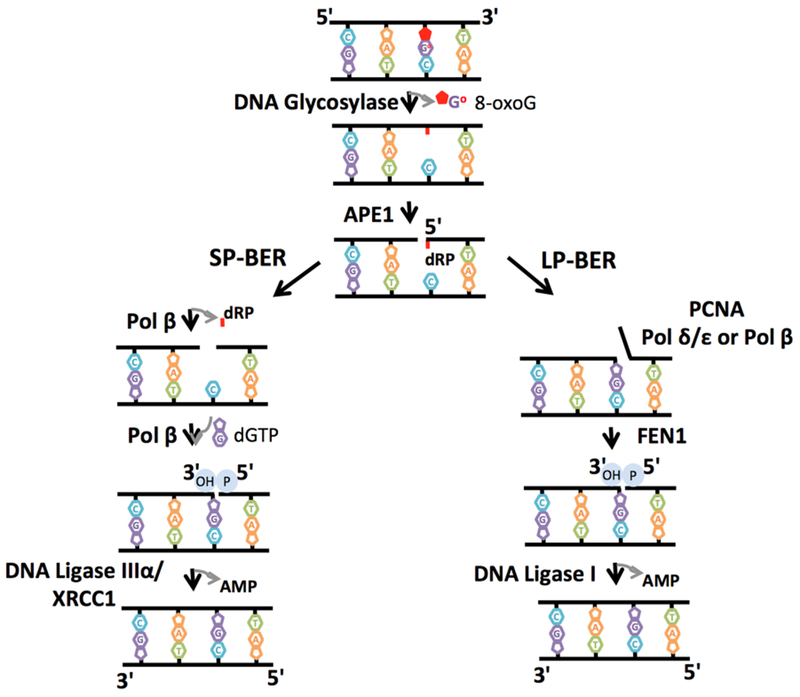
The scheme represents base excision repair (BER) pathway that involves several sequential enzymatic steps and hand off between BER proteins during short-patch BER (SP-BER) and long-patch BER (LP-BER) subpathways. A single DNA base damage (i.e., 8-oxoG) is initially removed by a DNA glycosylase, leaving an abasic site that is then processed by AP endonuclease 1 (APE1), leaving 3′-OH and 5′-deoxyribose phosphate (dRP) groups. In the SP-BER subpathway, DNA polymerase (pol) β removes the 5′-dRP group and fills the gap by inserting a single nucleotide (i.e., dGTP). In the LP-BER subpathway, if the dRP group is modified, proliferating cell nuclear antigen (PCNA)-dependent DNA synthesis of 2–13 nucleotides by pol δ/ε or pol β and their removal by Flap Endonuclease 1 (FEN1) occur. DNA ligase IIIα/XRCC1 complex and DNA ligase I joins 3′-OH and 5′-P DNA termini to complete the SP-BER and LP-BER subpathways, respectively.
Double-strand breaks (DSBs) are the most dangerous type of DNA damage because the integrity of both strands of the DNA duplex is lost [51]. DSBs can be generated either by environmental toxicants, ionizing radiation, and reactive oxygen species (ROS) or during cellular mechanisms such as DNA replication or meiosis [52]. DSBs are repaired through two pathways known as homology-based error-free homologous recombination (HR) and error-prone nonhomologous end-joining (NHEJ), which have different impacts on the fidelity and efficiency of DSB repair [53]. As a predominant DSB repair mechanism, classic or canonical NHEJ (cNHEJ) requires the recruitment of the DSB repair protein complex. This complex of core NHEJ factors includes the Ku 70 and 80 heterodimers, DNA Protein Kinase catalytic subunit (DNAPKcs), X-ray Cross-Complementing 4 (XRCC4), DNA ligase IV, XRCC Like Factor (XLF) and Artemis [54,55]. In contrast to DNA ligases I and DNA ligase IIIα/XRCC1, which play roles in several DNA repair pathways as well as DNA replication, DNA ligase IV functions only in the repair of DSBs during NHEJ, which is mediated by its interaction with XRCC4 [56]. However, it has been shown that alternative end-joining activities including either DNA ligase I or DNA ligase IIIα/XRCC1 can substitute for DNA ligase IV in cells deficient in core cNHEJ factors, and all three DNA ligases may compete for the final ligation step during chromosome translocation [57].
Substrate channeling during single- and double-strand break repair
Based on the product-complex recognition model supported by biochemical and structural studies, the BER pathway proceeds in an orderly fashion through several enzymatic steps and protein-protein interactions (Figure 3). The steps of the repair pathway involve substrate channeling mechanism [58,59]. The importance of this mechanism is the coordination of the substrate-product hand off between BER enzymes that coordinate with one another to receive the DNA substrate and efficiently pass the resulting repair product along to the next enzyme [60,61]. This hand off mechanism prevents the accumulation of toxic DNA strand break intermediates that could trigger cell cycle arrest, necrotic cell death, or apoptosis as well as harmful nuclease activities or recombination events [62]. The reconstitution of human BER with purified repair proteins, from the initial removal of a damaged base by a DNA glycosylase to the final ligation step by a DNA ligase, in a highly regulated manner has been shown in many studies [63–66]. This part of the review focuses on the substrate channeling from pol β to DNA ligase (DNA ligase I or DNA ligase IIIα/XRCC1 complex) during BER pathway (Figure 4). It has been shown through affinity chromatography developed by crosslinking with an antibody against pol β that the BER proteins pol β and DNA ligase I are coimmunoprecipitated from bovine testis nuclear extracts [67]. Both BER proteins are crucial components of a multiprotein complex that repairs single-base DNA lesions in vitro. In another study using purified repair proteins, it was shown that the completely ligated product was observed when pol β and DNA ligase I were preincubated with a singlenucleotide gap including a DNA substrate with a 5′-dRP group. However, the repair intermediate after the pretreatment of the enzymes with trap DNA containing the 5′-P group resulted only in the formation of a ligated trap itself [64]. This finding indicates that the DNA is channeled from pol β to DNA ligase I only when these BER enzymes are prebound to the initial single-nucleotide BER intermediate. Additionally, the channeling of substrates was shown using the pol β affinity-capture fractions in the cell extracts isolated from pol β-null mouse embryonic fibroblasts [60].
Figure 4. Functional interaction between DNA polymerase β and DNA ligases during base excision repair.
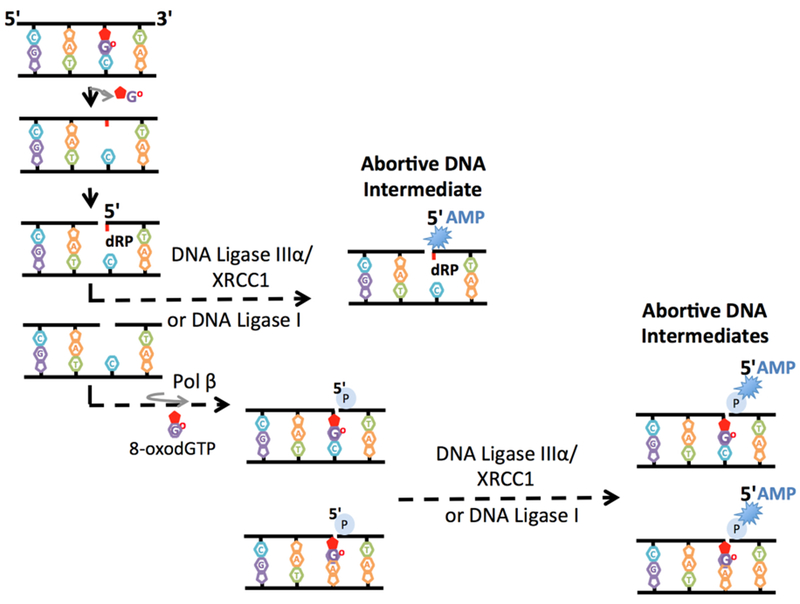
The scheme represents the two possible enzymatic steps in which abortive DNA repair intermediates can be formed as a result of the lack of substrate channeling from DNA polymerase (pol) β to DNA ligase I or DNA ligase IIIα/XRCC1 complex. DNA ligase reaction may abort, and the BER intermediate could become 5′-adenylated, yielding the abortive DNA intermediate with the 5′-AMP-dRP group. Alternatively, pol β oxidized nucleotide (i.e., 8-oxodGTP) insertion opposite the template base A or C may lead to DNA ligase failure and formation of an abortive ligation product with a 3′-inserted oxidized base and a 5′-adenylate group (5′-AMP) on the repair intermediate.
The assembly of multiprotein complexes and the formation of protein-protein interactions on DNA ends also play a crucial role in NHEJ during DSB repair (Figure 5). The DNA ends at most DSBs usually require processing prior to the last DNA ligation step catalyzed by DNA ligase IV/XRCC4 complex [68]. This DNA end processing includes DNA synthesis by DNA polymerase (pol) μ, pol λ, or terminal deoxynucleotidyl transferase (TdT) to fill the gaps generated during DNA end alignment [69,70]. However, the involvement of pol β in the repair of DSBs has also shown [71]. In a recently reported study, the hand off of ribonucleotide-containing repair intermediates from pol μ-mediated nucleotide insertion to subsequent DNA ligation through “sequential coupled strand break repair reactions” during cellular NHEJ has been suggested in an in vivo model system [72].
Figure 5. Substrate channeling of DNA repair intermediates and functional interaction between DNA polymerase μ and DNA ligase IV during double-strand break repair.
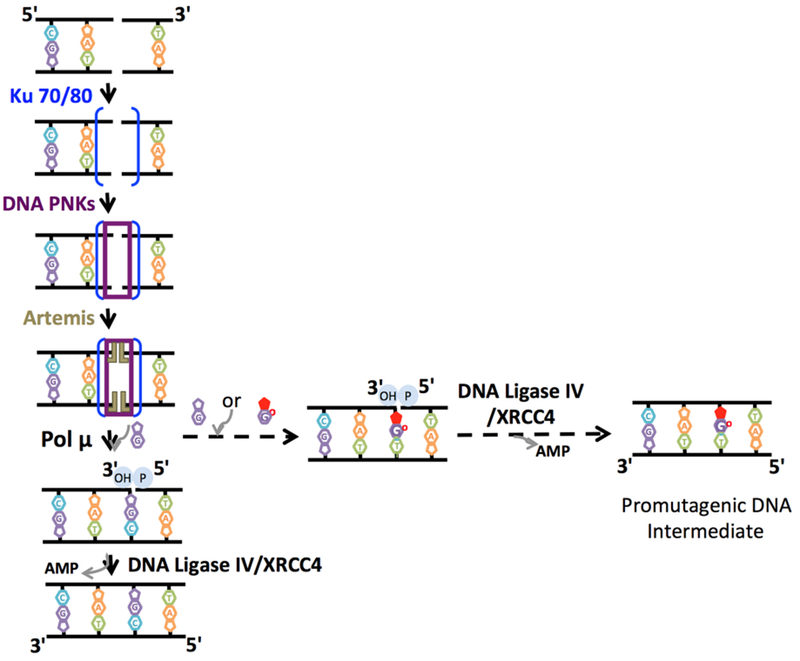
The scheme represents the recruitment of multi-protein complex and hand off between DSB repair proteins during non-homologous end-joining pathway. DNA polymerase μ-mediated dG:T mismatch (dGTP or 8-oxodGTP insertion opposite template base T) insertion may lead to efficient DNA ligation and formation of promutagenic DNA repair intermediate.
Effect of DNA polymerase nucleotide insertion on the fidelity of DNA ligation
The channeling of the pol β product to the next DNA ligation step enables the recognition of the DNA repair intermediate and its hand off from pol β to DNA ligase during the repair pathway [59,60,64,73]. I previously demonstrated that BER DNA ligases, namely, DNA ligase I or the DNA ligase IIIα/XRCC1 complex, cannot ligate an abasic site in the absence of 5′-dRP group removal [74,75]. This inability results in uncoordinated DNA ligase activity that could lead to the “5′-adenylate block” of the dRP-containing BER intermediate (i.e., 5′-adenylated-dRP or 5′-AMP-dRP) (Figure 4). The ligation of this intermediate by BER DNA ligases is highly sensitive to ATP and Mg2+concentrations in the ligation reaction [74]. In another study, using a coupled repair assay that enables the simultaneous observation of DNA polymerase-mediated nucleotide insertion coupled to DNA ligation, I demonstrated that pol β 8-oxodGTP insertion opposite the template base A or C leads to ligase failure and interrupts the BER pathway due to a lack of substrate channeling from pol β to DNA ligase [76,77]. This interruption results in the formation of an abortive ligation product with a 3′-inserted oxidized base and a 5′-adenylate group on the repair intermediate (Figure 4). In this study, I also reported that the extent of ligation failure is significantly sensitive to both the type of oxidized base inserted by pol β and the nature of the template base on the repair intermediate to be transferred to DNA ligase. Moreover, I demonstrated that BER DNA ligases respond to changes in the active site residues that make unique contacts in the pol β precatalytic ternary substrate complex by comparing the base pairing of an incoming nucleotide 8-oxodGTP base with A vs C, which showed different ligation efficiencies. Overall, the findings indicate that the functional interaction between pol β and BER DNA ligases (DNA ligase I or DNA ligase IIIα/XRCC1 complex) could determine the fate of the repair at the last step of the repair pathway. The loss of this interaction could lead to the formation of abortive DNA intermediates that could become persistent DNA breaks, leading to genome instability, deleterious mutagenesis, and cytotoxicity [73].
All three human DNA ligases are very selective for the chemical characteristics of base pairs and show different abilities to join nicks, including 3′-mismatches [12,13]. It has been known that DNA polymerases with no proofreading capacity, such as pol β, can incorporate mismatch nucleotides during DNA repair [78]. Accordingly, the fidelity of human DNA ligases in terms of the ligation efficiency of joining 5′-P and 3′-mismatch-containing ends at the nick has been investigated. For example, studies using purified human DNA ligases showed that DNA ligase III distinguishes correctly paired bases from mismatches at the 3′-terminus of the nick more efficiently than DNA ligase I [79,80]. In another study, it was reported that both human DNA ligases ligate 3′-dA more efficiently than the 3′-dC terminus paired to 8-oxodG on a template position [81]. However, more biochemical studies are required to further understand the channeling of the repair intermediate after pol β-mediated mismatch insertion coupled to DNA ligation during the BER pathway.
Furthermore, I investigated the NHEJ factors, including the DNA ligase IV/XRCC4 complex and pol μ, in a coupled repair assay and demonstrated that pol μ-mediated dG:T mismatch (dGTP or 8-oxodGTP insertion opposite template base T) insertion facilitates the channeling of the DSB repair intermediate to the next ligation step during NHEJ [82] (Figure 5). However, the DNA ligase IV/XRCC4 complex is itself not able to join the 3′-end with the Watson-Crick wobble base mismatch [83]. Similarly, a recently published study has reported DNA ligation facilitated by pol μ ribonucleotide incorporation in mouse embryonic fibroblast cells expressing neither pol μ norTdT [72]. The observed differences between pol β-BER DNA ligases and pol μ-DSB repair ligase could be due to either the unique features of pol μ or DNA ligase IV structure. For example, unlike other X-family DNA polymerases such as pol β and λ, pol μ undergoes no large-scale active site conformational changes upon nucleotide binding where the catalytic intermediates with a transient third metal ion were observed [84,85]. Similarly, the crystal structure of the catalytic core of human DNA ligase IV revealed dynamic and extensive interdomain interactions with the nicked DNA in which the active site undergoes a transition between open and closed conformations for efficient DNA nick sealing [86,87]. For example, it has been shown that DNA ligase IV undergoes conformational remodeling and end-alignment configuration for the ligation of diverse DNA end structures on chromosomal breaks, suggesting DNA ligase IV-mediated sensing of damaged and mismatched ends during cellular NHEJ [70]. On the other hand, additional X-ray crystallography studies are required to capture the DNA ligase/polymerase binary protein complex in action with a DNA repair intermediate containing noncompatible end structures.
Protein-protein interactions of DNA ligases and their impact on the fidelity of DNA ligation
ATP-dependent DNA ligases from bacteria to humans all share a conserved catalytic core structure that is responsible for nick sealing [88]. However, human DNA ligases have unrelated flanking N- and/or C-terminal regions that play roles in their specific protein-protein interactions with their partner proteins and thereby direct them to participate in different cellular functions [1,2,5,6,10].
Human DNA ligase I is required for the sealing of Okazaki fragments during discontinuous DNA replication and finalizes the last step of LP-BER [89]. The protein contains N-terminal region that has a nuclear localization signal and is responsible for interaction with other proteins, such as DNA-sliding clamps, proliferating cell nuclear antigen (PCNA), and pol β [90]. The interaction between PCNA and DNA ligase I occurs between the interdomain connector loop of PCNA and the PIP box of DNA ligase I (Figure 6a). The inactivation of the PCNA-binding site on the ligase completely abolishes the localization of DNA ligase I at the sites of DNA replication [91]. Moreover, PCNA improves the binding of DNA ligase I to nicked single-stranded DNA and stimulates ligation activity in vitro [92]. Biochemical studies have reported that the interaction between PCNA and DNA ligase I plays a key role in the coordination of the DNA synthesis and ligation steps during LP-BER. However, this interaction is not required to repair an abasic site through SP-BER [93,94]. PCNA cannot enhance ligation on a circular DNA substrate without the addition of replication factor C (RFC), a clamp loader responsible for loading PCNA onto DNA in an ATP-dependent reaction, and the RFC itself significantly inhibits the catalytic activity of DNA ligase I [95]. Thermodynamics and domain mapping studies also showed that the N-terminal 8-kDa domain of pol β interacts with the N-terminal noncatalytic part of DNA ligase I (Figure 6a). This interaction is significantly abolished when the lysine residues (K35, K62, and K72) that are responsible for pol β dRP lyase activity (i.e., 5′-dRP group removal) are mutated to alanine residues, and the C-terminal part of the protein with DNA synthesis activity has no effect on this interaction [67,96]. Overall, it seems that the distinct interplay between DNA ligase and its partner protein when bound to a repair intermediate with an incompatible end could affect the activity of DNA ligases. Additional biochemical studies are needed to understand the functional importance of the physical interaction between pol β and DNA ligase I and the effect of this interaction on the substrate channeling of repair intermediates during the LP-BER subpathway.
Figure 6. Protein partners of nuclear human DNA ligases.
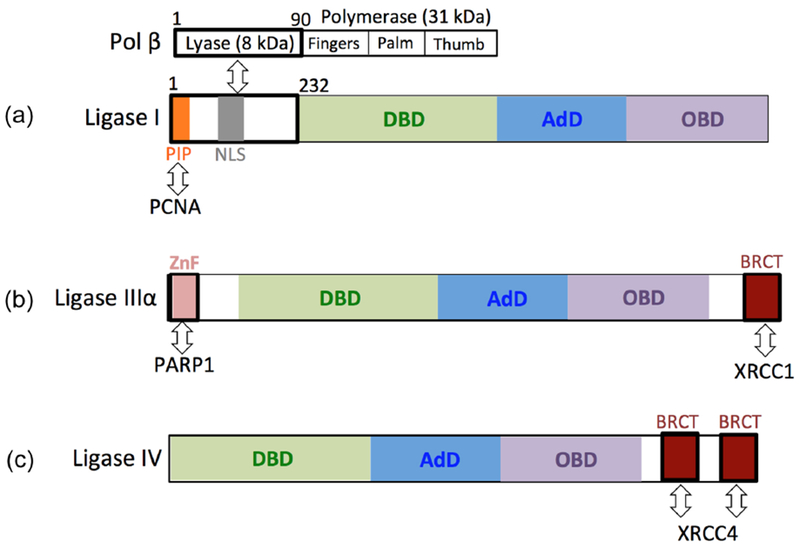
(a) The N-terminal domain of DNA ligase I, including the first 232 amino acid residues of the protein, interacts with the N-terminal part of DNA polymerase β harboring an 8 kDa lyase domain that is responsible for 5′-dRP removal. The PIP box of DNA ligase I mediates its interaction with proliferating cell nuclear antigen (PCNA). (b) The ovarian cancer susceptibility protein 1 (BRCT) and zincbinding (ZnF) domains of DNA ligase IIIα mediates protein-protein interactions with X-ray repair cross-complementing protein 1 (XRCC1) and Poly(ADP-Ribose) Polymerase 1 (PARP1), respectively. (c) DNA ligase IV forms a functional complex with X-ray repair crosscomplementing protein 4 (XRCC4) through its two BRCA1 C-terminal (BRCT) domains.
The DNA ligase that plays a role in the SP-BER subpathway, DNA ligase IIIα, contains a C-terminal region with a breast and ovarian cancer susceptibility protein 1 (BRCT) motif that is involved in the interaction with its nuclear partner XRCC1 [97] (Figure 6b). As a scaffold protein, XRCC1 has no catalytic activity but interacts with protein partners that are involved in all enzymatic steps of SP-BER, such as APE1 and pol β [98]. On the other hand, no stimulatory role of XRCC1 on nick sealing by DNA ligase IIIα has been reported [99,100]. Mammalian DNA ligase IIIα also has an N-terminal zinc-binding domain, termed a zinc finger, that is not required for catalytic activity; however, it serves as a nick sensor and bridges the two ends of DNA strand breaks during NHEJ [101–103]. As an important binding partner of XRCC1 and DNA ligase IIIα, PARP1 (Poly(ADP-Ribose) Polymerase 1) mediates the recruitment of both proteins to DNA damage sites in vivo, relying heavily on NAD-dependent-poly (ADP-ribose) synthesis [104]. The N-terminal region of DNA ligase IIIα adjacent to its zinc finger is responsible for its interaction with PARP1 (Figure 6b). Biochemical in vitro studies showed increased DNA end joining activity of DNA ligase IIIα in the presence of PARP1, and this stimulation is highly dependent on the presence of NAD+ when PARP-1 becomes poly(ADP-ribosyl)ated [104]. Time-lapse single-molecule studies using fluorescently labeled DNA ligases in vivo revealed that DNA ligase IIIα in complex with XRCC1 accumulates at sites of oxidative DNA damage before DNA ligase I is recruited through its interaction with PCNA [105].
DNA ligase IV interacts with its protein partner XRCC4 through the linker sequence between two BRCT domains of the protein (Figure 6c). Many physical and functional interactions have been reported between components of the NHEJ core complex [106,107]. DNA ligase IV plays a central role and is involved in a series of species-specific pairwise protein-protein interactions that coordinate the DNA end processing and ligation reactions that complete the repair of DSBs by NHEJ [53–56]. Some of these interactions also affect the ligation of noncompatible DNA ends by DNA ligase IV. For example, a stimulatory role of XRCC4 on the ligation of double-strand DNA ends with fully incompatible short 3′-overhang configurations that present no potential for base pairing has been reported [108]. Moreover, DNA-PKcs interacts with and promotes intermolecular joining by the DNA ligase IV/XRCC4 complex [86].
Oxidative stress and DNA ligation: New cancer therapeutic targets
DNA ligases are promising candidates for the development of small-molecule inhibitors for anticancer therapeutic purposes because of their crucial roles in multiple DNA repair pathways, replication, and recombination [109–111]. DNA ligase inhibitors have been developed using a computer-aided rational drug design strategy based on the crystal structure of human DNA ligase I [112]. For this approach, a less conserved DBD of the DNA ligase is targeted over the OBD and AdD domains, including a common active site lysine and catalytic residues [113]. Studies have shown that the efficacy or specificity of DNA damage-inducing anticancer drugs in chronic myeloid leukemia, breast cancer and neuroblastoma cells can be increased by using DNA ligase inhibitors [114–119]. For example, the DNA ligase I-specific inhibitor L87-G17 increased cytotoxicity in MCF7 breast cancer cells when combined with DNA-damaging agents such as the DNA alkylating agent methyl methanesulfonate (MMS) and decreased colon cancer cell survival under ionizing radiation with no observed effect on sensitivity in the cell lines from normal cells [120]. Similarly, enhanced cell growth arrest in HeLa cells was recently shown in response-combined treatment with the DNA alkylating agent temozolomide and the DNA ligase I, III and IV inhibitor L189. Moreover, biopsies taken from estrogen receptor- and progesterone receptor-positive and tamoxifen- and aromatase-resistant derivatives of MCF7 breast cancer cells exhibited significantly increased sensitivity to a combination of PARP and DNA ligase III inhibitors [121,122].
Many types of cancer cells have increased levels of ROS [123–125]. These highly reactive hydroxyl radicals react with the components of DNA as well as with the nucleotide pool, leading to oxidative DNA damage (i.e., 8-oxoG) and an oxidized nucleotide pool (i.e., 8-oxodGTP) [126–129]. In fact, the nucleotide pool is more susceptible to oxidation than the DNA molecule itself because of the abundance of water, the highly reactive intracellular environment, and the lack of protective mechanisms, such as the histone-like proteins bound to DNA molecules [130–133]. MutT Homolog 1 (MTH1) is the nucleotide pool sanitization enzyme that detoxifies the oxidized nucleotide pool and prevents the incorporation of oxidized nucleotides into genomic DNA by repair DNA polymerases such as pol β, suggesting that this protein could be targeted as an anticancer therapeutic approach [134,135]. It seems that small-molecule inhibitors of MTH1 have strong effects on the cell death of various cancer types [136,137]. On the other hand, these reports were followed by other studies reporting that cellularly active MTH1 inhibitors have no anticancer efficacy either in monotherapy or in combination therapies [138–140]. All reports contribute to the understanding of the complex MTH1 biology, its role in carcinogenesis, and the utility of its inhibition to cure cancer patients.
I previously demonstrated that BER-proficient mouse embryonic fibroblast (MEF) cells overexpressing the pol β gene show more cytotoxicity and DNA DSBs in response to oxidative stress than BER-deficient MEF cells with pol β gene deletion [76]. This oxidative stress-induced cell killing was increased in MEF cells co-treated with MTH1 inhibitor or following mth1 gene deletion, suggesting the involvement of the oxidized nucleotide pool and their pol β-mediated insertion during the BER pathway. However, further investigation is required to elucidate the biological consequences of DNA ligase deficiency or ligation inhibition in MEF cells under oxidative stress conditions in vivo. Recently, reduced DNA ligation by DNA ligase III/XRCC1 complex and a significant defect in the repair of oxidative DNA damage through SP-BER in vivo were reported in motor neurons derived from ALS patient-derived fibroblast cells [141].
Conclusions and future prospects
DNA ligation is the last step of almost all DNA repair pathways, and DNA ligases are known as biomarkers of abnormal DNA repair. DNA ligation failure products could become persistent DNA strand breaks that threaten genome stability. DNA polymerase-mediated ligation failure could be a source of mutagenic and toxic repair intermediates. The functional protein partners of DNA ligases can be considered chemotherapeutic targets with the goal of identifying more selective inhibitors for DNA ligases. In this respect, the protein-protein interactions between DNA ligases and DNA polymerases, particularly through the repair of oxidative DNA damage during the BER pathway, could be considered a potent new class of chemotherapeutic targets to improve the activity and/or specificity of the current generation of small-molecule ligase inhibitors. Therefore, biochemical and structural studies are needed to elucidate the structure-activity relationships and identify important residue(s) that play critical roles in the DNA polymerase-DNA ligase protein-protein interaction. The expression levels of DNA ligases in cancer cells represent an area of investigation that could identify cancers that would be ideal targets for ligase inhibition. The efficacy of anticancer drugs is highly influenced by cellular DNA repair capacity. The toxicity of DNA-damaging drugs can be reduced by the activities of several overlapping DNA repair pathways that remove lesions before the onset of DNA replication. Therefore, DNA repair pathways modulate the efficacy of cancer therapy. Cancer cells under increased oxidative stress are likely to be more vulnerable to damage by further ROS insults. As the BER pathway is the major player in the repair of oxidative DNA damage, BER DNA ligases (DNA ligase I and DNA ligase IIIα) and their interaction partner pol β and the development of new therapeutic agents that target their protein-protein interaction hold promise for potentiating the effects of cancer treatments.
Revised Research Highlights:
The substrate channeling from DNA polymerase β to DNA ligase during base excision repair (BER) pathway could prevent the accumulation of toxic repair intermediates.
DNA ligases finalize the DNA repair, replication, and recombination and play critical role for maintaining genome stability.
DNA ligase catalyzes a phosphodiester bond formation between adjacent 3′-OH and 5′-P termini of the nick DNA.
The insertion of oxidized or mismatch nucleotide by a DNA polymerase could confound the next DNA ligation step during single-stranded and double-stranded DNA repair.
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of Environmental Health Sciences Grant 4R00ES026191-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tomkinson AE, A.E., S. Vijayakumar, J.M. Pascal, T. Ellenberger, DNA ligases: structure, reaction mechanism, and function, Chem. Rev. 106 (2) (2006) 687–699. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T, Barnes DE, Mammalian DNA ligases, Annu. Rev. Biochem. 61 (1992) 251–281. [DOI] [PubMed] [Google Scholar]
- 3.Soderhall S, Lindahl T, Mammalian DNA ligases. Serological evidence for two separate enzymes, J. Biol. Chem 250 (21) (1975) 8438–8444. [PubMed] [Google Scholar]
- 4.Tomkinson AE, Sallmyr A, Structure and function of the DNA ligases encoded by the mamamlian LIG3 gene, Gene 531 (2) (2013) 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doherty AJ, Suh SW, Structural and mechanistic conservation in DNA ligases, Nucleic Acids Res. 28 (21) (2000) 4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellenberger T, Tomkinson AE, Eukaryotic DNA Ligases: Structural and Functional Insights, Annu. Rev. Biochem. 77 (2008) 313–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shuman S, DNA ligases: progress and prospects, J. Biol. Chem 284 (26) (2009) 17365–17369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bray CM, Sunderland PA, Waterworth WM, West CE, DNA ligase-a means to an end joining, SEB Exp. Biol. Ser. 59 (2008) 203–217. [PubMed] [Google Scholar]
- 9.Timson DJ, Singleton MR, Wigley DB, DNA ligases in the repair and replication of DNA, Mutat. Res 460 (3–4) (2000) 301–318. [DOI] [PubMed] [Google Scholar]
- 10.Lehman IR, DNA ligase: structure, mechanism, and function, Science 186 (4166) (1974) 790–797. [DOI] [PubMed] [Google Scholar]
- 11.Pascal JM, O’Brien PJ. A.E. Tomkinson, T. Ellenberger, Human DNA ligase I completely encircles and partially unwinds nicked DNA, Nature 432 (7016) (2004) 473–478. [DOI] [PubMed] [Google Scholar]
- 12.Johnson A, O’Donnell M, DNA Ligase: Getting a grip to seal the deal, Curr. Biol. 15 (2005) R90–2. [DOI] [PubMed] [Google Scholar]
- 13.Cherepanov AV, Vries S, Dynamic mechanism of nick recognition by DNA ligase, Eur. J. Biochem. 269 (2002) 5993–5999. [DOI] [PubMed] [Google Scholar]
- 14.Dickson KS, Burns CM, Richardson JP, Determination of the free-energy change for repair of a DNA phosphodiester bond, J. Biol. Chem 275 (21) (2000) 15828–15831. [DOI] [PubMed] [Google Scholar]
- 15.Doherty AJ, Wigley DB, Functional domains of an ATP-dependent DNA ligase, J. Mol. Biol. 285 (1999) 63–71. [DOI] [PubMed] [Google Scholar]
- 16.Weiss B, Thompson A, Richardson CC, Ezymatic breakage and joining of deoxyribonucleic acid. VII. Properties of the enzyme-adenylate intermediate in the polynucleotide ligase reaction, J. Biol. Chem 243 (17) (1968) 4556–4563. [PubMed] [Google Scholar]
- 17.Singleton MR, Hakansson K, Timson DJ, Wigley DB, Structure of the adenylation domain of an NAD+-dependent DNA ligase, Structure 7 (1) (1999) 35–42. [DOI] [PubMed] [Google Scholar]
- 18.Tomkinson AE, Totty NF, Ginsburg M, Lindahl T, Location of the active-site for enzyme-adenylate formation in DNA ligases, Proc. Natl. Acad. Sci. USA 88 (2) (1991) 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang SW, Chan JY, Analysis of the formation of AMP-DNA intermediate and the successive reaction by human DNA ligases I and II, J. Biol. Chem 267 (12) (1992) 8117–8122. [PubMed] [Google Scholar]
- 20.Tomkinson AE, Levin DS, Mammalian DNA ligases, BioEssays 19 (1997) 893–901. [DOI] [PubMed] [Google Scholar]
- 21.Taylor MR, Conrad JA, Wahl D, O’Brien PJ, Kinetic mechanism of human DNA ligase I reveals magnesium-dependent changes in the rate-limiting step that compromise ligation efficiency, J. Biol. Chem 286 (26) (2011) 23054–23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohman GJS, Chen L, Evans TC, Kinetic characterization of single strand break ligation in duplex DNA by T4 DNA ligase, J. Biol. Chem 286 (51) (2011) 44187–44196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cherepanov AV, Vries S, Kinetics and thermodynamics of nick sealing by T4 DNA ligase, Eur. J. Biochem. 270 (2003) 4315–4325. [DOI] [PubMed] [Google Scholar]
- 24.McNally JR, O’Brien PJ, Kinetic analyses of single-strand break repair by human DNA ligase III isoforms reveal biochemical differences from DNA Ligase I, J. Biol. Chem 292 (38) (2017) 15870–15879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bentley DJ, Harrison C, Ketchen AM, Redhead NJ, Samuel K, Waterfall M, Ansell JD, Melton DW, DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability, J. Cell. Sci 115 (7) (2002) 1551–1561. [DOI] [PubMed] [Google Scholar]
- 26.Olive PL, The role of DNA single- and double-strand breaks in cell killing by ionizing radiation, Radiat. Res. 150 (1998) S42–51. [PubMed] [Google Scholar]
- 27.Khanna KK, Jackson SP, DNA double-strand breaks: signaling, repair and the cancer connection, Nat. Genet 27 (3) (2001) 247–254. [DOI] [PubMed] [Google Scholar]
- 28.Caldecott KW, Single-strand break repair and genetic disease, Nat. Rev. Genetics 9 (8) (2008) 619–631. [DOI] [PubMed] [Google Scholar]
- 29.Rass U, Ahel I, West SC, Molecular mechanism of DNA deadenylation by the neurological disease protein aprataxin, J. Biol. Chem. 283(49) (2008) 33994–34001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds JJ, El-Khamisy SF, Katyal S, Clements P, McKinnon PJ, Celdecott KW, Defective DNA ligation during short-patch single-strand break repair in ataxia oculomotor apraxia-1, Mol. Cell Biol. 29 (5) (2009) 1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitaker AM, Freudenthal BD, APE1: A skilled nucleic acid surgeon, DNA Repair, 71 (2018) 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andres SN, Schellenberg MJ, Wallace BD, Tubmale P, Williams RS, Recognition and repair of chemically heterogeneous structures at DNA ends, Environ. Mol. Mutagen. 56(1) (2015) 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor RM, Whitehouse J, Cappelli E, Frosina G, Caldecott KW, Role of the DNA ligase III zinc finger in polynucleotide binding and ligation, Nucleic Acids Res. 26 (21) (1998) 4804–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor RM, Whitehouse CJ, Caldecott KW, The DNA ligase III zinc finger stimulates binding to DNA secondary structure and promotes end joining, Nucleic Acids Res. 28 (18)(2000)3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang SW, Huang P, Plunkett W, Becker FF, Chan JY, Dual mode of inhibition of purified DNA ligase I from human cells by 9-b-D-Arabinofuranosyl-2-fluoroadenine triphosphate, J. Biol. Chem 267 (4) (1992) 2345–2349. [PubMed] [Google Scholar]
- 36.Yang SW, Becker FF, Chan JY, Inhibition of human DNA ligase I activity by zinc and cadmium and the fidelity of ligation, Environ. Mol. Mutagen 28 (1) (1996) 19–25. [DOI] [PubMed] [Google Scholar]
- 37.Shuman S, Vaccinia virus DNA ligase: specificity, fidelity, and inhibition, Biochemistry.34 (1995) 16138–16147. [DOI] [PubMed] [Google Scholar]
- 38.Lindahl T, Ljungquist S, Apurinic and apyrimidinic sites in DNA, Basic Life Sci. 5a (1975) 31–38. [DOI] [PubMed] [Google Scholar]
- 39.Dianov GL, Sleeth KM, Dianova II, Allinson SL Repair of abasic sites in DNA, Mutat. Res. 531(1–2) (2003) 157–163. [DOI] [PubMed] [Google Scholar]
- 40.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G, Base excision repair of DNA in mammalian cells, FEBS Lett. 476 (1–2) (2000) 73–77. [DOI] [PubMed] [Google Scholar]
- 41.Lindahl T, Keynote: past, present, and future aspects of base excision repair, Prog. Nucleic Acid Res. Mol. Biol. 68 (2001) xvii–xxx. [DOI] [PubMed] [Google Scholar]
- 42.Parikh SS, Mol CD, Tainer JA, Base excision repair enzyme family portrait: integrating the structure and chemistry of an entire DNA repair pathway, Structure. 5 (12)(1997)1543–1550. [DOI] [PubMed] [Google Scholar]
- 43.Klungland A, Lindahl T, Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1), EMBO J. 16 (11) (1997) 3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E, Type of DNA glycosylase determines the base excision repair pathway in mammalian cells, J. Biol. Chem 274 (21) (1999) 15230–15236. [DOI] [PubMed] [Google Scholar]
- 45.Li M, Wilson DM, Human apurinic/apyrimidinic endonuclease 1, Antioxid. Redox Signal. 20 (4) (2014) 678–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prasad R, Beard WA, Strauss PR, Wilson SH, Human DNA polymerase beta deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism, J. Biol. Chem 273 (24) (1998) 15263–15270. [DOI] [PubMed] [Google Scholar]
- 47.Beard WA, Prasad R, Wilson SH, Activities and mechanism of DNA polymerase beta, Methods Enzymol. 408 (2006) 91–107. [DOI] [PubMed] [Google Scholar]
- 48.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH, Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps, J. Biol. Chem 273 (33) (1998) 21203–21209. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH, DNA polymerase beta and Flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair, J. Biol. Chem 280 (5) (2005) 3665–3674. [DOI] [PubMed] [Google Scholar]
- 50.Sleeth KM, Robson RL, Dianov GL, Exchangeability of mammalian DNA ligases between base excision repair pathways, Biochemistry. 43 (40) (2004) 12924–12930. [DOI] [PubMed] [Google Scholar]
- 51.Ma Y, Lu H, Schwarz K, Lieber MR, Repair of double-strand DNA breaks by the human nonhomologous DNA end joining pathway: the iterative processing model, Cell Cycle. 4 (9) (2005) 1193–1200. [DOI] [PubMed] [Google Scholar]
- 52.Mehta A, Haber JE, Sources of DNA double-strand breaks and models of recombinational DNA repair, Cold Spring Harb. Perspect Biol. 6 (2014) a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deriano L, Roth DB, Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage, Annu. Rev. Genet 47 (2013) 433–455. [DOI] [PubMed] [Google Scholar]
- 54.Kuhfittig-Kulie S, Feldmann E, Odersky A, Kuliczkowska A, Goedecke W, Eggert A, Pfeiffer P, The mutagenic potential of non-homologous end joining in the absence of the NHEJ core factors Ku70/80, DNA-PKcs and XRCC4-LigIV, Mutagenesis. 22 (3) (2007) 217–233. [DOI] [PubMed] [Google Scholar]
- 55.Mahaney BL, Hammel M, Meek K, Tainer JA, Lees-Miler SP, XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair, Biochem. Cell Biol. 91 (1) (2013) 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sallmyr A, Tomkinson AE, Repair of DNA double-strand breaks by mammalian alternative end-joining pathways, J. Biol. Chem 293 (27) (2018) 10536–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu G, Duan J, Shu S, Wang X, Gao L, Guo J, Zhang Y, Ligase I and ligase III mediate the DNA double-strand break ligation in alternative end-joining, Proc. Natl. Acad Sci. USA 113 (5) (2016) 1256–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mol CD, Izumi T, Mitra S, Tainer JA, DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination, Nature. 403 (6768) (2000) 451–456. [DOI] [PubMed] [Google Scholar]
- 59.Prasad R, Shock DD, Beard WA, Wilson SH, Substrate channeling in mammalian base excision repair pathways: passing the baton, J. Biol. Chem 285 (52) (2010) 40479–40488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prasad R, Williams JG, Hou EW, Wilson SH, Pol beta associated complex and base excision repair factors in mouse fibroblasts, Nucleic Acids Res. 40 (22) (2012) 11571–11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moor NA, Vasileva IA, Anarbaev RO, Antson AA, Lavrik OI Quantitative characterization of protein protein complexes involved in base excision DNA repair, Nucleic Acids Res. 43 (12) (2015) 6009–6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson SH, Kunkel TA, Passing the baton in base excision repair, Nat. Struct. Biol. 7 (2000) 176–178. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Prasad R, Beard WA, Kedar PS, Hou EW, Wilson SH, Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta, J. Biol. Chem 282 (18) (2007) 13532–13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prasad R, Beard WA, Batra VK, Liu Y, Shock DD, Wilson SH, A review of recent experiments on step-to-step “hand-off” of the DNA intermediates in mammalian base excision repair pathways, Mol. Biol 45 (4) (2011) 586–600. [PMC free article] [PubMed] [Google Scholar]
- 65.Schermerhorn KM, Delaney SA, Chemical and kinetic perspective on base excision repair of DNA, Acc. Chem. Res. 47 (2014) 1238–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dianov GL, Lindahl T, Reconstitution of the DNA base excision repair pathway, Curr. Biol. 4 (1994) 1069–1076. [DOI] [PubMed] [Google Scholar]
- 67.Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkinson AE, Wilson SH, Specific interaction of DNA polymerase β and DNA ligase I on a multiprotein base excision repair complex from bovive testis, J. Biol. Chem 271 (27) (1996) 16000–16007. [DOI] [PubMed] [Google Scholar]
- 68.Waters CA, Strande NT, Pryor JM, Strom CN, Mieczkowski P, Burkhalter MD, et al. , The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining, Nat. Commun. 5 (2014) 4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA, Nonhomologous end joining: a good solution for bad ends, DNA Repair (Amst). 17 (2014) 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conlin MP, Reid DA, Small GW, Chang HH, Watanabe G, et al. , DNA Ligase IV guides end-processing choice during nonhomologous end joining, Cell Rep. 20 (12) (2017) 2810–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ray S, Breuer G, DeVeaux M, Zelterman D, Bindra R, Sweasy JB, DNA polymerase beta participates in DNA end-joining, Nuc. Acids Res. 46 (1) (2018) 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pryor JM, Conlin MP, Carvajal J, Luedeman ME, Luthman AJ, Small GW, Ramsden DA, Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining, Science. 361 (6407) (2018) 1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Çağlayan M, Wilson SH, Oxidant and environmental toxicant-induced effects compromise DNA ligation during base excision DNA repair, DNA Repair. 35 (2015) 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Çağlayan M, Batra VK, Sassa A, Prasad R, Wilson SH, Role of polymerase β in complementing aprataxin deficiency during abasic-site base excision repair, Nat. Struct. Mol. Biol 21 (5) (2014) 497–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Çağlayan M, Horton JK, Prasad R, Wilson SH, Complementation of aprataxin deficiency by base excision repair enzymes, Nucleic Acids Res. 43 (4) (2015) 2271–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Çağlayan M, Horton JK, Dai DP, Stefanick DF, Wilson SH, Oxidized nucleotide insertion by pol β confounds ligation during base excision repair, Nat. Commun 8 (2017)14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Çağlayan M, Wilson SH, Role of DNA polymerase β oxidized nucleotide insertion in DNA ligation failure, J. Radiat. Res 58 (5) (2017) 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beard WA, Shock DD, Vande Berg BJ, Wilson SH, Efficiency of correct nucleotide insertion governs DNA polymerase fidelity, J. Biol. Chem 277 (49) (2002) 47393–47398. [DOI] [PubMed] [Google Scholar]
- 79.Husain I, Tomkinson AE, Burkhart WA, Moyer MB, Ramos W, Mackey ZB, Besterman JM, Chen J, Purification and characterization of DNA ligase III from bovine testes. Homology with DNA ligase II and vaccinia DNA ligase, J. Biol. Chem 270 (16) (1995) 9683–9690. [DOI] [PubMed] [Google Scholar]
- 80.Bhagwat AS, Sanderson RJ, Lindahl T, Delayed DNA joining at 3′ mismatches by human DNA ligases, Nuc. Acids Res. 27 (20) (1999) 4028–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hashimoto K, Tominaga Y, Nakabeppu Y, Moriya M, Futile short-patch DNA base excision repair of adenine:8-oxoguanine mispair, Nuc. Acids Res. 32 (19) (2004) 5928–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.M Çağlayan, S.H Wilson, Pol μ dGTP mismatch insertion opposite T coupled with ligation reveals a promutagenic DNA intermediate during double strand break repair, Nat. Comm 9 (1) (2018) 4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.I.J. Kimsey, E.S. Szymanski, W.J Zahurancik, A Shakya, Y Xue, et al. , Dynamic basis for dG•dT misincorporation via tautomerization and ionization, Nature. 554 (7691) (2018) 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moon AF, Pryor JM, Ramsden DA, Kunkel TA, Bebenek K, Pedersen LC, Sustained active site rigidity during synthesis by human DNA polymerase μ, Nat. Struc. Mol. Biol 21 (3) (2014) 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jaemsen JA, Beard WA, Pedersen LC, Shock DD, et al. , Time-lapse crystallography snapshots of a double-strand break repair polymerase in action, Nat. Comm 8 (1) (2018) 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen L, Trijillo K, Tomkinson AE, Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase, J. Biol. Chem 275 (34) (2000) 26196–26205. [DOI] [PubMed] [Google Scholar]
- 87.Kaminski AM, Tumbale PP, Schellenberg MJ, Williams RS, et al. , Structures of DNA-bound human ligase IV catalytic core reveal insights into substrate binding and catalysis, Nat. Comm 9 (1) (2018) 2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pergolizzi G, Wagner GK, Bowater RP, Biochemical and structural characterization of DNA ligases from bacteria and archaea, Biosci. Rep 36 (5) (2016) e00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Howes TRL, Tomkinson AE, DNA ligase I, the replicative DNA ligase, Subcell Biochem. 62 (2012) 4572–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petrini JH, Xiao Y, Weaver DT, DNA ligase I mediates essential functions in mammalian cells, Mol. Cell Biol 15 (1995) 4303–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levin DS, McKenna AE, Motycka TA, Matsumoto Y, Tomkinson AE, Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair, Curr. Biol 10 (15) (2000) 919–922. [DOI] [PubMed] [Google Scholar]
- 92.Tom S, Henricksen LA, Park MS, Bambara RA, DNA ligase I and proliferating cell nuclear antigen form a functional complex, J Biol. Chem 276 (27) (2001) 24817–24825. [DOI] [PubMed] [Google Scholar]
- 93.Levin DS, Bai W, Yao N, O’Donnel M, Tomkinson AE, An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining, Proc. Natl. Acad. Sci. U S A 94 (24) (1997) 12863–12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Montecucco A, Rossi R, Levin DS, Gary R, PArk MS, et al. , DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: identification of a common targeting mechanism for the assembly of replication factories, EMBO J. 17 (13) (1998) 3786–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Levin DS, Vijayakumar S, Liu X, Bermudez VP, Hurwitz J, Tomkinson AE, A conserved interaction between the replicative clamp loader and DNA ligase in eukaryotes. Implications for okazaki fragment joining, J. Biol. Chem 279 (53) (2004) 55196–55201. [DOI] [PubMed] [Google Scholar]
- 96.Dimitriadis EK, Prasad R, Vaske MK, Chen L, Tomkinson AE, Lewis MS, Wilson SH, Thermodynamics of human DNA ligase I trimerization and association with DNA polymerase β, J. Biol. Chem 273 (32) (1998) 20540–20550. [DOI] [PubMed] [Google Scholar]
- 97.Caldecott KW, McKeown CK, Tucker JD, Ljungguist S, Thomspson LH, An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III, Mol. Cell Biol. 14 (1) (1994) 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.KW. Caldecott, XRCC1 and DNA strand break repair, DNA Repair (Amst). 2 (2003) 955–969. [DOI] [PubMed] [Google Scholar]
- 99.Caldecott KW, Tucker JD, Stanker LH, Thompson LH, Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells, Nucleic Acids Res. 23 (23) (1995) 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K, Frosina G, Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair, J. Biol. Chem. 272 (38) (1997) 23970–23975. [DOI] [PubMed] [Google Scholar]
- 101.Caldecott KW, Aoufouchi S, Johnson P, Shall S, XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nicksensor’ in vitro, Nucleic Acids Res. 24 (2) (1996) 4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kukshal V, Kim IK, Hura GL, Tomkinson AE, Tainer JA, Ellenberger T, Human DNA ligase III bridges two DNA ends to promote specific intermolecular DNA end joining, Nucleic Acids Res. 43 (14) (2015) 7021–7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cotner-Gohara E, Kim IK, Hamel M, Tainer JA, Tomkinson AE, Ellenberger T, Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states, Biochemistry. 49 (29) (2010) 6165–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leppard JB, Dong Z, Mackey ZB, Tomkinson AE, Physical and functional interaction between DNA ligase IIIα and Poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair, Mol. Cell Biol. 23 (16) (2003) 5919–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mortusewicz O, Rothbauer U, Cardoso MC, Leonhardt H, Differential recruitment of DNA ligase I and III to DNA repair sites, Nuc. Acids Res. 34 (12) (2006) 3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, Lieber MR, Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells, Nature. 388 (6641) (1997) 492–495. [DOI] [PubMed] [Google Scholar]
- 107.Modesti M, Hesse JE, Gellert M, DNA binding of XRCC4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity, EMBO J. 18 (7)(2008) 2008–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gu J, Lu H, Tippin B, Shimazaki N, Goodman MF, Lieber MR, XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J. 26 (4) (2017) 1010–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kelley MR, Logsdon D, Fishel ML, Targeting DNA repair pathways for cancer treatment: what’s new?, Future Oncol. 10 (2014) 1215–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Singh DK, Krishna S, Chandra S, Shameem M, Deshmukh AL, Banerjee D, Human DNA Ligases: A comprehensive new look for cancer therapy, Med. Res. Rev 34 (3) (2014) 567–595. [DOI] [PubMed] [Google Scholar]
- 111.Tomkinson AE, Howes TR, Wiest NE, DNA ligases as therapeutic targets, Transl. Cancer Res. 2(3) (2013). [PMC free article] [PubMed] [Google Scholar]
- 112.Zhong S, Chen X, Zhu X, Dziegiekewska B, Bachman KE, Ellenberger, et al. , Identification and validation of human DNA ligase inhibitors using computer-aided drug design, J. Med. Chem 51 (15) (2008) 4553–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen X, Zhong S, Zhu X, Dziegiekewska B, Ellenberger T, et al. , Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair, Cancer Res. 68 (9) (2008) 3169–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, et al. , An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression, Cell. 151 (7) (2012) 1474–1487. [DOI] [PubMed] [Google Scholar]
- 115.Sun D, Urrabaz R, Nguyen M, Marty J, Stringer S, Cruz E, Medina L, Weitman S, Elevated expression of DNA ligase I in human cancers, Clin. Cancer Res. 7 (12) (2001)4143–4148. [PubMed] [Google Scholar]
- 116.Sallmyr A, Tomkinson AE, Rassool FV, Up-regulation of WRN and DNA ligase lllalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks, Blood. 112 (4) (2008) 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tobin LA, Robert C, Rapoport AP, Gojo I, Baer MR, Tomkinson AE, Rassool FV, Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias, Oncogene. 32 (14) (2013) 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fan J, Li L, Small D, Rassool F, Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy, Blood. 116 (24) (2010) 5298–52305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tobin LA, Robert C, Nagaria P, Chumsri S, Twaddell W, et al. , Targeting abnormal DNA repair in therapy-resistant breast cancers, Mol. Cancer Res. 10 (1) (2012) 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Howes TRL, Sallmyr A, Brooks R, Greco GE, Jones DE, Matsumoto Y, Tomkinson AE, Structure-activity relationships among DNA ligase inhibitors: Characterization of a selective uncompetitive DNA ligase I inhibitor, DNA Repair. 60 (2017)29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Toma M, Eradication of LIG4-deficient glioblastoma cells by the combination of PARP inhibitor and alkylating agent, Oncotarget. 9 (2018) 36867–36877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jahagirdar D, Combinatorial use of DNA ligase inhibitor L189 and Temozolomide potentiates cell growth arrest in HeLa, Current Cancer Therapy Reviews. 14 (2018) 1–11. [Google Scholar]
- 123.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB, Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 49 (11) (2010) 21603–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y, Du Y, Le W, Wang K, Kieffer N, Zhang J, Redox control of the survival of healthy and diseased cells, Antioxid. Redox Signal. 15 (11) (2011) 22867–2908. [DOI] [PubMed] [Google Scholar]
- 125.Rudd SG, Valerie NCK, Helleday T, Pathways controlling dNTP pools to maintain genome stability, DNA Repair. 44 (2016) 2193–204 [DOI] [PubMed] [Google Scholar]
- 126.Evans MD, Dizdaroglu M, Cooke MS, Oxidative DNA damage and disease: induction, repair, and significance, Mutat. Res 567 (1) (2004) 1–61. [DOI] [PubMed] [Google Scholar]
- 127.Nakabeppu Y, Sakumi K, Sakamoto K, Tsuchimoto D, Tsuzuki T, Nakatsu Y, Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids, Biol. Chem 387 (4) (2006) 373–379. [DOI] [PubMed] [Google Scholar]
- 128.Sekiguchi M, T, Tsuzuki, Oxidative nucleotide damage: consequences and prevention, Oncogene. 31 (2) (2002) 8895–8904. [DOI] [PubMed] [Google Scholar]
- 129.Kamiya H, Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: approaches using synthetic oligonucleotides and nucleotides, Nucleic Acids Res. 31 (2) (2003) 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ventura I, Russo MT, De Luca G, Bignami M, Oxidized purine nucleotides, genome instability and neurodegeneration, Mutat. Res 703 (1) (2010) 59–65. [DOI] [PubMed] [Google Scholar]
- 131.Nakabeppu Y, Sakumi K, Sakamoto K, Tsuchimoto D, Tsuzuki T, Nakatsu Y, Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids, Biol. Chem 387 (4) (2006) 373–379. [DOI] [PubMed] [Google Scholar]
- 132.Rai P, Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: Defective bricks build a defective house, Mutat. Res 703 (1) (2010) 71–81. [DOI] [PubMed] [Google Scholar]
- 133.Rai P, Onder TT, Young JJ, McFaline JL, Pang B, Dedon PC, Weinberg RA, Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence, Proc. Natl. Acad Sci. USA. 106 (1) (2008) 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nakabeppu Y, Kajitani K, Sakamoto K, Yamaguchi H, Tsuchimoto D, MTH1, an oxidized purine nucleoside triphosphatase, prevents the cytotoxicity and neurotoxicity of oxidized purine nucleotides, DNA Repair. 5 (7) (2006) 761–72. [DOI] [PubMed] [Google Scholar]
- 135.Nakabeppu Y, Oka S, Sheng Z, Tsuchimoto D, Skumi K, Programmed cell death triggered by nucleotide pool damage and its prevention by MutT homolog-1 (MTH1) with oxidized purine nucleoside triphosphatase, Mutat. Res 703 (1) (2010) 51–58. [DOI] [PubMed] [Google Scholar]
- 136.Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Strom CE, Svensson LM, Schultz N, et al. , MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool, Nature. 508 (7495) (2015) 215–221. [DOI] [PubMed] [Google Scholar]
- 137.Huber KV, Salah E, Radic B, Gridling M, Etkins JM, Stukalov A, Jemth AS, Gokturk C, et al. , Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy, Nature. 508 (7495) (2014) 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Berglund U, Sanjiv K, Gad H, Kalderen C, Koolmeister T, Pham T, Gokturk C, et al. , Validation and development of MTH1 inhibitors for treatment of cancer, Ann. Oncol 27 (12) (2016) 2275–2283. [DOI] [PubMed] [Google Scholar]
- 139.Ellermann M, Eheim A, Rahm F, Viklund J, Guenther J, Anderson M, Ericsson U, et al. , Novel class of potent and cellularly active inhibitors devalidates MTH1 as broad-spectrum cancer target, ACS Chem. Biol. 12 (8) (2017) 1986–1992. [DOI] [PubMed] [Google Scholar]
- 140.Kettle JG, Alwan H, Biasta M, Breed J, Davies NL, Eckersley K, et al. , Potent and selective inhibitors of MTH1 probe its role in cancer cell survival, J. Med. Chem 59 (6) (2016) 2346–2361. [DOI] [PubMed] [Google Scholar]
- 141.Wang H, Guo W, Mitra J, Hegde PM, Vandoome T, Eckelmann BJ, et al. , Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Leteral Sclerosis, Nat. Comm 9 (1) (2018) 3683. [DOI] [PMC free article] [PubMed] [Google Scholar]