

Here we describe the complete genome of a new ebolavirus, Bombali virus (BOMV) detected in free-tailed bats in Sierra Leone (species: Chaerephonpumilus and Mops condylurus). The bats were found roosting inside houses, indicating the potential for human transmission. We also show that the viral glycoprotein can mediate entry into human cells, though further studies are required to test whether exposure has actually occurred or if BOMV is pathogenic in humans.
Ebolaviruses (family: Filoviridae) are non-segmented, negative-sense, single-stranded RNA viruses. Five species have been described to date, for which the prototypic viruses are Zaire virus (EBOV), Bundibugyo virus (BDBV), Sudan virus (SUDV), Taï Forest virus (TAFV), and Reston virus (RESTV)1. With the exception of RESTV, all have been associated with severe disease in humans. EBOV was the first ebolavirus described and since 1976 more than 25 outbreaks have been recognized2. The most significant outbreak occurred in Guinea, Sierra Leone and Liberia in 2013–2016 where an estimated 28,000 humans were infected and 11,325 died3. The most recent EBOV outbreak is ongoing in the Democratic Republic of the Congo.
Despite more than 40 years of research and continued outbreaks, the reservoirs of EBOV and the other ebolaviruses remain unknown. Current evidence points to bats4–9, though failure to isolate a virus or recover a complete genome means that no ebolavirus has been conclusively linked to any particular bat species. We therefore initiated a survey in Sierra Leone to identify hosts of EBOV as well as any additional filoviruses that might be circulating in wildlife.
Between March and September 2016, 1278 samples were collected from 535 animals (244 bats, 46 rodents, 240 dogs, 5 cats) from 20 locations in Sierra Leone (Supplemental Figure 1). Three oral and two rectal swabs from four insectivorous bats were positive using a broadly reactive filovirus ‘family level’ cPCR assay (4/244). The resulting 680 bp fragment showed 75% nucleotide identity to other known ebolaviruses. Rectal swabs for two of the four positive bats were also positive using a separate ebolavirus ‘genus level’ cPCR assay. The resulting 187 bp fragment showed 83% nucleotide identity to known ebolaviruses. All samples collected from dogs, cats, and rodents were negative by both assays. Given the 2013 Ebola virus disease outbreak, we also screened all samples for EBOV using specific real-time PCR; however, all samples, including from bats, were negative.
All bats (n = 240) were barcoded to confirm species (Table 1). Of the four positive bats, three were identified as Little free-tailed bats (Chaerephonpumilus) based on 98% sequence identity in the Cytb gene and 99% in the CO1gene. The fourth bat was identified as an Angolan freetailed bat (Mops condylurus) based on 98% identity in the Cytb gene and 99% in the CO1 gene. These bats co-roost and are widely distributed across Western and Sub-Saharan Africa (Supplemental Figure 1). The four positive bats were adult females sampled between May 21st- 28th 2016, at three different sites within 20 km of each other in the Bombali District (Supplemental Figure 1). They were sampled inside human dwellings in small villages, where animals (poultry, goats, sheep) and crops (fruit, vegetables, oil tree) were raised for local consumption and sale (Supplemental Table 1).
Table 1.
Summary of bats tested for Filoviruses and BOMV.
| Bat Species | Bat Family | No. Tested |
No. Positive by Filovirus cPCR |
No. Positive by Ebolavirus genus PCR |
No. Positive by qRT- PCR for BOMV |
|---|---|---|---|---|---|
| Insectivorous Bats | |||||
| Chaerephon atsinanana | Molossidae | 1 | |||
| Chaerephon pumilus | Molossidae | 55 | 3 | 2 | 4 |
| Glauconycteris poensis | Vespertillionidae | 1 | |||
| Hipposideros abae | Hipposiderdae | 7 | |||
| Hipposideros jonesi | Hipposiderdae | 4 | |||
| Hipposideros larvatus | Hipposiderdae | 1 | |||
| Hipposideros ruber | Hipposiderdae | 50 | |||
| Mops condylurus | Molossidae | 52 | 1 | 1 | |
| Myotis bocagii | Vespertillionidae | 3 | |||
| Neoromicia rendalli | Vespertillionidae | 2 | |||
| Nycteris hispida | Nycteridae | 1 | |||
| Pipistrellus nanulus | Vespertillionidae | 3 | |||
| Rhinolophus fumigatus | Rhinolophidae | 3 | |||
| Rhinolophus landeri | Rhinolophidae | 1 | |||
| Rhinopoma microphyllum | Rhinopomatidae | 1 | |||
| Scotophilus viridis | Vespertillionidae | 26 | |||
| Unidentified Molossid bat | Molossidae | 1 | |||
| Unidentified Nycterid bat | Nycteridae | 3 | |||
| Fruit Bats | |||||
| Eidolon helvum | Pteropodidae | 2 | |||
| Epomophorus gambianus | Pteropodidae | 1 | |||
| Epomophorus labiatus | Pteropodidae | 2 | |||
| Epomophorus minor | Pteropodidae | 3 | |||
| Epomops buettikoferi | Pteropodidae | 2 | |||
| Micropteropus pusillus | Pteropodidae | 2 | |||
| Myonycteris angolensis | Pteropodidae | 12 | |||
| Myonycteris torquata | Pteropodidae | 5 | |||
| Total | 244 | 4 | 2 | 5 | |
Using unbiased high throughput sequencing, 98% of the genome was recovered from the oral swab of the Angolan free-tailed bat with an average depth of 12x. Using VirCapSeq, 42% of the genome was recovered with an average depth of 5x. Gene-walking using PCR and Sanger sequencing was used to obtain a second genome from the rectal swab of a Little free-tailed bat. The termini for both sequences were then verified using Rapid Amplification of cDNA ends to generate two complete BOMV genomes (Genbank accession numbers MF319185 and MF319186). The two genomes share 99.1% sequence identity to each other
Phylogenetic analyses showed that BOMV is sufficiently distinct to represent the prototypic strain of a new species within the Ebolavirus genus (Figure 1; Supplemental Figure 2). We suggest the species be named Bombali ebolavirus to reflect the location of first detection, as is consistent with the naming of other ebolavirus species. Assessment using NCBI’s PASC tool supported this new species assignment and it meets all criteria for a novel virus species suggested by Bao et al.10 (Supplemental Figure 3). Overall, the virus showed 55–59% nucleotide identity (64–72% amino acid) to other ebolaviruses, though areas of high sequence conservation and high variability were identified throughout the genome (Supplemental Figure 4). No evidence of recombination was observed. Selection analysis indicated that all genes were undergoing purifying selection; however, several individual residues did show evidence of positive selection (Table 2).
Figure 1.
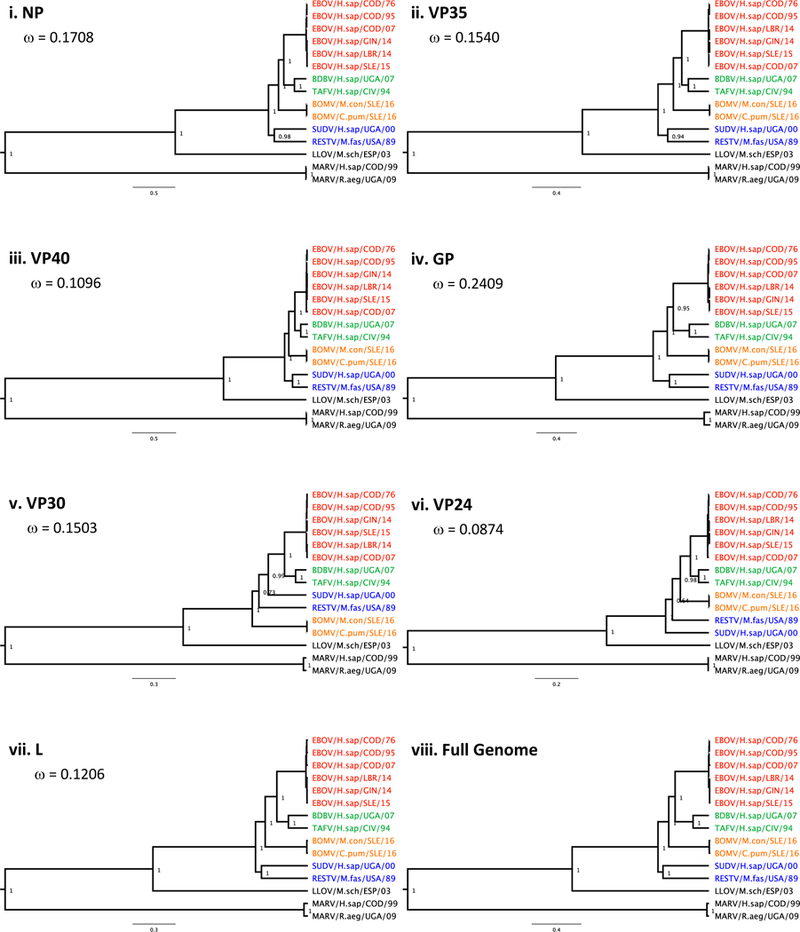
Phylogenetic tree comparing the relationship of BOMV to other known filoviruses. Each protein presented separately (i-vii) and as a complete genome (viii). Genbank accession numbers for reference sequences included in Supplemental Data.
Table 2.
Amino acids indicated to be under positive selection for at least 2 of the 4 datamonkey algorithms tested: SLAC, FEL, MEME, and FUBAR.
| Gene | Position |
|---|---|
| NP | 3(4), 11(2), 108(2), 502(2), 553(2), 577(2), 627(2) |
| VP35 | 63(2) |
| VP40 | 67(2) |
| GP1,2 | 310(4), 318(3), 321(3), 332(3) |
| VP30 | 276(2) |
| VP24 | None |
| L | 202(2), 1661(2), 1731(2), 1733(2), 1737(2), 1752(2), 1774(2), 2171(2) |
Numbering is according to EBOV.
Number in parentheses is the number of tests that indicated positive selection.
Sites indicated by 3 or more methods are bolded.
A BOMV-specific real-time PCR assay was used to re-screen all samples and to quantify the viral load of positive samples. This assay detected down to 10 genome copies with 91% efficiency and did not cross-react with Marburg virus (MARV. Genus: Marburgvirus, Family: Filoviridae), Lloviu virus (LLOV. Genus: Cuevavirus, Family: Filoviridae), or other known ebolaviruses (EBOV, SUDV, TAFV, BDBV, RESTV). Viral load in the four positive animals varied from 10,000 to 4 genome copies/ul. A rectal swab from one additional Little free-tailed bat was found to be weakly positive with approximately 3 genome copies/ul.
Given that Little and Angolan free-tailed bats are insectivorous, we considered the possibility that insects or other arthropods could be the source of this virus. However, sequences of arthropod mitochondrial DNA were only obtained from one of the positive samples - the oral swab of the Angolan free-tailed bat. Sequences of two different arthropods were detected: Eupleao core (butterfly) and Fujientomon dicestum (protura). BOMV load in this specimen was approximately 2,800 genome copies. By comparison, a rectal swab from a Little free-tailed bat, had an estimated 10,000 genome copies but no arthropod DNA. Therefore, despite previous suggestions that insects may be reservoir hosts or vectors of ebolaviruses11,12, we found no correlation between the presence of insect DNA and BOMV. These data suggest BOMV was not present merely as a component of the bat diet.
Free-tailed bats have been previously implicated as hosts of ebolaviruses. Both little and Angolan free-tailed bats were shown to survive experimental infection with EBOV (human Kikwit variant11), while Angolan free-tailed bats were suggested as the source of the 2013 Ebola virus disease outbreak in Western Africa3. Angolan free-tailed bats were also shown to have antibodies against EBOV, or a related virus5. The discovery of BOMV further supports their role as hosts of ebolaviruses, though additional surveillance will be required to determine if BOMV is distributed throughout their range and whether these bats sustain BOMV transmission over time (i.e., whether they are true reservoirs of BOMV).
Given that BOMV was found in close proximity to humans, we tested whether the BOMV GP1,2 could mediate virus entry into human cells. We generated a recombinant vesicular stomatitis virus (rVSV) encoding the BOMV GP gene (Figure 2, panel A), and showed that the rVSV-BOMV GP was infectious in human (293FT) cells (Figure 2, panel B) as well as in African green monkey kidney (Vero) cells (data not shown). These data indicate that the BOMV GP1,2 is fully competent to mediate viral entry. Entry and infection of rVSV-BOMV GP was also completely dependent on NPC1 (Figure 2, panel B), providing additional evidence that NPC1 is a universal receptor for filoviruses13,14. Sequence analysis showed that BOMV GP1,2 shares 92% of the known NPC1-interacting residues found in other ebolaviruses, with only two unique mutations identified at the binding interface, P146S and A148E (Figure 2, panel C)15–18. The corresponding NPC1 residues found within 5 Å of P146S and A148E were conserved between humans and free-tailed bats (both have D502 and V505). Neither of these mutations were predicted to interfere with GP-NPC1 recognition to block binding (Figure 2, panel D), which was supported by our experimental data.
Figure 2.
BOMV GP-mediated entry and infection is NPC1-dependent. (A) BOMV GP1,2 is incorporated in the rVSV particles detected by immunoblotting using ebolavirus anti-GP1 (B) Infectivity of rVSVs bearing BOMV, EBOV or VSV GP1 on WT or NPC1-KO U2OS cells complemented with or without human NPC1 cDNA. (C) GP1 alignment of the known human- infecting ebolaviruses (EBOV, SUDV, RESTV, BDBV, TAFV) and BOMV. Displayed regions pertain to the GP1 interface based on the GP1-human NPC1 crystal structure (PDB: 5F1B). Conserved residues in blue; viral-specific residues in yellow. Squared positions correspond to residues whose side chain heavy atoms are within 5 Â of any heavy atom in the human NPC1 receptor. (D) Left panel: Atomic representation of the interaction between the human NPC1 (red) and the EBOV GPi protein (blue) (PDB: 5F1B). Middle panel: Close-up view of the interface. Right panel: Close-up view of the modeled interface between the human NPC1 crystal structure (red) and the BOMV GP1 atomic model (blue). Displayed viral residues (in yellow) correspond to interfacial positions with different amino acids in the BOMV GP1 protein. Displayed residues on the human NPC1 (in white) correspond to residues with side chain heavy atoms within 5 Å of residues 146 and/or 148 in the EBOV or BOMV GP1.
We acknowledge that binding is not the only determinant of host susceptibility; however, it does represent the first critical step in spillover. Further, even if BOMV is able to establish a productive infection, it is not known whether the virus is virulent in humans as RESTV can also infect human cells but does not cause disease19,20. Data on proinflammatory cytokine expression in human macrophages or the degree to which BOMV antagonizes the human interferon response could help to clarify the pathogenic potential of this virus. Certain key motifs in BOMV VP35 (interferon induction) and VP24 (interferon signaling)19,21–23 are more similar to EBOV, while others are more similar to RESTV (Supplemental Figure 4). Thus, predictions of pathogenicity in humans cannot be made from the sequence alone.
While the pathogenic potential of BOMV is unknown, our data on cell entry suggests the virus could infect humans. Evidence of ebolavirus-reactive antibodies in humans prior to the 2013 outbreak24 suggests that an ebolavirus was already circulating in humans in this area. We suggest it is unlikely a virulent pathogen such as EBOV would circulate in humans without causing disease. Given also the cross-reactivity between ebolaviruses (Supplemental Table 2, Supplementary Figure 5), and that BOMV was discovered in bats inside houses, it is possible that BOMV or some other potentially non-pathogenic ebolavirus has already spilled over. Serosurveys of humans in contact with Little and Angolan free-tailed bats would help to confirm whether exposure has occurred.
Our study contributes to a better understanding of the diversity and ecology of ebolaviruses. First, our data provide strong evidence that bats serve as hosts for ebolaviruses and that additional unknown ebolaviruses may exist in wildlife. Identifying these viruses and testing their capacity for human infection would greatly enhance our understanding of ‘pre-emergent’ viral diversity. Second, it suggests that insectivorous bats play an important role in the ecology of ebolaviruses. To date, surveys have tended to focus on fruit bats, and while they do seem to be important hosts4,5,25,26, we support the previous suggestion by Saez et al. that future surveillance should be expanded to include insectivorous bats.
Finally, we stress that our study is not meant to create alarm or incite the retaliatory culling of bats. While bats have been implicated as reservoirs for a number of other infectious pathogens, they are also important insectivores, pollinators, and seed dispersers. Previous studies have shown that killing or disturbing bats in their natural habitat does not reduce the risk of transmission; rather, it can increase the number of susceptible bats and enhance disease transmission27. While BOMV has the potential to infect human cells, there is currently no evidence that the virus causes disease. Nonetheless, local community engagement is ongoing to explain the current state of understanding regarding BOMV.
Methods
Animal sampling
Oral and rectal swabs, and whole blood when possible, were collected into Trizol, frozen in liquid nitrogen, and stored at −80°C until analysis. Bat host species identification was confirmed by DNA barcoding of the cytochrome b (Cytb) and cytochrome oxidase subunit 1 (CO1) mitochondrial genes28. The presence of invertebrate DNA in BOMV positive samples was examined by PCR for a fragment of the CO1 gene29 (up to 48 clones sequenced from each).
Viral discovery and sequencing
Total RNA was extracted using Direct-Zol RNA columns (Zymo Research Corp), and cDNA prepared using Superscript III (Invitrogen). Samples were screened using three assays: 1) a nested filovirus ‘family level’ consensus PCR (cPCR) targeting a 680 bp fragment of the filovirus L gene, 2) an Ebolavirus ‘genus level’ cPCR targeting a 187 bp fragment of the NP gene30; and 3) a real-time PCR specific for the EBOV virus, targeting the L-gene31. Primer sequences for the cPCR filovirus assay were: Round 1: Filo-MOD-FWD: TITTYTCHVTICAAAAICAYTGGG, FiloL.conR: ACCATCATRTTRCTIGGRAAKGCTTT; Round 2: Filo-MOD-FWD: TITTYTCHVTICAAAAICAYTGGG, Filo-MOD-RVS: GCYTCISMIAIIGTTTGIACATT. For quantification of BOMV load, a quantitative real-time PCR was designed. Primers and probe sequences were: Filo_UCD_qFor: TCTCGACGAAGGTCATTAGCGA, Filo_UCD_qRev: TTGCTCTGGTACTCGCTTGGT, Filo_UCD_probe: FAM-TGCTGGGATGCTGTCTTTGAGCCT-BHQ.
Libraries for genome sequencing were generated with the Kapa Hyper Library kit (Kapabiosystems, Roche)32 and with VirCapSeq-VERT33, and sequenced on the Illumina Miseq platform. Contigs and unique singletons were assembled as described previously32. A second genome was generated by PCR walking using gene-specific primers. The termini were amplified using Rapid Amplification of cDNA ends (RACE) using anchor and virus specific primers. Host NPC1 sequences were generated by mapping unassembled singletons onto a reference NPC1 gene.
Phylogenetic analyses
Sequences were edited using Geneious (version 9.1.7) and aligned with ClustalW. Bayesian coalescent phylogenetic analysis was implemented using BEAST. Nucleotide substitution models were chosen using jModelTest and a Yule process speciation model. Each analysis was run for 1,000,000 generations. Maximum clade credibility trees were generated using the TreeAnnotator program in BEAST and edited using FigTree. Alignments and trees were created separately for each gene and a concatenation was used for the complete genome. Sequences were also analyzed using the PASC tool (NCBI) to classify sequences taxonomically10. The nucleotide alignment of the ebolavirus genomes was screened for recombination using the seven algorithms in the Recombination Detection Program (RDP version 4.87). The ebolavirus nucleotide alignment was analyzed for evidence of selection using the SLAC, FEL, MEME, and FUBAR algorithms, executed in datamonkey (http://datamonkey.org/) and with the M7 and M8 codon models in codeml (PAML package).
BOMV- GP1,2 Interaction with Human NPC1
WT (wild-type), NPC1-KO (NPC1 knockout) and NPC1-KO human osteosarcoma (U2OS) cells complemented with human NPC1 cDNA were cultured as described previously34. The recombinant vesicular stomatitis viruses (rVSV) bearing EBOV GP1,2 and VSV G have been described previously35.
Sequence encoding the full-length BOMV GP1,2 from the Angolan free-tailed bat was cloned between MluI and NotI restriction sites into the pVSV vector to replace the VSV G open reading frame35. The resulting plasmid was used to rescue rVSV-BOMV GP virus using the plasmid- based infectious VSV rescue system on 293FT cells as described previously36,37. The rescued virus was expanded on Vero cells and BOMV GP1,2 sequence was verified by RT-PCR followed by Sanger sequencing. Incorporation of BOMV GP1,2 into the VSV particles was detected by immunoblotting using a rabbit antiserum specific for ebolavirus GP1 residues 86–97 (EBOV GP numbering)38,39.
Monolayers of WT, NPC1-KO or NPC1-KO U2OS cells complemented with human NPC1 cDNA U2OS cells were infected with serial log dilutions of rVSVs expressing eGFP and bearing EBOV, BOMV or VSV glycoproteins for 1 hr at 37°C. Ammonium chloride at a final concentration of 20 mM was added at 1 hr post-infection to prevent subsequent rounds of infection. Infections were enumerated by counting eGFP-positive cells at 12–14 hrs postinfection and expressed as infectious units per ml.
Protein structure modeling
A sequence alignment between EBOV GP1,2 and the BOMV GP1,2 was carried out with T-coffee. Interfacial residues were identified using the crystal structure of the EBOV GP1,2 protein bound to human NPC140. BLAST was used for template search and alignment, while NEST was used to model the structure of the BOMV GP1,241. A non-redundant set of sequences was assembled, corresponding to proteins in the NCBI Protein Data Bank, using a sequence identity cutoff of 1.0 with CD-HIT42. A single iteration of BLAST was run against this dataset, and the template and alignment with the lowest e-value was selected (PDB: 5FHC; e-value: 1.4 e−139). The interaction of the human NPC1 protein with the BOMV GP1,2 was assessed using a structural alignment of the GP1,2 atomic model to the crystallized human NPC1-EBOV GP1,2 protein complex41 with the SKA program.
Peptide ELISA Assay
We designed and synthesized a series of peptides with increasing specificity for BOMV GP1, including one BOMV peptide with high sequence similarity with the other ebolaviruses (GP- 100); one that shares sequence similarity with some, but not all, other ebolavirus (GP-270); and one BOMV peptide that shows no sequence homology with the other ebolaviruses (GP-471, Supplemental Figure 5). Our rationale was to demonstrate decreasing cross-reactivity as a function of sequence variation. Peptides with high sequence similarity for EBOV (GP-313) and TAFV (GP-378) were designed to demonstrate specificity of peptides with known sequence homology to other ebolaviruses. ELISA was performed as described by King et al.43 with slight modifications. We coated plates overnight with each peptide (6ug/ml) or recombinant EBOV glycoprotein (0.5ug/ml, IBT Bioservices), blocked (1% BSA), and used 100 ul primary rabbit polyclonal antibodies (1:1000 dilution) against EBOV (eEnzyme LLC), SUDV (IBT Bioservices), BDBV (Sino Biological Inc.), TAFV (Alpha Diagnostic International Inc.) spiked in 1% dog serum followed by 100 ul secondary goat anti-rabbit antibodies (ImmunoReagents Inc.) (1:2000 dilution) conjugated to HRP and OPD substrate. Optical densities were read at 490 nm and signal was considered to be positive when the absorbance was at least 3 times higher than background.
Supplementary Material
Acknowledgements
We thank the government of Sierra Leone for permission to conduct this work; the Sierra Leone district and community stakeholders for support and for allowing us to perform sampling in their districts and communities; the Bombali Ministry of Health and Sanitation and Ministry of Agriculture district officers, field teams and regional lead including M LeBreton, F Jean Louis,
K Kargbo, LAM Kenny, V Lungay, W Robert, E Amara, D Kargbo, V Merewhether-Thompson, M Kanu, E Lavallie, A Bangura, M Turay, FV Bairoh, M Sinnah, and S Yonda for performing sample collection; Yongai Saah Bona for administrative and logistic support; laboratory staff for assistance with processing the samples, including M Coomber and O Kanu (University of Makeni) and V Ontiveros (UC Davis); T O’Rourke, D O’ Rourke (Metabiota) and D Greig (UC Davis) for assistance with data entry, B Lee for bioinformatics assistance, and J Morrison and A Rasmussen for technical guidance (Columbia University); N Randhawa for map graphics (UC Davis); and W Karesh and J Epstein (EcoHealth Alliance) for global input into study design. This study was made possible by the generous support of the American people through the United States Agency for International Development (USAID) Emerging Pandemic Threats PREDICT project (cooperative agreement number GHN-A-OO-09–00010-00), and by support from the National Institutes of Health (GM030518, S10OD012351 and S10OD021764). All animal sampling activities were conducted with permission from The Ministry of Agriculture, Forestry and Food Security and under the Institutional Animal Care and Use Committee at the University of California, Davis (protocol number: 16048). Inactivated samples in Trizol were shipped to the One Health Institute Laboratory, University of California, Davis for analysis under the PHS permit no. 2016–06-092.
References
- 1.Burk R et al. FEMS Microbiol Rev. 40, 494–519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saéz MA et al. EMBO Mol Med. 7, 17–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lo TQ, Marston BJ, Dahl BA & De Cock KM Annu Rev Med. 68, 359–370 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Leroy EM et al. Nature 438, 575–576 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Pourrut X et al. J Infect Dis. 196, S176–83 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Hayman DTS et al. Emerg Infect Dis. 18, 1207–1209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan J et al. Virol J. 9, 236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Negredo A et al. PLoS Pathog. 7, e1002304 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayme SI et al. Virol J. 12, 107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bào Y et al. Viruses 9, 106 (2017). [Google Scholar]
- 11.Swanepoel R et al. Emerg. Infect Dis. 2, 321–325 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leendertz SA J. Viruses 8, 30 (2016). [Google Scholar]
- 13.Carette JE et al. 477, 340–343 (2001). [Google Scholar]
- 14.Côté M et al. Nature 477, 344–348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller EH et al. EMBO J 31, 1947–1960 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng M et al. eLife 4, e11785 (2015). [Google Scholar]
- 17.Bornholdt ZA et al. mBio 7, e02154–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H et al. Cell 164, 258–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pappalardo M et al. Sci Rep. 6, 23743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miranda ME. & Miranda NL J Infect Dis. 204, S757–760 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Bale S et al. J Virol. 87, 10385–10388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reid SP et al. J Virol. 80, 5156–5167 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volchkov VE, Blinov VM & Netesov SV FEBS Lett. 305, 181–184 (1992). [DOI] [PubMed] [Google Scholar]
- 24.Schoepp RJ, Rossi CA, Khan SH, Goba A & Fair JN Emerg Infect Dis. 20, 1176–1182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Towner JS et al. PLoS One 2, e764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang X et al. Emerg Infect Dis. 23, 482–486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amman BR et al. Emerg Infect Dis. 20, 1761–1764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townzen JS, Brower AV & Judd DD Med Vet Entomol. 22, 386–393 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Folmer O et al. Mol Mar Biol Biotechnol. 3, 292–299 (1994). [PubMed] [Google Scholar]
- 30.Towner JS et al. J Virol. 78, 4330–41 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaaskelainen AJ et al. J Clin Virol. 67, 56–58 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Anthony SJ et al. mBio 8, e00373–17 (2017). [Google Scholar]
- 33.Briese T et al. mBio 6, e01491–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spence JS, Krause TB, Mittler E, Jangra RK & Chandran K mBio 7, e01857–15 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong AC, Sandesara RG, Mulherkar N, Whelan SP & Chandran K J Virol 84, 163–175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whelan SPJ, Ball LA, Barr JN & Wertz GT W. Proc. Natl. Acad. Sci. USA 92, 8388–8392 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleinfelter LM et al. mBio 6, e00801–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandran K, Sullivan NJ, Felbor U, Whelan SP & Cunningham JM Science 308, 1643–1645 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng M et al. Virology 468–470, 637–646 (2014). [Google Scholar]
- 40.Wang H et al. Cell 164, 258–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrey D et al. Proteins 53, S430–435 (2003). [Google Scholar]
- 42.Li W, Jaroszewski L & Godzik A Bioinformatics 17, 282–283 (2001). [DOI] [PubMed] [Google Scholar]
- 43.King DP et al. 2001 Vet Microbiol 80, 1–8 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.