

ABSTRACT
Pre-pregnancy obesity is an established risk factor for adverse sex-specific cardiometabolic health in offspring. Epigenetic alterations, such as in DNA methylation (DNAm), are a hypothesized link; however, sex-specific epigenomic targets remain unclear. Leveraging data from the Newborn Epigenetics Study (NEST) cohort, linear regression models were used to identify CpG sites in cord blood leukocytes associated with pre-pregnancy obesity in 187 mother-female and 173 mother-male offsprings. DNAm in cord blood was measured using the Illumina HumanMethylation450k BeadChip. Replication analysis was conducted among the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort. Associations between pre-pregnancy obesity-associated CpG sites and offspring BMI z-score (BMIz) and blood pressure (BP) percentiles at 4–5-years of age were also examined. Maternal pre-pregnacy obesity was associated with 876 CpGs in female and 293 CpGs in male offspring (false discovery rate <5%). Among female offspring, 57 CpG sites, including the top 18, mapped to the TAPBP gene (range of effect estimates: −0.83% decrease to 4.02% increase in methylation). CpG methylation differences in the TAPBP gene were also observed among males (range of effect estimates: −0.30% decrease to 2.59% increase in methylation). While technically validated, none of the TAPBP CpG sites were replicated in ALSPAC. In NEST, methylation differences at CpG sites of the TAPBP gene were associated with BMI z-score (cg23922433 and cg17621507) and systolic BP percentile (cg06230948) in female and systolic (cg06230948) and diastolic (cg03780271) BP percentile in male offspring. Together, these findings suggest sex-specific effects, which, if causal, may explain observed sex-specific effects of maternal obesity.
KEYWORDS: DNA methylation, epigenome-wide association study, maternal obesity, offspring body mass index, offspring blood pressure, cardiometabolic health, ALSPAC, NEST
Background
More than 9% of preschool-aged children 2–5 years of age are classified as obese, as are 18% of children ages 6–11 years and 21% of 12–19 years of age in the U.S [1]. Although the prevalence of childhood obesity may be plateauing, the prevalence in males and ethnic minorities continues to increase [2]. Childhood obesity is associated with higher blood pressure (BP), recurrent wheezing, and metabolic disease, which are consistent risk factors for common chronic diseases and conditions in later life, including type 2 diabetes, coronary heart disease, some cancers, and life-long obesity [3–10].
Data from the last 15 years have demonstrated that the first 1,000 days, the period from conception to two years of age, represents a critical developmental window in which exposures can play a major role in the programming of physiological systems involved in growth, energy metabolism, adipogenesis, appetite and glucose-insulin homeostasis of the developing offspring [11]. Both pre-pregnancy obesity and excessive gestational weight gain (GWG) independently contribute to cardiometabolic function, including obesity, in offspring [3–5]. With 25% of conceptions occurring among obese women in the U.S [12]., maternal obesity before and during pregnancy is perhaps one of the most common intrauterine risk factors for childhood obesity for which we have limited mechanistic insights.
While mechanisms by which maternal obesity is linked to childhood obesity are still unclear, mounting evidence from animal and human studies supports that a wide range of in utero exposures [10,13–17], including pre-pregnancy obesity and gestational weight gain [18–22], are associated with epigenetic modifications, including DNA methylation of cytosine-phosphate-guanine (CpG) sites, which can in turn alter the transcriptional capacity of genes important in metabolism. A recent meta-analysis comprising 19 studies with >9,000 participants with DNA methylation measured in umbilical cord blood leukocytes using CpG methylation arrays identified 86 maternal obesity-associated CpG sites; however, there was no evidence of enrichment for chromosomal regions or disease pathways [19]. In addition, sex-specific data relating maternal obesity to CpG methylation is lacking and limited research exists with follow-up into childhood to estimate the impact of pre-pregnancy related CpG methylation on cardiometabolic health [18]. To fill this gap, we conducted an epigenome-wide association analysis to determine whether associations between maternal pre-pregnancy obesity and CpG methylation, measured in umbilical cord blood leukocytes, varied by offspring sex, and whether maternal pre-pregnancy obesity-associated CpG methylation was associated with offspring cardiometabolic phenotypes, body mass index (BMI) and BP at 4–5 years of age.
Results
The distribution of maternal and offspring characteristics for 360 mother-offspring pairs are summarized in Table 1. African-American women comprised almost half (48%) of the NEST study sample. Participating women were approximately 25 years of age and older (72%), educated beyond high school (70%), and reported not smoking during pregnancy (68%). Approximately 48% had a pre-pregnancy BMI less than 25kg/m2, 21% were overweight (BMI 25–29.99 kg/m2), and 31% were obese (BMI ≥30 kg/m2). Among the obese, 12% were Class III obese (≥40kg/m2).
Table 1.
Selected baseline characteristics of study population (n = 360) mother-offspring pairs), Newborn Epigenetics STudy (NEST).
| N | % or Mean | SD | |
|---|---|---|---|
| Maternal age | |||
| <25 years | 101 | 28.1 | |
| 25–29 years | 88 | 24.4 | |
| ≥30 years | 171 | 47.5 | |
| Maternal ethnicity | |||
| African American | 172 | 47.8 | |
| European | 159 | 44.2 | |
| Other | 29 | 8.1 | |
| Maternal educational attainment | |||
| Less than high school | 32 | 8.9 | |
| High school graduate | 77 | 21.4 | |
| More than high school | 251 | 69.7 | |
| Maternal cigarette smoking | |||
| Smoked during pregnancy | 115 | 32.2 | |
| Did not smoke during pregnancy | 242 | 67.8 | |
| Maternal BMI before pregnancy | |||
| <25kg/m2 | 173 | 48.1 | |
| 25–29.9 kg/m2 | 75 | 20.8 | |
| ≥30kg/m2 | 112 | 31.1 | |
| Child sex | |||
| Female | 187 | 51.9 | |
| Male | 173 | 48.1 | |
| Birth weight | |||
| <2500g | 62 | 17.2 | |
| ≥2500g | 298 | 82.8 | |
| Breastfeeding status in first 3 months | |||
| Never breastfed | 69 | 19.2 | |
| Exclusively breastfed | 56 | 15.6 | |
| Bottle and breastfed | 109 | 30.3 | |
| Child BMI ≥85th percentile (age 4–5) | |||
| Not overweight/obese | 160 | 67.2 | |
| Overweight/obese | 78 | 32.8 | |
| Child BMI ≥95th percentile (age 4–5) | |||
| Not obese | 194 | 81.5 | |
| Obese | 44 | 18.5 | |
| Child Elevated BP (age 4–5) | |||
| No | 137 | 85.6 | |
| Yes | 23 | 14.4 | |
| Gestational age at delivery, months | 360 | 38.3 | 2.3 |
| Birth weight, grams | 359 | 3108.2 | 667.5 |
| BMI for age z score (age 4–5) | 238 | 0.34 | 1.66 |
| SBP percentile | 160 | 57.98 | 27.65 |
| DBP percentile | 160 | 67.83 | 22.91 |
Pre-pregnancy obesity and CpG methylation
Among female offspring, pre-pregnancy obesity was associated with differential methylation at 6,148 CpG sites (FDR <0.05; Supplemental Table 1), corresponding to 1,034 genes. Adjustment for cell proportion reduced the number of significant CpG sites to 876 corresponding to 101 genes (Supplemental Table 2; Figure 1). Among male offspring at birth, 781 CpG sites were associated with pre-pregnancy obesity (FDR <0.05); Supplemental Table 3). Adjustment for estimated cell proportions reduced the number of significant CpG sites to 293 corresponding to 31 genes (Supplemental Table 4; Figure 2). After adjusting for cell type estimates, lambdas (λ) were close to one for the mother-female offspring model (λ = 0.95) and mother-male offspring models (0.78). In a sensitivity analysis, we examined the association between maternal pre-pregnancy obesity and cell type proportions, where we observed significant positive associations among female offspring (Supplemental Table 5). Specifically, maternal obesity was associated with increased CD4+T-cells, B-cells, and granulocytes. We did not observe any associations in male offspring.
Figure 1.
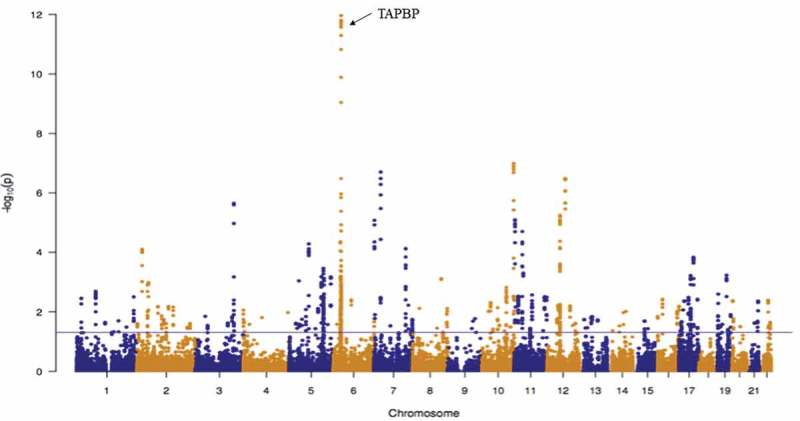
Manhattan plot for the associations between maternal pre-pregnancy obesity and offspring cord blood DNA methylation among females (n = 187) after adjustment for estimated cell proportions. A total of 876 CpGs had an FDR-adjusted P-value <0.05 (solid horizontal blue line).
Figure 2.

Manhattan plot for the associations between maternal pre-pregnancy obesity and offspring cord blood DNA methylation among males (n = 173) after adjustment for estimated cell proportions. A total of 293 CpGs had an FDR-adjusted P-value <0.05 (solid horizontal blue line).
Table 2 provides the top 20 genes with the most robust methylation differences for female offspring of obese and non-obese pregnant women before and after adjusting for cell proportions. After adjustment for cell proportion, the most robust methylation differences that were associated with maternal obesity corresponded to TAPBP (57 CpGs), RP11 (13 CpGs), NFE2L3 (6 CpGs), GLIPR1L2 (11 CpGs), and MFSD1 (10 CpGs). Of the 57 CpG sites annotated to the TAPBP gene, 48 were associated with higher offspring methylation (range of effect estimates: 0.83% to 4.02% increase in methylation; range of associated p-values: 1.31 × 10−10 to 1.61 × 10−12).
Table 2.
Top 20 genes with differentially methylated CpGs in offspring cord blood associated with maternal obesity before and after adjustment for cell proportions, among females (n = 187).
| Unadjusted for cell proportion |
Adjusted for cell proportion |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Annotated Genea |
# of CpG sites | Chr. | Median Methylation Differenceb |
P-valuec (min, max) |
Annotated Genea |
# of CpG sites |
Chr. | Median Methylation Differenceb |
P-valuec (min, max) |
| TAPBP | 30 | chr6 | 0.76% | 3.39e−26, 2.65e−02 | TAPBP | 57 | chr6 | 0.91% | 1.09e−12, 4.24e−02 |
| HMHB1 | 7 | chr5 | 2.83% | 7.40e−10, 2.26e−03 | RP11-109A6.3 | 13 | chr10 | −0.90% | 1.04e−17, 1.18e−02 |
| WT1 | 25 | chr11 | −1.93% | 1.57e−07, 2.64e−02 | NFE2L3 | 6 | chr7 | 4.40% | 2.00e−07, 3.69e−05 |
| PSMB9 | 11 | chr6 | −1.41% | 1.73e−06, 1.80e−02 | GLIPR1L2 | 11 | chr12 | 2.96% | 3.30e−07, 3.48e−06 |
| DDR1 | 30 | chr6 | −1.48% | 2.31e−06, 4.33e−02 | MFSD1 | 10 | chr3 | −1.12% | 2.26e−06, 3.00e−02 |
| PCDHGA4 | 38 | chr5 | −1.52% | 4.01e−06, 4.97e−02 | HOXC4 | 22 | chr12 | −1.78% | 5.71e−06, 4.30e−02 |
| PPT2 | 42 | chr6 | −0.97% | 4.02e−06, 4.41e−02 | LSP1 | 14 | chr11 | 0.56% | 8.30e−06, 1.89e−02 |
| GLIPR1L2 | 11 | chr12 | 3.20% | 4.10e−06, 7.31e−06 | WT1 | 16 | chr11 | −1.89% | 2.01e−05, 3.60e−02 |
| BRCA1 | 26 | chr17 | 0.94% | 4.72e−06, 3.80e−02 | DDR1-006 | 14 | chr6 | −1.70% | 4.49e−05, 4.52e−02 |
| FLJ12825 | 8 | chr12 | −1.80% | 5.37e−06, 2.01e−02 | CKMT2 | 9 | chr5 | −1.54% | 5.23e−05, 3.70e−02 |
| FBXO27 | 9 | chr19 | −2.3% | 6.22e−06, 1.44e−02 | MEST | 15 | chr7 | 1.33% | 7.55e−05, 4.51e−02 |
| LYPD3 | 8 | chr19 | −1.57% | 6.68e−06, 1.13e−02 | APOB | 12 | chr2 | −2.09% | 7.97e−05, 2.08e−03 |
| TMEM187 | 12 | chrX: | −1.65% | 9.09e−06, 4.66e−03 | LINC00336 | 9 | chr6 | 2.25% | 9.39e−05, 1.12e−03 |
| CSNK1E | 11 | chr22 | −1.82% | 9.17e−06, 3.17e−02 | C17orf64 | 9 | chr17 | −2.06% | 1.50e−04, 1.25e−03 |
| GRB7 | 13 | chr17 | −1.10% | 1.04e−05, 2.06e−02 | PCDHB15 | 4 | chr5 | −2.41% | 3.47e−04, 3.11e−03 |
| BAT1 | 29 | chr6 | 1.13% | 1.06e−05, 1.70e−02 | PRR5L | 5 | chr11 | 3.64% | 4.97e−04, 6.38e−04 |
| EIF2C2 | 5 | chr8 | 1.78% | 1.07e−05, 1.33e−05 | HOXB5 | 3 | chr17 | 1.51 | 6.07e−04, 1.18e−02 |
| MFSD1 | 10 | chr3 | −1.05% | 1.13e−05, 3.64e−02 | SEPT8-003 | 10 | chr5 | −1.63% | 6.64e−04, 2.36e−02 |
| GPSM3 | 10 | chr6 | 1.68% | 1.39e−05, 2.60e−02 | STK10 | 4 | chr5 | −1.45% | 6.75e−04, 1.17e−03 |
| ATF7 | 1 | chr12 | 7.16% | 1.69e−05 | PCDHGB8P-002 | 7 | chr5 | −2.88% | 4.20e−04, 2.48e−02 |
aAnnotated genes shown according to FDR-corrected p-value in ascending order
bMedian Methylation difference across all significant CpG sites annotated to denoted gene
cMinimum and maximum p-value for significant CpG sites annotated to denoted gene
Table 3 summarizes the 20 genes with the most robust methylation differences among male offspring of obese versus non-obese pregnant women. Prior to adjustment for cell proportion, the most robust differences were at CpG sites in or near GFI1 (9 CpGs), HOXA3 (15 CpGs), and WDFY4 (17 CpGs). After adjustment for cell proportion, robust differences remained at CpG sites in or near GFI1 (10 CpGs). Notably, CpG methylation differences in the TAPBP gene (17 CpGs) were also observed among male offspring. Of the 17 CpG sites that mapped to the TAPBP gene, 15 were associated with higher methylation; however, the magnitude of male-specific associations was smaller than those for female offspring (range of effect estimates: 0.79% to 2.59%; range of associated p-values: 5.57 × 10−3 to 6.07x10−3; Supplemental Table 2).
Table 3.
Top 20 genes with differentially methylated CpGs in offspring cord blood associated with maternal obesity before and after adjustment for cell proportions, among males (n = 173).
| Unadjusted for cell proportion |
Adjusted for cell proportion |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Annotated Genea |
# of CpG sites | Chr. | Median Methylation Differenceb |
P-valuec (min, max) |
Annotated Gene |
# of CpG sites | Chr. | Median Methylation Difference |
P-value (min, max) |
| GFI1 | 9 | chr1 | 3.74% | 3.48e−10, 1.04e-°3 | CYP26C1 | 10 | chr10 | 1.73% | 1.15e−08, 9.35e−03 |
| HOXA3 | 15 | chr7 | 1.37% | 2.15e−9, 3.84e−02 | GFI1 | 10 | chr1 | 2.42% | 1.15e−08, 2.92e−02 |
| WDFY4 | 17 | chr10 | −1.94% | 2.43e−08, 3.55e−02 | STMN1 | 16 | chr1 | −1.49% | 1.15e−08, 2.95e−02 |
| AGPAT1 | 13 | chr6 | −1.09% | 2.67e−08, 4.38e−06 | CPT1B | 14 | chr22 | 1.76% | 3.29e−05, 4.40e−03 |
| RNF5 | 2 | chr6 | −0.70% | 2.67e−08, 2.67e−08 | HOXA5 | 33 | chr7 | 1.14% | 3.29e−05, 4.26e−02 |
| RNF5P1 | 22 | chr6 | −1.02% | 6.54e−08, 1.90e−04 | ZNF135 | 10 | chr19 | 1.47% | 2.07e−04, 5.94e−03 |
| STMN1 | 15 | chr1 | −1.50% | 1.41e−07, 3.90e−02 | MOV10L1 | 8 | chr22 | 1.33% | 7.80e−04, 9.00e−04 |
| GABBR1 | 18 | chr6 | 1.59% | 1.61e−07, 3.18e−03 | PIGZ | 3 | chr3 | 2.84% | 1.15e−03, 3.12e−03 |
| CYP26C1 | 9 | chr10 | 1.64% | 3.15e−06, 3.84e−02 | HOXA | 8 | chr7 | 1.21% | 2.53e−03, 7.84e−03 |
| FOXK2 | 6 | chr17 | 3.10% | 7.97e−06, 1.56e−05 | C17orf98 | 8 | chr17 | −2.28% | 3.12e−03, 4.41e−02 |
| TLR5 | 5 | chr1 | 2.18% | 9.32e−06, 4.97e−03 | TAPBP | 17 | chr6 | 1.22% | 4.01e−03, 3.90e−02 |
| CUX2 | 8 | chr12 | −2.28% | 1.57e−05, 4.07e−05 | TLR5 | 5 | chr1 | 1.35% | 4.20e−03, 3.73e−02 |
| CMYA5 | 12 | chr5 | 3.2% | 2.15e−05, 4.79e−04 | CMYA5 | 12 | chr5 | 2.78% | 5.23e−03, 2.97e−02 |
| C1orf210 | 8 | chr1 | 1.52% | 2.44e−05, 2.09e−02 | POU2AF | 3 | chr11 | 1.50% | 5.23e−03, 3.72e−02 |
| PIGZ | 3 | chr3 | 3.05% | 7.96e−05, 2.55e−04 | KIAA0101 | 9 | chr15 | −0.87% | 5.41e−03, 7.07e−03 |
| CSNK1E | 10 | chr22 | −1.52% | 9.77e−05, 1.52e−03 | PTPN3 | 2 | chr9 | 0.65% | 5.41e−03, 5.76e−03 |
| PCDHB3 | 1 | chr5 | −6.60% | 1.15e−04 | BRSK2 | 2 | chr11 | 5.4% | 6.25e−03, 8.78e−03 |
| CPT1B | 14 | chr22 | 1.65% | 1.62e−4, 1.41e−02 | RNU6 | 5 | chr1 | 2.35% | 7.26e−03, 2.32e−02 |
| KIAA0101 | 9 | chr15 | −1.25% | 1.75e−04, 2.70e−04 | TMEM72 | 4 | chr10 | 3.47% | 7.31e−03, 1.10e−02 |
| BRD2 | 12 | chr6 | 1.23% | 1.78e−04, 3.39e−02 | AGPAT1 | 21 | chr6 | −0.78 | 7.82e−03, 4.78e−02 |
aAnnotated genes shown according to FDR-corrected p-value in ascending order
bMedian Methylation difference across all significant CpG sites annotated to denoted gene
cMinimum and maximum p-value for significant CpG sites annotated to denoted gene
Validation study
To determine whether findings identified using the Illumina HumanMethylation450k BeadChip (HM450k) array could be replicated in an independent dataset within the same cohort, we used more accurate pyrosequencing to quantify methylation at the TAPBP gene among male and female offspring born to obese (n = 20) and non-obese women (n = 24). We found that the CpG sites that flanked and included cg14419102 in TAPBP gene had ~2% higher methylation in offspring of obese women when compared to non-obese women (vs. 3% from the array). The relationship between pre-pregnancy obesity and higher methylation persisted at 311 bp chr6:28,943,509–28,943,820 region in female offspring at sequence regions flanking cg18353226 and cg06375761 near TAPBP, where significant methylation differences were the least robust (<2%), chr6:33,312,579–33,312,730.
Replication study – ALSPAC
HM450K methylation array data from 751 mother-offspring pairs in the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort from Bristol, UK was used as our replication dataset. Prior to multiple testing correction, maternal pre-pregnancy obesity was associated with differential methylation at 10 CpGs of TAPBP gene among female offspring and at 12 CpGs of the same gene among males after adjusting for cell proportion. Among the 10 CpGs of TAPBP gene identified in female offspring, only two overlapped with those identified in the NEST cohort, of which one (cg07500019) showed a positive association in both ALSPAC and NEST. Of the 12 CpGs of the TAPBP gene identified in males, none overlapped with those identified in NEST. Also, none of the CpGs of the TAPBP gene identified in ALSPAC survived correction for multiple testing in female or male offspring (Bonferroni correction for 227 tests – total CpGs that mapped to TAPBP gene – P-value<0.00022).
Associations between differential methylation of TAPBP and offspring cardiometabolic outcomes.
We evaluated whether methylation at CpGs identified in NEST near TAPBP were also associated with offspring BMI z-score and systolic and diastolic BP percentiles at 4–5 years of age. In female offspring, we found evidence for association between two pre-pregnancy obesity-related TAPBP CpG sites and BMI z-score [cg23922433: β = 1.58, 95% CI: 0.73, 0.14; cg17621507: β = −1.18, 95% CI: −2.22, −0.16], but not BP (Table 4). Another two TAPBP CpG sites were associated with systolic BP percentile [cg06230948: β = 9.54, 95% CI: 1.16, 17.93; cg07955457: β = 23.49, 95% CI: 0.03, 46.96]. Among male offspring, one CpG site was associated with higher systolic BP percentile [cg06230948: β = 8.11, 95% CI: 0.29, 15.95] and one CpG site was associated with lower diastolic BP percentile [cg0378027: β = −23.70, 95% CI: −41.17, −6.23].
Table 4.
Associationsa of TAPBP DNA methylation sitesb with child BMI z-score and elevated blood pressure at 4–5 years of age, among 238 female and male offspring in NEST cohort.
| Females |
Males |
|||||
|---|---|---|---|---|---|---|
| TAPBP genec, d | BMI z-score β (95% CI) |
Systolic Blood Pressure Percentile β (95% CI) |
Diastolic Blood Pressure Percentile β (95% CI) |
BMI z-score β (95% CI) |
Systolic Blood Pressure Percentile β (95% CI) |
Diastolic Blood Pressure Percentile β (95% CI) |
| cg23922433 | 1.58 (0.73, 0.14) | 4.23 (−22.46, 30.92) | 10.51 (13.64, 34.66) | – | – | – |
| cg16632096 | 0.28 (−1.08, 1.63) | −10.24 (−35.84, 15.37) | −11.44 (−34.65, 11.77) | – | – | – |
| cg17621507 | −1.18 (−2.22, −0.16) | −2.77 (−25.12, 19.59) | 2.67 (−17.64, 22.97) | – | – | – |
| cg03780271 | 0.21 (−1.22, 1.63) | 2.64 (−22.29, 27.56) | 4.29 (−18.34, 26.91) | −0.32 (−1.19, 0.55) | −5.78 (−30.15, 15.59) | −23.70 (−41.17, −6.23) |
| cg12589538 | −0.09 (−1.21, 1.03) | 0.70 (−20.09, 21.50) | 5.63 (−13.22, 24.48) | 0.04 (−0.81, 0.89) | 3.55 (−20.11, 27.22) | −8.59 (−26.19, 9.00) |
| cg11796996 | −0.08 (−2.51, 0.87) | −2.10 (−33.46, 29.25) | 3.65 (−.24.82, 32.12) | −0.59 (−1.69, 0.51) | 11.01 (−20.65, 42.68) | −14.60 (−38.11, 8.92) |
| cg02863594 | 0.59 (−0.95, 2.12) | −23.56 (−50.58, 3.46) | 6.15 (−18.82, 31.11) | −0.58 (−1.71, 0.56) | 22.31 (−8.27, 52.90) | −5.05 (−28.18, 18.08) |
| cg01253676 | −0.03 (−1.38, 1.31) | −9.21 (−33.86, 15.43) | 0.51 (−23.95, 22.97) | −0.21 (−1.22, 0.80) | 9.22 (−18.95, 37.40) | 0.41 (−20.70, 21.53) |
| cg20998791 | 0.25 (−0.84, 1.35) | −10.25 (−29.39, 8.89) | −1.06 (−18.56, 16.45) | −0.13 (−1.02, 0.77) | 9.41 (−14.18, 33.00) | −4.68 (−22.25, 12.99) |
| cg18353226 | 0.28 (−0.80, 1.35) | −6.19 (−25.22, 12.84) | −0.92 (−18.25, 16.41) | 0.12 (−0.68, 0.92) | 7.84 (−14.20, 29.88) | −3.01 (−19.52, 13.50) |
| cg14419102 | 0.32 (−0.57, 1.22) | −12.56 (−28.44, 3.31) | −0.07 (−14.58, 14.71) | 0.28 (−0.39, 0.95) | 5.60 (−12.82, 24.01) | −3.38 (−17.16, 10.39) |
| cg06375761 | 0.26 (−0.46, 0.97) | −9.30 (−22.28, 3.68) | −2.58 (−14.51, 9.34) | 0.12 (−0.41, 0.64) | 7.60 (−6.55, 21.75) | −2.47 (−13.11, 8.17) |
| cg26083458 | 0.05 (−0.85, 0.95) | −7.73 (−24.27, 8.81) | 2.66 (−12.44, 17.75) | 0.26 (−0.41, 0.93) | 6.29 (−11.56, 24.13) | −3.56 (−16.92, 9.80) |
| cg13638257 | −0.30 (−1.49, 0.89) | −0.84 (−21.85, 20.16) | 5.82 (−13.21, 24.86) | −0.08 (−0.92, 0.77) | 15.05 (−7.33, 37.44) | 0.59 (−16.32, 17.51) |
| cg05700142 | 0.21 (−0.47, 0.89) | −5.47 (−18.17, 7.23) | 3.65 (−7.91, 15.21) | 0.05 (−0.44, 0.53) | 9.61 (−3.29, 22.51) | −0.77 (−10.54, 9.00) |
| cg01654446 | 0.06 (−0.68, 0.81) | −2.96 (−17.24, 11.32) | 3.64 (−9.31, 16.60) | −0.28 (−0.74, 0.17) | 4.56 (−8.46, 17.57) | 4.15 (−5.56, 13.86) |
| cg18930100 | −0.24 (−1.44, 0.97) | −3.94 (−26.46, 18.58) | 3.09 (−17.37, 23.55) | −0.58 (−1.47, 0.31) | 24.21 (−1.16, 49.57) | 7.93 (−11.37, 27.22) |
| cg06230948 | −0.13 (−0.60, 0.34) | 9.54 (1.16, 17.93) | 0.38 (−7.47, 8.23) | 0.17 (−0.13, 0.47) | 8.11 (0.29, 15.95) | 3.87 (−2.07, 9.81) |
| cg08814531 | −0.43 (−2.07, 1.21) | 3.80 (−26.09, 33.69) | 20.99 (−5.77, 47.75) | −0.01 (−1.11, 1.09) | 4.76 (−23.05, 32.57) | 0.15 (−20.65, 20.95) |
| cg03591496 | −1.26 (−2.61, 0.28) | 8.55 (−18.60, 35.70) | 14.88 (−9.63, 39.38) | −0.28 (−1.28, 0.73) | 18.04 (−6.87, 42.96) | 7.18 (−11.61, 25.98) |
| cg20973878 | 0.92 (−0.72, 2.56) | −13.86 (−43.11, 15.40) | −20.93 (−47.25, 5.38) | – | – | – |
| cg27020289 | −0.78 (−2.26, 0.70) | 16.46 (−12.31, 45.23) | 1.23 (−25.11, 27.57) | – | – | – |
| cg00274587 | −0.29 (−1.60, 1.03) | −3.05 (−24.47, 18.37) | −5.59 (−25.02, 13.84) | – | – | – |
| cg00558147 | 0.09 (−1.49, 1.68) | 6.69 (−19.27, 32.65) | 9.95 (−13.56, 33.47) | – | – | – |
| cg00792719 | 0.92 (−0.59, 2.43) | 11.61 (−16.31, 39.53) | −16.99 (−42.17, 8.20) | – | – | – |
| cg07500019 | 0.00 (−1.76, 1.76) | 1.67 (−30.75, 34.09) | −8.62 (−38.01, 20.77) | – | – | – |
| cg07955457 | −0.12 (−1.48, 1.25) | 23.49 (0.03, 46.96) | 18.71 (−2.72, 40.15) | – | – | – |
| cg27385940 | −0.69 (−3.58, 2.20) | −1.88 (−13.38, 9.62) | −3.68 (−14.10, 6.74) | – | – | – |
a Adjusted for maternal age at delivery, race/ethnicity, maternal pre-pregnancy obesity, and smoking status during pregnancy.
bAll CpG sites listed are located in Chromosome 6.
cCpG sites listed were all associated with maternal pre-pregnancy BMI after accounting for multiple testing (q < 0.05). Associations between TAPBP gene CpGs and offspring BMI z-score and elevated blood pressure were not statistically significant after applying the Bonferroni test for multiple testing (female offspring p < 0.002 for 28 tests; male offspring p < 0.003 for 17 tests).
dCpGs were all located in the gene body of the TAPBP gene
Bold represents associations with p < 0.05.
Assessment of functional importance
To further examine the functional significance of the chromosome 6 genomic region containing pre-pregnancy obesity associated CpGs mapping to TAPBP (28 CpG sites in female and 18 CpG sites in male offspring), we used the UCSC Genome Browser to visualize the ~700bp region of statistically significant differential methylation identified by Illumina HM450K Methylation arrays (Chr6: 28,911,468– 28,912,166, GRCh37/hg37; Supplemental Figure 1). This region has 12 consecutive CpG sites with p-values <5x10-5 and contains another 13 CpG sites not included in the HM450K methylation array. This region contains the noncoding RNA LINC01556, and ENCODE data shows H3K27 methylation and partial overlap with two DNase I hypersensitive sites that also contain binding sites for more than 30 transcription factors, man of them related to cardiometabolic risk. The total extent of the DNase hypersensitive sites, transcription factor binding regions, and histone H3K27 methylation is 1600bp, centered on the sequence region of differential methylation we found.
In order to determine the potential functional significance of genes corresponding to differentially methylated CpGs in males (31 significant genes) and females (101 significant genes), we utilized the Database of Genotypes and Phenotypes (dbGaP) within the Disease/Drugs category of Enrichr [23,24]. The three most significantly associated categories in males were narcolepsy, body weight, and atherosclerosis, and in females, lymphocytes, ulcerative colitis, and hypertension. However, after p-value adjustment using the Benjamini-Hochberg method, these results were not significant. As a comparative measure, we also analyzed the 77 genes in the PACE study [19] using dbGaP. The three most significantly associated categories were insulin resistance, insulin, and lipoproteins, however, after p-value adjustment, these results were not significant. Given that our analysis was sex-specific whereas the PACE study was not, we note the limitation in our functional comparisons between these studies. We do highlight that indicators of cardiometabolic function (atherosclerosis, body weight, hypertension, and insulin resistance) were identified as potential categories of importance in both studies.
Discussion
To our knowledge, this is the first epigenome-wide analysis to examine sex-specific associations between maternal pre-pregnancy obesity, offspring DNA methylation in cord blood, and offspring BMI and BP. We identified multiple differentially methylated CpG sites in umbilical cord blood among male and female offspring of obese mothers compared with non-obese mothers. A key finding of our study was CpGs in or near the TAPBP gene were highly associated with maternal obesity in both male and female offspring. Specifically, we identified 74 maternal obesity-related CpG sites (57 CpGs in female and 17 CpGs in male offspring) corresponding to the TAPBP gene on chr6, located in the group of genes involving the class 1 major histocompatibility complex. Associations were most striking among female offspring, where methylation differences of approximately 5% were detected in 21 consecutive CpG sites within a genomic region spanning 700bp. The number and density of the significant CpG sites of the TAPBP gene identified were suggestive of a regulatory region warranting further investigation. Therefore, the findings were validated by pyrosequencing supporting the hypothesis that the vulnerability of fetal homeostasis differs by offspring sex. We further evaluated the association between pre-pregnancy maternal obesity-related CpG sites of TAPBP gene with offspring cardiometabolic outcomes and found links to BMIz and BP percentile in female offspring and systolic and diastolic BP percentile in male offspring.
While many differentially methylated CpGs of TAPBP gene were identified in relation to maternal pre-pregnancy obesity in the NEST cohort, none were replicated in the ALSPAC cohort. Reasons for lack of replication are unclear, however, we note that the prevalence and severity of pre-pregnancy maternal obesity were very different between the two cohorts – this might account for the discrepant findings. Among women participating in NEST, the mean BMI was 29 kg/m2, with an upper limit of 65 kg/m2, and the prevalence of pre-pregnancy maternal obesity was 31%. ALSPAC cohort had a lower mean BMI (22 kg/m2) and prevalence of maternal obesity (5%). It also is possible that differences in confounding structures across the two populations could account for our results not replicating among the ALSPAC cohort. Differences in ethnic distributions across the NEST and ALSPAC cohorts may also contribute to the lack of replication – ALSPAC cohort comprised ~96% European ethnicity, while NEST comprised 48% African American and 44% European Americans. CpG sites of TAPBP were also not identified in the larger Pregnancy and Childhood Epigenetics (PACE) consortium, a study of 23 independent cohorts [19]. The PACE consortium conducted an epigenome-wide DNA methylation study to identify CpG sites associated with maternal BMI at the start of pregnancy, where maternal overweight/obesity was defined as BMI ≥ 25 kg/m2. Differential methylation at CpG sites of TAPBP gene may only be associated with pre-pregnancy obesity among individuals with more severe levels of obesity and in a sex-specific manner, yet associations by sex were not examined. Lastly, we did not identify any CpG sites that overlapped with the 86 significant CpGs in the PACE consortium, which is also likely due to the heterogeneity across studies. Therefore, we suggest future studies replicate our findings in cohorts with similar population characteristics.
The significance of pre-pregnancy obesity associated methylation levels observed in TAPBP gene is unclear and requires gene- and network-specific functional experiments to elucidate mechanisms underlying these associations. However, hypermethylation of multiple CpG sites in TAPBP at birth was recently shown to be associated with increased risk of cardiometabolic endpoints (e.g., insulin sensitivity) among five-year old Australian children [25]. Our data contributes to the novel findings that hypermethylation of regulatory sequences of TAPBP may be an important link between maternal obesity and offspring cardiometabolic dysfunction.
Sex-specific physiological and behavioral responses to periconceptional obesity have been repeatedly demonstrated in animal models (e.g., fed high-fat diets before conception) and observational studies of paternal obesity [26–28]. However, sex-specific human data linking maternal obesity to periconceptional or prenatal obesity epigenetic response are limited. Obesity in the male or female parent at conception has been associated with methylation differences at birth of regulatory sequences of genes known to increase obesity risk in human offspring, including those targeting BDNF and IGF2, or identified from methylation arrays [10,13,14,29,30].
In our sex-specific replication analysis of CpG sites of the TAPBP gene and maternal pre-pregnancy obesity, initial statistically significant associations for many CpG sites did not survive multiple comparisons correction. TAPBP encodes the transporter associated with tapasin antigen processing (TAP) binding protein, a transmembrane glycoprotein that mediates the interaction between newly formed MHC class 1 molecules and TAP, an interaction essential for optimal peptide loading onto the MHC and antigen display. [31]. While mechanisms linking maternal obesity to sex-specific epigenetic response are still unknown, higher methylation levels of TAPBP such as those found in our study might decrease tapasin via decreased transcriptional activity, leading to impaired immune responses in offspring. Decreased tapasin has been shown to lower CD8 + T-cell responses in vitro [32–35]. Indeed, tapasin has also been associated with the autoimmune disease rheumatoid arthritis [36–38], and like obesity, this condition is also associated with chronic inflammation. Both excess adipose tissue and endothelial dysfunction are associated with elevated levels of pro-inflammatory cytokines, such as Interleukin-6 (IL-6) and Tumor Necrosis Factor-alpha (TNF-α). In mice, tapasin is activated by the cytokines IFN-γ and IFN-β, and to a lesser extent, TNF-α [39]. Taken together, our results are consistent with regulatory function for the identified TAPBP CpG sites with further experimental studies needed to confirm these findings. In addition to further interrogating the 700bp chr6 region contained within the HLA complex, evaluating whether these differentially methylated genes serve as biomarkers of immune dysfunction or as early indicators of an already altered state of inflammation might contribute to early obesity risk assessment [40,41].
In addition to the TAPBP gene, multiple CpG sites (ranging from 2–33 per gene) were also robustly associated with maternal pre-pregnancy obesity in female (e.g., GLIPR1L2, DDR1, and genomically-imprinted WT1) and male (e.g., STMN1, CYP26C1, TLR5, CMYA5, PIGZ, and genomically-imprinted GFI1) offspring. These genes are posited to have multiple annotated functions including cellular processes such as cell proliferation, apoptosis, differentiation and oncogenesis. Several also play major roles in activation of immune response and inflammation. DDR1 has been shown to contribute to the development of Th17-dependent inflammatory diseases [42,43]. Other genes, such as TLR5 and STMN1, are involved in pathways linked to obesity and obesity-related cancer [44]. TLR5 is a member of the toll-like receptor family which has been linked to inflammation, obesity and metabolic dysfunction in both humans and animal models [42,43,45–47]. Further, the gut microbiome has been shown to be impacted by TLR5 transcriptional activity in mice, which resulted in metabolic syndrome – hyperlipidemia hypertension, insulin resistance, and increased adiposity. STMN1, also known as stathmin 1, may have a role in cell cycle progression and cell migration [48,49]. Expression of STMN1 has been shown to interact with BMI to influence colorectal cancer outcomes [44]. Further work is warranted to examine the potential mechanisms by which perturbation of these genes might mediate the observed associations between maternal obesity and offspring cardiometabolic dysfunction outcomes such as blood pressure and obesity.
This prospective study, conducted in an ethnically and socioeconomically diverse population, has several strengths that include: the use of an established, highly reproducible methylation array; a comprehensive comparison of both maternal and childhood obesity by infant sex; and prospective follow-up of offspring into early childhood at 4–5 years of age. Despite these strengths, study limitations exist. First, the functional assessment included a small number of genes as inputs in the functional analysis tool, which limited the interpretation of the findings from this analysis. Second, pre-pregnancy obesity-associated CpG methylation was measured in cord blood leukocytes, yet the cell fractions driving these associations remain unknown, and should be investigated in the future. Third, maternal pre-pregnancy weight was based on self-reported data, which may have resulted in exposure misclassification. Fourth, with CpG methylation data available only at birth, we cannot exclude the possibility that postnatal modifications alter cardiometabolic status in children. Finally, although paternal obesity is correlated with maternal obesity, and paternal obesity is associated with altered CpG methylation in offspring [29,49], paternal anthropometric data were available on only 92 of the 360 children with DNA methylation measurements in NEST [50], precluding paternal sex-specific analyses.
In summary, we found multiple CpG sites that were differentially methylated at birth in relation to maternal obesity, the most striking of which was the TAPBP that was also associated with either weight status or elevated blood pressure, depending on the sex of the offspring. While multiple other CpG sites were also significantly associated with maternal obesity, the TAPBP gene is associated with immune-response and supports one mechanism by which maternal obesity increases the risk of offspring obesity and other cardiometabolic phenotypes may be via epigenetic modification of immune response before birth. Our data highlight the importance of this region for future interrogation to gain mechanistic insights linking pre-pregnancy obesity with the development of obesity in children.
Methods
Study population
Study participants for the current analysis were enrolled as part of the Newborn Epigenetics STudy (NEST), a pre-birth cohort study designed to improve our understanding of the role of environmental influences on epigenetic responses and phenotypes in children. Between 2005 and 2009, pregnant women receiving prenatal care at Duke Maternal Fetal Medicine Clinic who intended to use Duke Hospital for their obstetrics care were enrolled using a recruitment protocol whose details have been described previously [51,52]. Inclusion criteria were age 18 years or older, English speaking, and intent to use Duke University Medical Center for obstetric care for the index pregnancy. From these, women intending to move before the first birthday of the offspring, relinquish custody of the index child, or who had confirmed human immunodeficiency virus (HIV) infection, were excluded. A total of 1,101 eligible women were approached, 895 (81%) consented and were enrolled, and umbilical cord blood was successfully collected from 741 infants. The current study is limited to the 360 mother-child pairs with pre-pregnancy BMI, DNAm, and sex data; these pairs were comparable to the remainder of the cohort, with respect to maternal age at delivery, race/ethnicity, BMI, offspring weight, although these women had a higher education than the remainder of the cohort (p < 0.05). The study protocol was approved by the Duke University and Durham Regional Hospital Institutional Review Boards.
Data collection
Upon enrollment, participants completed a questionnaire, which queried women on sociodemographic, lifestyle and health characteristics. At delivery, parturition data were obtained from medical records, and infant cord blood specimens were collected in EDTA-treated tubes, and subsequently centrifuged using standard protocols to allow for collection of plasma and buffy coat for DNA extraction (Qiagen, Valencia, CA, USA) and stored at −80°C until required. Offspring were followed annually and at each follow-up, caretakers completed a caretaker-administered questionnaire regarding the feeding, sleep, and social and physical activity habits of the child. Medical records were also used to augment follow-up data.
Assessment of maternal body size
The enrollment questionnaire solicited information on maternal pre-pregnancy anthropometric measurements, including current height in meters (m) and usual pre-pregnancy weight in kilograms (kg) from which BMI was calculated and expressed as kg/m2. In these analyses, participants were defined as non-obese if their BMI was less than 30 kg/m2, and obese if their BMI was equal to or greater than 30 kg/m2, according to the World Health Organization classifications [53].
Assessment of childhood cardiometabolic outcomes
Data to compute offspring BMI z-score and BP percentiles were obtained from medical records and directly measured height and weight. Sex-specific body mass index (BMI) percentiles and BMI z-score were computed based on the Centers for Disease Control and Prevention protocols [54]. BP percentiles were calculated using the norms recommended by the National Education Program Working Group on High Blood Pressure in Children and Adolescents [55].
Assessment of covariates
Women self-reported race/ethnicity, level of education, and cigarette smoking status at enrollment. At delivery, medical records were abstracted to obtain maternal age at delivery and parturition data including mode of delivery, infection in labor, gestational age at birth, and infant birth weight and sex. In addition to offspring weight and height, follow-up questionnaires also queried mothers on breastfeeding for each of the 12 months following delivery, and breastfeeding duration was dichotomized at three months. A priori, we considered infant sex as a potential modifier of the association between maternal obesity and offspring methylation as emerging data suggest epigenetic perturbations may be likely sex-specific [26,56]. Therefore, all CpG analyses were stratified by infant sex.
DNA methylation
Methods for DNAm analyses have been described in detail elsewhere [57,58]. Briefly, cord blood was collected at delivery and genomic DNA was extracted from buffy coat specimen based on standard precipitation procedure. Bisulfite conversion was performed using the EZ-96 DNAm kit (Zymo Research Corporation, Irvine, CA, USA) according to manufacturer instructions. Illumina’s GenomeStudio Methylation module version 1.0 (Illumina, Inc.) was used to calculate the methylation fractions as the beta-value [β = M/(M + U + 100)], where M is the intensity of the methylated allele and U is the intensity of the unmethylated [59]. These beta values were log transformed to obtain the log ratio (log[β/(1 – β)]) and the detection p-value for each value, which represents a statistical test for the difference between the signal for each CpG and background noise.
Data pre-preprocessing and normalization were preformed using the minfi package in R [60]. Data verification plots (q-q plots) were created for all children and for each sex. A MethylSet object containing only methylated and unmethylated signals was created using the preprocessRaw function and beta-value densities were inspected to assess sample quality. We performed quantile normalization using preprocessQuantile function from the minfi package. The normalization procedure was applied to the methylated and unmethylated intensities separately. Since probe types and probe regions were confounded and DNA methylation varies across regions we set the stratify options to true. This normalization step also addressed outlier samples and removed poorly performing samples of both methylated and unmethylated channels when small intensities were close to zero. Batch normalization was preformed using ComBat [61] function from sva R package using plate as batch variable. A detection p-value is returned for every genomic position in every sample. A small p-value indicates a good position and significant above the background level. We excluded probes with a detection p-value > 0.01 in more than 20% of the samples. Potential outliers were removed based on the values beyond the lower and upper outer fences (values < 25th percentile minus 3 x interquartile range and values > 75th percentile + interquartile range) based on PACE guidelines [19]. To pursue CpG methylation marks responsive to pre-pregnancy obesity, we also excluded probes that overlap with SNPs using dropLociWithSnps function in minfi package in R, as SNPs inside probe bodies or at nucleotide extensions can alter data interpretation from downstream analysis. The missing data were imputed using champ.impute function from the ChAMP [62] R package using combine method. A total of 486,427 probes on the Illumina Methylation 450k array comprising type I probes (N = 135,476), type II probes (N = 350,036), control probes (N = 850) and SNP probes (N = 65) were assessed. After removal of control probes, probes with a detection p-value > 0.01 in more than 20% of the samples, and those mapping to SNPs, 422,916 CpGs remained from the robust linear regression in 360 umbilical cord blood samples comprised of 173 males and 187 females.
DNA methylation validation
Targeted pyrosequencing among an independent sample of NEST offspring, who were randomly selected from obese and non-obese mothers, was employed to validate two sequence regions. Assays were designed to flank both CpG sites of the most robust CpG methylation differences (p-values = 1e−12) in TAPBP and those of weaker association (p-values = 1e−5) from the HM450K methylation array in chromosome 6 (Chr6), referred to here as region 1 and region 2. Primers targeting Chr6 maternal obesity region 1, (Hg38) chr6:28,943,503–28,943,827, are Chr6Ob1F – 5ʹ AGG AAA ATA GAA GAG GAA TTT GT-3ʹ; Chr6Ob1R – 5ʹ [Btn]CAA AAA ATA CTA AAA AAA AAA CC-3ʹ; Chr6Ob1S – 5ʹ AAT ATT TTA TAT TGG TAG-3ʹ, primers to Chr6 maternal obesity region 2–2, (Hg38) chr6:33,312,586–33,312,726, are Chr6Ob2-2F – 5ʹ cac CAG TGT AAT AGG AGT TAT TTA ATT T-3ʹ; Chr6Ob2-2R – 3ʹ [Btn]ctg aAA AAC AAA AAC TCT TAC ACT C-3ʹ. Bases in lowercase for ChrOb2-2 primers are non-genomic sequences added in order to optimize primer annealing temperatures. Amplification conditions were as follows: 95⁰C – 5ʹ; 94⁰C – 20s, 55⁰C – 20s, 68⁰C – 40s, repeated 45X; 94⁰C – 3ʹ. For pyrosequencing, primer Chr6Ob1S was used for Chr6Ob1 amplicons, primer Chr6Ob2-2F was used for Chr6Ob2-2 amplicons. These assays were used to analyze 25ng bisulfite converted leukocyte DNA of n = 44 offspring, 20 of them born to obese women. Validation of DNAm quantitation used genomic DNA controls with 0% and 100% methylation (Qiagen, Inc.) mixed to generate methylation at 0%, 20%, 40%, 60%, 80%, and 100%.
Statistical analysis
Multivariable linear regression analyses were utilized to estimate the association between pre-pregnancy obesity (BMI≥30 vs. BMI<30 kg/m2) and CpG site methylation. These analyses were adjusted for maternal age, maternal cigarette smoking during pregnancy, ethnicity/race, parity. Cell type proportion were also adjusted for using the estimateCellCounts function in the minfi R package and the Reinius et al. reference dataset [63,64]. The Houseman blood reference dataset was utilized to estimate cell type proportion to compare our findings to existing studies that also used the Houseman reference (i.e., PACE consortium). We also adjusted for multiple comparisons using a false discovery rate (FDR) [65]. As a sensitivity analysis, we examined the sex-specific associations between maternal pre-pregnancy obesity and cell type estimates. The most robustly associated CpG sites, based on number of CpG sites with differential methylation and location within the gene, were then examined for associations with offspring BMI z-score and BP percentiles at age 4–5 years using multivariable linear regression analyses adjusted for maternal age at delivery, race/ethnicity, maternal pre-pregnancy obesity, and smoking status during pregnancy.
DNA methylation replication
Replication was accomplished by analyzing HM450K methylation array data using an identical analysis plan and covariate adjustment set in 751 mother-infant pairs (n = 367 mother-male pairs and n = 384 mother-female pairs) in the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort, despite the lower prevalence of maternal obesity than NEST participants (ALSPAC = 5% vs. NEST = 31%). ALSPAC is a general population pregnancy cohort that initially recruited 14,541 pregnancies with a due date between April 1991 and December 1992 in Avon, UK [66,67]. The study website contains details of all the data that is available through a fully searchable data dictionary (http://www.bris.ac.uk/alspac/researchers/data-access/data-dictionary). Written informed consent was obtained for all ALSPAC participants and ethical approval was granted from the ALSPAC Law and Ethics Committee and the local Research Ethics Committees. Methylation data were generated as part of the Accessible Resource for Integrated Epigenomics Studies (ARIES) project as described previously [68]. Data were normalized using the functional normalization method within the R package meffil [69]. CpG methylation was also measured in mixed cord blood leukocytes.
Funding Statement
The NEST project was conducted and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK: R01DK085173) and National Institute of Aging (NIA: R21AG041048) in collaboration with NEST investigators. This work was also funded by the National Institute of Environmental Health Sciences (grant number T32ES007018) and the National Institute for Minority Health and Health Disparities (grant number K99MD012808).. The UK Medical Research Council and Wellcome (Grant ref.: 102215/2/13/2) and the University of Bristol provide core support for ALSPAC. This publication is the work of the authors and GCS will serve as a guarantor for the contents of this paper. ALSPAC methylation data was generated through grants from the British Biological Sciences Research Council (BB/I025751/1 and BB/I025263/1). GCS and CLR work at the MRC Integrative Epidemiology Unit at the University of Bristol (grant numbers MC_UU_12013/2). The Bioinformatics analysis was supported in part by NIEHS under award number P30ES025128. The funders have no role in the research question, study design, data collection, statistical analysis, or preparation of the manuscript;National Institute of Environmental Health Sciences [T32ES007018];National Institute of Environmental Health Sciences [P30ES025128];National Institute on Aging [R21AG041048];National Institute on Minority Health and Health Disparities [K99MD012808];MRC Integrative Epidemiology Unit, The UK Medical Research Council and Wellcome [102215/2/13/2];University of Bristol [MC_UU_12013_1, MC_UU_12013_2].
Acknowledgments
We are thankful of the participants of the NEST study project and families of the ALSPAC study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material for this article can be accessed here.
References
- [1].Ogden CL, Carroll MD, Lawman HG, et al. Trends in obesity prevalence among children and adolescents in the United States, 1988–1994 Through 2013-2014. JAMA. 2016. June 7;315(21): 2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ogden CL, Carroll MD, Kit BK, et al. Prevalence of obesity and trends in body mass index among US children and adolescents, 1999–2010. JAMA. 2012;307(5):483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fuemmeler BF, Wang L, Iversen ES, et al. Association between prepregnancy body mass index and gestational weight gain with size, tempo, and velocity of infant growth: analysis of the newborn epigenetic study cohort. Child Obes [Internet] 2016. June [cited 2018 May 7];12(3): 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Oken E, Kleinman KP, Belfort MB, et al. Associations of gestational weight gain with short- and longer-term maternal and child health outcomes. Am J Epidemiol [Internet] 2009. July 15 [cited 2018 May 7];170(2): 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Taveras EM, Rifas-Shiman SL, Belfort MB, et al. Weight status in the first 6 months of life and obesity at 3 years of age. Pediatrics [Internet] 2009. April 1 [cited 2018 May 7];123(4): 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barker DJ. Fetal origins of coronary heart disease. BMJ [Internet] 1995. July 15 [cited 2018 May 7];311(6998): 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Claesson I-M, Sydsjö G, Olhager E, et al. Effects of a gestational weight gain restriction program for obese pregnant women: children’s weight development during the first five years of life. Child Obes [Internet] 2016. June [cited 2018 May 7];12(3): 162–170. [DOI] [PubMed] [Google Scholar]
- [8].Gregory EF, Goldshore MA, Henderson JL, et al. Infant growth following maternal participation in a gestational weight management intervention. Child Obes [Internet] 2016. June [cited 2018 May 7];12(3): 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tanvig M. Offspring body size and metabolic profile - effects of lifestyle intervention in obese pregnant women. Dan Med J [Internet] 2014. July [cited 2018 May 7];61(7): B4893. [PubMed] [Google Scholar]
- [10].Dolinoy DC, Weidman JR, Waterland RA, et al. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect [Internet] 2006. April [cited 2018 May 7];114(4): 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Woo Baidal JA, Locks LM, Cheng ER, et al. Risk factors for childhood obesity in the first 1,000 days. Am J Prev Med [Internet] 2016. June [cited 2018 May 7];50(6): 761–779. [DOI] [PubMed] [Google Scholar]
- [12].Branum AM, Kirmeyer SE, Gregory ECW. Prepregnancy body mass index by maternal characteristics and state: data from the birth certificate, 2014. Natl Vital Stat Rep [Internet] 2016. August [cited 2018 May 7];65(6): 1–11. [PubMed] [Google Scholar]
- [13].Waterland RA. Do maternal methyl supplements in mice affect DNA methylation of offspring? J Nutr [Internet] 2003. January 1 [cited 2018 May 7];133(1): 238; author reply 239. [DOI] [PubMed] [Google Scholar]
- [14].Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA [Internet] 2008. November 4 [cited 2018 May 7];105(44): 17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, et al. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. Zhang C, editor PLoS One. 2009. November 16 [cited.2018 May 7];4(11): e7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hoyo C, Daltveit AK, Iversen E, et al. Erythrocyte folate concentrations, CpG methylation at genomically imprinted domains, and birth weight in a multiethnic newborn cohort. Epigenetics [Internet] 2014. August 6 [cited 2018 May 7];9(8): 1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vidal AC, Murphy SK, Murtha AP, et al. Associations between antibiotic exposure during pregnancy, birth weight and aberrant methylation at imprinted genes among offspring. Int J Obes (Lond) [Internet] 2013. July 28 [cited 2018 May 7];37(7): 907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sharp GC, Lawlor DA, Richmond RC, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the avon longitudinal study of parents and children. Int J Epidemiol. 2015;44(4):1288–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sharp GC, Salas LA, Monnereau C, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet [Internet] 2017;26(20): 4067–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Michels KB, Harris HR, Barault L. Birthweight, maternal weight trajectories and global DNA methylation of LINE-1 repetitive elements. Fugmann SD, editor PLoS One. 2011. September 28 [cited.2018 May 7];6(9): e25254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Herbstman JB, Wang S, Perera FP, et al. Predictors and consequences of global DNA methylation in cord blood and at three years. El-Maarri O, editor PLoS One. 2013. September 4 [cited 2018 May 7];8(9): e72824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu X, Chen Q, Tsai HJ, et al. Maternal preconception body mass index and offspring cord blood DNA methylation: exploration of early life origins of disease. Environ Mol Mutagen. 2014;55(3):223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen EY, Tan CM Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; April 15; 14:128. [DOI] [PMC free article] [PubMed]
- [24].Kuleshov MV, Jones MR Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016. July 8; 44(W1):W90-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van Dijk SJ, Peters TJ, Buckley M, et al. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int J Obes [Internet] 2017;42(1): 28–35. [DOI] [PubMed] [Google Scholar]
- [26].Maloney CA, Hay SM, Young LE, et al. A methyl-deficient diet fed to rat dams during the peri-conception period programs glucose homeostasis in adult male but not female offspring. J Nutr [Internet] 2011. January 1 [cited 2018 May 8];141(1): 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mueller BR, Bale TL. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol Behav [Internet] 2007. May 16 [cited 2018 May 8];91(1): 55–65. [DOI] [PubMed] [Google Scholar]
- [28].Bruce-Keller AJ, Fernandez-Kim S-O, Townsend RL, et al. Maternal obese-type gut microbiota differentially impact cognition, anxiety and compulsive behavior in male and female offspring in mice. Rosenfeld CS, editor PLoS One [Internet] 2017. April 25 [cited 2018 May 8];12(4): e0175577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Soubry A, Schildkraut JM, Murtha A, et al. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC Med [Internet] 2013. February 6 [cited 2018 May 8];11(1): 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Soubry A, Murphy SK, Wang F, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes (Lond) [Internet] 2015. April 25 [cited 2018 May 8];39(4): 650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ortmann B, Copeman J, Lehner PJ, et al. A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP complexes. Science [Internet] 1997. August 29 [cited 2018 May 8];277(5330): 1306–1309. [DOI] [PubMed] [Google Scholar]
- [32].Tang Y, Wang J, Zhang Y, et al. Correlation between low tapasin expression and impaired CD8+ T‑cell function in patients with chronic hepatitis B. Mol Med Rep [Internet] 2016. October [cited 2018 May 8];14(4): 3315–3322. [DOI] [PubMed] [Google Scholar]
- [33].Shionoya Y, Kanaseki T, Miyamoto S, et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology [Internet] 2017. February 8 [cited 2018 May 8];6(2): e1274476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sokol L, Koelzer VH, Rau TT, et al. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J Transl Med [Internet] 2015. August 27 [cited 2018 May 8];13(1): 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jiang Q, Pan H, Ye D, et al. Downregulation of tapasin expression in primary human oral squamous cell carcinoma: association with clinical outcome. Tumour Biol [Internet] 2010. October 8 [cited 2018 May 8];31(5): 451–459. [DOI] [PubMed] [Google Scholar]
- [36].Bukulmez H, Fife M, Tsoras M, et al. Tapasin gene polymorphism in systemic onset juvenile rheumatoid arthritis: a family-based case-control study. Arthritis Res Ther [Internet] 2005. [cited 2018 May 8];7(2): R285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lee H-M, Sugino H, Aoki C, et al. Abnormal networks of immune response-related molecules in bone marrow cells from patients with rheumatoid arthritis as revealed by DNA microarray analysis. Arthritis Res Ther [Internet] 2011. June 16 [cited 2018 May 8];13(3): R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xiao X, Hao J, Wen Y, Wang W, Guo X Zhang F.. Genome-wide association studies and gene expression profiles of rheumatoid arthritis: an analysis. Bone Joint Res 2016. July;5(7):314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Abarca-Heidemann K, Friederichs S, Klamp T, et al, Regulation of the expression of mouse TAP-associated glycoprotein (tapasin) by cytokines. Immunol Lett [Internet] 2002. October 1 [cited 2018 May 8];83(3): 197–207. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12095710 [DOI] [PubMed] [Google Scholar]
- [40].Mattos RT, Medeiros NI, Menezes CA, et al. Chronic low-grade inflammation in childhood obesity is associated with decreased IL-10 expression by monocyte subsets. Richard Y, editor PLoS One. 2016. December 15 [cited 2018 May 8];11(12): e0168610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pachón-Peña G, Serena C, Ejarque M, et al. Obesity determines the immunophenotypic profile and functional characteristics of human mesenchymal stem cells from adipose tissue. Stem Cells Transl Med [Internet] 2016. April [cited 2018 May 8];5(4): 464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].El Azreq M-A, Kadiri M, Boisvert M, et al. Discoidin domain receptor 1 promotes Th17 cell migration by activating the RhoA/ROCK/MAPK/ERK signaling pathway. Oncotarget [Internet] 2016. July 19 [cited 2018 May 8];7(29): 44975–44990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kadiri M, El Azreq M-A, Berrazouane S, et al. Human Th17 migration in three-dimensional collagen involves p38 MAPK. J Cell Biochem [Internet] 2017. September [cited 2018 May 8];118(9): 2819–2827. [DOI] [PubMed] [Google Scholar]
- [44].Ogino S, Nosho K, Baba Y, et al. A cohort study of STMN1 expression in colorectal cancer: body mass index and prognosis. Am J Gastroenterol. 2009;104(8):2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pekkala S, Munukka E, Kong L, et al. Toll-like receptor 5 in obesity: the role of gut microbiota and adipose tissue inflammation. Obesity. 2015;23(3):581–590. [DOI] [PubMed] [Google Scholar]
- [46].Kim JY, Campbell LE, Shaibi GQ, et al. Gene expression profiling and association of circulating lactoferrin level with obesity-related phenotypes in Latino youth. Pediatr Obes [Internet] 2015. October [cited 2018 May 8];10(5): 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vijay-Kumar M, Aitken JD, Carvalho FA, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science [Internet] 2010. April 9 [cited 2018 May 8];328(5975): 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rubin CI, Atweh GF. The role of stathmin in the regulation of the cell cycle. J Cell Biochem. 2004;93(2):242–250. [DOI] [PubMed] [Google Scholar]
- [49].Soubry A, Hoyo C, Jirtle RL, et al. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. Bioessays [Internet] 2014. April [cited 2018 May 8];36(4): 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Soubry A, Guo L, Huang Z, et al. Obesity-related DNA methylation at imprinted genes in human sperm: results from the TIEGER study. Clin Epigenetics [Internet] 2016. December 6 [cited 2018 May 8];8(1): 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].McCullough LE, Mendez MA, Miller EE, et al. Associations between prenatal physical activity, birth weight, and DNA methylation at genomically imprinted domains in a multiethnic newborn cohort. Epigenetics [Internet] 2015. July 3 [cited 2018 May 7];10(7): 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hoyo C, Murtha AP, Schildkraut JM, et al. Folic acid supplementation before and during pregnancy in the Newborn Epigenetics STudy (NEST). BMC Public Health [Internet] 2011. January 21 [cited 2018 May 7]; 11(1): 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].World Health Organization. Obesity: preventing and managing the global epidemic. Report of a WHO Consultation of Obesity. Geneva, 3-5 June 1997. [PubMed] [Google Scholar]
- [54].Kuczmarski RJ, Ogden CL, Guo SS, Flegal KM, Mei Z, Wei R, Curtin LR, Roche AF, Johnson CL 2000. CDC growth charts for the United States: Methods and development. National Center for Health Statistics. Vita Health Stat. 2000; 11(246) [PubMed] [Google Scholar]
- [55].National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2):555–576. [PubMed] [Google Scholar]
- [56].Murphy SK, Adigun A, Huang Z, et al. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene [Internet] 2012. February 15 [cited 2018 May 8];494(1): 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98(4):680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Joubert BR, Håberg SE, Nilsen RM, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect [Internet] 2012. July 31 [cited 2018 May 8];120(10): 1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics [Internet] 2011. October [cited 2018 May 8];98(4): 288–295. [DOI] [PubMed] [Google Scholar]
- [60].Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics [Internet] 2014. May 15 [cited 2018 May 8];30(10): 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Leek JT, Johnson WE, Parker HS, et al. Sva: surrogate variable analysis. R Package Version 3300. 2018. [Google Scholar]
- [62].Tian Y, Morris TJ, Webster AP, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33(24):3982–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics [Internet] 2012. May 8 [cited 2018 May 8];13(1): 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Reinius LE, Acevedo N, Joerink M, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. Ting AH, editor PLoS One. 2012. July 25 [cited 2018 May 8];7(7): e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing [Internet]. J R Stat Soc Series B Stat Methodol. WileyRoyal Statistical Society; 1995. [cited 2018];57: 289–300. [Google Scholar]
- [66].Boyd A, Golding J, Macleod J, et al. Cohort Profile: the “children of the 90s”–the index offspring of the avon longitudinal study of parents and children. Int J Epidemiol [Internet] 2013. February [cited 2018 May 8];42(1): 111–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fraser A, Macdonald-Wallis C, Tilling K, et al. Cohort profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int J Epidemiol [Internet] 2013. February 1 [cited 2018 May 8];42(1): 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Relton CL, Gaunt T, McArdle W, et al. Data resource profile: accessible Resource for Integrated Epigenomic Studies (ARIES). Int J Epidemiol [Internet] 2015. August [cited 2018 May 8];44(4): 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Min J, Hemani G, Davey Smith G, et al. Meffil: efficient normalisation and analysis of very large DNA methylation samples. Bioinformatics. 2018. December 1; 34(23):3983-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.