

ABSTRACT
Parkinson’s Disease (PD) is a common neurodegenerative disorder currently diagnosed based on the presentation of characteristic movement symptoms. Unfortunately, patients exhibiting these symptoms have already undergone significant dopaminergic neuronal loss. Earlier diagnosis, aided by molecular biomarkers specific to PD, would improve overall patient care. Epigenetic mechanisms, which are modified by both environment and disease pathophysiology, are emerging as important components of neurodegeneration. Alterations to the PD methylome have been reported in epigenome-wide association studies. However, the extent to which methylation changes correlate with disease progression has not yet been reported; nor the degree to which methylation is affected by PD medication.
We performed a longitudinal genome-wide methylation study surveying ~850,000 CpG sites in whole blood from 189 well-characterized PD patients and 191 control individuals obtained at baseline and at a follow-up visit ~2 y later. We identified distinct patterns of methylation in PD cases versus controls. Importantly, we identified genomic sites where methylation changes longitudinally as the disease progresses. Moreover, we identified methylation changes associated with PD pathology through the analysis of PD cases that were not exposed to anti-parkinsonian therapy. In addition, we identified methylation sites modulated by exposure to dopamine replacement drugs.
These results indicate that DNA methylation is dynamic in PD and changes over time during disease progression. To the best of our knowledge, this is the first longitudinal epigenome-wide methylation analysis for Parkinson’s disease and reveals changes associated with disease progression and in response to dopaminergic medications in the blood methylome.
KEYWORDS: Parkinson’s disease, DNA methylation, longitudinal, blood epigenome, disease progression, biomarkers, dopamine replacement therapy, one-carbon metabolism
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disorder of the elderly, currently affecting ~2% of the population over 60 y of age [1]. PD presents clinically as a progressive movement disorder with resting tremor and postural instability, and it is characterized neuropathologically by intracytoplasmic α-synuclein (α-syn) aggregates in Lewy bodies [2]. Neurodegeneration occurs primarily in dopaminergic neurons of the substantia nigra, but Lewy body pathology occurs in limbic and cortical areas as PD progresses [3].
PD is a multifactorial disease where environmental and genetic factors are intricately associated. In idiopathic PD, 60–70% of dopaminergic neurons have already been lost by the time someone presents clinical symptoms sufficient for a diagnosis [4]. Multiple pre-motor biomarkers are actively being investigated for their potential to identify early-stage PD or patients at risk for developing PD [5,6], including clinical measures (rapid eye movement behaviour disorder (RBD), olfactory deficits, mood disorders); molecular measures (α-syn in cerebrospinal fluid and blood [7,8]); and brain imaging.
Epigenetic mechanisms are emerging as important factors in the molecular aetiology of neurodegenerative diseases, including PD [9,10]. Hypomethylation of the α-synuclein gene (SNCA) promotor region has been reported in substantia nigra of PD patients [11,12]. Moreover, this SNCA promotor hypomethylation has been shown to increase SNCA protein expression in cell culture, possibly contributing to the pathology of PD. Interestingly, L-dopa therapy has been associated with hypermethylation of the SNCA promotor, suggesting that current PD therapy may alter methylation [13]. While results on altered SNCA methylation in PD have not been replicated by other studies using smaller cohorts [14,15], epigenomic changes associated with other genes including hypomethylation of NPAS2 [16] and CYP2E1 [17]; and hypermethylation of PGC1-α [18] and the H1 haplotype of Tau (MAPT) [19], have also been implicated in PD.
We previously demonstrated alterations in the intracellular localization of DNA methyltransferase 1 (DNMT1), which catalyses the addition of methyl groups to the DNA, and that appeared retained in the cytoplasm in neurons from PD patients [20], due to interaction with misfolded α-syn. These observations provided a potential mechanistic explanation for the hypomethylation of multiple genes in PD and suggested global alterations in the methylome of PD patients. Indeed, we subsequently identified methylation changes in brain and blood samples from PD cases in comparison to control subjects in a pilot study using a small cohort. Notably, we identified concordant methylation changes in matching brain and blood samples, thus supporting the idea that blood may serve as a surrogate tissue for brain methylation analyses in PD [21]. Subsequently, Moore et al. [22] reported a subset of CpG sites with altered methylation in PD and confirmed two CpG sites via bisulphite sequencing in the second cohort of 219 PD patients versus 223 control individuals. Most recently, an epigenome-wide association study (EWAS) using 335 PD and 237 control blood DNA samples identified 82 CpG sites of altered DNA methylation in PD patients, also using the previous generation Infinium 450K HumanMethylation beadchip [23].
These studies provide indications that there are significant alterations to the methylome of PD patients in both brain and blood tissues. Noteworthy, these previous studies were based on single-time point samples, hindering the observation of potential epigenomic changes associated with disease progression. Here we report results of the first longitudinal analysis of genome-wide methylation surveying over 850,000 CpG sites in 189 PD patients and 191 controls enrolled in the Harvard Biomarkers Study at both baseline and a follow-up visit that was ~2 y later. We present evidence of specific methylation changes associated with PD status, disease progression, and PD medication.
Results
Our study included 380 participants from the Harvard Biomarker Study [24,25], 189 patients with PD (31% females; average age of 68 at enrollment visit) and 191 control subjects (34% females; average age of 69 at baseline, Table 1). From the 380 participants, 313 self-reported as white non-Hispanic; 40 as white Hispanic/Latino; 1 as Asian non-Hispanic; 2 as African–American Hispanic/Latino. Data was not available for the remaining 24 subjects. Levels of education did not significantly vary between cases and controls, and overall 82% of participants attended college. There were no significant differences in red or white blood cell count or in cognitive status (as per Mini-Mental State Examination, MMSE) between cases and controls at baseline or follow-up. We profiled samples obtained at the enrollment visit (baseline point) and a second longitudinal corresponding sample, which was collected on average 2.2 y later (SD = 0.85) and used as the follow-up point. The time elapsed between visits ranged between 0.8 and 11.98 y, and the mean of distributions was similar between controls and PD cases (p = 0.2206 as per unpaired t-test). We selected PD cases with longitudinally confirmed clinical diagnoses and with Hoehn and Yahr scale scores ≤3 at baseline, representing early-stage or mild PD. Disease duration was similar between female and male PD patients, with an average duration of 3.7 y (SD = 3.97) at baseline. During the period elapsed between baseline and follow-up visits no changes in cognition were observed in controls, while PD patients showed decay in MMSE performance, which reached only significance for males (Table 1). Smoking has been largely reported as a protective factor and inversely correlated with the occurrence of PD [26]. In our study, 41.8% of PD cases and 29.8% of controls reported to have previously smoked. The number of active smokers was low with only one PD case being an active smoker in comparison to seven control subjects still smoking (Table 1). Smoking status did not change between baseline and follow up visits and was included as a covariate in our analysis. Overall, PD patients showed a worsening in clinical manifestations as per HY scores, which changed significantly between baseline and follow up visits (p < 0.0001, as per paired t-test), indicating disease progression. Similarly, the Unified Parkinson’s Disease Rating Scale (UPDRS) scores increased between baseline and follow-up visits at an average rate of 3.05 points/year (p < 0.0001, as per paired t-test), consistent with disease worsening and reported progression rates.
Table 1.
Clinical and demographic characterization of the HBS study cohort.
| Phenotype | PD | Control |
|---|---|---|
| Female/Male | 59/130 | 64/127 |
| Age at baseline visit 1 | 67.31 (6.95)/68.08 (7.05) | 66.83 (2.63)/70.89 (6.16) *** |
| Age at follow-up visit | 69.58 (7.34)/70.33 (7.05) | 69.05 (2.54)/72.98 (6.03) ** |
| Education at baseline 2 | 15.25 (1.57)/15.22 (1.93) | 15.63 (1.18)/14.98 (2.07) |
| Ever Smoked | 27/53 | 20/37 |
| Current Smoker (baseline) | 1/0 | 2/5 |
| Current Smoker (follow-up) | 1/0 | 2/5 |
| Disease Duration at baseline3 | 3.73 (3.99)/3.72 (3.97) | |
| HY 4 (baseline) | 2.00 (0.48)/2.01 (0.42) | |
| HY (follow-up) | 2.33 (0.60)/2.20 (0.42) *** | |
| t-test HY 5 | 0.001717/0.0005481 | |
| MMSE 6 (baseline) | 29.02 (1.03)/28.98 (1.08) | 29.26 (0.92)/28.84 (1.17) |
| MMSE (follow-up) | 28.71 (1.95)/28.73 (1.61) | 29.22 (1.24)/28.96 (1.27) |
| WBC 7 (baseline) | 6.28 (1.33)/6.70 (1.59) | 6.11 (1.53)/6.41 (1.75) |
| WBC (follow-up) | 6.48 (1.54)/6.54 (1.55) | 6.15 (1.71)/6.40 (1.65) |
| RBC 8 (baseline) | 4.56 (1.23)/4.70 (0.42) | 4.45 (0.34)/4.75 (0.42) |
| RBC (follow-up) | 4.37 (0.39)/4.58 (0.52) | 4.44 (0.36)/4.69 (0.42) |
| De novo 9 (baseline) | 5/14 | |
| De novo (follow-up) | 1/2 | |
| on L-dopa/carbidopa or entacapone medication10 (baseline) | 38/82 | |
| on L-dopa/carbidopa or entacapone medication (follow-up) | 50/108 |
Values are expressed means with (SD) *** indicates p value <0.001 and ** indicates p value <0.01 for inter-group comparisons as per Student’s t test. (1) Age expressed in years. (2) Education expressed in years. College or above = 16; High school = 12; elementary school = 5. (3) Disease duration is calculated in years since diagnosis. A value of 0 is assigned is at baseline if the patient has received a diagnosis of PD during the same year of enrolling in the study. (4) Modified Hoehn and Yahr scale for clinical staging of Parkinson’s disease [46]. (5) Indicates p value of Welch two-sided two-sample t-test comparing the indicated category between enrollment and follow-up visits. Female and Male groups were analysed separately. Only provided for significant differences. (6) Mini-Mental State Examination. (7) White blood cells count. (8) Red blood cells count. (9) De novo patients that yet did not receive any type of anti-parkinsonian medication. (10) Based on Parkinson’s disease treatment that may affect one-carbon metabolism as defined in our study, including Sinemet; Comtan and Stalevo. Data was not available for: HY enrollment 2 cases; HY follow-up 13 PD cases; MMSE enrollment 61 CT cases; MMSE follow-up 67 CT and 2 PD; WBC/RBC enrollment 10 CT and 6 PD; WBC/RBC follow-up 48 CT and 50 PD cases.
Estimation of blood cell composition using methylation data
We used whole blood DNA to profile methylation; therefore, different lymphocyte cell type distributions between cases and controls may confound the analysis. We used distinctive cell-specific methylation profiles to estimate the proportional abundance of blood cell types and to evaluate whether alterations in white blood cell composition may be associated with PD pathology and have the potential to drive differential methylation between cases and controls. We applied the ‘estimate-CellCounts’ function in minfi [27] to estimate the proportional abundance of blood cell types in our study samples based on the intensity of specific probes present in the EPIC array. We observed that granulocytes (as a group, including neutrophils) were the most abundant cells in blood, as expected (Figure 1). Overall blood cell composition varied between control and PD groups. At baseline, PD patients showed higher estimated levels of granulocytes (p = 4.0E-6, as per t-test) and lower estimated B-cells (p = 0.0019) and NKs (p = 0.00055) in comparison to controls. These differences only persisted for granulocytes, which were higher in PD cases (p = 0.0066) and natural killers, which were lower in PD (p = 0.00065) in the follow-up visit. Intra-group analysis showed that only granulocytes (p = 0.00063) changed longitudinally in control subjects, while no changes were observed in PD cases between the time points analysed (Figure 1).
Figure 1.
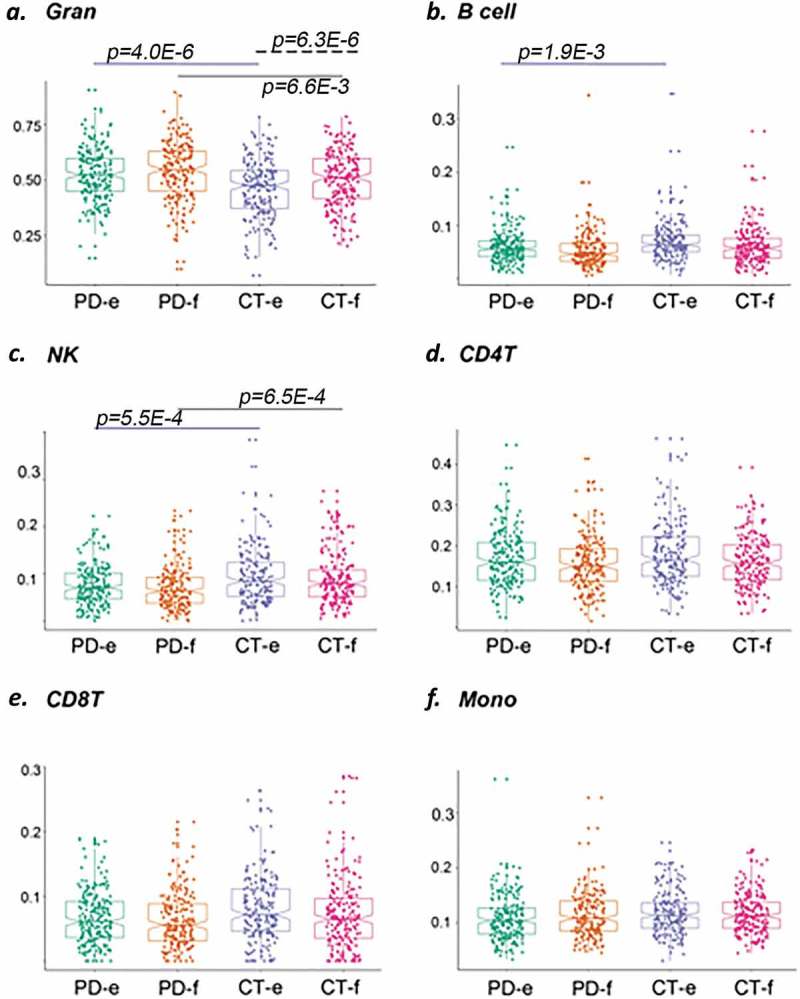
Comparison of individual cell type across control (CT) and PD groups at enrollment (e) and follow-up (f). Abundance of specific blood cell types was estimated based on unique methylation markers for cell identity. Shown in (a) granulocytes, in (b) B cells, in (c) natural killer cells, in (d) CD4T cells, in (e) CD8T cells, and in (f) monocytes. Blue solid line indicates comparison between PD cases vs. CT subjects at enrollment; black solid line indicates comparison between PD cases vs. CT subjects at follow-up; dash blue line indicates comparison between PD cases at follow-up vs. enrollment time points; dash black line indicates comparison between CT subjects at follow-up vs. enrollment time points. p-Value for the differences in cell composition estimates across groups as per Wilcoxon test after correction for multiple observations is indicated.
Methylation changes associated with Parkinson’s disease: cross-sectional analysis
We first conducted a cross-sectional analysis by comparing methylation profiles between cases and controls regardless of time of visit to define disease-associated changes in methylation, while accounting for repeated measures, using limma. We identified 7 probes showing differential methylation (DMPs) in PD with genome-wide scale significance at a p-value <5.0E-7 (adjusted p-value<0.05) and another 46 DMPs with marginal significance of adj. p < 0.2 (Figure 2; Table 2 and Sup. Table 1). Overall changes in methylation were modest, with |log2 FC| <0.6 (in M-values). Among the top changes in this group, we identified multiple CpGs clustering around 200 bp of the TSS of the Lamin Tail Domain Containing 1 (IFLTD1/LMNTD1), an intermediate filament protein, and Delta-Like Non-Canonical Notch Ligand 1 (DLK1), a transmembrane protein involved in differentiation of multiple cell types. Additional functional categories represented in this list (Table 2) include microtubule-associated proteins Doublecortin Like Kinase 1 (DCLK1: p = 3.19E-6) and Dynein Cytoplasmic 1 Heavy Chain 1 (DYNC1H1: p = 3.24E-6); the transcription factor LIM Domain Only 3 (LMO3: p = 3.68E-6), and a neurotransmission regulator synaptotagmin 12 (SYT12: p = 2.79E-7). These represent diverse cellular processes, some of which would not be expected to be important in blood cells (LMO3, which is brain specific; SYT12, which has a neuron-specific function). Thus, these findings may be reflective of similar alterations in the brain. This is supported by the finding of CYP2E1 in this study (Table 2) and previous reports of altered CYP2E1 methylation in the PD brain [17].
Figure 2.
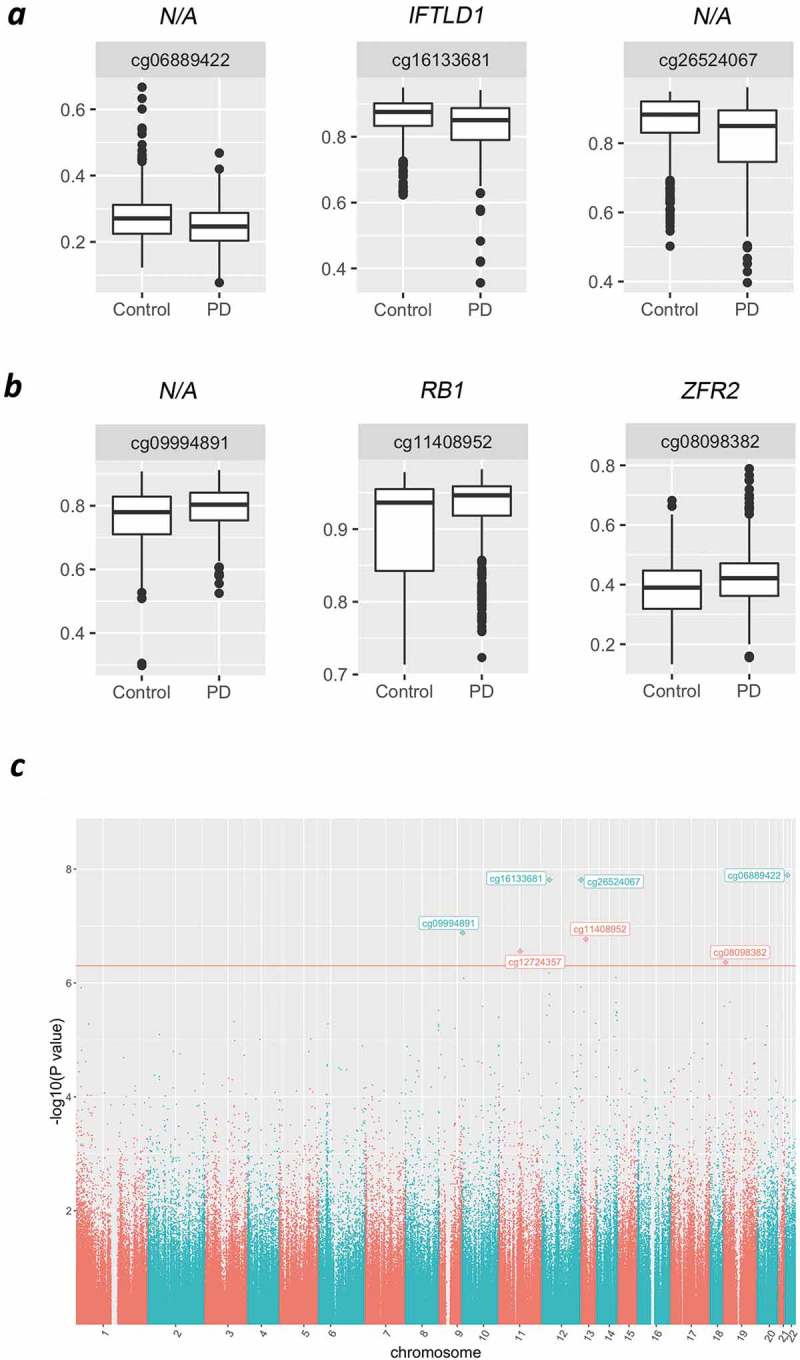
Case versus control comparisons in the HBS study. Representative box plots of top significant DMPs showing decreased (a) or increased (b) methylation in PD cases in comparison to control subjects. (c) Manhattan plot compiling genome-wide methylation sites and highlighting significant DMPs for the cross-sectional comparison of PD vs. CT. Red line indicates significance cut-off at FRD<0.05.
Table 2.
Top differentially methylated probes (DMPs) in PD in the cross-sectional analysis.
| Name | Chr | Position | Relation to TSS | Gene ID | logFC | P.Value | FDR |
|---|---|---|---|---|---|---|---|
| cg06889422 | chr22 | 24627294 | Body | N/A | −0.28384 | 1.28E-08 | 0.003907 |
| cg16133681 | chr12 | 25801621 | TSS200 | IFLTD1 | −0.38619 | 1.54E-08 | 0.003907 |
| cg26524067 | chr12 | 133003928 | Open Sea | N/A | −0.49670 | 1.55E-08 | 0.003907 |
| cg09994891 | chr10 | 2173024 | Open Sea | N/A | 0.31875 | 1.31E-07 | 0.024816 |
| cg11408952 | chr13 | 48892244 | Body | RB1 | 0.48620 | 1.72E-07 | 0.025987 |
| cg12724357 | chr11 | 66790285 | TSS1500 | SYT12 | 0.24073 | 2.79E-07 | 0.035123 |
| cg08098382 | chr19 | 3869345 | TSS1500 | ZFR2 | 0.30684 | 4.35E-07 | 0.046984 |
| cg23979954 | chr12 | 25801601 | TSS200 | IFLTD1 | −0.30554 | 6.65E-07 | 0.062331 |
| cg18279536 | chr14 | 101194748 | Body | DLK1 | 0.18266 | 8.04E-07 | 0.062331 |
| cg10405605 | chr10 | 6188149 | TSS200 | PFKFB3 | 0.15204 | 8.24E-07 | 0.062331 |
| cg04741728 | chr12 | 133003907 | Open Sea | N/A | −0.59444 | 1.17E-06 | 0.076787 |
| cg20787649 | chr1 | 17636898 | Body | N/A | 0.52986 | 1.22E-06 | 0.076787 |
| cg03681383 | chr12 | 25801522 | TSS200 | IFLTD1 | −0.30440 | 1.56E-06 | 0.090741 |
| cg12342048 | chr19 | 11465311 | Body | AC024575.1 | −0.24711 | 2.16E-06 | 0.108120 |
| cg19628497 | chr14 | 101194267 | Body | DLK1 | 0.20548 | 2.22E-06 | 0.108120 |
| cg13211181 | chr12 | 25801455 | 1stExon | IFLTD1 | −0.26773 | 2.52E-06 | 0.108120 |
| cg00515755 | chr19 | 1005248 | Body | N/A | 0.20412 | 2.57E-06 | 0.108120 |
| cg11469325 | chr10 | 75012359 | 1stExon | MRPS16 | −0.29904 | 2.57E-06 | 0.108120 |
| cg04224786 | chr8 | 144222401 | Open Sea | N/A | 0.26984 | 3.03E-06 | 0.109568 |
| cg08766508 | chr13 | 36430582 | TSS1500 | DCLK1 | 0.31522 | 3.19E-06 | 0.109568 |
| cg25588820 | Chr12 | 108070383 | Open Sea | N/A | −0.27810 | 3.19E-06 | 0.109568 |
| cg12127149 | chr14 | 102487020 | Body | DYNC1H1 | 0.09433 | 3.24E-06 | 0.109568 |
| cg05763097 | chr14 | 103569340 | Body | EXOC3L4 | 0.29651 | 3.33E-06 | 0.109568 |
| cg01181415 | chr12 | 16757954 | 5ʹUTR | LMO3 | −0.13991 | 3.68E-06 | 0.112632 |
| cg18121862 | chr14 | 101195312 | Body | N/A | 0.15255 | 3.72E-06 | 0.112632 |
| cg13315147 | chr10 | 135341528 | Body | CYP2E1 | 0.35550 | 4.02E-06 | 0.117046 |
| cg18397450 | chr14 | 105830631 | Body | PACS2 | −0.38021 | 4.56E-06 | 0.122011 |
| cg21435367 | chr3 | 133574742 | Body | RAB6B | −0.25162 | 4.80E-06 | 0.122011 |
| cg15756507 | chr17 | 65471461 | Body | N/A | 0.13906 | 5.05E-06 | 0.122011 |
Name indicates probe designation at Illumina EPIC human methylation array; Chr is chromosome location of the CG; position refers to Genome Reference Consortium Human Build 37 (GRCh37) hg19; N/A indicates no annotated gene associated with the probe.
Noteworthy, many DMPs associated with PD present intermediate levels of methylation (β values>0.2 and <0.8), which represent the more dynamic portion of the methylome and more likely to respond to environmental/physiological cues, and which may reflect intrinsic alterations due to disease progression.
Identification of differentially methylated regions (DMRs)
DNA methylation is influenced by CpG topology and methylation in one site is dependent on the methylation status of nearby CpGs by potential cooperation in recruiting methylating/demethylating factors, extending their activity in wider local DNA domains [28]. CpG clusters showing concerted changes in methylation are deemed highly relevant in the modulation of transcription. We searched for differentially methylated regions (DMRs), or groups of at least four CpGs within proximal genomic locations using DMRCate [29], a stringent model that links proximal sites after testing their significance as individual DMPs (Figure 3, Table 3). Among the top DMRs in PD, we identified CYP2E1, with 13 CGs altered (p = 1.22E-22). This gene was one of the first reported as differentially methylated in PD brains [17]. Therefore, our results in whole blood are consistent with this previous finding and support a role for methylation in regulating this gene, which contributes to cholesterol and lipid metabolism, pathways that are altered in PD, also highlighting the utility of whole blood profiling. Another notable DMR is LY6G5C, with five CGs altered (p = 3.92E-9), which has recently been identified as a brain-specific epigenetic marker of schizophrenia [30].
Figure 3.
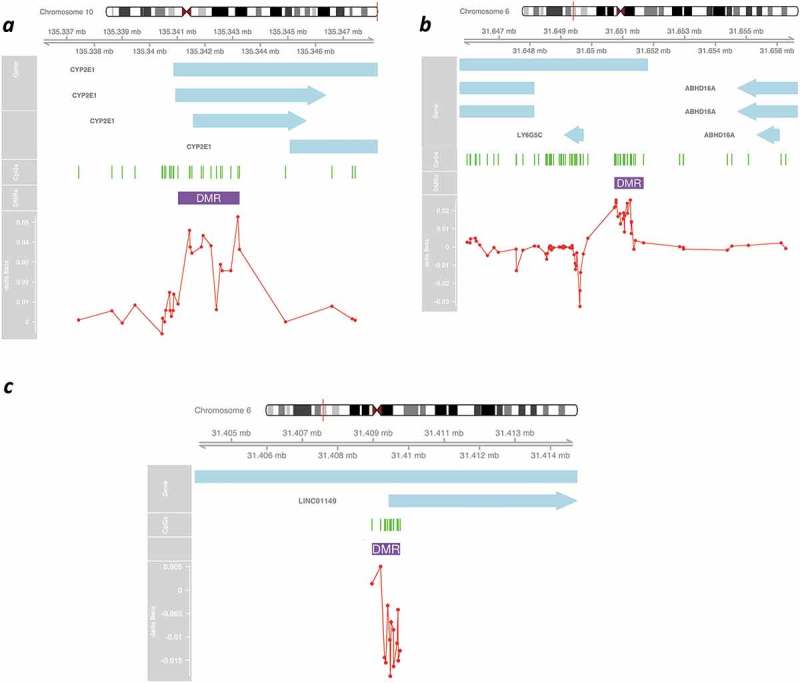
Graphical representation of differentially methylated regions (DMRs) in PD cases in the cross-sectional study. Representative schematics for top DMRs (Table 3) associated with CYP2E1 (a); LY6G5C (b) and CCDC89 (c). Genomic location is indicated by chromosome position (based on Genome Reference Consortium Human Build 37 (GRCh37) hg19). Transcripts are indicated by light blue arrows. CGs appear as green lines. Red line represents Delta Beta of PD vs CT comparison for all the CGs constituting the significant region.
Table 3.
Differentially methylated regions (DMRs) associated with PD.
| Chr | start | end | #CGs | minfdr | Stouffer | maxbetafc |
Overlapping promoters |
|---|---|---|---|---|---|---|---|
| Regions with increased methylation in PD cases | |||||||
| chr10 | 135341025 | 135343248 | 13 | 1.22E-22 | 0.00004 | 0.06768 | CYP2E1 |
| chr19 | 54945959 | 54946993 | 6 | 4.14E-15 | 0.00019 | 0.05752 | AC008746.3 |
| chr18 | 72916776 | 72917390 | 6 | 1.14E-10 | 0.03901 | 0.04425 | ZADH2 |
| chr8 | 144222015 | 144222455 | 5 | 2.54E-18 | 0.00001 | 0.04395 | N/A |
| chr1 | 153599479 | 153600156 | 15 | 1.05E-12 | 0.10454 | 0.04178 | S100A1 |
| chr11 | 2889840 | 2891360 | 35 | 1.04E-20 | 0.81658 | 0.03802 | KCNQ1DN |
| chr14 | 101194145 | 101195312 | 4 | 9.83E-17 | 0.00000 | 0.03520 | DLK1 |
| chr6 | 31650786 | 31650930 | 5 | 3.92E-09 | 0.38577 | 0.03252 | LY6G5C |
| chr6 | 28983835 | 28984341 | 5 | 6.20E-12 | 0.19530 | 0.03166 | N/A |
| chr17 | 65471303 | 65471507 | 5 | 2.45E-09 | 0.02194 | 0.02850 | N/A |
| chr6 | 29599012 | 29599390 | 9 | 1.19E-11 | 0.23989 | 0.02814 | GABBR1 |
| chr10 | 6187854 | 6188415 | 5 | 2.87E-14 | 0.00086 | 0.02319 | PFKFB3 |
| chr15 | 91473059 | 91473569 | 10 | 2.38E-11 | 0.02671 | 0.02146 | UNC45A |
| chr6 | 32144667 | 32146779 | 35 | 8.45E-25 | 0.05366 | 0.01962 | AGPAT1 |
| chr16 | 3114847 | 3115809 | 12 | 7.41E-17 | 0.00671 | 0.01755 | IL32 |
| Regions with decreased methylation in PD cases | |||||||
| chr6 | 28945189 | 28945507 | 7 | 2.77E-13 | 0.00338 | −0.04644 | RN7SL471P |
| chr8 | 57350735 | 57351067 | 5 | 1.06E-10 | 0.06139 | −0.04393 | PENK |
| chr6 | 164506692 | 164507305 | 9 | 3.77E-12 | 0.03187 | −0.04304 | RP1 |
| chr12 | 25801455 | 25801945 | 6 | 9.60E-29 | 0.00000 | −0.04099 | IFLTD1 |
| chr13 | 111521981 | 111522651 | 5 | 2.12E-12 | 0.00725 | −0.04075 | LINC00346 |
| chr10 | 77542302 | 77542585 | 9 | 1.28E-12 | 0.01495 | −0.03571 | LRMNDA |
| chr2 | 48844369 | 48845068 | 10 | 8.32E-17 | 0.04500 | −0.03301 | GTF2A1L |
| chr6 | 31409319 | 31409757 | 12 | 1.48E-10 | 0.22543 | −0.02132 | LINC01149 |
| chr5 | 138210632 | 138211184 | 12 | 4.46E-12 | 0.05387 | −0.01719 | LRRTM2 |
Start and end indicate with the genomic coordinates for location of the defined DMR based on Genome Reference Consortium Human Build 37 (GRCh37) hg19. # CG indicates how many CGs were included in the DMR; minfdr is the minimum adjusted p-value from the CGs constituting the significant region; Stouffer is the Stouffer transformation of the group of FDRs for individual CGs at the DMR; maxbetafc is the maximum absolute beta fold change within the region. N/A indicates no annotated gene associated with the probe.
Methylation changes associated with PD progression: longitudinal analysis
The main goal of our study was to investigate whether blood DNA methylation changes as PD pathology progresses. To address this, we fit linear models by robust regression using an M estimator using the rlm() function in the R MASS package on the all the PD samples over time regardless of medication status to identify genomic sites where methylation varies over time in PD cases. We identified 138 DMPs that significantly changed over time in PD cases only at p < 1.0E-7 with a rate of change ranging from a 0.8% increase and a 0.6% decrease in methylation per year and supporting dynamic methylation changes in the blood methylome associated with disease progression (Table 4 and Sup. Table 2). Ageing is an important determinant of DNA methylation [31,32]. Although we controlled for age in our models, we also calculated the association of methylation changes with age to rule out that ageing was the main driver of the longitudinal epigenetic changes we observed in PD. We observed a significant association with age for only 2/138 probes changing in PD over time (Sup. Table 2). Furthermore, none of these longitudinal DMPs overlaps with probes composing the epigenetic clock from Horvath [33], further supporting that the observed changes are due to disease progression. We used Ingenuity Pathway analysis to interrogate pathway enrichment in the differentially methylated probes. For this analysis, we applied a less stringent cut-off criteria at p < 1.0E-6 (886 DMPs and 534 mapped genes entered into the analysis). Notably, the top category enriched in the Disease and Function annotation was neurological disorders; including 13 genes associated with Alzheimer’s disease (p = 1.08E-2). Among enriched canonical pathways, biotin-carboxyl carrier protein showed a significant p-value of 2.25E-3, including DMPs mapping to Acetyl-CoA carboxylase alpha (ACACA), the rate limiting enzyme in the synthesis of long-chain fatty acids. Interestingly, decreased long-chain acylcarnitines have been recently proposed as potential early diagnostic markers for PD [34].
Table 4.
Top longitudinal changes in methylation in PD cases.
| Probe ID | Chr | Position | Gene ID | Change rate PD |
Pvalue PD |
Change rate Controls | Pvalue Controls |
|---|---|---|---|---|---|---|---|
| Methylation decreasing over time | |||||||
| cg26275301 | chr17 | 81041828 | METRNL | −0.604 | 7.56E-08 | −0.00026 | 6.93E-01 |
| cg07856430 | chr18 | 42339206 | N/A | −0.525 | 8.67E-09 | −0.00058 | 2.85E-01 |
| cg13153353 | chr7 | 4175865 | SDK1 | −0.524 | 5.43E-08 | 0.00244 | 2.30E-05 |
| cg03565777 | chr12 | 125028339 | NCOR2 | −0.508 | 8.40E-08 | −0.00276 | 1.38E-06 |
| cg24136431 | chr19 | 47016623 | N/A | −0.392 | 6.40E-08 | −0.00045 | 2.93E-01 |
| cg16655626 | chr19 | 45886078 | N/A | −0.334 | 2.46E-09 | 0.00152 | 4.38E-01 |
| cg04843111 | chr1 | 156617074 | BCAN | −0.328 | 1.00E-09 | −0.00088 | 6.64E-01 |
| cg08988821 | chr7 | 74075293 | GTF2I | −0.326 | 4.22E-08 | −0.00107 | 6.71E-01 |
| cg01791421 | chr1 | 19996240 | N/A | −0.309 | 8.38E-08 | −0.00105 | 2.19E-03 |
| cg13994376 | chr13 | 112554122 | LINC00354 | −0.292 | 1.25E-08 | 0.00005 | 8.71E-01 |
| cg15987655 | chr5 | 139196457 | N/A | −0.278 | 5.86E-08 | −0.00285 | 1.42E-01 |
| cg03389720 | chr16 | 8780048 | ABAT | −0.277 | 2.00E-08 | −0.00091 | 1.88E-03 |
| cg15765398 | chr21 | 46409994 | LINC00163 | −0.272 | 5.57E-09 | −0.00114 | 4.16E-01 |
| cg09750643 | chr11 | 1718086 | KRTAP5 | −0.257 | 8.04E-09 | −0.00022 | 4.07E-01 |
| Methylation increasing over time | |||||||
| cg17046825 | chr13 | 21081216 | N/A | 0.834 | 5.35E-08 | −0.00311 | 6.60E-04 |
| cg26126295 | chr4 | 119095240 | N/A | 0.775 | 1.78E-08 | 0.00030 | 9.44E-01 |
| cg06688960 | chr20 | 1504932 | N/A | 0.713 | 1.04E-09 | −0.00053 | 8.49E-01 |
| cg25979148 | chr4 | 871629 | N/A | 0.210 | 2.55E-08 | −0.00001 | 9.95E-01 |
| cg15540764 | chr7 | 36919658 | ELMO1 | 0.140 | 7.33E-08 | −0.00040 | 9.61E-03 |
| cg05933219 | chr2 | 240099221 | HDAC4 | 0.134 | 5.61E-08 | −0.00058 | 4.87E-01 |
| cg14946911 | chr20 | 56772325 | N/A | 0.128 | 2.24E-09 | 9.91E-06 | 9.90E-01 |
| cg09593391 | chr8 | 144737908 | ZNF623 | 0.126 | 9.53E-08 | −0.00017 | 8.51E-01 |
| cg18496624 | chr17 | 38094692 | N/A | 0.123 | 5.44E-09 | −0.00005 | 7.14E-01 |
| cg10401356 | chr8 | 140712424 | N/A | 0.123 | 1.11E-08 | 0.00037 | 6.62E-01 |
| cg08561469 | chr16 | 81944150 | PLCG2 | 0.116 | 3.38E-09 | 0.00002 | 8.52E-01 |
| cg15475168 | chr7 | 101860421 | N/A | 0.115 | 1.16E-08 | −0.00023 | 7.69E-01 |
| cg20397902 | chr16 | 88624807 | N/A | 0.109 | 1.06E-08 | −0.00023 | 7.43E-01 |
| cg01202950 | chr15 | 74943647 | EDC3 | 0.106 | 6.20E-09 | −0.00027 | 6.90E-01 |
Name indicates probe designation at Illumina EPIC human methylation array; Chr is chromosome location of the CG; position refers to Genome Reference Consortium Human Build 37 (GRCh37) hg19; N/A indicates no annotated gene associated with the probe. Rate of change indicates percentage methylation change/year.
Longitudinal methylation changes associated with PD medication
Dopamine replacement is the standard of clinical care for Parkinson’s disease, and the vast majority of patients receive dopamine precursors, like levodopa/carbidopa (commercialized as Sinemet or Stalevo) and/or inhibitors of Catechol-O-methyltransferase (COMT; commercial name Comtan). Importantly, the metabolism of these compounds directly impact the one-carbon pathway, potentially affecting the supply of methyl-group donor molecules and the activity of DNA methyltransferases (Figure 4). Although current levodopa products are formulated to prevent break-down in the gastrointestinal tract and systemic circulation, the potential impact of these drugs on blood methylation has not been explored at genome-wide scale before. Therefore, we analysed the effect of anti-parkinsonian therapy on DNA methylation by defining the category ‘PD Medication’ (which included L-dopa and entacapone). PD cases receiving carbidopa; levodopa and/or entacapone (Sinemet; Stalevo and/or Comtan), either as single drugs or in combination at any dose and at any time point were categorized as the PD medication group. PD cases that did not take any of these drugs by the time of blood sampling were coded as PD NOT medicated. According to this classification, 69 patients were grouped as ‘PD NOT Medicated’, from which 27 remained unexposed to these drugs at the follow-up visit.
Figure 4.
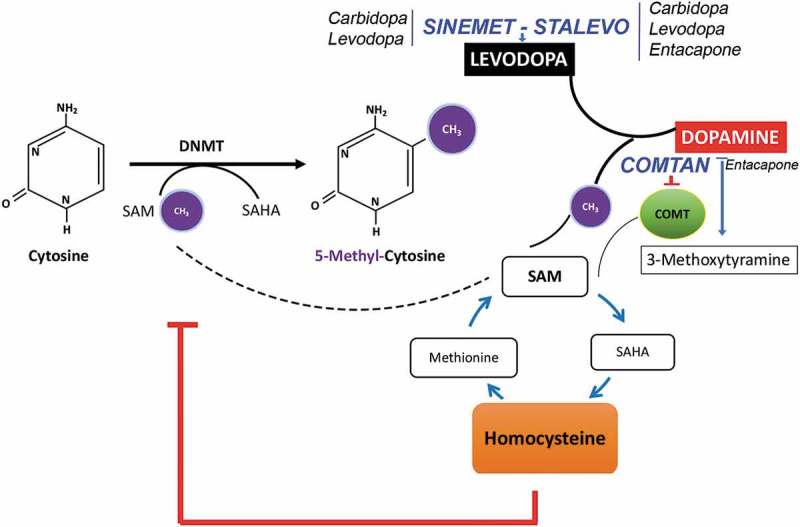
Cross-talk between one-carbon metabolism and Levodopa catabolism. Diagram depicting the effects of dopamine metabolism on the one-carbon metabolic pathway. Conversion of levodopa to dopamine requires the breakage of S-adenosyl methionine as methyl group donor which is the same source of methyl groups used by DNA methyltransferases (DNMTs) to methylation cytosine residues. In addition, homocysteine produced by the conversion levodopa-dopamine is an inhibitor of DNMTs activity and may alter DNA methylation. COMT further mediates the conversion of dopamine into O-methyl derivatives (3-methoxytyramine) by consuming methyl groups from SAM. The potential interaction with PD medication is indicated, with Sinemet and Stalevo increasing levodopa levels and COMTAN inhibiting COMT activity.
We first analysed longitudinal changes in methylation in the group of PD cases receiving PD medication. We identified 237 probes showing significant changes of methylation over time at a p < 1.0E-7 and showing modest changes in methylation ranging in the order of 0.63% reduction and 0.86% increase in methylation/year (Table 5; Figure 5 and Sup. Table 3). Longitudinal methylation in these sites did not change significantly in control subjects. Only 23/237 of the probes changing in the PD medicated group were significantly associated with ageing (Sup. Table 3), the remaining 214 CpG sites showed longitudinal changes in patients taking medication, which are likely involved in both the effect of medication and PD progression over time. Genes tagged by these CpGs (Table 5) function as transcription factors (ZNF544, ZNF623, GTF2I), extracellular matrix proteins (BCAN), non-coding RNAs (Y_RNA, LINC00163), neural cell adhesion (PCDH1), and synaptic transmission (RIMBP2), once again suggesting that signals related to central nervous system dysfunction can be detected in peripheral blood DNA.
Table 5.
Top Longitudinal changes in methylation in PD cases receiving L-dopa/entacapone.
| Probe ID | Chr | Position | Gene ID |
Change rate PD Med |
Pvalue PD Med |
Change rate Controls |
Pvalue Control |
|---|---|---|---|---|---|---|---|
| Methylation decreasing over time | |||||||
| cg13643943 | chr5 | 138629019 | MATR3 | −0.0632 | 7.25E-08 | −0.00417 | 2.26E-01 |
| cg08988821 | chr7 | 74075293 | GTF2I | −0.0396 | 7.24E-09 | −0.00107 | 6.71E-01 |
| cg16655626 | chr19 | 45886078 | N/A | −0.0373 | 2.20E-08 | 0.00152 | 4.38E-01 |
| cg14386312 | chr19 | 58740861 | ZNF544 | −0.0346 | 2.92E-08 | −0.00204 | 2.90E-01 |
| cg15765398 | chr21 | 46409994 | LINC00163 | −0.0344 | 1.11E-11 | −0.00114 | 4.16E-01 |
| cg15987655 | chr5 | 139196457 | N/A | −0.0329 | 1.98E-08 | −0.00285 | 1.42E-01 |
| cg04843111 | chr1 | 156617074 | BCAN | −0.0327 | 8.26E-08 | −0.00088 | 6.64E-01 |
| cg14240188 | chr1 | 11621054 | N/A | −0.0281 | 1.77E-08 | 0.00038 | 8.03E-01 |
| cg04551581 | chr18 | 44226771 | N/A | −0.0273 | 5.48E-08 | −0.00150 | 3.08E-01 |
| cg15298173 | chr15 | 29903900 | RP11-300A12 | −0.0262 | 7.18E-08 | −0.00297 | 5.48E-02 |
| cg15202607 | chr5 | 149520120 | PDGFRB | −0.0234 | 4.44E-08 | −0.00151 | 1.89E-01 |
| cg10429957 | chr5 | 141245719 | PCDH1 | −0.0197 | 8.39E-08 | −0.00139 | 2.38E-01 |
| Methylation increasing over time | |||||||
| cg26126295 | chr4 | 119095240 | N/A | 0.0862 | 8.67E-08 | 0.00030 | 9.44E-01 |
| cg25979148 | chr4 | 871629 | N/A | 0.0252 | 4.47E-09 | −0.00001 | 9.95E-01 |
| cg10224806 | chr12 | 131188144 | RIMBP2 | 0.0221 | 9.04E-08 | 0.00156 | 1.16E-01 |
| cg18629514 | chr7 | 5388895 | N/A | 0.0178 | 8.06E-08 | 0.00025 | 7.94E-01 |
| cg03501539 | chr11 | 114760598 | N/A | 0.0166 | 6.57E-08 | 0.00014 | 8.87E-01 |
| cg15008072 | chr17 | 7440546 | Y_RNA | 0.0166 | 7.51E-08 | 0.00113 | 2.20E-01 |
| cg05933219 | chr2 | 240099221 | HDAC4 | 0.0162 | 1.37E-09 | −0.00058 | 4.87E-01 |
| cg18763089 | chr1 | 1683738 | NADK | 0.0155 | 1.10E-08 | 0.00067 | 3.78E-01 |
| cg05120150 | chr14 | 105912598 | MTA1 | 0.0143 | 3.80E-08 | 0.00006 | 9.39E-01 |
| cg09593391 | chr8 | 144737908 | ZNF623 | 0.0139 | 8.19E-08 | −0.00017 | 8.51E-01 |
| cg10401356 | chr8 | 140712424 | N/A | 0.0138 | 3.24E-09 | 0.00037 | 6.62E-01 |
| cg22511774 | Chr13 | 114746915 | N/A | 0.0137 | 3.27E-08 | 0.00036 | 6.52E-01 |
Name indicates probe designation at Illumina EPIC human methylation array; Chr is chromosome location of the CG; position refers to Genome Reference Consortium Human Build 37 (GRCh37) hg19. Rate of change indicates percentage methylation change/year. N/A indicates no annotated gene associated with the probe.
Figure 5.
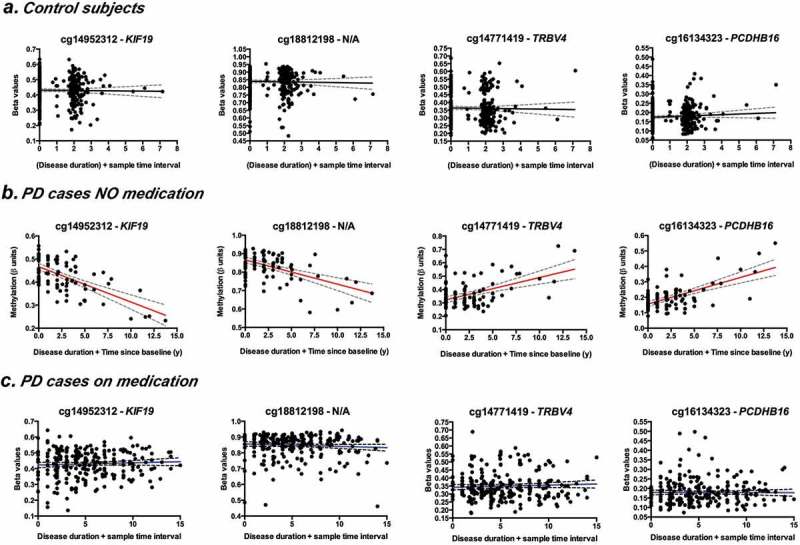
Longitudinal changes in the blood methylome associated with PD progression are compensated by PD medication. Representative plots showing regression of methylation (individual β-values) as a function of disease duration and time between baseline and follow-up samples. Comparison of top changing probes between controls (a); PD cases NOT receiving medication (b) and PD cases on medication (c). Coefficient of change as determined by mixed linear models (Tables 5 and 6) and corresponding p-values are indicated.
Longitudinal methylation changes in the absence of L-dopa or entacapone and associated solely with PD progression
One of the most valuable analyses in our study is the possibility of exploring the blood methylome in PD patients that did not yet receive L-dopa/entacapone. Despite the limitation of a reduced cohort size (N = 27), this comparison may unveil epigenomic alterations only due to PD systemic pathophysiology. We thus analysed changes in methylation over time in patients not receiving L-dopa/entacapone (PD NOT medicated; according to the criteria described in the previous section). We identified 24 probes showing significant changes of methylation over time at a p < 1.0E-7. Notably these sites showed the largest size effects compared to the other analysis groups, with methylation changes ranging from a 1.5% reduction to a 1.7% increase in methylation/year (Table 6 and Figure 5). Longitudinal methylation in these sites did not change significantly in control subjects and none of the sites showed association with age. Genes showing differential methylation in this group (Table 6) include those involved in cytoskeletal functioning, like microtubules (KIF19, KIF22, TAOK2, MAPK13, CEP70) and actin (SWAP70). In addition, genes involved in immune response are also noted (RAIT1E and TRBV4). Interestingly, methylation the changes observed in this group are different than the alterations identified inpatients receiving L-dopa/entacapone (Table 5), therefore supporting that: 1) PD progression alters the blood methylome per se and 2) L-dopa/entacapone treatments induce changes in blood methylation.
Table 6.
Longitudinal changes in methylation in PD cases NOT receiving L-dopa/entacapone.
| Probe ID | Chr | Position | Gene ID |
Change rate PD No Med |
Pvalue PD |
Change rate Controls |
Pvalue Controls |
|---|---|---|---|---|---|---|---|
| Methylation decreasing over time | |||||||
| cg14952312 | chr17 | 72350710 | KIF19 | −1.537 | 2.41E-09 | −0.00156 | 6.41E-01 |
| cg18812198 | chr11 | 68715658 | N/A | −1.324 | 7.66E-08 | 0.00114 | 6.95E-01 |
| cg15969149 | chr10 | 77352287 | N/A | −1.054 | 1.61E-08 | 0.00087 | 7.34E-01 |
| cg15531997 | chr10 | 34999169 | N/A | −0.981 | 5.39E-12 | 0.00023 | 8.71E-01 |
| cg19174044 | chr20 | 18446362 | DZANK1 | −0.914 | 3.71E-08 | 0.00091 | 6.32E-01 |
| cg13937758 | chr6 | 150217448 | RAET1E | −0.711 | 3.55E-08 | −0.00227 | 1.83E-01 |
| Methylation increasing over time | |||||||
| cg14771419 | chr7 | 142012988 | TRBV4 | 1.713 | 1.03E-09 | −0.00114 | 7.31E-01 |
| cg16134323 | chr5 | 140562034 | PCDHB16 | 1.507 | 7.50E-10 | −0.00042 | 8.29E-01 |
| cg13252209 | chr16 | 51603716 | N/A | 1.279 | 1.78E-08 | −0.00192 | 5.70E-01 |
| cg03700944 | chr6 | 46703468 | PLA2G7 | 1.093 | 8.90E-09 | −0.00262 | 2.94E-01 |
| cg06688960 | chr20 | 1504932 | N/A | 0.988 | 3.41E-08 | −0.00053 | 8.49E-01 |
| cg26421310 | chr1 | 25257058 | RUNX3 | 0.093 | 2.43E-08 | −0.00055 | 3.31E-03 |
| cg27380788 | chr16 | 4526765 | NMRAL1 | 0.073 | 9.24E-08 | −0.00046 | 2.78E-03 |
| cg11107196 | chr16 | 71918265 | ZNF821 | 0.065 | 5.36E-09 | −0.00019 | 1.37E-01 |
| cg09585751 | chr6 | 24646282 | KIAA0319 | 0.056 | 9.21E-08 | −0.00021 | 4.51E-02 |
| cg14934866 | chr16 | 29985156 | TAOK2 | 0.053 | 7.30E-08 | −0.00023 | 7.87E-02 |
| cg21645604 | chr6 | 36098567 | MAPK13 | 0.043 | 3.31E-08 | −0.00014 | 5.70E-02 |
| cg07038191 | chr16 | 29801882 | KIF22 | 0.041 | 5.83E-08 | −0.00028 | 3.27E-04 |
| cg23993697 | chr11 | 9685562 | SWAP70 | 0.04 | 7.05E-08 | −0.00012 | 2.22E-01 |
| cg17129217 | chr9 | 91933717 | SECISBP2 | 0.039 | 6.52E-08 | −0.00022 | 5.45E-03 |
| cg15193793 | chr14 | 90422250 | EFCAB11 | 0.038 | 8.37E-08 | −0.00012 | 2.83E-01 |
| cg19116545 | chr3 | 138313166 | CEP70 | 0.037 | 3.02E-08 | −0.00022 | 8.33E-03 |
| cg18104674 | chr18 | 60190218 | ZCCHC2 | 0.036 | 5.58E-08 | −0.00011 | 9.65E-02 |
| cg20390702 | chr12 | 8850385 | RIMKLB | 0.036 | 6.54E-08 | −0.00019 | 1.85E-02 |
Name indicates probe designation at Illumina EPIC human methylation array; Chr is chromosome location of the CG; position refers to Genome Reference Consortium Human Build 37 (GRCh37) hg19; N/A indicates no annotated gene associated with the probe. Rate of change indicates percentage methylation change/year. N/A indicates no annotated gene associated with the probe.
Discussion
Specific whole blood methylation changes correlate with Parkinson’s disease
Epigenetic changes are emerging as contributing factors to PD and other neurodegenerative diseases. To identify a comprehensive set of differentially methylated sites associated with PD, we performed a case versus control comparison on extremely well characterized and phenotyped patient samples acquired by the Harvard Biomarkers Study (HBS). Leveraging the full cohort size of 792 samples, a comparison of PD versus control groups identified 7 DMPs significantly associated with PD, many of them tagging genetic loci with interesting functional consequences with respect to pathways and cellular functions previously implicated in PD.
Analysis of neighbour sites with consistent alterations in methylation or differentially methylated regions (DMRs), more likely to affect gene expression, identified altered methylation in 13 CpG sites (p = 1.22E-22) at the cytochrome P450 2E1 (CYP2E1) locus in whole blood of PD patients. Interestingly, CYP2E1, which has been reported to be hypomethylated in PD brain samples [17], encodes a member of the cytochrome P450 mixed-function oxidase system responsible for metabolizing environmental toxins. Since environmental factors and external toxins contribute to PD vulnerability [35], altered regulation of CYP2E1 may signal the response to environmental cues that associate or contribute to PD onset. We note, however, that previous reports describe hypomethylation of the CYP2E1 locus in PD brain. Here we find consistent increased methylation in several CpG sites across the 5ʹ region of the gene in the blood (Figure 3(a) and Table 3). Reasons for this tissue-specific difference remain to be clarified, but identification of this locus across independent studies and separate tissues suggests an important role in PD. Another notable DMR includes 1-acylglycerol-3-phosphate O-acyltransferase 1 (AGPAT1: 35 CGs hypermethylated; p = 8.45E-25). AGPAT1 catalyses the conversion of lysophosphatidic acid (LPA) into phosphatidic acid (PA). LPA is required for dopamine neuron development and, in the 6-OHDA model of PD, reduced LPA has implicated in dopamine neuron degeneration through activity at the LPA1 receptor [36]. In addition, among the DMRs identified was one in the lymphocyte antigen 6 family member G5C (LY6G5C: 5 CGs hypermethylated; p = 3.92E-9), recently identified as a brain-specific epigenetic marker of schizophrenia [30]; and clustering in the major histocompatibility complex (MHC) class III region of chromosome 6, encoding genes with critical functions in immunity. Genetic variation of MHC associates with sporadic PD [37], suggesting that altered immunity in PD may be trackable via epigenetic changes in blood.
Specific blood cell types are altered in Parkinson’s disease
Blood methylation profiles have been used to estimate the relative abundances of specific blood cell types in Parkinson’s disease [38]. In the HBS cohort, we observed similar increases in granulocytes of PD patients and lower estimated B-cells and NKs in comparison to controls, with granulocytes and NK remaining altered in the follow-up visit. The finding of increased granulocytes in PD confirms prior reports [38]. However, we also observed significant and persistent reductions to NK cells in PD, whereas no changes were previously observed for this blood cell type. While the mechanistic explanations for these changes will require additional study, it is tempting to speculate that changes in these immune-related cells could reflect ongoing inflammatory responses that occur in the brain during PD.
Longitudinal analyses reveal methylome changes over time in PD patients
Analysis of samples from patients at baseline and follow-up, which averaged 2.2 y later, provided the opportunity to interrogate the extent to which methylation profiles of PD patients change over time. During the sampling interval, PD cases showed modest but highly significant disease progression as assessed by both HY (p < 0.0001, as per paired t-test) and UPDRS scores (p < 0.0001; see also Table 1).
Single locus hits showing longitudinal methylation changes in PD with the highest statistical significance (Table 4) highlight pathways of potential importance to nervous system function. These include 4-aminobutyrate aminotransferase (ABAT: p = 2.0E-8), which is responsible for breaking down the neurotransmitter γ-aminobutyric acid (GABA), and Enhancer of mRNA Decapping 3 (EDC3: p = 6.2E-9); EDC3 promotes removal of the 5ʹ cap structure of mRNAs during their degradation. Recessive mutations in EDC3 cause intellectual disability [39], suggesting an impairment of neurological functions linking mRNA decapping to normal cognition. In addition, multiple cytoskeletal and extracellular matrix-associated proteins showed differential methylation over time in PD, including Keratin-Associated Protein 5–5 (KRTAP5: p = 8.04E-9); Engulfment And Cell Motility 1 (ELMO1: p = 7.33E-8); Brevican (BCAN: p = 1.0E-9); and Sidekick Cell Adhesion Molecule 1 (SDK1: p = 5.43E-8). Lastly, Nuclear Receptor Corepressor 2 (NCOR2: p = 8.4E-8) and Histone Deacetylase 4 (HDAC4: p = 5.61E-8) were hypo- and hyper-methylated, respectively, in PD patients at the follow-up visit relative to baseline, suggesting potential alterations to the epigenetic machinery itself. This could be due either to PD progression or, potentially, to the administration of PD medications that alter one-carbon metabolism pathways (see below).
Dopamine replacement therapies alter the methylome in PD patients
Dopamine replacement therapies directly impact one-carbon metabolism, consuming methyl groups that are required for DNA methylation and increasing homocysteine levels, which directly inhibits the activity of DNA-methyltransferases (Figure 4). Determining the extent to which common PD medications impact methylation profiles is, therefore, critical for studying differential methylation in the context of PD. The HBS contains a number of PD patients that were not taking any medication associated with PD treatment at the time of enrollment. Samples from 25 of these patients were included in our study. In addition, we were particularly interested in exploring a potential interaction between levodopa and COMT-inhibitors on DNA methylation profiles, therefore we grouped all PD cases that ever received Sinemet, Stalevo and Comtan as ‘PD Medication’.
Sinemet contains two active ingredients: levodopa and carbidopa. Levodopa is a dopamine agonist. Carbidopa is a peripheral inhibitor of DOPA-decarboxylase, preventing the conversion of Levodopa to dopamine outside the brain. Stalevo is a combination of carbidopa, levodopa, and entacapone- an inhibitor of catechol-O-methyltransferase (COMT). Comtan is the commercial denomination for entacapone.
The conversion of levodopa to dopamine consumes methyl-groups from the donor (SAM) used by DNMTs to methylate DNA. COMT further metabolizes dopamine into 3-methoxytyramine at expense of SAM. Therefore, the three drugs may alter the availability of SAM and the levels of homocysteine, which in turn inhibits DNMTs.
The majority of patients were taking Sinemet during the study (n = 104 at baseline and n = 140 at follow-up); from which 13 cases at baseline and 15 at follow-up were also on Comtan. Comtan was never administered alone or in combination with Stalevo. Use of Stalevo was reported for 21 cases at baseline and 26 at follow-up. The cases using Sinemet + Comtan may have equivalent effects to those on Stalevo. All patients taking any combination or formulation of these drugs are receiving levodopa/carbidopa.
While we recognize that the 25 non-medicated PD cases are a relatively small sample size, preliminary analysis of longitudinal changes in methylation in patients naïve to these drugs identified 217 DMPs. These DMPs may denote true PD associations separate from any modifications that may be imposed by dopamine replacement medications. To the best of our knowledge, this is also the first genome-wide analysis of blood methylation in PD patients that did not receive L-dopa/entacapone. Importantly, the comparison between medicated versus not medicated patients showed larger changes in methylation longitudinally, suggesting that medication modifies the epigenome. Therefore, as the methylome of naïve cases better resembles the epigenetic profiles of the disease, they provide additional value for early diagnosis. Study of prodromal cases before phenoconversion and of a properly powered naïve cohort will be fundamental for the appraisal of methylation as an early PD classifier tool.
One important observation from the longitudinal analysis is the overall damping effect that PD medication has on the blood methylome, illustrated in the group comparison of top DMPs changing longitudinally in Figure 5. On one hand, as our cohort is dominated by PD cases receiving medication, the changes in methylation are smaller when using the entire cohort for the analysis. On the other hand, the larger changes in methylation in PD NOT medicated cases supports the utility of blood methylation as an early disease classifier and a potential indicator of disease progression and, eventually, drug efficacy/history. Additional studies including more subjects are needed to corroborate these findings, despite the scarcity of samples from non-medicated/naïve patients.
One of the limitations of our analysis is the number of time points investigated. Not only having only two data sets per subject restricts the analysis, but also the average longitudinal time-lapse of ~2 y may not be sufficient to detect significant changes in methylation beyond the high variability of the population. UPDRS total scores are estimated to increase by 4.7 points per year in PD patients not taking medication [40]. In contrast, we observed only 3.05 points/year increase in UPRDS in cases, suggesting slow disease progression in our study cohort, an effect likely due to 79.9% of PD cases under anti-parkinsonian treatment at the time of the study. While the correlates between clinical motor scales and molecular mechanisms like DNA methylation are not determined, the slow disease progression may result in smaller changes in methylation. Future studies including additional longitudinal points and spanning longer periods of time may detect additional epigenetic changes relevant to pathology.
In summary, we present evidence demonstrating that changes to the methylome in PD are detectable in blood; change over time; and in many cases reflect cellular processes implicated in ongoing neurodegeneration in the brains of PD patients. In particular, the longitudinal sampling of our study emphasizes that DNA methylation is dynamic in PD and that common PD medication, including levodopa formulations and COMT-inhibitors impact methylation. Taken in all, these studies support the potential of blood DNA methylation as an epigenetic biomarker of disease, although additional profiling of large longitudinal cohorts is needed to complete the characterization of DNA methylation changes during the onset and progression of Parkinson’s disease.
Materials and methods
1. Study cohort
This longitudinal case-control study is nested within the Harvard Biomarkers Study (HBS) [7,24,25,41]. The HBS is an ongoing case-control study including individuals with PD, Alzheimer’s disease, and controls without neurologic disease, and collecting high-quality biosamples and high-resolution clinical phenotypes longitudinally over a five-year period (under funding from the Harvard NeuroDiscovery Center). Clinical characteristics of patients with PD enrolled in HBS that were selected for this study are shown in Table 1. Individuals with early-stage PD and controls (CT) were enrolled into HBS from Massachusetts General Hospital and Brigham & Women’s Hospital. Inclusion criteria for cases with PD were age ≥21; diagnosis of PD according to UK brain bank criteria; MMSE score >21. Main exclusion criteria for cases with PD were the diagnosis of a blood or bleeding disorder, known haematocrit <30. Cases for the nested longitudinal case-control methylation study were selected from the HBS population based on additional criteria that included the availability of follow-up visit(s), age ≥55, baseline Hoehn and Yahr stage ≤3, baseline MMSE > 27. Sex-matched and age-similar controls were selected who had a baseline MMSE > 27 and available follow-up visit(s). Cases and controls with a past medical history of cancer (that might affect methylation status) were excluded. To increase diagnostic certainty, subjects with a diagnosis change on longitudinal follow-up were excluded from the current study. PD cases carrying a known G2019S LRRK2 mutation were also excluded. HBS and the use of HBS biosamples and data for the current study were approved by the Institutional Review Board of Partners HealthCare. All HBS participants signed informed consent forms.
2. Array processing
Genomic DNA (1 µg) samples received from HBS were coded and randomized with respect to disease status. DNA was bisulphite converted (EZ DNA Methylation kits, Zymo Research, D5003) per Illumina’s recommendation. The samples were processed and hybridized to Infinium MethylationEPIC BeadChip (Illumina, WG-317–1002) and signal was scanned with Illumina’s iScan. Longitudinal sample pairs corresponding to the same subjects were run on the same chip to avoid batch effects. Raw IDAT files were exported for processing in R.
3. Data normalization and quality control
The study was conducted at the laboratories of Dr Dunckley at Arizona State University (ASU) and Dr Desplats at University of California San Diego (UCSD). Both laboratories applied unified standard operating procedures (SOPs) according to Illumina’s recommendations. We performed careful quality control and pre-processing steps using the R Bioconductor package Minfi v. 1.22.1 [42]. Minfi Detection P values were calculated (detP). No samples had mean detP value >0.05. Sex prediction was performed and eight samples with discordant calls were removed from the analysis. Subject identity in paired-samples was determined using SNPs-matching probes contained in the EPIC array [43]. Samples with ratios of non-methylated/methylated sites (uMeth/mMeth) <10.5 were also removed (Sup. Figure1(a)). The call rate was calculated as the proportion of probes in each sample with a detP of <0.01 (Sup. Figure1(c)). We ran technical replicates across sites and batches for control. Replicates were removed by taking the sample with the highest call rate. As a result, a total of 36 samples were removed from downstream analyses and the remaining 792 samples (two time-point longitudinal samples from 197 PD cases and 199 controls) were normalized using ssNoob, a method recommended for EPIC array data processing [44] (Sup. Figure 1(b)). After normalization, probes were removed that failed in one or more samples (detP >0.01), were located on sex chromosomes, had SNPs at the CpG site, or documented to be cross-reactive from Pidsley et al. (2016) [45], leaving 755,625 probes for analysis. In addition, we evaluated the correlation of intensities between sample replicates and longitudinal pairs repeated across the arrays to verify association (Sup. Figure 1(d)).
4. Methylation data analysis: PD vs control with repeated measures
All probes were used to build multi-dimensional scale plots to visualize the variation in the data.
Probe-wise differential methylation analysis was performed with the Bioconductor package limma. Beta values were converted to M-values for statistical analysis. Consensus correlation was calculated for longitudinal samples using the limma duplicateCorrelation function using 10,000 randomly selected probes and patient ID as the block parameter. The model design for the longitudinal analysis was ~0+ condition_visit + batch_site + age + sex + smoke + duration_at_baseline + CD8T+CD4T+NK+Bcell+Mono+Gran, and was adjusted as needed to test PD vs CT . The limma function lmFit including patient ID as the block variable and the consensus correlation from duplicate-Correlation() function was run with the specified design on M-values, followed by fitting the desired contrasts and running the limma function eBayes to calculate differentially methylated probes. The corresponding Beta value is also included in tables and was used to calculate differential methylation as Delta Beta between the indicated comparisons. Differentially methylated regions were analysed using DMRcate [29].
5. Longitudinal analysis
Longitudinal analysis was performed on different subsets of the data (Control only, PD only, Medication true only and Medication false only). On each of these datasets, we fit a linear model by robust regression using an M estimator with the R function rlm in the R package MASS. The model is outlined below and includes the covariates baseline age (blineAge), sex, cell type composition, site, smoking status (smoked) and duration at baseline plus time between visits (duration_plus_time). The term ‘time’ in the model is composed by the sum of disease duration at baseline (which = 0 in control subjects or PD cases diagnosed less than one year before baseline sample) and the time interval between baseline and follow-up samples.
Rlm (x~ blineAge+Sex+CD8T+CD4T+NK+Bcell+Mono+Gran+Site+Smoked+duration plus time -1, maxit = 100).
Availability of data
Array data obtained from this study is deposited at the Parkinson’s Disease Biomarkers Program (PDBP) database (https://pdbp.ninds.nih.gov) as PDBP-STUDY0000249.
Funding Statement
This work was supported by the Michael J. Fox Foundation for Parkinson’s Research [11097];Michael J. Fox Foundation for Parkinson’s Research [11097.2];National Institutes of Health [U01NS082157, U01NS095736, and U01NS100603].
Acknowledgments
This study was supported by the Michael J Fox Foundation (MJFF) under grants # 11097.2 to T.D. and # 11097 to P.D.; a gift from the KiMe Fund of the East Tennessee Foundation to T.D.; with additional funding from the American Parkinson Disease Association (to C.R.S.) and NIH grants U01NS082157, U01NS095736, and U01NS100603 to C.R.S.; and partially funded by grant UL1TR001442 of CTSA to K.F. The Harvard Biomarkers Study is supported by the Harvard NeuroDiscovery Center (HNDC), MJFF, the Parkinson’s Disease Biomarkers Program (PDBP) under grants U01 NS082157, U01NS100603 of the NINDS, and the Massachusetts Alzheimer’s Disease Research Center (ADRC) under grant P50 AG005134 of the National Institute on Aging.
We thank all study participants, their families, and friends for their support and participation, and our study coordinators. We thank the following Studies and Investigators:
Harvard Biomarkers Study. Co-Directors: Harvard NeuroDiscovery Center: Clemens R. Scherzer, Bradley T. Hyman, Adrian J. Ivinson; Investigators and Study Coordinators: Harvard NeuroDiscovery Center: Daly Franco, Frank Zhu; Brigham and Women’s Hospital: Lewis R. Sudarsky, Michael T. Hayes, Chizoba C. Umeh, Reisa Sperling; Massachusetts General Hospital: John H. Growdon, Michael A. Schwarzschild, Albert Y. Hung, Alice W. Flaherty, Deborah Blacker, Anne-Marie Wills, U. Shivraj Sohur, Nicte I. Mejia, Anand Viswanathan, Stephen N. Gomperts, Vikram Khurana, Mark W. Albers, Maria Alora-Palli, Alireza Atri, Scott McGinnis, Nutan Sharma, Randy Buckner, Thomas Byrne, Maura Copeland, Bradford Dickerson, Matthew Frosch, Teresa Gomez-Isla, Steven Greenberg, James Gusella, Trey Hedden, E Tessa Hedley-Whyte, Keith Johnson, Aaron Koenig, Marta Marquis-Sayagues, Gad Marshall, Olivia Okereke, Anat Stemmer-Rachaminov, Jessica Kloppenburg; University of Ottawa: Michael G. Schlossmacher; Scientific Advisory Board: Massachusetts General Hospital: John H. Growdon; Brigham and Women’s Hospital: Dennis J. Selkoe, Reisa Sperling; Harvard T.H. Chan School of Public Health: Alberto Ascherio; Data Coordination: Harvard NeuroDiscovery Center: Thomas Yi, Massachusetts General Hospital: Joseph J. Locascio, Haining Li; Biobank Management Staff: Harvard NeuroDiscovery Center: Gabriel Stalberg, Zhixiang Liao.
Disclosure statement
CRS has collaborated with Pfizer, Biogen, Sanofi, Opko, Proteome Sciences, Genzyme Inc., and Lysosomal Therapies; has consulted for Genzyme; has served as Advisor to the Michael J. Fox Foundation, NIH, Department of Defense; is on the Scientific Advisory Board of the American Parkinson Disease Association; has received funding from the NIH, the U.S. Department of Defense, the Harvard NeuroDiscovery Center, the Michael J. Fox Foundation, American Parkinson Disease Association, and the M.E.M.O. Hoffman Foundation.
All other authors declare no conflict of interest related to this study.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Dauer W, Przedborski S.. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. [DOI] [PubMed] [Google Scholar]
- [2].Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. [DOI] [PubMed] [Google Scholar]
- [3].Mattila PM, Rinne JO, Helenius H, et al. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 2000;100(3):285–290. [DOI] [PubMed] [Google Scholar]
- [4].Ferrer I, Martinez A, Blanco R, et al. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm. 2011;118(5):821–839. [DOI] [PubMed] [Google Scholar]
- [5].Haas BR, Stewart TH, Zhang J. Premotor biomarkers for Parkinson’s disease – a promising direction of research. Transl Neurodegener. 2012;1(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zheng B, Liao Z, Locascio JJ, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2(52):52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Locascio JJ, Eberly S, Liao Z, et al. Association between alpha-synuclein blood transcripts and early, neuroimaging-supported Parkinson’s disease. Brain. 2015;138(Pt 9):2659–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Scherzer CR, Grass JA, Liao Z, et al. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc Natl Acad Sci U S A. 2008;105(31):10907–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mittal S, Bjornevik K, Im DS, et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science. 2017;357(6354):891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pavlou MAS, Outeiro TF. Epigenetics in Parkinson’s disease. Adv Exp Med Biol. 2017;978:363–390. [DOI] [PubMed] [Google Scholar]
- [11].Jowaed A, Schmitt I, Kaut O, et al. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci. 2010;30(18):6355–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Matsumoto L, Takuma H, Tamaoka A, et al. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One. 2010;5(11):e15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schmitt I, Kaut O, Khazneh H, et al. L-dopa increases alpha-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov Disord. 2015;30(13):1794–1801. [DOI] [PubMed] [Google Scholar]
- [14].Guhathakurta S, Evangelista BA, Ghosh S, et al. Hypomethylation of intron1 of alpha-synuclein gene does not correlate with Parkinson’s disease. Mol Brain. 2017;10(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Richter J, Appenzeller S, Ammerpohl O, et al. No evidence for differential methylation of alpha-synuclein in leukocyte DNA of Parkinson’s disease patients. Mov Disord. 2012;27(4):590–591. [DOI] [PubMed] [Google Scholar]
- [16].Lin Q, Ding H, Zheng Z, et al. Promoter methylation analysis of seven clock genes in Parkinson’s disease. Neurosci Lett. 2012;507(2):147–150. [DOI] [PubMed] [Google Scholar]
- [17].Kaut O, Schmitt I, Wullner U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics. 2012;13(1):87–91. [DOI] [PubMed] [Google Scholar]
- [18].Su X, Chu Y, Kordower JH, et al. PGC-1alpha promoter methylation in Parkinson’s disease. PLoS One. 2015;10(8):e0134087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Coupland KG, Mellick GD, Silburn PA, et al. DNA methylation of the MAPT gene in Parkinson’s disease cohorts and modulation by vitamin E in vitro. Mov Disord. 2014;29(13):1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Desplats P, Spencer B, Coffee E, et al. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem. 2011;286(11):9031–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Masliah E, Dumaop W, Galasko D, et al. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics. 2013;8(10):1030–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Moore K, McKnight AJ, Craig D, et al. Epigenome-wide association study for Parkinson’s disease. Neuromolecular Med. 2014;16(4):845–855. [DOI] [PubMed] [Google Scholar]
- [23].Chuang YH, Paul KC, Bronstein JM, et al. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017;9(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol. 2016;80(5):674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu G, Locascio JJ, Corvol JC, et al. Prediction of cognition in Parkinson’s disease with a clinical-genetic score: a longitudinal analysis of nine cohorts. Lancet Neurol. 2017;16(8):620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016;15(12):1257–1272. [DOI] [PubMed] [Google Scholar]
- [27].Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lovkvist C, Dodd IB, Sneppen K, et al. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016;44(11):5123–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Peters TJ, Buckley MJ, Statham AL, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wockner LF, Morris CP, Noble EP, et al. Brain-specific epigenetic markers of schizophrenia. Transl Psychiatry. 2015;5:e680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23(8):413–418. [DOI] [PubMed] [Google Scholar]
- [32].Horvath S, Zhang Y, Langfelder P, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13(10):R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saiki S, Hatano T, Fujimaki M, et al. Decreased long-chain acylcarnitines from insufficient beta-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci Rep. 2017;7(1):7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Helley MP, Pinnell J, Sportelli C, et al. Mitochondria: a common target for genetic mutations and environmental toxicants in Parkinson’s disease. Front Genet. 2017;8:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang XY, Zhao EY, Zhuang WX, et al. LPA signaling is required for dopaminergic neuron development and is reduced through low expression of the LPA1 receptor in a 6-OHDA lesion model of Parkinson’s disease. Neurol Sci. 2015;36(11):2027–2033. [DOI] [PubMed] [Google Scholar]
- [37].Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42(9):781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Horvarth SaR BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY). 2015;7(12):1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ahmed I, Buchert R, Zhou M, et al. Mutations in DCPS and EDC3 in autosomal recessive intellectual disability indicate a crucial role for mRNA decapping in neurodevelopment. Hum Mol Genet. 2015;24(11):3172–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Holden SK, Finseth T, Sillau SH, et al. Progression of MDS-UPDRS scores over five years in De Novo Parkinson disease from the Parkinson’s progression markers initiative cohort. Mov Disord Clin Pract. 2018;5(1):47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ding H, Dhima K, Lockhart KC, et al. Unrecognized vitamin D3 deficiency is common in Parkinson disease: harvard biomarker study. Neurology. 2013;81(17):1531–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Heiss JA, Just AC. Identifying mislabeled and contaminated DNA methylation microarray data: an extended quality control toolset with examples from GEO. Clin Epigenetics. 2018;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fortin JP, Triche TJ Jr., Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33(4):558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pidsley R, Zotenko E, Peters TJ, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Goetz CG, Poewe W, Rascol O, et al. Movement disorder society task force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord. 2004;19(9):1020–1028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Array data obtained from this study is deposited at the Parkinson’s Disease Biomarkers Program (PDBP) database (https://pdbp.ninds.nih.gov) as PDBP-STUDY0000249.