

Abstract
Nitrosomonas europaea cytochrome c-552 (Ne c-552) variants with the same His/Met axial ligand set but different EPR spectra are characterized structurally to aid the understanding of how molecular structure determines heme electronic structure. Visible light absorption, Raman, and resonance Raman spectroscopy of the protein crystals is performed along with structure determination. The structures solved are of Ne c-552, which displays a “HALS,” or highly anisotropic axial low-spin EPR spectrum, and the deletion mutant Ne N64Δ, which has has a rhombic EPR spectrum. Two X-ray crystal structures of wild-type Ne c-552 are reported; one is of the protein isolated from N. europaea cells (Ne c-552n, 2.35-Å resolution) and the other is of recombinant protein expressed in E. coli (Ne c-552r, 1.63-Å resolution). Ne N64Δ crystallized in two different space groups, and two structures are reported (monoclinic, 2.1-Å resolution; hexagonal, 2.3-Å resolution). Comparison of the structures of the wild-type and mutant proteins reveals that heme ruffling is increased in the mutant; increased ruffling is predicted to yield a more rhombic EPR spectrum. The 2.35-Å Ne c-552n structure displays 18 molecules in the asymmetric unit; analysis of the structure is consistent with the population of more than one axial Met configuration, as seen previously by NMR. Finally, the mutation is shown to yield a more hydrophobic heme pocket and expel waters from near the axial Met. These structures reveal that heme pocket residue 64 plays multiple roles in regulating the axial ligand orientation and the interaction of water with the heme. These results support the hypothesis that more ruffled hemes lead to more rhombic EPR signals in cytochromes c with His/Met axial ligation.
Keywords: crystal structure, cytochrome c, electronic structure, EPR, heme ruffling
Graphical Abstract
Many roles for one residue: X-ray crystal structures of N. europaea cytochrome c-552 and a single deletion mutant demonstrate that one heme pocket residue influences axial ligand conformation, heme conformation, and access of water to the heme, with significant consequences for electronic structure.
Introduction
Low-spin hemes in proteins play crucial roles in biological energy transduction by performing electron transfer. Fulfilling this function requires that the heme-interacting polypeptide: (i) tunes the heme to the appropriate redox potential; (ii) modulates heme coupling to electron donors and acceptors; (iii) and minimizes reorganization energy. These properties allow efficient electron transfer to occur at the low driving forces typical of biological electron transfer reactions.[1] The heme iron in electron-transfer proteins is typically ligated by two protein-donated axial ligands to maintain a low-spin state and minimize redox-linked structural changes in the coordination environment; His/His and His/Met are the most common axial ligand sets for electron-transfer heme proteins.[2] The heme electronic structure is the key link between active-site structure and function, and thus significant efforts have been made to identify how the heme, its ligands, and the heme environment determine electronic structure in heme proteins. In the case of heme with His/His axial ligation, the relationship between the heme coordination environment and electronic structure is well understood. Oxidized hemes with a S=1/2 (dxy)2(dxz, dyz)3 ground-state configuration have been classified as Type I or Type II according to the properties of their EPR spectra.[3] Type I hemes, also called highly axial low-spin or highly anisotropic low-spin (HALS), are characterized by a large gmax value (> 3.3). Type II hemes have a gmax value < 3.2 and a rhombic EPR spectrum. The structural basis for the variation in the EPR spectra of the bis-His heme proteins is driven by the angle formed by the two axial His imidazole planes; perpendicular His ligands result in high gmax values (Type I), while parallel His ligands yield low gmax values (Type II).[4] The basis for this relationship is that perpendicular His ligands provide an axial coordination environment. In contrast with the His/His case, for hemes with His/Met axial ligation, no such relationship between ligand orientations and g values has been identified.
His/Met heme axial ligation is most commonly seen in cytochromes c (cyts c), which are widely distributed electron transfer proteins that to serve as important model systems for understanding how proteins control redox potential,[6] electronic coupling,[7] and reorganization energy.[8] Furthermore, studies of cyts c have been vital to our understanding of heme electronic structure,[9] organismal evolution,[10] and for developing new approaches to determine three-dimensional structure.[11] Cyts c contain a heme c that is covalently bound to the polypeptide via two Cys residues, one of which is adjacent to the axial His. The axial His is constrained along the heme α,γ-meso axis by its proximity to the heme-bound Cys residue,[12] but the axial Met can adopt a range of different orientations (Figure 1A, B). In some cases, fluxionality of the heme axial Met – in which it inverts at the sulfur rapidly on the NMR time scale – has been observed.[13] The value of gmax in cyts c shows significant variation between the HALS and rhombic types, as shown in Figure 2. However, no relationship between the relative orientations of the axial ligands and gmax has been identified.[14] In an earlier EPR and NMR study of cyts c, the magnitude of the heme out-of-plane distortion known as ruffling (Figure 1C) and variations in the axial ligand bond strength were put forward as possible factors determining gmax values.[15] A subsequent investigation demonstrated that heme ruffling indeed impacts gmax. This analysis examined cyt c variants and demonstrated that those with the more ruffled hemes show greater rhombicity and lower gmax values.[16] A similar relationship has been noted in cyanide derivatives of globins.[17] However, for a given system, the changes in gmax brought about by changes in heme ruffling are relatively small, and may not account for the range of gmax values observed.[16]
Figure 1.

A) Heme, axial His, axial Met, and Asn-64 are shown for Ne c-552r (PDB: 4JCG), showing Met in the S configuration, and pyrrole labels. B) Heme, axial His, and axial Met are shown for Ne N64Δ (PDB: 3ZOX), showing Met in the R configuration. C) View of the highly ruffled heme in horse cyt c (PDB: 1HRC).[5]
Figure 2.

The X-band (~ 9.66 GHz) EPR spectra of cytochrome c from B. pasteureii c-553, N. europaea c-552r, H. thermophilus c-552, P. aeruginosa c-551, N. europaea mutant N64Δ and P. aeruginosa mutant N64V. The spectra were recorded at T = 10 K with an applied microwave power of 1.0 mW, modulation amplitude 0.75 mT, modulation frequency 100 KHz, 55 dB gain, sweep time 168 s, time constant 82.92 msec and 4–6 scans signal accumulation and averaged. Data from [14–16,18]
Recent studies utilizing Pseudomonas aeruginosa cyt c551 (Pa c-551) and Nitrosomonas europaea cyt c552 (Ne c-552) as model systems to investigate the structural basis for variations in EPR spectra of heme proteins with His/Met axial ligation showed that selected point mutations introduced in the heme pocket triggered large changes in the EPR spectra.[14–15] In particular, while the gmax values for Pa c-551 and Ne c-552 are 3.20 and 3.34, respectively, the Pa N64V and Ne N64Δ variants have respective gmax values of 3.05 and 3.13 (Table 1, Figure 2).[15] In Pa c-551 and Ne c-552, Asn64 is within hydrogen-bonding distance of the axial Met δS (Figure 1A), but axial ligation is not changed in these mutants. Such large changes in gmax values caused by the introduction of a single point mutation or deletion of a residue other than an axial ligand are unprecedented and provide an opportunity to investigate the structural factors that determine g values for ferric heme with His/Met axial ligation.
Table 1.
Values of gmax and magnitude of out-of-plane distortion along the ruffling coordinate (B1u) for selected His/Met cyts c
| Protein[a] | gmax | B1u (Å)[b] | PDB, resolution[c] |
|---|---|---|---|
| B. pasteureii c-553 | 3.36[18] | 0.35 | 1C75 (0.97 Å)[19] |
| Ne c-552r | 3.34[15] | 0.62 | 4JCG (1.63 Å) |
| Ht c-552 | 3.23[20] | 0.62 | 1YNR (2.00 Å)[21] |
| Pa c-551 | 3.20[15] | 0.49 | 351C (1.60 Å)[22] |
| Ne N64Δ | 3.13[15] | 0.71[a] | 3ZOx (2.1 Å) |
| Horse cyt c | 3.06[15] | 1.00 | 1HRC (1.90 Å)[5] |
Average of values for the 4 monomers for the monoclinic crystal form.
To identify a structural basis for variations in EPR spectra of His/Met-ligated hemes, we have determined X-ray crystal structures of Ne c-552, which has a Type I spectrum, and Ne N64Δ, a deletion mutant with a Type II spectrum. Two structures for Ne c-552 are reported here; one is of Ne c-552 isolated from N. europaea (Ne c-552n) and the other is of recombinant Ne c-552 expressed in E. coli (Ne c-552r). Ne N64Δ crystalized in two different space groups and structures of both are reported here. It is important to underline that Ne c-552n and Ne c-552r feature indistinguishable EPR envelopes and g-tensor resonances, including for the 57Fe-enriched Ne c-552.[15, 18] Furthermore, a similar alteration of the gmax signal line-shape has been observed in both Ne c-552n and Ne c-552r in response to pH alterations of the medium. Analysis of the heme conformation observed in the structures yields support for the proposed relationship between heme ruffling and gmax values. Finally, the structure of Ne c-552n shows more than one axial Met conformation, consistent with Met fluxionality, a phenomenon previously identified by NMR.[13]
Results and Discussion
X-ray Diffraction Experiment and Structure Refinement
Our initial crystallization screening for Ne c-552r and Ne N64Δ revealed that these samples could be crystallized under several different conditions (Table S1, S2). Some of the conditions gave poorly diffracting crystals, while others gave acceptable X-ray diffraction. The four c-552 variants whose crystal structures are presented here were crystallized under different conditions, and belong to different crystal forms with different unit cells (Table 2).
Table 2.
Crystal data, data collection and refinement statistics
| Ne c-552 | WT(r) | N64Δ(monoclinic) | N64Δ(hexagonal) | WT(n) |
|---|---|---|---|---|
| Crystal Data | ||||
| Space group | P6522 | C2 | P6522 | P212121 |
| a , b, c (Å) | 60.4 / 60.4 / 109.1 | 138.5 / 81.2 / 42.3 | 137.7 / 137.7 / 88.8 | 59.5 / 137.9 / 204.7 |
| αβγ(°) | 90.0 / 90.0 / 120.0 | 90.0 / 105.2 / 90.0 | 90.0 / 90.0 / 120.0 | 90.0 / 90.0 / 90.0 |
| Crystal size (μm3) | 300 × 300 × 500 | 100 × 90 × 15 | 100 × 20 × 20 | N.A. |
| Data Collection | ||||
| X-ray source | CHESS-F1 | ESRF-ID29 | ESRF-ID29 | EMBL-X11 |
| Wavelength (Å) | 0.9180 | 0.9760 | 0.9760 | 0.8496 |
| Detector | Q270 ADSC | 6M Pilatus | 6M Pilatus | MARCCD |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Beam size (μm2) | 40×40 | 50×30 | 30×30 | N.A. |
| Flux (photons/sec) | 2.8·1010 | 3.1·1011 | 1.3·1011 | N.A. |
| Absorbed dose (MGy) | N.A. | 6.4 | 13.1 | N.A. |
| Resolution range (Å) | 26.4−1.63 / 1.7−1.63 | 40.8−2.1 / 2.2−2.1 | 68.8−2.3 / 2.4−2.3 | 38.8−2.35 / 2.48−2.35 |
| Completeness (%)[a] | 98.0 / 83.0 | 94.3 / 94.0 | 99.5 / 99.6 | 97.1 / 89.1 |
| Redundancy (%)[a] | 6.2 / 3.5 | 2.4 / 2.4 | 5.6 / 5.7 | 3.3 / 2.8 |
| I/sd(I)[a] | 37.8 / 2.7 | 4.7 / 2.3 | 9.0 / 3.7 | 13.6 / 3.2 |
| Rsym[a,b] | 4.0 / 37.8 | 15.0 / 49.7 | 15.8 / 57.3 | 5.8 / 30.3 |
| Total observations | 317320 | 61280 | 126058 | 227534 |
| Unique reflections | 15211 (27203)[c] | 24887 | 22416 | 68939 |
| Mosacity | 0.50 | 0.46 | 0.45 | 0.60 |
| Refinement Statistics | ||||
| Rwork (%)[d] | 19.8 | 21.2 | 17.5 | 20.1 |
| Rfree (%)[e] | 23.4 | 27.4 | 22.6 | 25.8 |
| Mean overall isotropic B-factor (Å2) | 36.6 | 18.7 | 12.2 | 27.3 |
| Ramachandran plot: most favored / allowed regions / generously allowed regions (%) | 97.5 / 2.5 / 0.0 | 91.5 / 8.5 / 0.0 | 91.5 / 8.5 / 0.0 | 88.9 / 10.6 / 0.4 |
| Estimated overall coordinate error based on Rwork/Maximum Likelihood (Å) |
N.A. / 0.16 | 0.21 / 0.14 | 0.21 / 0.12 | 0.38 / 0.19 |
| Rmsd bonds (Å)/angles (°): |
0.016/1.63 | 0.017/1.87 | 0.024/1.91 | 0.015/1.85 |
| Solvent content (%) | 61.0 | 59.5 | 61.4 | 51.9 |
| Molecules per asu | 1 | 4 | 4 | 18 |
| Added waters | 138 | 160 | 194 | 216 |
| Volume not occupied by model (%) | N.A. | 49 | 52 | 40 |
| Unmodelled residues | none | none | 2× Lys81 | 1–9 chain P |
| PDB code | 4JCG | 3ZOX | 3ZOY | 3ZOW |
The value before the backslash is for all data, and the value after the backslash is for the data in the highest resolution shell
Rsym = ∑|/-</>| / ∑/
The parenthetical value denotes the separate treatment of F+ and F− for anomalous scattering calculations.
Rwork = ∑ (|Fobs| − |Fcalc|) / ∑|Fobs|
Rfree is the Rcryst calculated on the 5% reflections excluded for refinement.
Ne c-552r produced crystals that diffracted to the highest resolution and yielded a refined structure to 1.63 Å with one molecule per asymmetric unit. By contrast crystals of Ne c-552n contained 18 molecules per asymmetric unit, which were refined to 2.35 Å resolution. The Ne N64Δ crystals were solved in two different crystal forms, each containing 4 molecules in the asymmetric unit, that were refined to 2.1 and 2.3 Å resolution, respectively. All structures were determined by molecular replacement and yielded final R-values between 17.5–21.2% (R-free between 22.6–27.4%) with reasonable geometry (Table 2). In general, the polypeptide chains are enveloped continuously in electron density maps with no visible breaks in the main chain; both the N-terminal and C-terminal residues are visible for most of the protein subunits in the respective asymmetric units (Figure 3). One exception is the subunit corresponding to chain R in Ne c-552n structure, which exhibits poorly defined electron density. The heme groups are well ordered, and the structures well refined as demonstrated by limited negative and positive electron density in the (Fo-Fc) difference maps shown for the heme regions in Figure 3. The location and identity of iron within the heme group is corroborated by a representative 14 σ anomalous difference Fourier map from the Ne c-552r structure (purple mesh in Figure 3a) in which the signal exceeded 26 σ The overall structures are very similar with relatively low RMSD differences. A superposition of the Ne c-552r and Ne N64Δ structures produced RMSD values between 0.5–0.6 Å for backbone Cα atoms. Among the 18 monomers of Ne c-552n, the RMSD values are 0.3–0.8 Å; here, subunits corresponding to chains M and R contributed to the higher values. By contrast, comparisons among the Ne N64Δ subunits produced RMSD values of 0.2–0.3 Å, which is on the order of the coordinate error.
Figure 3.

The heme pocket of the different Ne c-552 structures shown with the electron densities: A) Ne c-552r at 1.63 Å resolution (4JSC), B) Ne c-552n (chain B) at 2.35 Å resolution (3ZOW), C) Ne c-552n (chain G) at 2.35 Å resolution (3ZOW), D) Ne N64Δ hexagonal crystal form (chain B) at 2.3 Å resolution (3ZOY), E) Ne N64Δ monoclinic crystal form (chain A) at 2.3 Å resolution (3ZOX). The 2Fo-Fc electron density maps (wheat) are contoured at 1σ/1.2σ, while the Fo-Fc electron density difference maps are contoured at +3σ/+3.25σ (green) and −3σ/−3.25σ (red) for BCDE/A, respectively. For A the anomalous map is included countoured at 14σ (purple).
Light absorption and Raman spectroscopy on Ne c-552r and Ne N64Δ crystals
Light absorption spectra of both Ne c-552r and Ne N64Δ crystals show that the crystals contain the protein in the ferric Fe(III) state because of the observation of bands at around 690 nm as well as the absence of bands at 552 and 520 nm (Figure 4).[23] Due to the thickness of the crystals and the large absorption below 600 nm, the single-crystal spectra are easily saturated. This saturation results in poorly determined spectra in the 400–600 nm region for cytochromes with this absorption profile. The small difference in the peaks at 690 nm for Ne c-552r and Ne N64Δ will be addressed and analyzed in forthcoming work. The resonance Raman and Raman spectra of Ne c-552r and Ne N64Δ crystals, respectively, are also in accordance with a ferric state of the crystals with the ν4 mode observed at 1372 cm−1 (Figure S1).[24]
Figure 4.

Single-crystal light absorption spectra of Ne c-552r (dark green) and Ne N64Δ (dark red) in the oxidized state, and of Ne c-552r (green) and Ne N64Δ (red) crystals after exposure to X-rays corresponding to collection of diffraction data.
The crystal structures presented here have received a lower X-ray dose (Table 2) than the Henderson/Garman limit of 20/30 MGy, which is used to limit general radiation damage of protein crystals to an acceptable level.[25] However, metal sites are especially vulnerable to electrons generated during the X-ray induced radiation damage, since they are susceptible to photoreduction.[26] It has been shown that this often occurs on a much shorter time scale than other types of damage.[27] Therefore, both single-crystal light absorption and Raman spectroscopy have become essential tools complementing crystal structure determinations of redox proteins and metalloproteins.[28] Here, light absorption spectra of the Ne c-552r and Ne N64Δ crystals were measured before and after exposure to X-rays for collection of diffraction data. As shown in Figure 4, X-ray exposure changes the absorption spectra of Ne c-552r and Ne N64Δ. For both Ne c-552r and Ne N64Δ, the bands characteristic of the ferric state at around 690 nm as well as at 650 and 565 nm lose intensity upon X-ray exposure. Loss of the band at around 690 nm indicates the reduction of the oxidized protein or the loss of axial Met ligation to Fe(III).[29] Appearance of a band at around 613 nm suggests the presence of high-spin ferric species, which may be caused by the loss or the weakening of the axial Met-Fe(III) bond. Furthermore, for Ne N64Δ, an increase in absorbance at 550 and 520 nm indicates reduction to form a low-spin Fe(II) heme.[29] Thus, data collection affects heme redox state and possibly axial ligation, which must be considered when interpreting results. Since the X-ray induced reduction occurs in the frozen state at 100 K, large structural changes cannot occur, however, small structural changes can occur as well as changes to the electronic structure. For heme proteins, the distance from the heme iron to ligands has been observed to increase in crystals upon exposure to X-rays. For example, for Fe-O in compound II in cytochrome c peroxidase and myoglobin, a linear lengthening of the bond with X-ray exposure was observed.[30]
Heme Axial Met Orientations
The heme axial Met may take on different orientations in different cyts c, and sometimes within the same protein. The two orientations most commonly observed are illustrated in Figure 1A,B. They differ from each other by inversion at the Met sulfur and are referred to as R and S according to the sulfur chirality. This inversion results in a change in the orientation of the sulfur p orbital relative to the iron and heme orbitals by ~ 60°. In the S form, the axial Met ε-CH3 toward pyrrole A, and in the R form, it points toward pyrrole B. This difference in orientation changes relative energies of the iron dπ (dxz and dyz) orbitals. The dπ orbital with the higher energy primarily contains the unpaired electron, resulting in large Fermi contact shifts for heme substituents aligned with its axis.[31] This orbital also is the primary location of the orbital hole, and the corresponding axis of the heme thus will contain the orbital hole. When the significant difference between the chemical shifts on the x and y axes of the heme of horse cyt c was first noted decades ago, it was hypothesized that it may reflect variances in electronic coupling to redox partners.[32] Recently it was demonstrated experimentally that changing the location of the orbital hole by perturbing interactions with the heme indeed influences electron transfer rates, in accordance with this hypothesis.[33]
The first report of the NMR spectrum of oxidized Ne c-552 revealed that, unlike other cyts c characterized previously, Ne c-552 has very similar hyperfine shifts for its four heme methyls, with the resonances falling within 4 ppm of each other.[34] In contrast, other cyts c typically exhibit a spread of heme methyl shifts on the order of 20 ppm.[31a] Later, it was found that the unusual pattern observed for Ne c-552 came about because of fluxionality of the axial Met by inversion through sulfur, resulting in an average of the shift patterns expected for the R and S configurations, which yields similar shifts for all heme methyls.[13a] This phenomenon also has been observed for Hydrogenobacter thermophilus cytochrome c-552 (Ht c-552)[13b] and was shown to enhance protein stability by increasing folded state conformational entropy.[35] However, the X-ray crystal structure of reduced Ht c-552 shows the Met in the R configuration,[21] and the NMR structure of reduced Ne c-552 shows the Met in the S configuration,[36] as does the high-resolution crystal structure of oxidized Ne c-552r reported here (Figure 5a). However, we see indications of axial Met fluxionality in the crystal structure of Ne c-552n, as explained below.
Figure 5.

The heme pocket of the different Ne c-552 structures shown with the individual molecules (monomers) in the asymmetric unit overlaid. A) Ne c-552r (Chain A), B) Ne c-552n (chain A-R), C) Ne N64Δ hexagonal (chain A-D), D) Ne N64Δ monoclinic (chain A-D). Water molecules are shown as red spheres. Alignment of the heme pocket of the monomers was generated in PyMOL.
The Ne c-552n structure contains 18 molecules in the asymmetric unit (chains A – R), giving the possibility of trapping different conformations in the different monomers (Figure 3B,C). For the monomers (chains) ABCDEFIJKLMNPQ the Met is in the S configuration, with the ε-CH3 oriented toward pyrrole A. This is confirmed by flipping Met to the R configuration and re-refining the structure. This results in negative electron density for the R configuration, while positive electron density appears for the S configuration (Figure S2B), showing that only one orientation is acceptable. For chains GHOR the structure can be refined with either Met orientation, without appearance of any positive and negative electron density (Figure S2C). These monomers therefore have no preferred Met orientation in the crystal structure. This finding could indicate fluxionality of the Met in Ne c-552n (Figure 5B), however, it also could be a result of disorder in the crystal. Therefore, in accordance with the Ne c-552r structure reported here and the NMR structure of reduced Ne c-552, the orientation toward pyrrole A, corresponding to the S configuration at sulfur, is the preferred Met orientations in Ne c-552 structures. Nevertheless, indications for the existence of alternate Met orientations are observed, in agreement with the behavior of the oxidized protein as assessed by NMR spectroscopy.[13a] It is important to note that the different orientations observed in the crystals also may be a result of the effects of X-rays on the oxidation state and/or structure. Furthermore, cryo cooling of the samples for X-ray diffraction experiments may deplete conformers present at room temperature.[37]
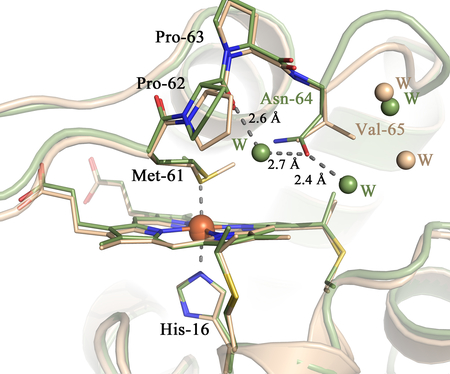
Ne N64Δ, in contrast with native, shows a single Met orientation with a Met sulfur in the R configuration (Figure 5C,D), in agreement with analysis of this variant by NMR.[15] The existence of the Met in a single R configuration is strongly supported by the crystal structures because it is observed in two different crystal forms as well as in all of the 4 monomers in the asymmetric unit of the two structures. Flipping the Met side chain from R to S configuration and re-refining the structure results in negative electron density for the S configuration, while positive electron density appears for the R configuration, showing that only one orientation is acceptable (Figure S2A). The change in orientation and configuration in the mutant supports the hypothesis that the residues on the Met-bearing loop on the distal side of the heme play a key role in orienting the axial Met. In Figure 6, the structure of the Ne N64Δ variant is compared with Ne c-552r. The overall protein fold and the heme axial His orientation remain nearly unchanged upon the introduction of the deletion mutation (Figure 6A). The deletion of Asn64 leads to a shortening of the axial Met-bearing loop with Val65 replacing the position of Asn-64, and reorientation of Asn66, while the structure and position of the residues before Pro63 and after Val67 do not change significantly (Figure 6B).
Figure 6.

Structure overlay of the Ne c-552r (green) and one monomer of Ne N64Δ (light pink): A) Overall structure showing the Asn-64/deletion with shortening of loop 3, B) Residues 61–67 including the deletion shown as sticks C) heme pocket shown with key residues and the closest waters to Asn-64 in c-552r and Val-65 in N64Δ (seen from side), D) heme pocket shown with key residues and the closest waters to Asn-64 in c-552r and Val-65 in N64Δ (seen from above). Alignment of the Ne c-552r and Ne N64Δ monoclinic (in A,B) /hexagonal (in C,D) monomer was generated in PyMOL.
The change in orientation of the Ne c-552 axial Met upon deletion of Asn64, which places a Val residue in the position previously occupied by Asn, is consistent with the finding that mutation of Gln64 to Val in Ht c-552 locks the fluxional Met into the “R” configuration.[20] Asn in this position favors the “S” configuration seen in Pa c-551 and in Ht Q64N; in these proteins Asn64 hydrogen bonds with the axial Met δS and the Met ε-CH3 points away from residue 64, consistent with a 3.6-Å distance from Met δS to Asn δN and with the observation of a Fermi contact shift for Asn64 δNH2 protons in the ferric state.[38] The hydrophobic Val, in contrast, interacts with the Met ε-CH3, which orients it toward pyrrole B, with the γC positioned 3.6 Å from Met εC. Ne c-552 is unusual among these protein variants for having an Asn64 that appears to hydrogen-bond with a fluxional axial Met. The introduction of a Asn δN-to-Met δS H-bond in Ht Q64N was given as the basis for suppressing Met fluxionality,[38] but other factors must contribute. The longer Met-bearing loop in Ne c-552 relative to Pa c-551 alone is not a reason for Met fluxionality in Ne c-552, as fluxionality is maintained in V65Δ mutants of Ne c-552.[14] Although the basis for and functional implications of axial ligand fluxionality in these proteins are not fully understood, the ability to change axial Met orientations and dynamics in these proteins by protein engineering provides a valuable resource for testing how spin density distribution on hemes influences electron transfer to redox partners.[33]
Heme Conformation
When bound to a protein, heme is rarely planar, but rather displays out-of-plane distortions. The most commonly observed distortions in proteins are saddling, doming, and ruffling.[39] Hemes c are usually ruffled as a result of the covalent attachment,[40] although the amount of ruffling varies significantly among different cyts c (Table 1). Variations in ruffling have been shown to alter spin density distribution on ferric heme[20, 31a, 41] and redox potential.[20] Analysis of EPR spectra of cyt c variants with different amounts of heme ruffling supports the model that increasing ruffling raises the dxy orbital energy relative to the dπ orbitals, increasing rhombicity.[16] Thus, cyts c with more ruffled hemes are predicted to have more rhombic EPR spectra, corresponding to lower gmax values. In agreement with this finding is the observation that cyanide complexes of some globins display HALS EPR signals for species with relatively planar hemes.[17] The determination of crystal structures of Ne c-552 variants here allows us to further investigate the relationship between heme conformation and electronic structure. The amount of out-of-plane distortion along the ruffling coordinate has been determined for Ne c-552r and Ne N64Δ using a normal coordinate structural decomposition procedure (Table 1).[42] We find that Ne N64Δ has a more ruffled heme than wild-type, in accord with expectation from its rhombic EPR spectrum (Figure 2). The variants shown in Table 1 show a general trend of increasing ruffling with decreasing gmax, although Pa c-551 deviates from this correlation. Clearly, ruffling is not the only factor determining gmax values, but it appears to play an important role.
The EPR spectra of a number of cyts c, including Ne c-552,[18] have been shown to exhibit both major and minor species, often in a pH-dependent manner.[43] The two species are typically a “rhombic” and a “HALS” component. A possible origin of these two components is the existence of an equilibrium between conformers favoring a more planar and a more ruffled heme. Other evidence of this behavior is seen by resonance Raman specroscopy of Rhodobacter capsulatus cyt c2. Although the X-ray crystal structure of R. capsulatus cyt c2 reveals a nearly planar heme,[44] resonance Raman studies of this protein detect two components, one with significant heme ruffling, and the other with a more planar heme.[45] The basis for the sensitivity of this phenomenon to pH may be the influence of pH on hydrogen bonding within the Cys-X-X-Cys-His heme attachment motif, which modulates heme ruffling; this pH effect on heme conformation has been observed for heme c peptide fragments (microperoxidases).[40]
Heme Pocket Water Molecules
The results reported herein indicate that residue 64 not only influences the axial Met orientation, but also controls access of water to the heme, which is expected to affect redox potential and reorganization energy for electron transfer. The backbone of residue 64 is oriented toward the protein surface, while its side chain usually points toward the axial Met (in Ht c-552. However, the side chain points toward solvent).[21] The Ne N64Δ mutation effectively places a Val in place of Asn, creating a more hydrophobic heme environment and expelling water molecules from the heme pocket (Figure 6C,D). Comparison of the heme environments of Pa c-551 (PDB: 351C),[22] Ht c-552 (PDB: 1YNR)[21] and horse cyt c (PDB: 1HRC)[5] is shown in Figure S3. Both Pa c-551 and Ht c-552 have water molecules close to the heme site, while in horse cyt c waters are located slightly farther from the heme, with access blocked by Phe82. The solvent exposure of the heme, or the polarity of the heme surroundings, is an additional factor that may account for differences in electronic structure seen between different hemes. The waters may directly influence the heme electronic structure by changing polarity. Alternatively, greater exposure of the heme to solvent may reflect a more flexible heme pocket, which has been correlated to a more planar heme. The basis for this relationship is that mobility of the heme attachment site in cyts c allows the heme to relax into a planar conformation.[46]
Conclusions
In this paper we report the first X-ray crystal structures of Ne c-552, along with the structure of the Ne N64Δ mutant. Spectra of the crystals confirm that the heme is oxidized at the start of data collection, but show evidence of photoreduction and other changes to the heme environment during data collection. The structure of recombinant Ne c-552r is particularly high-quality, with a 1.63-Å resolution. Analysis of these structures reveals that the mutation influences heme ruffling, which provides a basis for the altered electronic structure of this mutant. Furthermore, evidence for the population of more than one axial Met conformation is observed in the structure of Ne c-552n. These results confirm that heme conformation plays a role in determining electronic structure of cytochromes. Future studies of the effects of X-rays on the samples and also of the electronic structure of these proteins are underway.
Experimental Section
Protein Sample Preparation
Purification of Ne c-552n from N. europaea:
The Ne c-552n was a gift from Alan B. Hooper (University of Minnesota) and isolated from N. europaea as previously described.[23b]
Expression and purification of Ne c-552r and Ne N64Δ:
Expression plasmids for recombinant Ne c-552 (pSNEC) and Ne N64Δ (pSNECN64Δ) prepared previously were used in this study.[13a, 15, 47] Ne c-552 and Ne N64Δ were expressed and purified using the same procedure. The proteins were expressed by growth in BL21(DE3)* cells under the control of the T7 promoter. BL21(DE3)* cells harboring pSNEC (Ampr) or pSNECN64Δ (Ampr), along with pEC86 (Cmr)[48] were grown in LB medium supplemented with 50 μg/ml Amp and 50 μg/ml Cm. Initial 25-mL cultures were grown at 37 °C and 175 rpm shaking for ~ 8 hours, and then added to 1 L of LB medium in a 4-L flask and grown at 37 °C and 110 rpm for ~ 16 hours. Harvested cells from each 0.5 L of growth medium were lysed in 20 mL of lysis buffer (10 mM pH 8 TRIS buffer including 4 mg/ml lysozyme, 1 μL/mL Triton X-100, 1 μg/ml DNase I). After a 1-hr incubation at 30 °C, the contents of the lysis reaction were centrifuged and the pink supernatant was collected. The pH of the supernatant was decreased to 4 with acetic acid and the supernatant was centrifuged following a 10-minute incubation at 45 °C. Initial purification of the resultant supernatant was performed on a CM Sepharose Fast Flow (Sigma) cation exchange column equilibrated with 10 mM sodium acetate buffer at pH 4.5; elution of the protein was achieved with a 0–200 mM sodium chloride gradient. Concentrated protein solution was exchanged into 10 mM sodium acetate buffer at pH 4.5 and oxidized with 5-fold excess of K[Co(EDTA)] for at least 12 hr. The oxidized protein was further purified on a Fast Protein Liquid Chromatography (FPLC) system equipped with a Mono-S 10/10 column (GE Healthcare Lifesciences) in 10 mM sodium acetate buffer, pH 4.5, and a 0–18 mM sodium chloride gradient. Only the central portion of the protein band was collected, and purity was verified by NMR spectroscopy. No phosphate ions or residual phosphate-contaminated items were used during the purification steps to avoid any complications in structure refinement steps due to phosphate ions. Because previous cytochrome c crystallization attempts that included imidazole in crystallization solutions resulted in imidazole binding to the iron by displacing the axial Met, we avoided the use of any imidazole during the purification and sample preparation.
Protein Crystallization
Ne c-552r samples for crystallization were prepared in 50 mM Na-HEPES buffer at pH 7.0 and concentrated to 7–8 mM (ε409 = 116 mM−1 cm−1). Initial crystal-growth screening of Ne c-552r and Ne N64Δ was done by the hanging drop method with JCSG Core Suites I to IV (Qiagen Inc.) at 4 °C by use of a Mosquito liquid handling robot (TTP LabTech) in the Structural Biology and Biophysics Facility at the University of Rochester Medical Center (Table S1). Additional crystallization screening for Ne c-552r and Ne N64Δ was performed at the Regional Core Facility for Structural Biology and Bioinformatics at the South-Eastern Norway Regional Health Authority with an Orxy6 crystallization robot (Douglas Instruments Ltd) with JSCG+ (Molecular Dimension Ltd), Index (Hampton Research Corp.) and Sigma Basic+Extended Kit (Sigma-Aldrich Co. LLC) screens. Screening yielded several promising hits that were optimized (supporting material Table S2). The final crystallization condition used for Ne c-552r was 19% (w/v) PEG 6000, 0.2 M LiCl, 0.1 M Tris-HCl pH 7.4 with crystals grown by hanging drop at 4 °C. For Ne N64Δ the final conditions used for the monoclinic crystal form was 0.1 M CHES pH 9.5, 30% (w/v) PEG 8000, and for the hexagonal crystal form 0.1 M Tris-HCl pH 8.5, 3.1 M ammonium sulfate; both habits were grown by sitting-drop at 4 °C from 7.8 mM protein. The best Ne c-552r crystals grew from solutions of PEG and exhibited a rod-like hexagonal habit with c* along the major axis; crystals grew to a maximum size of 0.3 mm × 0.3 mm × 0.5 mm in 2 weeks. Similarly, screening of Ne N64Δ also resulted in high-quality crystals that were optimized at 4 °C by the sitting drop method. The best Ne N64Δ crystals were monoclinic and were prepared from PEG 8000; a high-salt hexagonal crystal form was also prepared from ammonium sulfate. Ne c-552n was crystallized according to previously reported conditions.[49]
X-ray Diffraction Experiment and Structure Refinement
Ne c-552r:
Single crystals were cryo-protected in mother liquor containing 5% to 20% (w/v) glycerol in place of water. Crystals were transferred for 3 min into each of four cryo-solutions, followed by capture in thin nylon loops (Hampton Research) that were flash-frozen in a stream of nitrogen gas at −173 °C. X-ray diffraction data were collected at beam line A1 of the Cornell High Energy Synchrotron Source (Ithaca, NY). Data were collected at −173 °C using a Q210 CCD detector (ADSC, CA). and reduced with HKL2000.[50] The hexagonal Ne c-552r structure had one molecule per asymmetric unit, and was solved by molecular replacement using the Pa c-551 structure (PDB entry 351C[22]) as a search model in PHASER.[51] Refinement was conducted in PHENIX,[52] and model building was conducted in COOT.[53] In the later stages, TLS refinement was introduced using the program defaults. Restraints were applied for the Fe-NHEME, Fe-NHIS, Fe-SMET or SCYS-CHEME distances.
Ne N64Δ and Ne c-552n:
For X-ray diffraction experiments of Ne N64Δ, single crystals were transferred into a cryo-solution at 4°C for 10–20 seconds before being flash-frozen in liquid nitrogen, held by nylon loops (Hampton Research). At higher temperatures the crystals seemed to dissolve. The cryo-solutions were made by including 30% glucose as a cryoprotectant in the respective crystallization solutions. For Ne N64Δ, the diffraction data were collected at beam line ID29 at the ESRF (European Synchrotron Radiation Facility), Grenoble, France. Data were collected at 100 K using a 6M Pilatus detector (DECTRIS Ltd.). The Ne c-552n data were collected at beam line X11 at EMBL at DESY (Deutsches Elektronen-Synchrotron), Hamburg, Germany, with a MARCCD detector at 100 K. The diffraction data were processed and scaled with MOSFLM[54] and SCALA[55] and the space group confirmed with POINTLESS through the CCP4 software suite.[56] The monoclinic Ne N64Δ structure with four molecules in the asymmetric unit was solved by molecular replacement with PHASER[51] A homolog model of Ne N64Δ modeled with SWISS-MODEL[57] from the Pa c-551 structure (PDB entry 351C)[22] was used as a search model in PHASER. The hexagonal Ne N64Δ with four molecules in the asymmetric unit, and the Ne c-552n with 18 molecules in the asymmetric unit were solved using the monoclinic Ne N64Δ as a search model in PHASER. Refinements included multiple cycles of restrained refinement in REFMAC[58] and model building and addition of waters in COOT.[53] In the later stages, TLS refinement was introduced based on the monomers. For the monoclinic Ne N64Δ structure medium NCS restraints were introduced. No restraints were used for the Fe-NHEME, Fe-NHIS, Fe-SMET or SCYS-CHEME distances. To calculate the absorbed X-ray dose (Gy = J kg−1) of the different crystals during crystallographic data collection the program RADDOSE[59] was used. Structure figures were made with PyMOL.[60] All backbone root mean square deviation (RMSD) values were calculated using PDBeFold (http://www.ebi.ac.uk).
Single-crystal light absorption spectroscopy:
Light absorption spectroscopy was carried out on single-crystals (frozen as described above) of Ne c-552r and Ne N64Δ at 100 K before and after exposure to X-rays. These experiments were performed at beamline BM01 SNBL (Swiss-Norwegian Beam Line) at ESRF, and at beamline X10SA and SLSpectroLAB at SLS (Swiss-Light-Source). At SNBL spectra was recorded with an online microspectrophotometer model XSPECTRA (4DX System AB) equipped with a halogen lamp and ORIEL CCD detector (Andor Technology). At X10SA spectra were recorded with an on-axis built microspectrophotometer system with an Andor 303i Czerny–Turner spectrograph and a Newton electron multiplying CCD (Andor Technology).[61]
Single-crystal (resonance) Raman spectroscopy:
Raman spectroscopy was carried out on single crystals of Ne c-552r and Ne N64Δ at 100 K. The resonance Raman spectrum (λex = 405 nm) of Ne c-552r was recorded at beamline X10SA and SLSpectroLAB at SLS with an on-axis built microspectrophotometer system equipped with a 405 nm laser (OMICRON Laserage, LDM405.400.CWA.L.W) and an Andor 303i Czerny–Turner spectrograph and a Newton electron multiplying CCD (Andor Technology).[61] Raman spectra were accumulated in the 600–2000 cm−1 range with ~1 mW laser intensity at the sample, 60-s exposure time and 2 accumulations. The Raman spectra (λex = 785 nm) of Ne N64Δ was recorded at SNBL at ESRF on a Renishaw inVia Raman microscope equipped with a 785 nm near-IR laser. All spectra were recorded online through backscattering at 100 K. Raman spectra were accumulated in the 200–2000 cm−1 range with ~10 mW laser intensity at the sample, 45-s exposure time and 5 accumulations. All spectra were baseline corrected in order to remove contributions from fluorescence and were slightly smoothed.
Supplementary Material
Acknowledgements
We thank Dr. J. Jenkins for technical assistance. K.L.B. acknowledges support from NIH/NIGMS R01-GM63170. This research was funded in part by a shared instrumentation grant from NIH S10-RR026501 to J.E.W. This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS). The Macromolecular Diffraction at CHESS (MacCHESS) facility is supported by NSF award DMR-0936384 and the NIH/NIGMS award GM103485. We thank Dr. S. Karlsen for crystallization and data collection of Ne c-552n, obtained fom Prof. Alan B. Hooper, and Dr. B. Dalhus for the crystallization screening at the Regional Core Facility for Structural Biology and Bioinformatics at the South-Eastern Norway Regional Health Authority (Grants no. 2009100 and 2011040). The K.K.A group acknowledges financial support from the Norwegian Research Council through projects 177661/V30 / 214239/F20 /218412/F50 and MLSUiO program for Molecular Life Science research at the University of Oslo through the PX-Oslo X-ray core facilities. G.Z. acknowledges a fellowship grant from EC FP7 People Marie Curie Actions PIEF 235237. We acknowledge financial support for synchrotron travel from the Norwegian Research Council through projects 138370/V30 /216625/F50 (K.K.A.), and from the European Community’s Seventh Framework Programme (FP7/2007–2013) under BioStruct-X (grant agreement N°283570) on project 1760 (K.K.A./H.P.H.). We gratefully acknowledge the ESRF (MX-1201) for providing beam time, the EMBL Hamburg at DESY for providing beam time, the Swiss–Norwegian Beam Line (SNBL) at ESRF (01-02-928) for providing beam time with online UV-vis and Raman spectroscopy, and SLS (20110052, 20110683) for providing beamtime with online microspectrophotometer. We thank the team at X11 EMBL Hamburg, the team at ID29 at ESRF, the team at SNBL and Dr. Wouter van Beek for the help with the online UV-vis and Raman setup, and the team at X10SA and Dr. Guillaume Pompidor for help with the on-axis online microspectrophotometer.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- [1].Marcus RA, Sutin N, Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar]
- [2].Reedy CJ, Elvekrog MM, Gibney BR, Nucleic Acids Res. 2008, 36, D307–D313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Walker FA, Inorg. Chem 2003, 42, 4526–4544. [DOI] [PubMed] [Google Scholar]
- [4].a) Teschner T, Yatsunyk L, Schunemann V, Paulsen H, Winkler H, Hu CJ, Scheidt WR, Walker FA, Trautwein AX, J. Am. Chem. Soc 2006, 128, 1379–1389; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walker FA, Chem. Rev 2004, 104, 589–615. [DOI] [PubMed] [Google Scholar]
- [5].Bushnell GW, Louie GV, Brayer GD, J. Mol. Biol 1990, 214, 585–595. [DOI] [PubMed] [Google Scholar]
- [6].Battistuzzi G, Borsari M, Sola M, Antioxid. Redox Signal 2001, 3, 279–291. [DOI] [PubMed] [Google Scholar]
- [7].Gray HB, Winkler JR, Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3534–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gray HB, Winkler JR, Ann. Rev. Biochem 1996, 65, 537–561. [DOI] [PubMed] [Google Scholar]
- [9].Banci L, Bertini I, Luchinat C, Pierattelli R, Shokhirev NV, Walker FA, J. Am. Chem. Soc 1998, 120, 8472–8479. [Google Scholar]
- [10].Bertini I, Cavallaro G, Rosato A, J. Inorg. Biochem 2007, 101, 1798–1811. [DOI] [PubMed] [Google Scholar]
- [11].Banci L, Bertini I, Bren KL, Cremonini MA, Gray HB, Luchinat C, Turano P, J. Biol. Inorg. Chem 1996, 1, 117–126. [Google Scholar]
- [12].Bowman SEJ, Bren KL, Nat. Prod. Rep 2008, 25, 1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Bren KL, Kellogg JA, Kaur R, Wen X, Inorg. Chem 2004, 43, 7934–7944; [DOI] [PubMed] [Google Scholar]; b) Zhong L, Wen X, Rabinowitz TM, Russell BS, Karan EF, Bren KL, Proc. Natl. Acad. Sci. U.S.A 2004, 101, 8637–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zoppellaro G, Bren KL, Ensign AA, Harbitz E, Kaur R, Hersleth H-P, Ryde U, Hederstedt L, Andersson KK, Biopolymers 2009, 91, 1064–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zoppellaro G, Harbitz E, Kaur R, Ensign AA, Bren KL, Andersson KK, J. Am. Chem. Soc 2008, 130, 15348–15360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Can M, Zoppellaro G, Andersson KK, Bren KL, Inorg. Chem 2011, 50, 12018–12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Van Doorslaer S, Tilleman L, Verrept B, Desmet F, Maurelli S, Trandafir F, Moens L, Dewilde S, Inorg. Chem 2012, 51, 8834–8841. [DOI] [PubMed] [Google Scholar]
- [18].Zoppellaro G, Teschner T, Harbitz E, Schuenemann V, Karlsen S, Arciero DM, Ciurli S, Trautwein AX, Hooper AB, Andersson KK, ChemPhysChem 2006, 7, 1258–1267. [DOI] [PubMed] [Google Scholar]
- [19].Benini S, Gonzalez A, Rypniewski WR, Wilson KS, Van Beeumen JJ, Ciurli S, Biochemistry 2000, 39, 13115–13126. [DOI] [PubMed] [Google Scholar]
- [20].Liptak MD, Wen X, Bren KL, J. Am. Chem. Soc 2010, 132, 9753–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Travaglini-Allocatelli C, Gianni S, Dubey VK, Borgia A, Di Matteo A, Bonivento D, Cutruzzolà F, Bren KL, Brunori M, J. Biol. Chem 2005, 280, 25729–25734. [DOI] [PubMed] [Google Scholar]
- [22].Matsuura Y, Takano T, Dickerson RE, J. Mol. Biol 1982, 156, 389–409. [DOI] [PubMed] [Google Scholar]
- [23].a) Yamanaka T, Shinra M, Journal of Biochemistry 1974, 75, 1265–1273; [DOI] [PubMed] [Google Scholar]; b) Arciero DM, Peng QY, Peterson J, Hooper AB, FEBS Lett. 1994, 342, 217–220. [DOI] [PubMed] [Google Scholar]
- [24].Larsen RW, Chavez MD, Nunez DJ, Davidson MW, Knaff DB, Krulwich TA, Ondrias MR, Arch. Biochem. Biophys 1990, 283, 266–270. [DOI] [PubMed] [Google Scholar]
- [25].a) Henderson R, Proceedings of the Royal Society B-Biological Sciences 1990, 241, 6–8; [Google Scholar]; b) Owen RL, Rudino-Pinera E, Garman EF, Proc. Natl. Acad. Sci. U.S.A 2006, 103, 4912–4917; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Garman EF, Acta Crystallogr. Sect. D-Biol. Crystallogr 2010, 66, 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Carugo O, Carugo KD, Trends Biochem. Sci 2005, 30, 213–219; [DOI] [PubMed] [Google Scholar]; b) Beitlich T, Kuehnel K, Schulze-Briese C, Shoeman RL, Schlichting I, J. Synchrotron Radiat. 2007, 14, 11–23; [DOI] [PubMed] [Google Scholar]; c) Hersleth H-P, Hsiao Y-W, Ryde U, Gorbitz CH, Andersson KK, Chem. Biodiversity 2008, 5, 2067–2089. [DOI] [PubMed] [Google Scholar]
- [27].Hersleth H-P, Andersson KK, Biochim. Biophys. Acta 2011, 1814, 785–796. [DOI] [PubMed] [Google Scholar]
- [28].a McGeehan JE, Bourgeois D, Royant A, Carpentier P, Biochim. Biophys. Acta 2011, 1814, 750–759; [DOI] [PubMed] [Google Scholar]; b) Ronda L, Bruno S, Bettati S, Mozzarelli A, Biochim. Biophys. Acta 2011, 1814, 734–741. [DOI] [PubMed] [Google Scholar]
- [29].Scott RA, Mauk AG, Cytochrome c: A Multidisciplinary Approach, University Science Books, Sausalito, CA, 1996. [Google Scholar]
- [30].a) Meharenna YT, Doukov T, Li H, Soltis SM, Poulos TL, Biochemistry 2010, 49, 2984–2986; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hersleth H-P, Andersen NH, Andersson KK, J. Biol. Inorg. Chem 2011, 16, S481. [Google Scholar]
- [31].a) Shokhirev NV, Walker FA, J. Biol. Inorg. Chem 1998, 3, 581–594; [Google Scholar]; b) Shokhirev NV, Walker FA, J. Am. Chem. Soc 1998, 120, 981–990. [Google Scholar]
- [32].a) Wüthrich K, Proc. Natl. Acad. Sci. U.S.A 1969, 63, 1071–1078; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Senn H, Keller RM, Wüthrich K, Biochem. Biophys. Res. Commun 1980, 92, 1362–1369. [DOI] [PubMed] [Google Scholar]
- [33].Liptak MD, Fagerlund RD, Ledgerwood EC, Wilbanks SM, Bren KL, J. Am. Chem. Soc 2011, 133, 1153–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Timkovich R, Cai ML, Zhang BL, Arciero DM, Hooper AB, Eur. J. Biochem 1994, 226, 159–168. [DOI] [PubMed] [Google Scholar]
- [35].Wen X, Patel KM, Russell BS, Bren KL, Biochemistry 2007, 46, 2537–2544. [DOI] [PubMed] [Google Scholar]
- [36].Timkovich R, Bergmann D, Arciero DM, Hooper AB, Biophys. J 1998, 75, 1964–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T, Proc. Natl. Acad. Sci. U.S.A 2011, 108, 16247–16252; [DOI] [PMC free article] [PubMed] [Google Scholar]; bb) Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber, Nature 2009, 462, 669–U149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wen X, Bren KL, Biochemistry 2005, 44, 5225–5233. [DOI] [PubMed] [Google Scholar]
- [39].Shelnutt JA, Song XZ, Ma JG, Jia SL, Jentzen W, Medforth CJ, Chem. Soc. Rev 1998, 27, 31–41. [Google Scholar]
- [40].Ma JG, Laberge M, Song XZ, Jentzen W, Jia SL, Zhang J, Vanderkooi JM, Shelnutt JA, Biochemistry 1998, 37, 5118–5128. [DOI] [PubMed] [Google Scholar]
- [41].Nakamura M, Coord. Chem. Rev 2006, 250, 2271–2294. [Google Scholar]
- [42].Jentzen W, Song XZ, Shelnutt JA, J. Phys. Chem. B 1997, 101, 1684–1699. [Google Scholar]
- [43].Brautigan DL, Feinberg BA, Hoffman BM, Margoliash E, Peisach J, Blumberg WE, J. Biol. Chem 1977, 252, 574–582. [PubMed] [Google Scholar]
- [44].Benning MM, Wesenberg G, Caffrey MS, Bartsch RG, Meyer TE, Cusanovich MA, Rayment I, Holden HM, J. Mol. Biol 1991, 220, 673–685. [DOI] [PubMed] [Google Scholar]
- [45].Othman S, Fitch J, Cusanovich MA, Desbois A, Biochemistry 1997, 36, 5499–5508. [DOI] [PubMed] [Google Scholar]
- [46].Michel LV, Ye T, Bowman SEJ, Levin BD, Hahn MA, Russell BS, Elliott SJ, Bren KL, Biochemistry 2007, 46, 11753–11760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ye T, Kaur R, Wen X, Bren KL, Elliott SJ, Inorg. Chem 2005, 44, 8999–9006. [DOI] [PubMed] [Google Scholar]
- [48].Arslan E, Schulz H, Zufferey R, Kunzler P, Thöny-Meyer L, Biochem. Biophys. Res. Commun 1998, 251, 744–747. [DOI] [PubMed] [Google Scholar]
- [49].Nagata C, Moriyama H, Fujiwara T, Fukumori Y, Tanaka N, J. Biochem 1994, 116, 946–947. [DOI] [PubMed] [Google Scholar]
- [50].Otwinowski Z, Minor W, Macromolecular Crystallography, Pt A 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [51].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, Acta Crystallogr. Sect. D-Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Emsley P, Lohkamp B, Scott WG, Cowtan K, Acta Crystallogr. Sect. D-Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Leslie AGW, Powell HR, in Evolving Methods for Macromolecular Crystallography, Vol. 245 (Eds.: Read RJ, Sussman JL), Springer; Netherlands, 2007, pp. 41–51. [Google Scholar]
- [55].Evans P, Acta Crystallogr. Sect. D-Biol. Crystallogr 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- [56].a) Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Acta Crystallogr. Sect. D-Biol. Crystallogr 2011, 67, 235–242; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Potterton E, Briggs P, Turkenburg M, Dodson E, Acta Crystallogr. Sect. D-Biol. Crystallogr 2003, 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- [57].a) Arnold K, Bordoli L, Kopp J, Schwede T, Bioinformatics 2006, 22, 195–201; [DOI] [PubMed] [Google Scholar]; b Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T, Nucleic Acids Res. 2009, 37, D387–D392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].a) Murshudov GN, Vagin AA, Dodson EJ, Acta Crystallogr. Sect. D-Biol. Crystallogr 1997, 53, 240–255; [DOI] [PubMed] [Google Scholar]; b) Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA, Acta Crystallogr. Sect. D-Biol. Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].a) Murray JW, Ravelli RBG, Garman EF, J. Appl. Crystallogr 2004, 37, 513–522; [Google Scholar]; b Paithankar KS, Garman EF, Acta Crystallogr. Sect. D-Biol. Crystallogr 2010, 66, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].The PyMOL Molecular Graphics System, Version 1.5, Schrödinger, LLC.
- [61].Owen RL, Pearson AR, Meents A, Boehler P, Thominet V, Schulze-Briese C, J Synchrotron Radiat. 2009, 16, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.