

Abstract
The objective of this comprehensive review is to summarize and discuss the available evidence of how adipose tissue inflammation affects insulin sensitivity and glucose tolerance. Low-grade, chronic adipose tissue inflammation is characterized by infiltration of macrophages and other immune cell populations into adipose tissue, and a shift towards more pro-inflammatory subtypes of leukocytes. The infiltration of pro-inflammatory cells in adipose tissue is associated with an increased production of key chemokines such as C-C motif chemokine ligand 2, pro-inflammatory cytokines including tumor necrosis factor α and interleukins 1β and 6, as well as reduced expression of the key insulin sensitizing adipokine, adiponectin. In both rodent models and humans, adipose tissue inflammation is consistently associated with excess fat mass and insulin resistance. In humans, associations with insulin resistance are stronger and more consistent for inflammation in visceral as opposed to subcutaneous fat. Further, genetic alterations in mouse models of obesity that reduce adipose tissue inflammation are – almost without exception - associated with improved insulin sensitivity. However, a dissociation between adipose tissue inflammation and insulin resistance can be observed in very few rodent models of obesity as well as in humans following bariatric surgery- or low-calorie diet-induced weight loss, illustrating that the etiology of insulin resistance is multifactorial. Taken together, adipose tissue inflammation is a key factor in the development of insulin resistance and type 2 diabetes in obesity, along with other factors that likely include inflammation and fat accumulation in other metabolically active tissues.
Introduction
The role of adipose tissue in whole-body metabolic homeostasis has gained appreciation in recent decades as a deeper understanding of the essential biological functions of this organ has developed. Adipose tissue was originally believed to serve simply as an inert energy storage reservoir; however, it is now known to also function as a major endocrine organ that secretes adipokines, cytokines, and chemokines (8). These signaling factors regulate diverse metabolic processes in many organs including liver, skeletal muscle, pancreas, and brain, and in adipose tissue itself (383). Concomitant with the global increase in obesity prevalence in recent decades, there has been an increase in prevalence of type 2 diabetes mellitus (T2DM; Table 1) (128). Substantial research efforts have been undertaken to understand the molecular and cellular basis of the association between excess adiposity and impaired glucose homeostasis that underlies T2DM, and several adipose-tissue centric mechanisms have been proposed as potential links. Of these, chronic, low-grade adipose tissue inflammation has received considerable attention since its initial characterization in obese mice and humans (188, 189). Furthermore, adipose tissue inflammation may be a common underlying contributor to some of the other proposed mechanisms mediating the development of insulin resistance in obesity.
Table 1.
List of abbreviations
| AGPAT | 1-acylglycerol-3-phosphate acyltransferase |
| AMPK | AMP-activated protein kinase |
| APPL1 | adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 |
| ATGL | adipose triglyceride lipase |
| ATM | adipose tissue macrophages |
| BAI | body adiposity index |
| BMI | body mass index |
| CCL | C-C motif chemokine ligand |
| CCR | C-C motif chemokine receptor |
| CLS | crown-like structures |
| COX | cyclooxygenase |
| CRP | C-reactive protein |
| CVD | cardiovascular disease |
| DAMP | damage-associated molecular pattern |
| DGAT | diacylglycerol acyltransferase |
| DNL | de novo lipogenesis |
| ER | endoplasmic reticulum |
| FFA | free fatty acids |
| GIR | glucose infusion rate |
| GPAT | glycerol-3-phosphate acyltransferase |
| GWAS | genome-wide association study |
| HFD | high-fat diet |
| HMW | high-molecular weight |
| HOMA | homeostatic model assessment |
| HSL | hormone sensitive lipase |
| IκK | inhibitor of κ kinase |
| IL | interleukin |
| IFN | interferon |
| JNK | c-Jun NH2-terminal kinase |
| KO | knockout |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| MHO | metabolically healthy obesity |
| MIP | macrophage inflammatory protein |
| NAFLD | non-alcoholic fatty liver disease |
| NLRP3 | nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing 3 |
| PAMP | pathogen-associated molecular pattern |
| PPAR | peroxisome proliferator-activated receptor |
| PRR | pattern recognition receptors |
| RBP | retinol binding protein |
| SAA | serum amyloid protein A |
| SAT | subcutaneous adipose tissue |
| SCD | stearoyl-CoA desaturase |
| SVC | stromavascular cell |
| T2DM | type 2 diabetes mellitus |
| TG | triglyceride |
| TLR | toll-like receptor |
| TNFα | tumor necrosis factor-α |
| TZD | thiazolidinedione |
| UPR | unfolded protein response |
| VAT | visceral adipose tissue |
| VSG | vertical sleeve gastrectomy |
| WC | waist circumference |
| WHR | waist to hip ratio |
| WHtR | waist to height ratio |
Here, we review the evidence from rodent and human studies on the role of chronic, low-grade adipose tissue inflammation in the development of insulin resistance and T2DM. We will discuss how adipose tissue inflammation may contribute to the development of insulin resistance and the increased risk of T2DM in obesity (Figure 1). We begin with an overview of the relationship between obesity and insulin resistance and factors that may mediate this association. We also highlight metabolically healthy obesity (MHO) and lipodystrophy, which are exceptions to the positive association between adiposity and insulin resistance. An overview of the current understanding of immune cell infiltration and associated downstream molecular events that commonly accompany chronic caloric excess and impair insulin signaling follows. Much of what is known regarding the temporal development of adipose tissue inflammation in obesity and associated insulin resistance has largely been gained from the study of rodent models under high-fat diet (HFD) feeding conditions. In humans, several cross-sectional studies that compare adipose tissue inflammation in non-obese versus obese and insulin sensitive versus insulin resistant subjects have been conducted and are informative of the association between inflammation and these common metabolic states. Data from both rodent and human studies are considered together in the discussion of whether and how adipose tissue inflammation may be a key mechanism driving obesity-associated insulin resistance.
Figure 1. Associations between obesity, insulin resistance, and adipose tissue inflammation.
Obesity is associated with both insulin resistance and adipose tissue inflammation in humans and rodent models. Adipose tissue inflammation and insulin resistance are also associated, but the direction of causality is controversial. This review will explore each of these relationships, highlighting evidence generated from studies conducted in both humans and rodents.
Teaching points: Obesity is associated with both insulin resistance and adipose tissue inflammation in humans and animal models. Adipose tissue inflammation and insulin resistance are also associated, but the direction of causality is controversial.
Background
The epidemics of obesity and type 2 diabetes mellitus
Since the 1970s, the United States has experienced an unprecedented increase in the prevalence of both obesity and T2DM. Based on the most recently published statistics, 38% of US adults were obese in 2013/2014, as defined by a body mass index (BMI) ≥ 30 kg/m2 (129). This is sharply increased from 13% in 1960 (128). This epidemic of obesity has been paralleled with an epidemic of T2DM. Among adults in the United States, National Health and Nutrition Examination Survey data estimated the prevalence of diabetes was 14.3% in 2011/12 (306). Intriguingly, 36% of those individuals who were found to be diabetic had not been previously diagnosed (306). Because type 1 diabetes accounts for only 3.6–6.0% of all diabetes cases (307), the prevalence of T2DM was at least 13.4%. Possibly as concerning is the prevalence of pre-diabetes among US adults, which was 38% in 2011/12 (306).
Aside from being a health concern, as outlined below, obesity is also a fiscal issue (125, 126, 465). Direct medical costs are estimated to be up to 45% higher for obese (BMI ≥ 30 kg/m2) individuals as compared to those for normal weight (BMI < 25 kg/m2) individuals, with a crude overall estimate of 30% higher costs (125, 434, 513). A 2016 meta-analysis of studies investigating the economic burden of obesity in the United States reported that the direct medical costs of obesity was $1,910 per person annually (234), translating to nearly $150 billion on the national level (234). For diabetes, healthcare costs in 2012 amounted to $245 billion, with on average $13,700 in medical expenditures annually for each patient with diabetes (19).
Associated health risks with overweight and obesity
Obesity is a serious health concern because it is associated with an increased risk of several health conditions including T2DM, hypertension, hyperlipidemia, cardiovascular disease (CVD), arthritis, gallbladder disease, certain cancers and non-alcoholic fatty liver disease (NAFLD) (Figure 2) (4, 513). In particular, the association between adiposity and T2DM is strong, as highlighted in a recent meta-analysis comprised of 18 prospective cohort studies, with a sample size of 590,251 individuals across a wide geographic range that included the United States, Asia-Pacific, and Europe (3). Overall, obese individuals had a seven fold higher risk of diabetes, and overweight individuals had a roughly three fold higher risk of T2DM, when compared to normal weight individuals (3). However, it should be noted that there were variations in the relative risks according to the study population characteristics including gender and study region, as well as study quality characteristics including sample size, method of diabetes assessment, and method of BMI ascertainment (3). Of note, obese women had a higher risk for T2DM with a relative risk of approximately eight compared to obese men who had a relative risk of T2DM of approximately six when compared to normal weight peers (3). Although the reasons for these sex differences remain unclear, it may be related to fat distribution and mass (3). Additionally, there remains uncertainty as to whether the relationship between obesity and T2DM is linear or whether there is a threshold effect, as will be discussed in more detail in later sections (3).
Figure 2. Causes of and health risks associated with obesity.
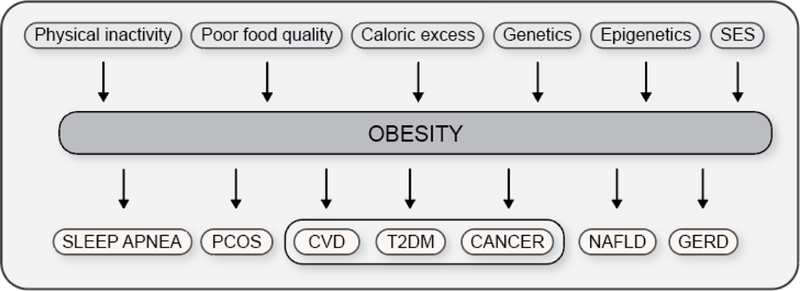
Several factors contribute to the development of obesity; these factors may have an environmental, biological, or genetic basis. Obesity subsequently increases the risk for the development of many diseases and disorders, including CVD, T2DM, and cancer, three of the top ten killers of adults in the United States. Abbreviations: SES, socioeconomic status; PCOS, polycystic ovary syndrome; CVD, cardiovascular disease; T2DM, type 2 diabetes mellitus; NAFLD, nonalcoholic fatty liver disease; GERD, gastroesophageal reflux disease.
Teaching points: The development of obesity is complex and many factors are known to contribute to it; these factors may have an environmental, biological, or genetic basis. Obesity increases the risk for the development of many other diseases and disorders, including CVD, T2DM, and cancer, three of the top ten killers of adults in the United States.
SES, socioeconomic status; PCOS, polycystic ovary syndrome; CVD, cardiovascular disease; T2DM, type 2 diabetes mellitus; NAFLD, nonalcoholic fatty liver disease; GERD, gastroesophageal reflux disease.
Obesity can be measured using a variety of tools including BMI, waist circumference (WC), waist to hip ratio (WHR), and more recently waist to height ratio (WHtR) or body adiposity index (BAI) (36, 240). BMI and BAI are indicators of overall mass, whereas WC and WHR are traditionally thought to capture abdominal obesity, where there are increased levels of visceral adipose tissue (VAT) (240). A meta-analysis by Kodama et al. (240) identified 15 prospective cohort studies that investigated the relationship between obesity measured as WHtR and at least one additional obesity indicator (BMI, WC, or WHR) and T2DM. The results showed that per one standard deviation increase in WHtR the relative risk for T2DM is 1.62 (240). Per one standard deviation increase in BMI, WC, and WHR, the relative risks for diabetes were 1.55, 1.63, and 1.52, respectively (240). These results also indicate that WC and WHtR may be better indicators of obesity-associated T2DM as compared to WHR or BMI. Overall, this suggests that the relationship between adiposity and T2DM may be stronger than is indicated by meta-analyses that rely on BMI alone as a measure of adiposity.
Mechanisms linking obesity to T2DM
Glucose intolerance and T2DM ensue whenever the homeostatic control of plasma glucose concentrations is impaired due to a combination of low glucose effectiveness and a decrease in insulin sensitivity that is not fully compensated by an increase in the amount of insulin produced by the pancreatic β-cell (216, 420).
Glucose effectiveness is the ability of glucose to stimulate its own disposal at higher than basal concentrations, in a manner that is independent of insulin (420). Glucose effectiveness may account for up to 50% of postprandial glucose disposal (420), and reduced glucose effectiveness seems to be similar in importance to reduced insulin sensitivity for T2DM risk (291). Even though there is some evidence to suggest that an increase in adiposity reduces glucose effectiveness in mice (11), it is unclear whether this finding extends to humans, and there is no indication that low-grade chronic adipose tissue inflammation affects glucose effectiveness. This manuscript will therefore not consider a potential link between adipose tissue inflammation and reduced glucose effectiveness, even though it is important to emphasize that the absence of data should not be equated to the absence of a relationship between adipose tissue inflammation and glucose effectiveness.
Plentiful data do exist, however, on the relationship between insulin resistance and both adiposity and adipose tissue inflammation, in both animal models of obesity as well as humans. Insulin resistance is a state characterized by a reduced response of insulin target cells, such as myocytes and adipocytes, to the binding of insulin to the insulin receptor. The principal effect of insulin in these target tissues is to stimulate glucose uptake in the postprandial phase, and to inhibit lipolysis in adipocytes. In hepatocytes, major functions of insulin are to inhibit gluconeogenesis and to stimulate de novo lipogenesis (DNL). Any degree of insulin resistance needs to be compensated by increased insulin production by the pancreatic β-cell in order to prevent hyperglycemia (216). Thus, in healthy, non-diabetic individuals, insulin production from the pancreas increases as insulin sensitivity decreases (Figure 3), such that the product of insulin sensitivity and β-cell function, the so-called disposition index, will remain constant even if insulin sensitivity were to change substantially. If the pancreatic β-cell fails to fully compensate for insulin resistance, a phenomenon called β-cell dysfunction, glucose intolerance and eventually T2DM ensues (216). The causes and contributing factors for pancreatic β-cell dysfunction are incompletely understood. While increased adiposity and adipose tissue inflammation may be a contributing factor in the etiology of β-cell dysfunction, this relationship is likely mediated through insulin resistance itself (158), or plausibly related to pancreatic steatosis due to increased flux of nonesterified or free fatty acids (FFA) (216), even though strong evidence for this is lacking. As with glucose effectiveness, data suggesting a direct impact of adipose tissue inflammation on the pancreatic β-cell are relatively sparse. Therefore, this manuscript will largely focus on the impact of adipose tissue inflammation on insulin resistance, as a suggested primary mechanism through which adipose tissue inflammation is likely to exert an effect on glucose tolerance and therewith T2DM risk.
Figure 3. Insulin sensitivity, pancreatic β-cell function, and glucose effectiveness in the regulation of glucose homeostasis.
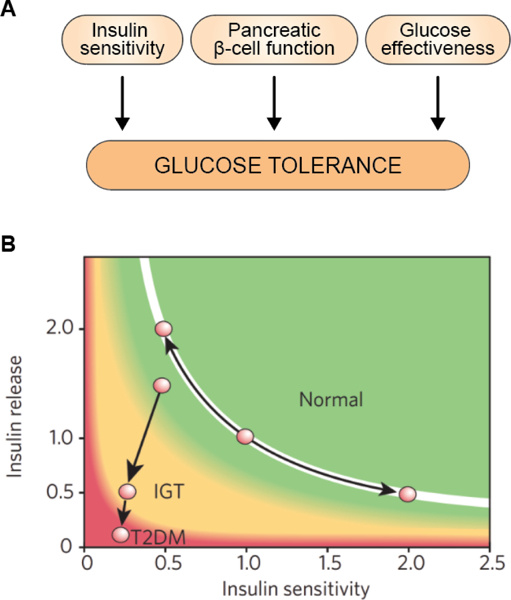
Glucose tolerance, i.e. the body’s ability to maintain glucose within a relatively narrow homeostatic range, is regulated by three key factors: insulin sensitivity, pancreatic β-cell function, and glucose effectiveness (A) (38, 215, 420). Insulin sensitivity is the responsiveness of liver and extrahepatic tissues, such as skeletal muscle and adipose tissue, to insulin (215). There are a number of physiological and pathophysiological mechanisms that affect insulin sensitivity, as we will discuss throughout this paper. In healthy, glucose tolerant individuals, as insulin sensitivity declines, the pancreatic β-cell will compensate by producing more insulin (B) (217). Only when the β-cell is unable to fully compensate for a given degree of insulin resistance will glucose intolerance ensue (217). This phenomenon, commonly called β-cell dysfunction, is a critical component of glucose homeostasis (217). In fact, in healthy, glucose tolerant individuals, the product of insulin sensitivity and pancreatic β-cell function, the disposition index, is constant as insulin sensitivity changes due to physiologic or pathophysiologic events (217). Put more simply, highly insulin sensitive individuals release little insulin in response to glucose stimulation, simply because more is not needed and would, in fact, be harmful, while less insulin sensitive individuals secrete more insulin to maintain normal glucose homeostasis. It is only when insulin production and secretion cannot fully compensate for insulin resistance that the disposition index declines and glucose intolerance and eventually T2DM ensues (217). The third key factor contributing to glucose homeostasis is glucose effectiveness, the ability of glucose to drive its own disposal and inhibit endogenous gluconeogenesis, in a manner independent of insulin (38, 420). Even though glucose effectiveness clearly exhibits substantial inter-individual variability (420), and reduced glucose effectiveness is as much a risk factor for T2DM as reduced insulin sensitivity (291), little is known about factors affecting glucose effectiveness. Glucose effectiveness is therefore often overlooked in studies of glucose homeostasis (101). Figure 3b reproduced from Kahn et al. (216), with permission.
Teaching points: Glucose tolerance is the body’s ability to maintain glucose within a relatively narrow homeostatic range and is regulated by three key factors: insulin sensitivity, pancreatic β-cell function, and glucose effectiveness (A) (38, 215, 420). Insulin sensitivity is the responsiveness of target cells to insulin signaling (215). In healthy, glucose tolerant individuals, a decline in insulin sensitivity is compensated for by an increase in insulin production and secretion (shown in green, normal glucose tolerance) (B) (217). Only when the pancreatic β-cells are unable to fully compensate for a decline in insulin sensitivity will glucose intolerance (shown in yellow, impaired glucose tolerance) and eventually T2DM (shown in red) ensue (217). Figure 3b reproduced from Kahn et al. (216), with permission.
Over the last decades, numerous mechanisms have been suggested to mediate the impact of expanded fat tissue on insulin sensitivity [reviewed in (216, 383)]. One of the earliest hypotheses for a link between obesity to insulin resistance and T2DM centered around elevated flux of FFA from the expanded adipose tissue to liver and muscle, which was hypothesized to affect insulin sensitivity in these tissues due to acute lipotoxicity and/or ectopic fat storage (383). FFA have also been shown to exert lipotoxicity on the pancreatic β-cell, suggesting that chronically elevated FFA concentrations, as in obesity, may play a role in β-cell dysfunction (216). The recognition of adipose tissue as an endocrine organ showed that some secreted hormones, such as adiponectin, could affect systemic insulin sensitivity. Specifically, the paradoxical finding of suppressed circulating adiponectin concentrations in obese individuals, together with the finding that adiponectin stimulates fat oxidation and insulin sensitivity in liver and muscle, gave rise to the hypothesis that hypoadiponectinemia could be one of the links between obesity and insulin resistance (532). Similarly, increased production of hormones such as resistin, retinol binding protein (RBP)-4, or pro-inflammatory cytokines in expanded adipose tissue may contribute to systemic insulin resistance (216, 383). Of particular relevance to this manuscript, many of the mechanisms known or hypothesized to link obesity and insulin resistance are associated with, or a direct consequence of, low-grade chronic adipose tissue inflammation. Specifically, insulin resistance in adipocytes induced by pro-inflammatory cytokines, including tumor necrosis factor-α (TNFα) and interleukin (IL)-6, attenuates the inhibitory effect of insulin on lipolysis and FFA release (216, 383). TNFα also directly inhibits adipocyte production of adiponectin (451), and pro-inflammatory signaling upregulates resistin production (2).
Objective of this paper
The main objective of this paper is to provide an overview of low-grade chronic adipose tissue inflammation, and its relationship with adiposity and insulin resistance, a key determinant of glucose intolerance. To provide context, we will initially review the relationship between obesity and insulin resistance, with a detailed description of the mechanisms linking increased adiposity to insulin resistance.
ADIPOSITY AND INSULIN RESISTANCE
Overweight and obesity develop as a result of chronic excess accumulation of energy in adipose tissue depots. Obesity is now widely regarded as one of the greatest risk factors for the development of insulin resistance. In this section, we review evidence that demonstrates a positive association between adiposity and insulin resistance (Figure 4), first from rodent models and then from human studies. We also discuss exceptions to and factors that modify the adiposity and insulin resistance association. Finally, we review proposed mechanisms that may underlie the association.
Figure 4. Obesity is associated with the development of insulin resistance.
The association between excess adiposity and insulin resistance is well established; however, there are exceptions to the relationship wherein obese individuals may be insulin sensitive and individuals with a deficit of adipose tissue, as in lipodystrophy, may be severely insulin resistant.
Teaching points: The association between excess adiposity and insulin resistance is well established in humans and animal models. However, there are known exceptions to this relationship wherein some obese individuals may be insulin sensitive while some individuals with a deficit of adipose tissue, as in lipodystrophy, may be severely insulin resistant.
Adipose tissue biology overview
A major physiological function of white adipose tissue is to store excess energy during times of caloric surplus, i.e., when exogenous fuel supply exceeds that which is required to support total energy expenditure. Triacylglycerols, or triglycerides (TG), are the predominant energy-storing lipid species in adipocytes, the parenchymal cells of adipose tissue. Triglycerides are hydrolyzed, and FFA and glycerol are released into the circulation for uptake and use by peripheral tissues during times of energy deficit. In addition to its central role in management of energy availability, adipose tissue exerts extensive control over systemic metabolic health largely through the production and secretion of adipokines by the adipocyte. Over the past 25 years, the discovery and study of these signaling molecules has revealed that they regulate diverse metabolic and physiologic processes including fatty acid oxidation, DNL, gluconeogenesis, insulin signaling, glucose uptake, food intake, and energy expenditure in metabolically active tissues such as liver, skeletal muscle, and brain (8). However, a role for adipose tissue in whole-body metabolic regulation was recognized even prior to the discovery of adipokines and their associated metabolic functions. Indeed, adverse health consequences associated with both an excess and a lack of fat tissue have been known for several decades.
TG are comprised of three fatty acyl chains esterified to a glycerol backbone; there are three major sources of fatty acids for TG synthesis. Exogenous dietary lipids and adipose-derived FFA are two of the major sources. The third pool are those that are de novo synthesized from carbohydrate precursors via the DNL pathway (445). DNL occurs in liver and adipose tissue when carbohydrates are available in excess of energy needs and hepatic glycogen capacity. In adipose tissue, TG synthesis occurs through the glycerolipid pathway (453). Fatty acids are sequentially esterified to a glycerol backbone by three enzymes, all of which have multiple isoforms: glycerol-3-phosphate acyltransferase (GPAT), 1-acylglycerol-3-phosphate acyltransferase (AGPAT), and diacylglycerol acyltransferase (DGAT) (424, 453). Insulin signaling regulates GLUT4 trafficking to the plasma membrane, thus regulating glucose uptake by the adipocyte. Insulin also regulates the expression and/or activity of several of the enzymes involved in both DNL and TG synthesis (453, 548). Lipolysis of adipose tissue TG results in the release of FFA and glycerol into the circulation for uptake and use by peripheral tissues. The fate of FFA upon uptake from the circulation largely depends on the tissue (e.g., liver versus skeletal muscle) and the physiological state (eg., fasted versus fed). The sequential hydrolysis of fatty acids in lipolysis occur largely through three different lipases: desnutrin/adipose TG lipase (ATGL), hormone sensitive lipase (HSL), and monoacylglycerol lipase (204). Lipolysis is normally suppressed by insulin signaling, where activity of both HSL and desnutrin/ ATGL are controlled by phosphorylation events downstream of insulin binding to its receptor (397).
A growing body of evidence suggests that adipose tissue DNL may play a role in maintenance of systemic insulin sensitivity in obesity. Expression of lipogenic genes and a master regulator of lipogenic gene expression, Srebf1, are reduced in epididymal adipose tissue of obese mice (334, 431) and in VAT and subcutaneous adipose tissue (SAT) of obese humans, and increase in SAT following bariatric surgery (105). Furthermore, expression of lipogenic genes in both VAT and SAT is positively correlated with measures of insulin sensitivity (105, 174).
Insulin resistance in genetic rodent models of obesity
There are several rodent models available for the study of obesity and related morbidities; these models have been comprehensively reviewed (220, 281, 388, 432). As it is beyond the scope of this review to discuss all models with an adiposity or body weight phenotype, we review representative and widely used genetic models of obesity. Spontaneous monogenic models of murine obesity provided early evidence suggesting adiposity is associated with impaired glucose homeostasis. Mutations in the leptin signaling pathway proved especially informative, with readily identifiable hallmark phenotypes including obesity, hyperphagia, hyperglycemia, and hyperinsulinemia (81). A recessive mutation causing extreme obesity was discovered in 1949 in the Jackson Laboratory colony and named Obese (also referred to as ob) (203). More than four decades later, the genetic defect was identified as a nonsense mutation in Lep that results in a truncated form of leptin and whole-body leptin deficiency (549). Profound insulin resistance in Lepob/ob mice was demonstrated by administration of extremely high insulin doses at levels that were lethal in non-obese mice but not in Lepob/ob animals. In addition, blood glucose levels remained over 200 mg/dL one hour post-insulin injection in the Lepob/ob group (298). Subsequent pair-feeding experiments conducted with wildtype and Lepob/ob mice suggested that the insulin resistance was directly associated with the obesity; blood glucose levels rapidly responded to exogenous insulin administration in wildtype and food-restricted, lean Lepob/ob mice but remained relatively elevated in an obese Lepob/ob group (33). Additional evidence for a link between adiposity and insulin resistance came from the study of Leprdb/db mice (195) homozygous for a recessive mutation in Lepr, which encodes the leptin receptor (74). Leprdb/db mice are obese and develop extreme hyperglycemia by six to eight weeks of age (195) while hyperinsulinemia is evident as early as three weeks (32). The Zucker rat model has also been widely used to study obesity and metabolic health since its discovery in 1963 (555), and subsequent work demonstrated that the model has a mutation in the leptin receptor (452). Like their murine counterparts, Zucker rats are hyperphagic, obese, hyperglycemic and hyperinsulinemic (554, 555).
Another common obesity mouse model is the Agouti mouse. In wildtype mice, the Agouti gene regulates coat color by inhibiting the production of black/brown pigments, to produce a red/yellow coat coloration. However, dominant Agouti mutations, such as the lethal yellow (Ay) and viable yellow (Avy) alleles, cause an obese phenotype in heterozygotes (220, 328). Agouti expression is largely restricted to the hair shaft and skin during neonatal development and in the testis in adults among wildtype mice, but Ay mice exhibit ectopic expression in a wide panel of tissues (55). The ectopic hypothalamic Ay expression directly contributes to the development of obesity through antagonism of melanocortin receptors 3 and 4, both of which are downstream of leptin signaling in the arcuate nucleus (326). Ay and Avy mice are hyperphagic and develop obesity by early adulthood and develop early onset hyperinsulinemia; Avy develop hyperglycemia and glucose intolerance (328).
With disrupted leptin signaling being the common root cause of obesity in these genetic models, an adiposity-independent effect of leptin in the regulation of insulin sensitivity cannot be ruled out. These rodent models of obesity therefore provide only suggestive evidence linking excess adiposity to the development of systemic insulin resistance. However, as we will see in the next section, obesity brought on by chronic caloric excess is characterized by a similar reduction in insulin sensitivity.
Insulin resistance in diet-induced rodent models of obesity
While there are many advantages to studying metabolic disturbances that accompany excessive adiposity in genetic mouse models of obesity (537), obesity induced via dietary manipulation is also a commonly used experimental approach. In contrast to monogenic models, diet-induced models of obesity may more closely reflect the natural development of adipose tissue accumulation as it occurs in most obese humans. In addition, the timing of onset can be readily controlled and developmental defects that may arise in genetic models are avoided. However, there is extensive heterogeneity in body weight response to various diets among different genetic mouse strains. For example, a study that included more than 100 inbred mouse strains revealed substantial variation in percent body fat in response to consumption of a standard low-fat chow diet and to a high-sucrose, HFD (32% kcal from fat), indicating strong genetic control over adiposity in response to dietary composition (370). Epigenetic modifications are also known to influence adiposity and weight gain in response to diet (293). Thus, our understanding of the relationship between these genetic or epigenetic factors that affect susceptibility to weight gain, diet composition, obesity, and associated metabolic disease may be hampered or skewed by studies that focus solely on animals that are either sensitive or resistant to the effects of a HFD on adiposity. Despite these issues, HFD feeding of mice remains a popular method to produce obese rodent for studies of metabolic dysfunction. The diet used for the control group is an important factor to consider in animal studies of diet-induced obesity, as the interpretation of study results can differ significantly depending on the control diet (35). Although standard laboratory chow is commonly used, a defined diet that differs from the HFD only in macronutrient composition (fat and carbohydrate) is the most appropriate control diet (501). The use of a defined low-fat control diet eliminates bioactive dietary components that are common in complex chow diets but not in HFD. C57BL/6 mice are a commonly used strain in such studies as they are highly susceptible to HFD-induced adiposity with body weight divergence from chow-fed controls as early as four weeks of HFD feeding, and they continue to gain weight under long-term feeding regimens (450). There is also variability in change in total adipose tissue mass among different depots and mouse strains in response to HFD (371, 509). The age of animals at the onset of HFD feeding is yet another factor that can influence weight gain (51, 246, 517). HFD feeding is commonly associated with the development of insulin resistance and impaired glucose homeostasis in many models; however, the duration of HFD feeding and diet composition, including dietary fat source, influence not only body weight but also the timing of onset and severity of disturbed glucose regulation (54). Rapid onset of insulin resistance and impaired glucose tolerance was demonstrated in a study with C57BL/6 mice in which measures of glucose homeostasis were assessed after three days, one, two, five, or ten weeks of HFD feeding (60% kcal from fat) (268). Concomitant with increased adiposity and adipocyte size, glucose intolerance, hyperinsulinemia, and systemic insulin resistance were already evident as early as three days of HFD; fasting glucose and insulin levels were more elevated and systemic insulin sensitivity was reduced with longer-term feeding, as demonstrated by comparison of three-day versus ten-week HFD feeding (268). However, even diets comprised of a lower fat content at a level of 25–30% kcal from fat can induce glucose intolerance and systemic insulin resistance in mice (132, 371). Parks et al. demonstrated that genetic background contributes significantly to the degree of insulin resistance induced by a HFD regimen (371). While percent body fat and depot-specific fat mass were positively associated with HOMA-IR in both male and female mice fed a high-sucrose, HFD for eight weeks (371), the substantial heterogeneity in insulin resistance at any level of adiposity in response to HFD feeding (Figure 5) suggests that factors other than fat mass per se play a major role in determining insulin sensitivity.
Figure 5. Variation in adiposity and insulin resistance among inbred strains of mice.
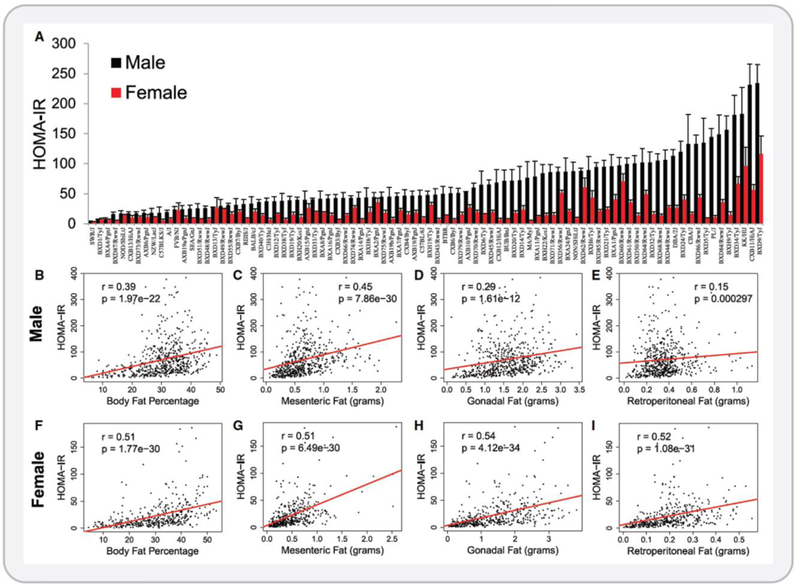
Males and females of more than 100 inbred strains of mice were fed a high-sucrose high-fat diet for eight weeks. Adiposity and systemic insulin resistance were highly variable in response to this diet. (A) HOMA-IR was determined in males and females. HOMA-IR was correlated with total body fat percentage (B and F) and with mesenteric (C and G), gonadal (D and H), and retroperitoneal (E and I) adipose depots of both male (B – E) and female (F – I) mice. The substantial variability in insulin resistance at any given level of adiposity can be appreciated in panels B – I. These data reveal that there is substantial genetic control over these metabolic phenotypes and that total adiposity may not be the primary factor driving insulin resistance. Reproduced from Parks et al. (371), with permission.
Teaching points: This large-scale animal study very clearly showed that genetic variation has a major impact on both adiposity and insulin sensitivity in response to a high calorie diet. In this study, over 100 inbred mouse strains were fed a high-sugar high-fat diet for eight weeks and fat mass, percent body fat, and insulin resistance were measured. This study design allowed the authors to assess the effect of genetics on these metabolic phenotypes. The substantial variability in adiposity and insulin sensitivity across the different mouse strains in response to the high-sugar high-fat diet is evident and indicates that genetic variation exerts significant control over these phenotypes. These results also support the idea that total adiposity may not be the primary factor driving the development of insulin resistance. Reproduced from Parks et al. (371), with permission.
Obesity and insulin resistance in humans
In the context of human obesity, Bierman et al. suggested as early as the 1960s that excess adipose tissue may be a root cause of insulin resistance and subsequent hyperinsulinemia (39). Since then, multiple observational studies have demonstrated a positive association between measures of adiposity and insulin resistance in humans.
A study of healthy men of European and Asian Indian descent demonstrated that total, subcutaneous, and visceral fat areas were all negatively associated with insulin sensitivity as assessed by glucose disposal rate in euglycemic-hyperinsulinemic clamp procedures (385). A separate cross-sectional study of non-diabetic South American adults found that both BMI and WHtR were negatively associated with Homeostatic Model Assessment (HOMA)-insulin sensitivity (calculated as inverse of HOMA-insulin resistance) (296). A cross-sectional study of lean and obese men from the United States reported that several indices of adiposity, including BMI, WC, and percent body fat, were all negatively associated with insulin sensitivity, as assessed by glucose infusion rate (GIR) from euglycemic-hyperinsulinemic clamps (367). A cross-sectional study of lean and obese, glucose tolerant individuals assessed the relationship between adiposity and insulin sensitivity, where euglycemic-hyperinsulinemic clamps were performed at three different insulin infusion rates (85). When normalized to lean tissue mass, basal hepatic, skeletal muscle, and adipose insulin sensitivity were all significantly greater in lean as compared to obese subjects. Collectively, these clinical studies demonstrate that regardless of the method used to assess adiposity or insulin sensitivity, a negative association between these two phenotypes is apparent.
It is also important to note that the relationship between adiposity and insulin sensitivity is modified by ethnicity (48, 385). The prevalence of both obesity and T2DM is higher in Hispanics and African Americans than Caucasians in the United States (65). Several studies have shown that for many ethnic groups, diabetes occurs at a lower BMI, and hence at a disproportionate rate as compared to Caucasian individuals of similar age (68, 294, 466). The Multiethnic Cohort study of over 180,000 individuals living in the United States revealed that the age-adjusted diabetes prevalence was higher in African American, Native Hawaiian, Japanese, and Latino populations as compared to Caucasians in every BMI category and among both men and women (294). Asian Indians also exhibit significantly lower insulin sensitivity as compared to Caucasians of similar age and BMI, which might be explained at least in part by the two-fold greater area of VAT in the Asian Indian group (385). Additional studies reported increased prevalence of diabetes at a lower BMI in Asian populations as compared to Caucasians (68, 466). Indeed, the current evidence demonstrating an increased risk for T2DM at a lower BMI in Asian Americans is so strong that the American Diabetes Association recommends diabetes testing for any adult in this subpopulation with a BMI ≥23 kg/m2 (191). Another study demonstrated that the relationship between adiposity and insulin sensitivity differs for Pima Indians when compared to Caucasians (48). In the Pima Indian group, there was a negative relationship between glucose disposal rate and percent body fat up to a level of 28% fat, and there was no further reduction in glucose disposal rate beyond that level of adiposity. However, in the Caucasian population, there was a significant linear negative association between adiposity and insulin sensitivity, without any evidence of a threshold effect (48). Another interesting finding of a disconnect between adiposity and T2DM risk was seen in Samoa, which has one of the highest rates of both obesity and diabetes in the world (82, 83). A recent study revealed that a variant of CREBRF common in the Samoan population is associated with an increased risk of obesity, and each copy of the variant is associated with an approximately 1.4 kg/m2 increase in BMI (314). Surprisingly, the variant was also associated with a significantly reduced risk for the development of T2DM, indicating that diabetes occurs at comparatively higher BMI values in carriers of this variant. Mechanisms through which this variant may confer protection against diabetes is not yet known. While this is not an extensive list of all ethnic groups that differ in their relationship between adiposity and insulin sensitivity, these examples strongly suggest that genetic factors modify the relationship between adiposity and insulin resistance in humans, and future work in this area should be carried out in ethnically diverse cohorts. Further, these differences across ethnicities may be better understood after there is a more comprehensive understanding of the mechanisms linking increased adiposity with decreased insulin sensitivity.
Variability in the association between adiposity and insulin resistance
The body of evidence generated from animal models and human studies described previously clearly demonstrates a positive association between adiposity and insulin resistance. However, there are exceptions to this relationship, wherein low adiposity may be met with extreme insulin resistance or morbid obesity may be free of metabolic dysfunction, with maintenance of normal glucose tolerance and insulin sensitivity. Examples of such individuals are evident in Figure 6, where there is a large amount of variability in GIR at any given BMI. The variation in GIR is perhaps most prominent in the obese group, as several individuals with extreme obesity (BMI >40 mg/kg2) exhibit high GIRs that are more characteristic of non-obese individuals. In addition, at the other end of the adiposity spectrum, several individuals in the non-obese group exhibit low GIR that are generally characteristic of the obese population. This overlap in insulin sensitivity between lean and even morbidly obese individuals suggests that increased fat mass is not likely the singular cause of insulin resistance but rather that more complex mechanisms underlie the association. Indeed, work conducted after the initial reports of the observed inverse association between fat mass and glucose tolerance and insulin sensitivity revealed that adipose tissue function, adipose tissue distribution, and age may be strong modulators of these associations (266, 362, 463). A relatively recently developed hypothesis by Dr. Roy Taylor at Newcastle University that provides an interesting perspective in this regard posits that individual fat thresholds determine the degree to which each person’s adipose tissue can safely store TG before ectopic fat storage and negative metabolic consequences manifest (461).
Figure 6. Insulin sensitivity and adiposity are negatively associated in humans.
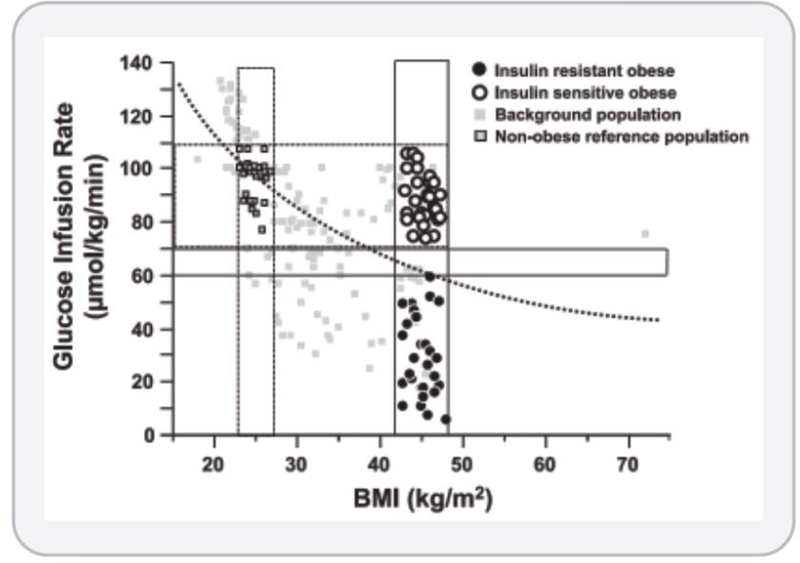
Insulin sensitivity was determined by glucose infusion rate from euglycemic-hyperinsulinemic clamps and plotted against adiposity as estimated by BMI. These data highlight the variability in insulin sensitivity at any given BMI. Although an inverse association between BMI and insulin sensitivity generally applies on a population level, however there is large variability in insulin sensitivity at any given BMI, particularly at among the overweight and obese BMI categories (>25 kg/m2). Of note, there are obese individuals who maintain a level of insulin sensitivity equivalent to those with BMI around 25 kg/m2. These individuals may be referred to as ‘metabolically healthy obese (MHO)’ and comprise approximately 10–30% of the obese population. These individuals remain free from the metabolic syndrome and glucose intolerance that would be expected based on their BMI. Reproduced from Kloeting et al. (238).
Teaching points: In this study, the authors used the euglycemic hyperinsulinemic clamp procedure to measure the glucose infusion rate (y-axis) needed to maintain glucose concentrations in plasma constant under high insulin concentrations, which is the gold-standard method for the determination of insulin sensitivity. A high glucose infusion rate is indicative of high insulin sensitivity. In non-obese to obese individuals (body mass index, BMI, on the x-axis), they found that adiposity and insulin sensitivity are negatively correlated. In addition, this study also highlights the variability in insulin sensitivity that exists at any given BMI, indicating that total fat mass is not the only determinant of insulin sensitivity. Indeed, there are obese individuals who maintain a level of insulin sensitivity equivalent to those with a non-obese BMI. These individuals may be referred to as ‘metabolically healthy obese (MHO)’ and are estimated to comprise approximately 10–30% of the obese population. These individuals do not develop metabolic syndrome, insulin resistance, and impaired glucose tolerance that would be expected based on their BMI. Figure reproduced with permission (Reproduced from Kloeting et al. (238).
Adipose tissue distribution and insulin sensitivity
In humans, SAT is estimated to account for up to 90% of total fat mass (270). Although VAT accounts for a relatively minor portion of total fat mass, approximately only 10–20% in obese and non-obese adults (1), the volume of this depot is recognized as one of the strongest predictors of insulin resistance (49). In 1956, Vague proposed that obesity could be categorized by the location of the excess adipose tissue, gynoid for lower body and android for upper body, and that android obesity was associated with a greater risk for development of T2DM (480). Evans et al. built upon this idea using simple anthropometric measurements as a proxy for central adiposity and discovered that WHR inversely associated with insulin sensitivity and glucose tolerance (114). Follow-up imaging-based studies sought to clarify whether the risk associated with central adiposity could be explained by the volume of SAT versus VAT. Bonora reported that glucose uptake was significantly inversely correlated with VAT, but not SAT, area in nondiabetic obese women (49). Other clinical studies using the euglycemic-hyperinsulinemic clamp method to assess insulin sensitivity reported similar results, with a significant association between insulin sensitivity and VAT, but not SAT, area (49). A study of over 500 Filipino, African American, and Caucasian women revealed that age-adjusted T2DM prevalence was highest among women in the highest tertile of VAT volume (24). Even within this highest VAT tertile, there were stark differences in T2DM prevalence by ethnicity, with rates of 46.6% (Filipino), 14.7% (African American), and 9.8% (Caucasian). Coincident with greater VAT volume, Filipino women had the highest overall T2DM prevalence at 32% although the African American group had a significantly higher BMI and greater SAT volume as compared to the Filipino and Cauasian groups (24). Together these data suggest that in addition to adipose tissue distribution, genetic or other factors also contribute to the risk for T2DM. Other studies suggest that central SAT mass or area may be an independent risk factor for insulin resistance (1, 146, 373). In a study of adult men with a range of adiposity, glucose disposal rate was more strongly correlated with abdominal SAT mass than with VAT mass, although both correlations were significant (1). However, after adjusting for total fat mass, the association between glucose disposal rate and VAT was lost and the association with SAT only trended towards significance (P=0.06) while the associations between glucose disposal rate and truncal skinfold thickness and total abdominal fat (SAT plus VAT) were preserved (1).
Other studies suggest that visceral adiposity may simply be a stronger predictor of metabolic dysfunction than total or subcutaneous adiposity, but not a causal factor. For example, hepatic fat content has been proposed to be a major driver of impaired insulin sensitivity, and this hypothesis is supported by clinical studies that have dissociated the effects of visceral adiposity and intrahepatic lipid accumulation on glucose homeostasis (116, 285). One study assessed multi-tissue insulin sensitivity in obese subjects who differed significantly either by liver fat content (3.6% vs. 25.3%) or visceral adiposity (766 cm3 vs. 1946 cm3) while matched for BMI, percent body fat, age, and sex (116). Significant differences in insulin sensitivity of liver, skeletal muscle, and adipose tissue were detected between the groups that differed by liver fat content while no differences in insulin sensitivity were detected between the groups that differed by VAT volume (116). In a comparison of insulin sensitivity between class I (BMI 30.0–34.9 kg/m2) and class III (BMI ≥ 40.0 kg/m2) obese individuals matched for liver fat, there were no differences in plasma glucose, insulin, or FFA, or in any measures of insulin sensitivity assessed by euglycemic-hyperinsulinemic clamps (285). A separate study of obese adults demonstrated a significant inverse correlation between liver fat content and adipose, liver, and skeletal muscle insulin sensitivity. In that study, liver fat was the strongest predictor of insulin resistance in all tissues as compared to several measures of adiposity, although VAT volume was also a significant predictor (244). However, in a study of over 350 obese or diabetic subjects with a wide range of visceral adiposity and liver fat content, Kotronen et al. demonstrated that both of these sites of fat accumulation are independent predictors of fasting serum insulin and hepatic insulin sensitivity (249). In contrast, only liver fat explained variation in fasting plasma glucose levels. This study suggests that while liver fat is a strong predictor of many components of metabolic dysfunction, visceral adiposity may also be a significant contributor (249). Taken together, these data suggest that the liver is an important site of lipid accumulation that may substantially alter whole-body glucose homeostasis through its effects on multi-organ insulin sensitivity, although the mechanisms through which the liver may influence extrahepatic insulin sensitivity are not yet understood.
There is also a growing body of evidence that suggests the accumulation of lower body SAT may be metabolically protective (288, 463). Thus, the detrimental effects of VAT could perhaps be partially offset by the beneficial effects of SAT, which could partly explain the inverse association between measures of adipose distribution, such as WHR, and insulin sensitivity. In addition, it is possible that the differential metabolic effects of distinct adipose depots could explain some of the variability in the association between BMI and insulin sensitivity. Subcutaneous fat in the lower body region, as compared to upper-body fat, is positively associated with insulin sensitivity and a slower rate of lipolysis and FFA release into the circulation (288). A cross-sectional study of overweight and obese men and women divided into two groups by high or low insulin-mediated glucose uptake found that after adjusting for sex and BMI, the insulin resistant group had significantly greater VAT while the insulin sensitive group had significantly more subcutaneous abdominal fat and thigh fat (301). More direct evidence of the protective effect of SAT came from mouse models of adipose tissue transplantation (470). Compared to a sham operated group, mice that received transplants of SAT into either the dorsal subcutaneous region or into the visceral epididymal region gained less body weight and fat mass and exhibited greater insulin sensitivity in the liver and the endogenous SAT several weeks post-surgery. However, the lower body weights seen in the SAT transplantation group were not controlled for, so it is not known whether the improvement in insulin sensitivity is at least partially explained by lower total body weight (470). Collectively, the body of evidence from humans and rodent models supports the hypothesis that fat distribution is an important regulator of insulin sensitivity and whole-body metabolic homeostasis.
Aging, adiposity, and insulin sensitivity
Aging is associated with a decline in function at the cellular, organ, and whole-body levels and thus increases the risk for the development of disease (131, 278, 341). T2DM is one of many aging-associated diseases; CVD, cancer, and neurodegeneration are also included in the list (341, 362). Current estimates of the prevalence of diabetes in the United States clearly demonstrate a positive association with age, as the rate in the 65+ years age group is substantially higher than that of the 45–64 age group (65) (Figure 7). This increase in prevalence occurs despite a reduction in the prevalence of overweight and obesity among adults 60+ years of age (352). Consistent with increased T2DM prevalence in the elderly, both reduced glucose tolerance and increased insulin resistance are associated with aging (124, 398). These age-associated changes in glucose metabolism may be largely explained by changes in adipose tissue distribution, with a shift from SAT to VAT (362, 498). Even after controlling for percent body fat, parity, and physical activity, an increase in visceral adiposity occurs after the 6th decade in women (197). Aging-related alterations in fat distribution are consistent with those described in the preceding section that are associated with detrimental effects on insulin sensitivity. The observed relationship between insulin resistance and aging in humans has been investigated in a systematic way: when total fat mass or VAT mass is controlled for, it becomes evident that adiposity, rather than aging itself, is the stronger predictor of insulin resistance (18, 31, 64, 86). For example, when young (24 – 47 years) and old (60 – 75 years) subjects were matched by either level of fitness or adiposity, there were no differences in insulin sensitivity (assessed by clamp) between younger and older subjects (18). In a study of young (mean 23.7 years) versus old (mean 70.1 years) subjects who differed significantly by BMI, percent body fat, and VAT mass, measures of adiposity were significant predictors of insulin sensitivity but age was not (31). Another study assessed the impact of age on the major determinants of glucose tolerance in BMI-matched young (27 years) and old (63 years) individuals with normal glucose tolerance (10). Using the frequently sampled intravenous glucose tolerance test, glucose effectiveness was significantly lower in the old group while neither insulin sensitivity nor first phase insulin secretion, a measure of pancreatic β-cell function, were different between the two groups. However, it should also be noted that in addition to changes in fat mass and distribution, aging is also accompanied by a loss of lean mass which may also contribute to reduced insulin sensitivity in the elderly (248, 308). Additional evidence for a role of VAT accumulation in aging-associated insulin resistance came from a rodent study by Gabriely et al. in which VAT or SAT was surgically removed from 15-month old rats and indices of insulin sensitivity were measured five months later (138). Glucose disposal rate was improved and hepatic glucose output was reduced in the VAT removal group as compared to the sham-operated and SAT removal groups (138). Furthermore, the level of insulin sensitivity in the VAT removal group was similar to that of healthy young (two-month old) rats, again supporting the concept that fat distribution is an important determinant of metabolic health in aging. Taken together, current evidence clearly indicates that aging is associated with insulin resistance that may be largely explained by altered adipose tissue mass and distribution.
Figure 7. Type 2 diabetes is an aging-associated disease.
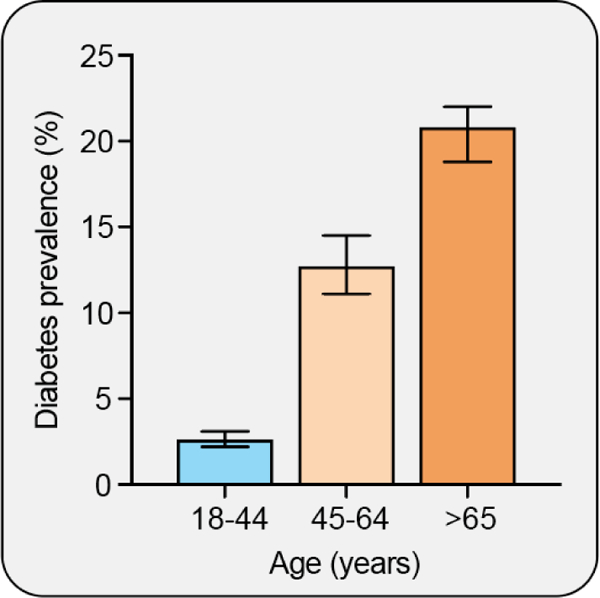
The prevalence of diabetes increases with age, and is substantially increased in the 65 and older population as compared to the 45 – 64 year old population (65).
Teaching points: The prevalence of diabetes increases with age and is nearly doubled in the 65 and older population as compared to the 45 – 64 year old population (65). Aging is also associated with insulin resistance that may be largely explained by a shift in adiposity from subcutaneous to visceral depots and an increase in ectopic fat accumulation.
Lipodystrophy and insulin sensitivity
As mentioned previously, and observed in Figure 6, there are individuals that are outliers to the generally observed inverse association between adiposity and insulin sensitivity. The study of these individuals and representative mouse models can offer unique insights into the role of adipose tissue in whole-body metabolic homeostasis, and will be reviewed in the following sections.
Lipodystrophy and obesity are opposite extremes on the spectrum of adiposity and the study of both conditions has provided great insight into the importance of the fat-storing and endocrine functions of adipose tissue in whole-body metabolic homeostasis. Lipodystrophies are heterogeneous in terms of etiology and severity, as they can be either congenital or acquired, with generalized or partial (regional) fat loss (193). Despite the dramatic difference in absolute fat mass of lipodystrophic as compared to obese individuals, the overlap in their metabolic phenotypes is striking (Figure 8). Most forms of human lipodystrophy are associated with some degree of insulin resistance, hepatic steatosis, and dyslipidemia (393). The extent of metabolic dysfunction is generally proportional to the level of adipose tissue deficit, such that more extreme fat loss, as seen in generalized lipodystrophies, is associated with more severe insulin resistance and development of T2DM (193). Genetic mouse models of lipodystrophy also exhibit severe insulin resistance (322, 426). Many of the known genes responsible for monogenic forms of lipodystrophy are directly involved in major functions of adipose tissue, including adipogenesis, TG synthesis, or lipid droplet formation, consistent with a central role for functional adipose tissue in whole-body metabolic homeostasis (193). Although it is not yet clear why some types of lipodystrophy affect certain depots and spare others, the adipose tissue pattern associated with this condition commonly presents a double metabolic insult, as subcutaneous gluteofemoral fat is generally lost while VAT expands (193).
Figure 8. Metabolic characterization of obesity, metabolically healthy obesity (MHO), and lipodystrophy.
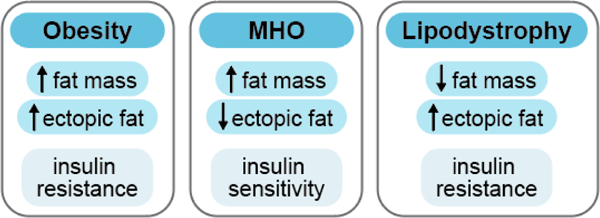
Metabolically functional, healthy adipose tissue may be a key determinant of overall metabolic health. Although all obese individuals are characterized by excess adiposity, not all obese individuals develop metabolic dysfunction. An estimated 10 – 30% of the obese population may be metabolically healthy and exhibit reduced ectopic fat accumulation and are more insulin sensitive compared to metabolically unhealthy obese individuals (44, 45). Lipodystrophy is characterized by reduced adiposity but is accompanied by metabolic dysfunction including ectopic fat deposition and severe insulin resistance (140). The metabolic differences among obese, MHO, and lipodystrophic populations reveal that total adiposity is not likely the major determinant of metabolic health but rather that metabolically functional adipose tissue that maintains insulin sensitivity is a major contributor to whole-body metabolic health.
Teaching points: The study of obesity, metabolically healthy obesity (MHO), and lipodystrophy reveals that healthy, functional adipose tissue may be a key determinant of overall metabolic health. Although all obese individuals are characterized by excess fat tissue, not all obese individuals develop metabolic dysfunction such as insulin resistance. A small proportion of the obese population maintains a state of metabolic health in which they remain more insulin sensitive as compared to metabolically unhealthy obese individuals (44, 45). Lipodystrophy, on the other hand, is characterized by a reduced amount of adipose tissue, but is usually accompanied by ectopic fat deposition and severe insulin resistance (140). These examples strongly suggest that total adiposity is not likely the primary determinant of metabolic health.
Metabolically healthy obesity in humans
Obese individuals with greater insulin sensitivity than would be predicted based on their BMI, as illustrated in Figure 6, are increasingly being viewed not as outliers but as a metabolically distinct group that comprise approximately 10–30% of the total obese population (44). These individuals maintain insulin sensitivity and glucose tolerance in the face of increasing BMI and offer an intriguing opportunity to learn more about the influence of adiposity on metabolic health. The risk for cardiovascular and metabolic diseases, such as T2DM, is lower in MHO individuals as compared to metabolically unhealthy individuals of similar BMI (44, 438). However, it is also hypothesized that because metabolic syndrome, insulin resistance, and fitness are strongly associated with age, MHO may be a temporary metabolic state that obese individuals transition through on their way to obesity-related insulin resistance (45). While the prevalence of MHO does substantially decrease with age, MHO individuals can be found in the oldest age categories in different cohorts (44, 485). Indeed, in a combined analysis of 10 cohorts which included 28,000 obese adults in Europe, the overall age-standardized prevalence of MHO was 12% among the obese population (485). Overall, our current knowledge of MHO is consistent with the findings discussed above that insulin sensitivity is more closely related to intraabdominal and ectopic fat deposition than to total body fat mass, i.e. MHO individuals may be the subpopulation of the obese who manage to expand their (peripheral) SAT and prevent excessive intraabdominal and ectopic fat deposition (44). Consistent with greater insulin sensitivity in MHO individuals as compared to the metabolically unhealthy individuals, levels of several circulating factors that are associated with insulin sensitivity are also improved in MHO. For example, RBP-4 is directly associated with visceral adiposity and inversely associated with insulin sensitivity (141, 152). In non-obese and obese women, serum RBP-4 was highest among those with visceral adiposity as compared to those without, and levels were not different between the non-obese and obese groups that did not have visceral adiposity (265). MHO individuals also have higher circulating levels of high-molecular weight (HMW) adiponectin (9, 103, 107) and lower FFA (340, 482) as compared to insulin resistant obese individuals. These factors may contribute to the protection from metabolic dysfunction in MHO despite having excess adiposity.
One limitation in this field is that consensus on the definition of MHO does not currently exist, but advocates of an official MHO classification have proposed to define metabolic health as the absence of the metabolic syndrome and maintenance of insulin sensitivity defined as HOMA-IR <2.5 (44). Alternatively, in some observational studies MHO individuals are defined as those with BMI >30.0 kg/m2 who do not have any metabolic disorders, including impaired glucose tolerance, dyslipidemia, or hypertension (44).
Adiposity and insulin sensitivity summary
Whole body adiposity is generally associated with insulin resistance in rodents and humans, but there is a substantial amount of residual variation likely explained by numerous factors that influence the relationship. The examples of obesity and lipodystrophy presented above, while differing in external presentation, share a common underlying metabolic phenotype: a relative deficit of healthy, functional adipose tissue commonly accompanied by whole-body metabolic dysfunction. Lipodystrophy is characterized by reduced total fat mass and altered adipose tissue topography while obesity is characterized by excess total fat mass. In contrast, the adipose tissue of MHO individuals likely maintains proper functioning capacity, with storage of neutral lipids and secretion of adipokines, despite increased total fat mass indicative of obesity, thereby preventing excessive ectopic fat storage. An understanding of adipose tissue function in these contexts provides further support for the hypothesis that absolute adipose tissue mass is not the primary factor driving the association between adiposity and the development of metabolic dysfunction (Figure 8). Rather, as mentioned above, the health of VAT and SAT depots and low levels of ectopic fat deposition are likely key determinants of whole-body insulin sensitivity.
Overview of mechanisms linking increased adiposity to insulin resistance
Efforts to understand the mechanisms underlying the association between adiposity and insulin resistance have been substantial, many mechanisms proposed (383). However, the heterogeneous metabolic phenotypes associated with obesity make it difficult to deconstruct the association and understand contributing components. This point is exemplified by studies demonstrating that the association between adiposity, insulin resistance, and impaired glucose homeostasis is not explained simply by absolute fat mass, as described in detail above. Accumulating evidence now clearly indicates that the functional capacity or ‘health’ of adipose tissue is a likely major determinant of whole-body metabolic homeostasis. In this section, we briefly describe several of the current leading hypothesized mechanisms that may mediate obesity-associated insulin resistance and supporting evidence from studies of MHO and lipodystrophy. Within the context of the proposed mechanisms, we also discuss functional and metabolic differences of adipose tissue depots that are consistent with adipose tissue distribution as an important determinant of metabolic health. As will become apparent later in the manuscript, all of these proposed mechanisms may partly function as mediators in the relationship between adipose tissue inflammation and systemic insulin resistance.
Elevated plasma free fatty acids
Adipose tissue is the primary source of circulating, albumin-bound FFA. Circulating FFA increase in concentration with fasting and become available for hepatic very low-density lipoprotein synthesis and for energy generation by peripheral tissues (148). Elevated systemic FFA are commonly cited as a hallmark characteristic of obesity and are hypothesized to contribute to obesity-associated insulin resistance. Although the rate of FFA release per kg fat mass declines with increasing adiposity, increased total fat mass in obesity leads to greater total FFA release (315). Elevated fasting FFA with increasing adiposity have been demonstrated in some (41, 206) but not all human studies, including two large epidemiological studies (224). Variability in plasma FFA has also been reported in lipodystrophic mouse models, as FFA were significantly elevated in one model (322) but not in a different model (426).
Relative to nondiabetic overweight subjects, plasma FFA were elevated over a 24-hour period in age and BMI-matched individuals with mild or severe T2DM (389). In a study that included lean and obese insulin sensitive and insulin resistant individuals, Ferrannini et al. found that plasma FFA differed significantly by insulin sensitivity but not by obesity (121). Consistent with these results, another cross-sectional study demonstrated that FFA were significantly lower in the MHO group as compared to the metabolically unhealthy obese group, but did not differ from the non-obese metabolically normal group (447). A five-year prospective study demonstrated that plasma FFA were highly predictive of risk for T2DM when percent body fat, sex, and insulin-stimulated glucose uptake were controlled for. Further, the incidence of T2DM was nearly doubled among individuals with high (90th percentile) as compared to individuals with low (10th percentile) plasma FFA (365). A reciprocal glucose fatty acid cycle was originally proposed by Randle over 50 years ago (387). This theory proposed that the elevated FFA in obesity and insulin resistance may exacerbate impaired glucose metabolism due to inhibitory effects of products of fatty acid oxidation on enzymes central to glucose catabolism (386, 387). Consistent with the Randle cycle hypothesis, studies of lipid bolus infusions revealed that elevated FFA are associated with many perturbations of glucose metabolism, including reduced whole body glucose uptake, reduced skeletal muscle glycogen synthesis and glycolysis, and increased hepatic glucose output (47). However, using magnetic resonance spectroscopy, Roden et al. demonstrated that FFA act at the site of glucose uptake and/or glucose phosphorylation, in contrast to Randle’s proposal that enzymatic inhibition is the driving mechanism in the glucose fatty acid cycle (395). More direct evidence that FFA directly impact glucose metabolism was demonstrated by pharmacological inhibition of overnight fasting-induced elevation of plasma FFA which was associated with improved insulin sensitivity and glucose tolerance in obese subjects with T2DM (408).
Elevated plasma and tissue ceramides
Ceramides belong to the class of sphingolipids, are comprised of a sphingosine and a fatty acid, and serve as the building block for more complex sphingolipid species (305). Early in vitro experiments demonstrated that ceramide-mediated disturbance of glucose metabolism occurs predominantly through the inhibition of Akt activity (449). This impairment in insulin signaling reduces GLUT4 protein at the plasma membrane and subsequently reduces glucose uptake in 3T3-L1 adipocytes (449). Results from animal and human studies implicate ceramides in the development of insulin resistance (72). For example, total and select individual plasma ceramide species were elevated in obese T2DM individuals as compared to control non-obese individuals, and insulin sensitivity was inversely correlated with total and individual plasma ceramides (171). However, the relative contribution of adiposity versus ceramides on insulin resistance was not addressed in this study. In other studies, ceramides were significantly elevated in SAT of obese diabetic as compared to obese nondiabetic individuals (42, 71) while ceramide content of VAT did not differ between these groups (71). More direct evidence of a role for ceramide in glucose homeostasis has been generated from several murine studies of altered ceramide metabolism (71, 150, 183, 384, 476, 522). Phenotypes of mice with reduced ceramide synthesis or increased ceramide catabolism are consistently characterized by improved glucose tolerance and insulin sensitivity (71, 150, 183, 476, 522).
Hypoadiponectinemia
The endocrine function of adipose tissue is now well appreciated with the discovery and functional characterization of many adipokines. Adiponectin was first identified in the mid-1990s (192, 283, 335, 414) and remains a highly studied adipokine, as it is well established as an insulin-sensitizing hormone that exerts control over several metabolic processes in a panel of different tissues (399, 475).
Adipocytes secrete adiponectin in low-molecular, mid-molecular and HMW complexes that are detected in human serum and in culture media conditioned by adipose tissue or mature adipocytes (214, 359, 414). Adiponectin is distinct from most other adipokines in that its expression and circulating levels are inversely related to adiposity (25, 228, 237). In both humans and mice, circulating adiponectin levels are higher in females than in males (80, 228, 359). Early evidence of a role for reduced levels of adiponectin in obesity-associated impaired glucose homeostasis came from studies that demonstrated total plasma adiponectin is directly correlated with insulin sensitivity, lower in diabetic as compared to nondiabetic patients, and increased with weight loss (25, 190, 510). A cross-sectional study of over 700 adults compared circulating adiponectin levels between BMI-matched metabolically healthy and metabolically unhealthy individuals in six different BMI strata. In each BMI category, three of which were obese classes, adiponectin was significantly higher in the metabolically healthy groups (7). Another large study of nearly 2500 individuals reported significantly higher plasma adiponectin levels among metabolically healthy non-obese and MHO groups when compared to the metabolically unhealthy non-obese and obese groups (9). In addition, adiponectin levels were not different between the metabolically healthy non-obese and obese groups even after controlling for age, sex, BMI, and hormone therapy. Given the results from these clinical studies, it is perhaps unsurprising that lipodystrophic humans exhibit very low circulating levels of adiponectin and the level of reduction in adiponectin tends to correlate with the severity of adipose tissue deficiency (140, 166). In a genetic mouse model of lipoatrophy that lacks nearly all abdominal white adipose tissue, serum adiponectin is undetectable and the mice present with systemic insulin resistance and ectopic fat deposition (532). Administration of recombinant adiponectin to these mice partially restored insulin sensitivity and reduced hepatic and skeletal muscle TG accumulation and circulating lipid levels. Overexpressing adiponectin in Lepob/ob mice completely prevented the expression of a diabetic phenotype, in spite of the fact that the adiponectin transgenic mice had greater total fat mass and had substantially lower physical activity (235). Interestingly, the expanded fat tissue in the adiponectin transgenic mice was characterized by smaller adipocytes and less inflammation, and the animals had lower liver fat content. As additional evidence for adiponectin’s insulin sensitizing effects, administration of recombinant adiponectin significantly improved glucose tolerance and insulin sensitivity in HFD and high carbohydrate diet-fed C57BL/6 and Leprdb/db mice (532). Maeda et al. demonstrated that treatment with the insulin-sensitizing thiazolidinedione (TZD) class of anti-diabetic medications increased plasma total adiponectin concentration in a dose-dependent manner in both humans and mice (284). Follow-up work by Pajvani et al. showed that the change in the ratio of HMW to total adiponectin was strongly and directly correlated to the change in insulin sensitivity following TZD treatment while there was no correlation between the change in total adiponectin and change in insulin sensitivity, suggesting the HMW multimers are the more metabolically potent, insulin-sensitizing form of adiponectin (360). In agreement with this hypothesis, Fisher et al. demonstrated that the ratio of HMW to total adiponectin correlated with glucose tolerance more strongly than did total adiponectin in a study of normal to obese subjects (127). As with total adiponectin, circulating levels of HMW adiponectin correlate inversely with adiposity; however, a study of aging in mice demonstrated that the association is not significant in animals of advanced age (311).
The metabolic effects of adiponectin are predominantly mediated through the two adiponectin receptors, AdipoR1 and AdipoR2 (530, 533). Activation of these receptors induces activation of the adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1), AMP-activated protein kinase (AMPK), peroxisome proliferator-activated receptor-α (PPARα), and p38 MAPK (mitogen-activated protein kinase) signaling pathways (399, 475). Downstream events of these active signaling pathways increase glucose uptake and fatty acid oxidation in skeletal muscle. Adiponectin enhances survival of the β-cells in the pancreas, and may regulate glucose-stimulated insulin secretion in some conditions. In the liver, adiponectin improves insulin sensitivity with an accompanying reduction in gluconeogenesis. In addition, work from Holland and colleagues revealed a direct role for adiponectin signaling in regulation of cellular ceramide levels (184, 185). These in vitro and in vivo studies revealed that both AdipoR1 and AdipoR2 exhibit ceramidase activity that is enhanced by adiponectin and reduces cellular ceramide concentration (184, 185). Consistent with these data, crystal structures of both AdipoR1 and AdipoR2 suggest they possess intrinsic ceramidase activity (487).
Adiponectin also exerts metabolic regulation through anti-inflammatory effects. In vitro experiments revealed that adiponectin treatment reduces the expression and secretion of several pro-inflammatory cytokines and chemokines including IL-6, IL-8, C-C motif chemokine ligand 2 (CCL-2, also called monocyte chemoattractant protein-1, MCP-1), and others (97, 553). Furthermore, adiponectin reduced secretion of the anti-inflammatory factors IL-10 and IL-1Ra from macrophages and dendritic cells (514). Evidence also suggests reciprocal regulation of adiponectin expression by pro-inflammatory factors. For example, in vitro experiments with 3T3-L1 adipocytes demonstrated that treatment with either IL-6 or TNFα reduces adiponectin expression and secretion (120, 223, 284).
Ectopic lipid accumulation
An essential function of adipose tissue is to store excess energy in the neutral lipid form of TG, which prevents ectopic lipid accumulation in tissues such as liver, skeletal muscle, and pancreas. The proper storage of fatty acids in adipose depots is crucial because the buildup of lipids in tissues whose primary function is not energy storage is strongly associated with the development of metabolic dysfunction. As discussed previously, lipid content in non-adipose tissues may be one of the strongest predictors of insulin resistance, in obese as well as non-obese individuals. For example, lipodystrophy is commonly associated with ectopic fat accumulation, particularly in the liver (140, 193, 375). Mouse models of lipodystrophy also exhibit significant hepatic fat accumulation (322, 426). Several studies have shown a negative association between hepatic lipid content and whole-body insulin sensitivity (116, 244, 285). In one study that included over 300 adults, liver fat content and percentage with fatty liver were both significantly lower in the insulin sensitive MHO group as compared to the obese insulin resistant group (439). In this study, intramyocellular lipid accumulation was also significantly lower in the MHO group; however, when the groups were separated by sex this trend was only observed among men (439). A separate study of non-diabetic lean adults demonstrated that intramyocellular lipid content was negatively associated with insulin sensitivity, and the association was independent of age, BMI, and fasting plasma glucose (255). Another study demonstrated similar results in a study of non-obese and obese Pima Indians, in which muscle lipid content, as measured in tissue biopsy, was negatively correlated with insulin sensitivity, again measured by clamp (363). More recent studies suggest that pancreatic fat may be an important contributor to pancreatic β-cell dysfunction (173, 442, 515).
Various factors may contribute to ectopic lipid accumulation. Circulating FFA derived from adipose tissue lipolysis are a major source of fatty acids for TG synthesis in non-adipose tissues. For example, one study of obese subjects with NAFLD estimated that circulating FFA provide nearly 60% of the fatty acids in hepatic TG (100). Reduced levels of circulating adiponectin and related downstream signaling in liver and muscle likely also contribute to the accumulation of lipids, as adiponectin is a major regulator of fatty acid oxidation and thus at least partially regulates lipid levels in these tissues (275, 399, 531). Genetic factors also affect ectopic fat distribution as differences in organ fat accumulation have been demonstrated among ethnic groups. In a study of obese Hispanic, African American, and Caucasian adolescents matched for body fat, age, and sex, hepatic fat was elevated in the Causcasian and Hispanic groups but was undetectable in the African American group (273). There was also differential accumulation of intramyocellular fat, which was significantly elevated in the Hispanic group and not different between the African American and Caucasian groups (273). Another study showed that among overweight Hispanic and African American young adults matched for age and BMI, pancreatic fat was significantly higher in the Hispanic group as compared to the African American group (263). Together, these studies suggest that inter-ethnic differences in lipid partitioning exist irrespective of obesity and may be important determinants of metabolic disease susceptibility.
Systemic inflammation
Obesity is commonly described as a state of chronic low-grade systemic inflammation, as excess adiposity is positively associated with mildly elevated levels of several circulating cytokines and acute phase response proteins in both mice and humans. IL-6 is a well characterized pro-inflammatory cytokine that is produced by certain immune cells, such as monocytes, macrophages, and T cells, and by non-immune cells such as fibroblasts, endothelial cells, and adipocytes (416, 493); both SAT and VAT depots secrete this cytokine (134). Serum IL-6 is elevated in diabetic and nondiabetic obese individuals as compared to non-obese controls (30, 151). Signaling of IL-6 occurs through a receptor complex comprised of IL-6R and glycoprotein 130 (gp130) on target tissues, such as the liver (416). Hepatic IL-6 signaling stimulates the expression of acute phase response proteins, including C-reactive protein (CRP) and serum amyloid protein A (SAA) (416). Consistent with elevated IL-6, systemic levels of both CRP and SAA are significantly elevated in obese subjects as compared to lean or overweight individuals (497, 534). SAA antagonizes insulin signaling in adipocytes and thus elevated levels may contribute to insulin resistance (123). Hepatic expression and serum levels of SAA are also induced in mice fed a HFD for 16 weeks, consistent with obesity-associated elevation of circulating IL-6 levels (413). Circulating levels of other inflammatory proteins, such as TNFα, MCP-1, macrophage inflammatory protein (MIP)-1α, IL-1, and IL-8 are also elevated in obesity (151, 233). Concurrent with severe insulin resistance, systemic inflammation and macrophage infiltration specifically of adipose tissue, but not in other tissues, occurs in lipodystrophic mice (176).
Adipose tissue inflammation
Several lines of evidence strongly suggest that low-grade chronic inflammation in expanded adipose tissue is a major contributor in the development of systemic insulin resistance. Early work demonstrated increased expression of TNFα in obese, insulin resistant adipose tissue of both mice and humans (188, 189). Serum IL-6 is elevated in diabetic and nondiabetic obese individuals as compared to lean controls (30), likely predominantly due to increased expression in expanded adipose tissue. Furthermore, adipose tissue levels of IL-6 inversely correlated with insulin sensitivity (30). These early studies and the body of follow-up work that continues today have shaped our understanding of the cellular crosstalk that exists between immune cells and adipocytes, illustrating that this crosstalk influences whole-body metabolic regulation. The known or suggested effects of adipose tissue inflammation on insulin resistance includes direct effects, i.e., the induction of adipocyte insulin resistance by pro-inflammatory cytokines, as well as indirect effects mediated in large part by elevated flux of FFA, hypoadiponectinemia, ectopic lipid accumulation that may result from increased FFA and low circulating adiponectin, and systemic inflammation (Figure 9). In other words, the mechanisms discussed above that are likely mediators between obesity and insulin resistance are all influenced to varying degrees by low-grade chronic inflammation in adipose tissue. For example, the pro-inflammatory cytokine TNFα is known to suppress adiponectin expression in adipocytes in vitro (284), which may explain in part the lower concentrations of adiponectin among obese individuals. Induction of insulin resistance in adipocytes is also known to increase the flux of FFA to ectopic tissues, due to an attenuation of the inhibitory effect of insulin on adipocyte lipolysis in the postprandial period (440). Activation of inflammatory signaling pathways is also linked with ceramide accumulation. Several studies have demonstrated that TNFα signaling regulates ceramide synthesis in vitro (175, 411, 524). In experiments conducted using mature adipocytes and chemical inhibition of caspase activity, ceramide production was shown to be an essential component of TNFα-mediated insulin resistance (156). Lipopolysaccharide (LPS) treatment of isolated skeletal muscle induces ceramide synthesis (182). In addition, palmitate stimulates ceramide accumulation in isolated muscles from wildtype mice but not in muscles from mice with defective signaling of the Toll-like receptor (TLR) 4, suggesting that saturated fatty acid-mediated accumulation of ceramides occurs through the TLR4 pathway (182). In the remainder of this manuscript, we will discuss in detail the known associations between adipose tissue inflammation and both adiposity and insulin resistance.
Figure 9. Adipose tissue inflammation may be a driving factor in the development of systemic insulin resistance (IR).
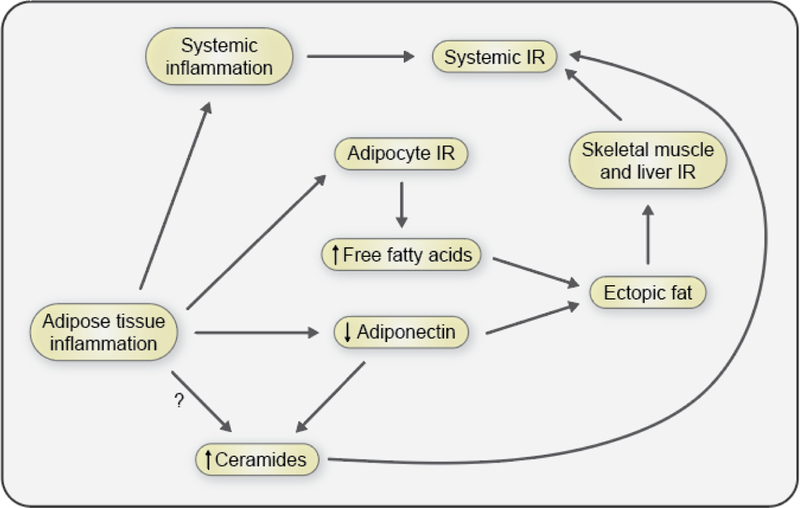
Chronic, low-grade adipose tissue inflammation is associated with the development of systemic IR in obesity. There are several mechanisms through which adipose tissue inflammation may contribute to IR: a) the secretion of cytokines by obese adipose tissue directly contributes to systemic inflammation which is associated with IR; b) adipose tissue-derived cytokines also impair local adipocyte insulin sensitivity which leads to increased lipolysis and secretion of free fatty acids; c) inflamed adipose tissue is also associated with reduced secretion of the insulin-sensitizing adipokine adiponectin. Adiponectin receptors contain intrinsic ceramidase activity, and reduced adiponectin as well as increased pro-inflammatory cytokine signaling may lead to increased levels of ceramides which are associated with adipose tissue inflammation and insulin resistance; d) elevated circulating free fatty acids and reduced adiponectin are associated with increased ectopic fat deposition. Hepatic and skeletal muscle fat accumulation are associated with impaired insulin signaling and systemic IR.
Teaching points: Chronic, low-grade adipose tissue inflammation is associated with the development of systemic IR in obesity. There are several potential mechanisms through which adipose tissue inflammation may contribute to the development of IR, including an increase in the production and secretion of pro-inflammatory cytokines that directly contributes to systemic inflammation and impairs adipocyte insulin sensitivity. IR at the level of the adipocyte results in increased lipolysis and secretion of free fatty acids. Adipose tissue inflammation is also associated with reduced secretion of the insulin-sensitizing adipokine adiponectin. Reduced adiponectin and increased pro-inflammatory cytokine signaling may lead to increased levels of ceramides which are also associated with adipose tissue inflammation and insulin resistance. Many of these mechanisms also contribute to the development of ectopic fat accumulation, in organs such as the liver and skeletal muscle, which is also strongly associated with systemic IR.
ADIPOSE TISSUE INFLAMMATION IN OBESITY
The development of adipose tissue inflammation in obesity and its potential role in driving insulin resistance are the major focus points of the remainder of this review. We discuss the diverse functions of adipose tissue immune cells and the evidence of whether adipose tissue inflammation directly contributes to the development of insulin resistance and T2DM in obesity. In this section, we review the accumulating data that demonstrate the development of chronic, low-grade inflammation in adipose tissue during obesity (Figure 10).
Figure 10. Obesity is associated with adipose tissue inflammation.
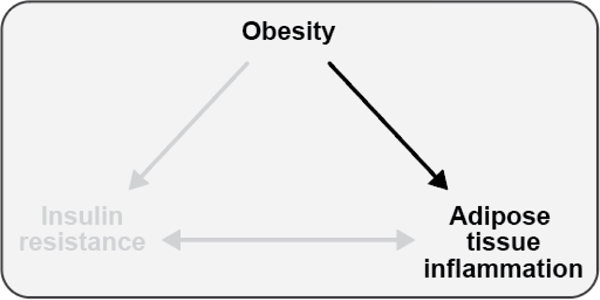
The development of chronic, low-grade adipose tissue inflammation is common in obesity.
Teaching points: The development of chronic, low-grade adipose tissue inflammation is common in obesity.
Overview of acute inflammation and inflammatory response
Inflammation is classically described as an acute response by the host organism to an infectious agent or tissue damage. Clinically, acute inflammation often presents as a combination of redness, swelling, pain, or increased temperature, either locally or systemically (i.e., fever) (261). The inflammatory process itself involves the focused delivery of humoral products and leukocytes to the site of inflammation, and is associated with an increase in the basal metabolic rate (78, 187, 261, 264, 304, 329). An acute inflammatory response involves the actions of surveilling innate resident immune cells, such as macrophages and mast cells. Upon exposure to a pathogen or necrotic tissue, these tissue-resident immune cells initiate an immune response by the release of numerous inflammatory mediators that enhance blood flow, increase blood vessel permeability, and facilitate the recruitment of leukocytes from systemic circulation. These secreted factors include cytokines such as TNFα, IL-1β, IL-6, and interferon (IFN) γ, chemokines such as CCL8 (IL-8) and CCL2 (MCP-1), adhesion molecules such as ICAM, E-selectin, and P-selectin, in addition to histamines, prostaglandins, leukotrienes, and many others (241, 261, 264, 304, 329, 336). Collectively, their actions serve to facilitate a rapid, short-term response designed to neutralize and/or remove the offending insult and restore tissue homeostasis. Of particular relevance to this paper, it is well known that acute infections trigger insulin resistance (541), plausibly in order to secure sufficient glucose supply for leukocytes involved in the inflammatory process.
Infiltrating neutrophils are the first responders to the site of acute inflammation and release their granule contents, a combination of reactive oxygen species, myeloperoxidase, and the proteases elastase, proteinase 3 and cathepsin G (241, 245, 261, 304, 336, 377). While designed to rid the host of invading microbes, these defense molecules can also damage surrounding host tissue (261, 304). Neutrophils also contribute to the recruitment, activation, and programming of the antigen presenting cells, such as macrophages and dendritic cells (336). One principal role of macrophages and dendritic cells is to take up microbes and present microbial antigens to cells of the adaptive immune system (333). Resident and recruited macrophages also aid in the removal of foreign particles and cellular debris, and once the pathogen is eliminated, initiate a repair phase. There is a switch from the production and secretion of pro-inflammatory prostaglandins to anti-inflammatory lipoxins during this macrophage-mediated resolution phase. Lipoxins halt the recruitment of neutrophils in favor of monocyte recruitment (69). Recruited monocytes differentiate into macrophages that then scavenge dead and dying neutrophils and initiate tissue repair (241, 261, 304, 336). Failure of the acute response to eliminate the pathogen results in a more chronic immune cell infiltration mainly comprised of macrophages and lymphocytes, and the transition of the immune response from the innate to the adaptive arm (333). Prolonged failure to resolve the inflammation, whether by the persistence of the pathogen, the presence of foreign bodies that cannot be broken down, or auto-immunity, results in a chronic inflammatory state characterized by the formation of tertiary lymphoid tissues and granulomas (304).
The signaling cascades at the heart of inflammatory processes include cell surface receptors for pro-inflammatory cytokines such as TNFα, ILs, and IFNs. There are also several different types of pattern recognition receptors (PRR). These receptors recognize and bind pathogen-associated molecular patterns (PAMPs) present on microorganisms or endogenous products of damaged tissue (i.e. dead and dying cells) that are released, also called damage-associated molecular patterns (DAMPs) (210). The PRR family includes TLRs, NOD (nucleotide-binding oligomerization-domain protein)-like receptors, and C-type lectin receptors. Activation of pro-inflammatory cytokine receptors and PRR signal through the c-Jun NH2-terminal kinase (JNK) and inhibitor of κ kinase (IκK)/nuclear factor-κB (NF-κB) pathways (295, 302, 356, 467).
Overview of adipose tissue inflammation
In comparison to a classical acute inflammatory response to a pathogen, obesity-associated inflammation differs with respect to its origin, intensity (often described as being low grade or sub-acute), and chronic persistence without resolution (154, 187). In addition, the metabolic rate normalized to lean body mass is unchanged in obesity (60), in contrast to the increase in an acute systemic inflammatory response. Obesity-associated inflammation afflicts many organs, including adipose tissue, skeletal muscle, liver, pancreas, and brain (154, 187, 302, 356, 464). Adipose tissue is now known to be a significant source of pro-inflammatory cytokines. Extensive research in rodent models suggests that the inflammation of adipose tissue may be a major factor in the development of metabolic disease. With the high prevalence of metabolic diseases, adipose tissue inflammation has become a major area of interest.
It should be emphasized that inflammation in tissues other than adipose, as well as ectopic fat deposition, are also important contributors to development of obesity-associated metabolic dysfunction. Genetic models of obesity present with whole-body metabolic dysfunction that may be due to a combination of metabolic disturbances that develop across several tissues. For example, in addition to massive adiposity, Lepob/ob, Leprdb/db, and Agouti obese models all develop hepatic steatosis (21, 469). Extensive inflammation and fibrosis also develop in the liver of Leprdb/db mice (406). These phenotypes, commonly present along with increased fat mass and adipose tissue inflammation, make it difficult or even impossible to disentangle the relative contributions of each factor in the development of insulin resistance. That is, most studies conducted in both rodents and humans do not allow a differentiation between the metabolic effects of inflammation in adipose tissue versus that in other tissues versus ectopic fat deposition on the development of systemic insulin resistance. Furthermore, little is known about the temporal sequence of the development of adipose tissue inflammation relative to these metabolic disturbances, and specifically insulin resistance in different insulin responsive tissues. Nevertheless, extensive evidence implicates a) the infiltration of immune cells, particularly macrophages, into adipose tissue; b) the activation of pro-inflammatory pathways leading to increased secretion of chemokines, cytokines, and other mediators of inflammation; and c) a range of molecular mechanisms such as endoplasmic reticulum (ER) stress, mitochondrial dysfunction, hypoxia, fibrosis, cellular senescence, and changes in lipid metabolism in linking obesity to T2DM. The following sections will summarize and discuss this evidence from animal and human studies.
Immune cell infiltration
The most salient feature in obesity-associated adipose tissue inflammation is the accumulation of pro-inflammatory cytokine-secreting immune cells in adipose tissue (356). Two seminal studies conducted in mice identified macrophages as the predominant immune cell type that accumulates in obese adipose tissue, accounting for 30–50% of the non-adipocyte cell fraction (504, 523). Most studies support the notion that VAT contains a greater number of macrophages than SAT in both mice (16, 332, 355) and humans (57, 168, 169), even though this was not seen in all studies (194). To address whether the increase in the number of adipose tissue macrophages (ATM) in obesity was due to expansion of the local resident population or due to recruitment of cells from non-adipose sources, Weisberg et al. lethally irradiated recipient mice that expressed the CD45.2 allele and then transplanted bone marrow from donor mice that expressed the CD45.1 allele (504). After six weeks of HFD feeding, 85% of the macrophages in the periepididymal adipose depot were CD45.1+, i.e., they were newly recruited from the circulation.
More recent studies, however, provide direct evidence in support of localized, resident cell proliferation. Specifically, Jenkins et al. observed that local resident tissue cells exhibit the ability to rapidly proliferate in response to increasing concentrations of the cytokine IL-4 (205). In addition, Schulz et al. identified two distinctive myeloid lineages, one that gives rise to bone marrow derived circulating monocytes, and the other arising from older yolk sac precursors that reside in tissues (419). At the same time, the relative importance of locally proliferated vs. newly recruited macrophages for adipose tissue inflammation and its downstream sequalae has as yet remained largely unclear. This was expanded upon, in part, by Davies et al. demonstrating that proliferating pro-inflammatory macrophages were bone marrow derived, and that resident tissue macrophages also have proliferative capability in response to inflammation (94). Finally, returning to the diet-induced obesity model, Haase et al. reported not only that the number of ATM in adipose tissue increases, particularly in association with the formation of crown-like structures (CLS) that surround necrotic adipocytes, but importantly that as much as 15% of these supposedly “recruited” ATM were in fact derived from proliferation of local resident cells (160). Existing evidence therefore would suggest that peripheral recruitment and to a somewhat lesser extent localized proliferation are both involved in the inflammatory processes driving the obesity-associated increase of macrophage numbers in adipose tissue. However, in human adipose tissue the relative proportion of recruited vs. locally proliferated macrophages in CLS remains unknown.
In conjunction with the increase in the number of macrophages in adipose tissue, the transcript levels of pro-inflammatory and macrophage-specific genes are upregulated with increasing adipocyte mass and adiposity (504, 523). A direct comparison of the adipocyte fraction, the stromavascular cell (SVC) fraction, and selectively isolated macrophages revealed that many of the pro-inflammatory factors, such as TNFα, IL-6, MCP-1 and others, were most abundantly expressed by macrophages (504, 523). Macrophage infiltration of SAT and VAT depots with increasing adiposity has also been demonstrated in humans (50, 56, 92, 93, 169, 545). Several studies have characterized the infiltrating ATM as pro-inflammatory as opposed to resident non-inflammatory macrophages in lean adipose tissue (61, 73, 78, 178, 280, 356, 460). Early work proposed that the pro-inflammatory macrophages accumulating in obese adipose tissue were similar to classically activated (“M1”) macrophages, while the non-inflammatory macrophages in lean adipose tissue were more similar to alternatively-activated macrophages (“M2”) (73, 280, 356). As will be discussed in subsequent sections in greater detail, it is now clear that pro-inflammatory ATM are not similar to M1 macrophages (253, 526).
Counterintuitively, the number of macrophages in adipose tissue does not immediately decrease with weight loss, and even increases in some studies, in both mouse models and humans. Kosteli and colleagues report that caloric restriction and weight loss lead to an initial increase in the number of ATM (in the first week) in mice, in both epidydimal (VAT) and inguinal (SAT) depots, and that ATM numbers only decline with the prolonged weight loss that occurs over several weeks (247). In that study, the number of ATM was also responsive to the dietary macronutrient composition, with lower ATM numbers on a calorically restricted high-carbohydrate vs. high-fat diet. Only partially consistent with these findings, Zamarron et al. fed obese mice a hypocaloric normal diet, and found that the number of ATMs in epidydimal but not inguinal adipose tissue was reduced (543). However, ATMs maintained a pro-inflammatory profile in the mice even after normalizing their body weight to that of lean, normal diet-fed mice, which was consistent with persistently high levels of insulin resistance in these animals (543). Taken together, this limited evidence from mouse models of obesity suggests that macrophage-driven adipose tissue inflammation is not attenuated in the initial and even advanced phases of weight loss.
There are also human data to suggest that weight loss does not always reduce ATM numbers in adipose tissue, and can even lead to an increase in the number of ATM, although not all studies are consistent. Substantial weight loss over 6–12 months following bariatric surgery, for example, was associated with an about three-fold increase in the number of ATMs (measured by flow cytometry) in SAT (162). Interestingly, in that and another study (254), we consistently observed a massive, several-fold increase in the number of neutrophils into SAT. This change was seen within two weeks of bariatric surgery and persisted until at least 6–12 months post bariatric surgery, suggesting that weight loss may be a pro-inflammatory stimulus in the adipose tissue. This is supported by data showing that very low-calorie induced rapid weight loss of ~10% over ~6 weeks led to a significant increase in the number of ATM, as assessed by IHC staining for the macrophage marker CD68 (14). More modest weight loss of 5–10% from either surgery or a lifestyle intervention does not acutely seem to reduce the number of ATM in SAT (250, 254), even though Kovacikova and colleagues nicely showed that ATM numbers are reduced after a subsequent weight loss maintenance phase (250). Contrasting data suggest an immediate reduction in SAT ATM numbers (CD68+-cells in IHC) in response to ~6% weight loss from a low-calorie diet in a 12-week intervention, a discreptancy that may be explainable by the slower rate of weight loss (27). One study by Cancello et al. found a substantial reduction of ~50% in the number of ATM (quantified by HAM56+/CD68+-staining in IHC) in SAT three months following Roux-en-Y gastric bypass surgery (56). While most studies assessed changes in inflammation and adipose tissue leuckoyte populations in response to weight loss only in SAT, one study is notable due to the fact that they made use of 55 individuals who had two different bariatric surgeries ~12 months apart that allowed them to collect both SAT and VAT (417). These authors found that sleeve gastrectomy-induced weight loss over 12 months led to a reduction in the number of VAT ATM only in ~60% of the population. Interestingly, metabolic benefits such as improvements in insulin sensitivity were seen in all individuals, including those in whom weight loss did not trigger a reduction in the number of omental ATM (417). One important caveat in these studies that needs to be taken into account is that bariatric surgery leads to numerous changes in the body, in addition to substantial weight loss, some of which have been described, while others may as yet be unknown. The relationship between adiposity, adipose tissue inflammation, and insulin resistance may therefore be confounded post bariatric surgery by any of these bariatric surgery-induced changes. Taken together, while the data are not fully conclusive, it is clear that weight loss in humans does not automatically trigger a reduction in adipose tissue inflammation or the number of ATM, and in some cases may even lead to an increase in ATM numbers.
The fact that macrophages accumulate in adipose tissue in the context of both caloric excess/obesity and fasting/weight loss illustrates that adipose tissue inflammation and macrophage accumulation in adipose tissue are not specific to obesity. While it is not currently clear whether these diverse exposures trigger adipose tissue inflammation through similar pathways, one may speculate that a common denominator may be elevated concentrations of nutrients such as fatty acids in adipose tissue. It is also worth pointing out that ATMs can become activated as a result of stimuli other than a disruption in energy homeostasis. For example, cold exposure potently upregulates IL-4 expression in ATMs (338), which mediates several of the adaptive responses to cold. While the resulting ATM phenotype is likely different from that of ATMs in the setting of weight gain/obesity or fasting/weight loss, the dependency of a physiological response on ATMs illustrates that the presence and activation of macrophages in adipose tissue is not per se a pathophysiological event.
While much of the initial work focused on macrophages, a growing body of evidence has now revealed that many types of immune cells infiltrate obese adipose tissue, and these infiltrating leukocyte populations also contribute to the inflammatory processes within adipose tissue, as noted in several recent reviews (78, 264, 295, 329, 460, 507).
As discussed earlier in the context of weight loss, neutrophils are the likely first responders to inflamed obese adipose tissue. In mice, HFD rapidly induces neutrophil accumulation in adipose tissue, with a significant increase observed within three days of HFD initiation (106, 161, 454). Current evidence suggests that this early infiltration of neutrophils is a transient phase that is quickly replaced by infiltration of macrophages and other leukocytes. However, there are currently no comparable data available from humans, and the limited data that do exist warrant further investigation. Nijhuis et al. noted an absence of neutrophils outside of the adipose tissue vasculature, based on IHC staining for myeloperoxidase (343). In contrast, we recently isolated and identified neutrophils, based on the co-expression of CD15 and CD16 by flow cytometry, from the SVC of adipose tissue, and reported that their numbers are substantially increased following weight loss (162, 254). Accumulation of several of the less abundant myeloid cell types, such as mast cells, dendritic cells, and eosinophils, is also influenced by increased adiposity. Murine models of diet-induced obesity exhibit increased infiltration of mast and dendritic cells (37, 274), but decreased numbers of eosinophils in adipose tissue (516). Evidence from human clinical studies also suggests that obese adipose tissue contains more mast and dendritic cells relative to lean adipose tissue (37, 274).
Lymphocytes are also present in adipose tissue, and their relative proportions change with increasing adiposity (20, 102, 236, 345, 512, 518). However, where inflammation of adipose tissue and classic inflammation may differ is in the temporal sequence of T cell infiltration, and to a lesser extent, function. In the classic setting, naïve lymphocytes become activated effector cells upon exposure to specific antigens displayed by antigen presenting cells. These activated cells then proliferate into CD8+ cytotoxic cells that aid in the removal of infected cells or CD4+ helper cells that enhance and regulate the immune response and confer long-term immunity (333). In contrast to infection-induced inflammation, in obesity-associated adipose tissue inflammation infiltration of T cells precedes that of pro-inflammatory macrophages and may be necessary for subsequent recruitment and activation of macrophages (236, 345). However, the temporal relationship of these events is not fully resolved. Lee et al. observed a significant increase in ATM infiltration in diet-induced obese lymphocyte-deficient Rag1-null mice, suggesting that lymphocytes are not necessary for increased infiltration of macrophages during adipose tissue inflammation (268). HFD feeding in mice is associated with a shift in the CD4+ effector T cell population away from the predominance of TH2 T cells, as in lean adipose tissue, and towards more TH1 and cytotoxic T cells (512). The change in the T cell population composition includes a relative loss of regulatory T cells (Tregs) with the onset of obesity, which may be a potential mechanism through which subacute chronic inflammation fails to resolve (122, 364, 512). As with neutrophils, the temporal sequence for T cell infiltration cannot readily be established in humans. However, like that of macrophages, T cell accumulation in human adipose tissue does correlate with adiposity (102, 236, 471, 546). There also remains uncertainty as to the role of Tregs in the inflammatory process in human adipose tissue. Feuerer and colleagues observed lower expression of the Treg-specific gene FOXP3 in VAT as compared to SAT of obese subjects (122). In addition, FOXP3 expression negatively correlated with BMI, suggesting that this regulatory immune cell population was reduced in the VAT of obese individuals; however, this study lacked non-obese controls (122). Conversely, two other studies reported that all T cell genes assayed were up-regulated, and correlated with obesity and inflammation, in both human SAT and VAT, with no significant reduction in Tregs (471, 546). Another T cell subtype, the invariant natural killer T-cell (iNKT), also exhibits reduced numbers in human adipose tissue as BMI increases (282). Finally, B cells, which produce antibodies against specific foreign antigens, also accumulate in adipose tissue soon after the initiation of HFD feeding in mice (511). B cells may also contribute to macrophage and T cell activation in the context of obese adipose tissue.
Pro-inflammatory mediator secretion
Many of the cytokines involved in a classic, infection-driven inflammatory response, including TNFα, IL-1β, IL-6, and IFNγ, as well as chemokines such as MCP-1 are also produced in expanded adipose tissue of obese rodents and humans. In general, infiltrating immune cells are the major sources of pro- and anti-inflammatory cytokines produced in adipose tissue, although adipocytes and other stromal cells also contribute (119, 467, 504, 523, 545). Hotamisligil and colleagues, in their 1993 landmark paper, were the first to describe that the gene expression and protein secretion of the key pro-inflammatory cytokine TNFα was elevated in the adipose tissue of four different rodent models of obesity (189). Weisberg et al., in their paper describing the presence of macrophages in expanded adipose tissue, were among the first to describe that TNFα is expressed almost exclusively in ATM, while other key cytokines such as IL-6 are partly expressed by other SVCs (504). Aside from TNFα and IL-6, the concentrations of other key pro-inflammatory cytokines including IL-1β and IL-18, both products of the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing 3 (NLRP3) inflammasome activation, are increased in the adipose tissue of obese, HFD-fed mice compared to leaner, chow-fed littermates (443). Along with the recognition that obesity is associated with macrophage infiltration into adipose tissue, it was recognized that chemokines such as MCP-1 play a key role in this process. In fact, MCP-1 is upregulated in the white adipose tissue of Lepob/ob, Leprdb/db, and HFD-fed wild-type mice, along with markers of macrophage infiltration (523). In the HFD-fed animals that continuously gained weight on this diet, a lasting increase in the expression of MCP-1 and markers of macrophage infiltration did not become apparent until week 11 (even though there was a small initial increase in MCP-1 expression three weeks after initiation of the HFD), suggesting that the onset of an inflammatory response was delayed (523). As with TNFα and IL-6, expression of MCP-1 was highest in the non-adipocyte fraction of adipose tissue (523). The adipose tissue expression of adiponectin was first reported to be suppressed in diet-induced obese mice by Yamauchi and colleagues (532), in line with the finding that adipocyte expression of adiponectin is attenuated by both TNFα and IL-6 [reviewed in (451)].
While most of the initial observations were made in mouse models of obesity, very similar changes in the expression of cytokines, chemokines, and adiponectin occur in human obese adipose tissue (379, 391). Also, systemic concentrations of many pro-inflammatory cytokines are elevated during obesity, although the elevation is moderate and remains lower than what is typically observed during an infection (29, 30, 53, 118, 229, 330, 459, 467, 474). Consistent with the reduced adipose tissue expression of adiponectin, the plasma concentration of adiponectin is inversely associated with body weight in both mice and humans [reviewed in (451)].
The corresponding cytokine and chemokine cell surface receptors and downstream signaling cascades are activated in cultured adipocytes and macrophages and in whole adipose tissue explants. These downstream signaling cascades include the JNK and NFκβ pathways. Another key pathway in the pathogen-driven immune response is the NLRP3 inflammasome, which also appears to be responsive to metabolic stimuli [reviewed in (391)]. The NLRP3 inflammasome is traditionally thought to be activated by the pore-forming exotoxins produced by Gram-positive bacteria (304), but is speculated to be activated by metabolic signals such as FFA in obesity [reviewed in (444)]. In addition, ATM NLRP3 expression is correlated with obesity in humans (112).
Molecular mechanisms underlying adipose tissue inflammation
Many factors likely contribute to the development of inflammation in adipose tissue. We will here discuss ER stress, hypoxia, adipocyte hypertrophy and death, mitochondrial dysfunction, fibrosis, fatty acid-induced activation of macrophages, and a possible role of TLRs, and cellular senescence as potential mechanisms contributing to adipose tissue inflammation. Of note, all of these should not be seen as mutually exclusive, or as likely primary causes, but rather as linking mediators between chronic caloric excess and adipose tissue inflammation, or as factors that may perpetuate chronic inflammation in the tissue. It is also important to emphasize that this list of putative mechanistic links is almost certainly not exhaustive, and that key stimuli for adipose tissue inflammation may not yet be known.
The ER is the central organelle coordinating the synthesis, processing, and trafficking of secretory and membrane proteins. Several protein chaperones exist within the lumen of the ER to facilitate the proper folding of proteins. The inappropriate accumulation of misfolded or unfolded proteins causes ER stress which triggers the unfolded protein response (UPR) (499). The UPR then activates specific signaling pathways that can facilitate protein folding, reduce protein synthesis, and increase protein degradation, with the goal of reducing the misfolded protein burden on the ER (155, 226, 499). There is significant crosstalk between ER stress and inflammation in that the UPR activates NFκB and JNK signaling, and pro-inflammatory cytokines can induce branches of the UPR (154, 358, 477, 499). In addition, failure of the UPR to resolve the stress initiates apoptosis and the generation of DAMPs, which promotes the pro-inflammatory state (154). Given the interaction between ER stress and inflammatory signaling pathways, it is not surprising that expression of markers of the UPR are increased in adipose tissue of obese humans and HFD-fed mice (227, 422).
During adipocyte hypertrophy, angiogeneis is also stimulated in order to provide oxygen to the expanding tissue. If adipose tissue expansion is too rapid, developing vasculature cannot keep up with oxygen demand and hypoxia occurs. Several studies have shown that obese adipose tissue is hypoxic as compared to lean adipose tissue (163, 186, 538). The hypoxia-inducible factor (HIF) family of transcription factors regulate the tissue response to a hypoxic environment. In obese mice, several studies have revealed the presence of hypoxic regions in adipose tissue, and increased expression of the oxygen sensing transcription factor HIF-1α (135, 186, 267, 538). Under normal tissue oxygen (PO2) concentrations, HIF-1α is continuously synthesized but degraded. However, low PO2 concentrations cause inhibition of hydroxylases that normally inactivate and target HIF-1α for degradation. Activated HIF-1α translocates to the nucleus, binds to hypoxia response elements, and induces expression of genes involved in angiogenesis, cell proliferation and survival, inflammation, and energy metabolism (472, 473). There is also evidence suggesting that activation of HIF-1 is necessary for myeloid cell infiltration (88), that hypoxia may polarize ATMs towards a pro-inflammatory phenotype (135), and that this can be enhanced by exposure to palmitate (429). In humans, CD14-positive cells isolated from VAT and exposed to hypoxic conditions exhibited enhanced secretion of pro-inflammatory cytokines (349). A separate study demonstrated that postprandial blood flow to adipose tissue is decreased by as much as 40% in obese insulin resistant individuals relative to lean insulin sensitive individuals (147), although whether this results in hypoxic adipose tissue is less clear (147, 372). Goossens et al. reported that obese insulin resistant subjects had 50% higher adipose tissue PO2 relative to leaner, more insulin sensitive controls (147). However, this study also noted a 60% reduction in adipose tissue O2 consumption among obese individuals, and that the expression of mitochondrial function markers was inversely correlated with adipose tissue PO2 (147). Nevertheless, the metabolic signature of anaerobic metabolism, with increased metabolism of lactate and pyruvate, has yet to be demonstrated in adipose tissue (179).
Adipocyte hypertrophy itself may contribute to adipose tissue inflammation. Adipocyte size correlates with measures of metabolic dysfunction and may be a determinant of adipose tissue insulin resistance. Cross-sectional studies have shown that adipocyte size in VAT negatively correlates with insulin sensitivity (167, 348) and positively with the degree of hepatic steatosis (348). The mean adipocyte size of VAT is significantly smaller in MHO insulin sensitive individuals as compared to insulin resistant obese individuals (238, 348). Consistent with these human data, mean adipocyte size of gonadal adipose tissue is significantly smaller in insulin sensitive adiponectin transgenic Lepob/ob mice despite overall greater fat mass compared to their Lepob/ob counterparts (235). Increased adipocyte size may be one factor that initiates an inflammatory response in obese adipose tissue, with the production and secretion of the chemokine MCP-1 that functions to attract monocytes to the tissue. Mean adipocyte size is significantly increased in Lepob/ob and Leprdb/db mice as compared to non-obese wildtype mice, and hypertrophied adipocytes are more susceptible to death than are small adipocytes (332). In adipose tissue, macrophages form CLS by surrounding necrotic adipocytes, and likely play a major role in taking up released lipids from dead adipocytes (79, 332). Although the number of CLS in humans may not be as high as that in obese mice, it is estimated that in both mice and humans, nearly 90% of all macrophages in obese adipose tissue are localized to such CLS (79).
Impaired mitochondrial function may play a role in adipose tissue inflammation in the context of chronic caloric excess. In mice, obesity is associated with altered mitochondrial morphology, reduced mitochondrial number, and impaired function and reduced oxygen consumption in adipose tissue (396). Evidence suggests a similar reduction in mitochondrial function of adipose tissue of obese humans (540). One byproduct of mitochondrial dysfunction is the production of reactive oxygen species. Systemic measures of oxidative stress correlate with adiposity in both humans and mice. Furukawa and colleagues measured lipid peroxidation in obese individuals and observed a significant positive correlation with BMI and WC (137). They also observed elevated lipid peroxidation in a mouse model of obesity and elevated levels of H2O2, both in circulation and in adipose tissue (137).
Structural and connective components comprise the extracellular matrix (ECM) of adipose tissue and serve an essential function of maintaining the basic tissue architecture. However, it is now clear that the ECM also plays an important role in adipose tissue function and related metabolic phenotypes. Remodeling of the ECM is essential for adipocyte expansion and contraction to accommodate changes in energy stores (400). In adipose tissue, the HIF-1α isoform exerts major control over the hypoxic response that may occur during adipose expansion and induces angiogenic, inflammatory, and fibrotic gene expression programs (90, 163). Thus, with activation of HIF-1α in obese adipose tissue, fibrosis develops due to excessive accumulation of ECM components. Direct evidence for a role of adipose tissue fibrosis in adipose tissue function, metabolic health, and glucose homeostasis has been generated from animal studies. Khan et al. demonstrated that collagen VI deficiency in Lepob/ob mice is associated with increased mean adipocyte cell size and improved insulin signaling and glucose tolerance (231). Adipose tissue-specific HIF-1α overexpression in HFD-fed wildtype mice or in Lepob/ob mice caused adipose tissue fibrosis, adipose tissue inflammation, and impaired glucose tolerance (163). In mice lacking one copy of matrix metalloproteinase (MMP)-14, ECM remodeling and collagen turnover are impaired and adipose tissue expansion is inhibited (77). A study of weight stable obese or lean subjects revealed that several genes encoding components of the ECM are significantly upregulated in SAT of obese as compared to lean individuals, and direct staining of collagen also demonstrated significantly greater SAT fibrosis in the obese subjects (172). Divoux et al. reported that VAT fibrosis was significantly greater in obese subjects as compared to lean controls (98). Although these studies demonstrate differences in fibrosis with adiposity, data implicating fibrosis in impairment of insulin signalling in human adipose tissue are mixed. In agreement with evidence from rodent models, obese insulin resistant adipose tissue had higher expression of CD68, likely reflective of a higher number of macrophages, higher expression of genes encoding ECM proteins (collagen V, MMP7, TSP1), HIF-1α and VEGFA as compared to insulin sensitive subjects matched for BMI (262). Other studies reported that compared to that from lean subjects, SAT from obese insulin resistant subjects was more fibrotic with more collagen VI and less elastin and collagen V (433). Tam et al. noted that the rapid onset of IR that accompanies acute overfeeding leads to transcriptional induction of ECM genes (457). However, other groups have not found a direct association between fibrosis and surrogate markers of insulin resistance. For example, Muir et al. reported that obese bariatric surgery patients with T2DM had less fibrosis in VAT compared to non-T2DM patients, and the absence of fibrosis was associated with greater adipocyte hypertrophy and adipose dysfunction (331). The authors speculated that perhaps fibrosis in the context of obesity places a limit on extreme adipocyte hypertrophy in an attempt to preserve normal adipocyte function. Furthermore, Lackey et al. observed increased expression of collagen VI (COL6A3) in VAT of MHO subjects as compared to obese subjects with metabolic syndrome and in healthy subjects as compared to those with T2DM (257).
Macrophages express TLR4 and its co-receptor CD14, which detects LPS, a major component of the outer membrane of Gram-negative bacteria. LPS exposure strongly induces a pro-inflammatory immune response (333, 526). Current evidence indicates circulating LPS is increased in diet-induced obese mice and in individuals with T2DM, and this LPS may be a contributing factor to obesity-associated inflammation (133). Proposed mechanisms for elevated LPS include increased gut permeability (58) and translocation of LPS or live gram-negative bacteria through the intestinal mucosa to adjacent mesenteric adipose tissue (17). To which degree humans are actually exposed to gut microbial LPS is uncertain. However, lipid species such as palmitate, a long-chain saturated fatty acid, and ceramides, which circulate at higher concentrations in obese individuals, can also activate macrophages (95, 182, 243, 494). Consistent with this, we have shown that exposure of macrophages in vitro to a cocktail of palmitate, glucose, and insulin induces a low-grade pro-inflammatory state that we termed ‘metabolic activation’, characterized by elevated cell surface expression of ABCA1 and CD36, which may provide a plausible mechanism through which ATM could become pro-inflammatory, activated cells in obesity (253, 264, 545, 547). It has been suggested that fatty acids activate macrophages by binding to TLR4 (339, 423). However, other evidence indicates that long chain saturated fatty acids are not direct ligands of TLR4 but rather that saturated fatty acid-induced inflammation requires an initial priming event of TLR4 (110, 258). Intriguingly, mice lacking TLR4 (a) are protected from the ability of a systemic lipid infusion to trigger insulin resistance, and (b) become obese when fed a HFD but remain relatively insulin sensitive with lower levels of inflammation in liver and adipose tissue (423). Thus, while fatty acid-induced pro-inflammatory activation of macrophages is a plausible mechanism underlying weight gain-induced adipose tissue inflammation, and while TLR4 likely plays a role in the etiology of adipose tissue inflammation, questions remain as to whether fatty acid binding to TLR4 is relevant in vivo.
One particular factor that may partly explain inter-individual variability in adipose tissue expandability may be a decline in tissue function and flexibility due to cellular senescence. Senescence of different cell types within adipose tissue is associated with obesity and biological age, both factors that are strong determinants of T2DM risk. Cellular senescence is characterized by a state of permanent growth arrest in mitotic cells. Data from mouse and human studies provide evidence that senescent cells accumulate in adipose tissue during obesity (412, 427, 462, 496) and insulin resistance (313, 412). The potential metabolic consequences of senescent cell accumulation in adipose tissue are diverse. For example, senescent preadipocytes negatively impact the adipogenic capacity of the depot, as fewer progenitor cells are capable of differentiation into mature, energy-storing adipocytes (316, 525). In endothelial cells, senescence reduces angiogenesis and nitric oxide synthase levels and activity (111, 312). Senescent CD4+T cells have also been detected in obese adipose tissue of HFD-fed mice (427). Although basic cellular functions are impaired during senescence (e.g., differentiation), senescent cells are metabolically active and adopt a senescent-associated secretory phenotype (SASP) wherein they secrete pro-inflammatory cytokines and signaling factors. Thus, senescence impairs normal adipose tissue function and likely directly contributes to the inflammatory tone that is characteristic of obese adipose tissue. Furthermore, work over the past decade has demonstrated an active role of immune cells in clearance of senescent cells (180). Components of the SASP, including cytokines and chemokines, specifically attract immune cells to the site of senescence. In the clearance of senescent tumor cells, it is known that neutrophils, macrophages and natural killer cells participate in a coordinated response (529), while clearance of pre-cancerous cells requires CD4+ T cells (222). Although very little is known about the role of ATM in clearance of senescent cells in adipose tissue, it is hypothesized that macrophages are important in this capacity in adipose tissue and that with aging, macrophages themselves may become senescent and dysfunctional which leads to an accumulation of senescent cells (164). While the existing data suggest a role for cellular senescence in adipose tissue loss of function (i.e., limitations in storage capacity) and inflammatory mediator production, numerous questions remain about the effect of obesity and aging on cellular senescence in adipose tissue, and about the relative contribution of cellular senescence to adipose tissue dysfunction and inflammation.
Obesity and adipose tissue inflammation: rodent models
Adipose tissue inflammation is a common phenotype in obese mice. Strong evidence of this association was generated in parallel studies by Xu et al. and Weisberg et al. (504, 523). Using genetic models of obesity such as Lepob/ob, Leprdb/db, and Ay/+, as well as HFD-fed wildtype mice, these papers demonstrated that adipose tissue inflammation develops with excess adiposity regardless of the underlying cause. Expression of pro-inflammatory cytokines and chemokines, such as TNFα, IL-6, MIP-1α, MCP-1, and macrophage markers, such as F4/80 and CD68, are significantly elevated in adipose tissue of these obese animals relative to lean, wildtype mice. These studies also were among the first to demonstrate that the SVC fraction, as compared to the adipocyte enriched fraction, was the predominant source of the cytokines and chemokines (504, 523). Increased macrophage infiltration of adipose tissue was also demonstrated by histological staining for F4/80+ in mice, which positively correlated with fat mass (504).
Further evidence of the strong link between increased adiposity and adipose tissue inflammation is provided by genetic mouse models that are protected against HFD-induced or genetic forms of obesity. One such model is the SCD1 knockout (KO) mouse. The stearoyl-CoA desaturase (SCD) family of enzymes synthesize monounsaturated fatty acids from saturated fatty acid precursors (317). The SCD1 isoform is expressed in metabolic organs including adipose tissue and liver (319, 550), and in mice exerts significant control over whole-body metabolic regulation. For example, SCD1 deficiency in Ay/a and HFD-feeding models protects against excessive adiposity, insulin resistance, and hepatic steatosis (318, 320, 347). Follow-up work revealed that SCD1 deficiency in both of these models is also associated with reduced adipose tissue inflammation, with lower expression of pro-inflammatory cytokines and chemokines and reduced macrophage infiltration into adipose tissue (276). Notably, these animals are hyperphagic on a HFD (320), which supports the hypothesis that adipose tissue inflammation is caused by excess adiposity rather than the HFD itself, or the excessive energy intake it triggers.
Taken together, strong and consistent evidence from HFD-induced obese as well as genetically obese mouse models support the notion that the accumulation of excessive adiposity is a key driver of adipose tissue inflammation in mice.
Obesity and adipose tissue inflammation: human studies
Several recent reviews have summarized the relationship between adipose tissue leukocyte infiltration and obesity in humans (264, 329). Despite some important differences, the overall association between obesity and adipose tissue inflammation appears to be as robust in humans as in mice. A summary of cross-sectional human studies that have assessed adipose tissue inflammation and adiposity is presented in Table 2. Even though many different measures have been used to assess and quantify the complex biological process of adipose tissue inflammation, the overall evidence is consistent that the number of immune cells and the expression of pro-inflammatory cytokines is higher in the adipose tissue of obese as compared to lean individuals. Specifically, with regard to leukocyte populations, as assessed by flow cytometry of the SVC fraction of SAT and/or VAT, investigators unanimously found positive associations between the numbers of macrophages (50, 92, 93, 471, 545), dendritic cells (37), and T-cells (both CD4- and CD8-positive) (102) and measures of adiposity. Similarly consistent associations were observed for the number of macrophages, as assessed by histological staining, and measures of adiposity (56, 169, 357, 504). The measurement of transcript levels of genes encoding key proteins in the inflammatory process revealed higher expression of TNFα (109, 194), IL-6 (109), and markers of T-cell infiltration (546), but lower expression of adiponectin (109) in adipose tissue from obese as compared to lean or non-obese individuals (Table 2).
Table 2.
Relationship between AT inflammation and obesity in humans: evidence from observational studies.
| Subjects | Methodology | Covariates | Groups | Outcome | Reference |
|---|---|---|---|---|---|
| n=50 (39F) 40±3.3 yrs 8 African American, 42 Caucasian |
Gene expression:
TNFA Tissue stimulation: TNFα, IL-6 |
None | BMI: <25 (n=9) 25–30 (n=9) 30–40 (n=17) >40 (n=15) |
No difference in AT TNFA mRNA
expression across BMI. AT secretion of TNFα, but not IL-6, was significantly lower among lean individuals compared to overweight and obese persons. |
(229) |
| n=65F Postmenopausal Caucasian |
Gene expression: ADIPOQ, IL6, TNFA | None | BMI: <25 (n=20) 25–30 (n=25) >30 (n=20) |
Adipose tissue IL6 and TNFA mRNA increased linearly with BMI and was significantly higher among obese compared to lean, while ADIPOQ mRNA was 1/3 lower among overweight and obese compared to lean women. | (109) |
| n=14 (8F) age: N/A race: N/A |
Gene expression:
CD68 IHC: CD68 |
None | None (BMI range: 19.4–60.1) |
Expression of CD68 mRNA and number of CD68+ cells was significantly higher in SAT of obese vs. lean subjects. | (504) |
| n=43 age: N/A race: N/A |
Flow cytometry: CD14, CD31 | None | SAT (gluteal: n=19 (mean BMI=25±1),
abdominal: n=7 (mean BMI=25.3±0.5)) VAT: n=17 (mean BMI=26.3±0.8) |
A significant positive correlation between BMI and the percentage of ATM (CD14+CD31+) in the stromavascular fraction isolated from adipose tissue, irrespective of depot. | (92) |
| n=24F 29–59 yrs Caucasian |
Gene expression: CCL2, HIF1A, CSF3,
PLAUR IHC: CD68, HAM56 |
None | BMI: <25 (n=7) >40 (n=17) |
Greater number of ATM (CD68+HAM56+) in SAT of obese vs. lean women. Levels of expression of macrophage attraction genes CCL2, CSF3, HIF1A, and PLAUR were significantly higher in SAT of obese vs. lean women. | (56) |
| n=21 (9F) 44.7±16.2 yrs ethnicity: N/A |
IHC: MAC2 | None | SAT (gluteal and abdominal) and VAT (n=28
tissue biopsies) BMI: <25 (n=7) 25–29.9 (n=2) >30 (n=12) |
Adipocyte death was positively correlated with obesity and mean adipocyte size in both SAT and VAT depots; MAC2+ ATM selectively CLS around dead adipocytes and rates of adipocyte death increase with obesity | (79) |
| n=50 (17F) 21–81 yrs ethnicity: N/A |
Gene expression: TNFA, IL6, ADIPOQ,
MIP1A, CCL2, IL8 Flow cytometry: CD14 |
None | VAT (mean BMI 25.4±1.4) | Percentage of CD14+ATM in VAT was positively correlated with BMI, CD14+ ATM expressed higher levels of CCL2, MIP1A, and IL8 than did mature adipocytes suggesting that the greater proportion of ATM in obesity is responsible for the elevated production of chemokines. | (93) |
| n=60 (30F) 29–82 yrs Caucasian |
Gene expression: CD68, CCL2,
CSF1 IHC: CD68 |
BMI: <25 (n=20) >28.5 (n=20 with IA obesity, n=20 with SC obesity) |
CD68+ ATM were more abundant in VAT vs. SAT in all groups, while lean controls had lower overall CD68+ ATM count in each depot, followed by SC obese and IA obese. IA obesity was associated with higher macrophage infiltration compared to SC obesity (both in number of CD68+ cells and CD68 mRNA); CCL2 and CSF1 mRNA were more abundant in VAT vs. SAT in all groups, while IA obese had the highest expression of these genes. | (169) | |
| n=29F 25–75 yrs ethnicity: N/A |
IHC: CD68 ELISA: CCL2, CSF1 |
None | VAT vs. SAT (paired samples, BMI: 32–57) | BMI was significantly correlated with number of macrophages in VAT but not SAT depot, protein levels of CCL2 and CSF1 were significantly increased in VAT compared to SAT | (169) |
| n=34 age: N/A ethnicity: N/A |
Gene expression: CCR5, RANTES, CD3, CD11B | None | SAT: lean (n=3) obese (n=10) SAT and VAT paired samples: morbidly obese (n=21) |
Obese subjects had significantly higher mRNA levels of RANTES and its receptor CCR5 in SAT than lean subjects, mRNA levels of RANTES and CCR5 in SAT were positively correlated with BMI, VAT from morbidly obese subjects had higher mRNA levels of RANTES and CCR5 than SAT, RANTES mRNA was strongly positively correlated with CD3 and CD11B mRNA in VAT. | (518) |
| n=20 age: N/A ethnicity: N/A |
IHC: CD14, CD206 Flow cytometry: CD14, CD206 |
None | SAT (n=14) VAT (n=6) |
Number of CD14+CD206+ ATM in SAT and VAT was significantly correlated with BMI. | (545) |
| n=78F 42±1 yrs ethnicity: N/A |
Flow cytometry: CD14, CD206 | None | None (BMI 20–30) |
Percentage of CD14+CD206+ ATM in SAT was significantly correlated with BMI. | (50) |
| n=40 (28F) 39.4±2.6 yrs Caucasian |
Gene expression: CD68,
TNFA IHC: CD206 |
Age, sex | BMI: <30 (n=20) >40 (n=20) |
Number of CD206+ ATM in SAT and VAT of obese subjects was 3-fold higher than lean controls. No significant difference in ATM number between VAT and SAT in obese subjects, mRNA expression of CD68 and TNFA was higher in VAT and SAT of obese vs. lean subjects. | (194) |
| n=54 (13F) 44–79 yrs ethnicity: N/A |
Gene expression: CD3, CD68,
IFNG IHC: CD4 |
None | None (BMI 22–48) |
In these diabetic patients, both CD3 and INFG mRNA expression in SAT was significantly correlated with WC, while CD68 was not (n=54). CD4+ T cell staining in VAT correlated with BMI (n=19). | (236) |
| n=26F 41.1±8.6 yrs Caucasian |
IHC: CD40, CD163, CD206 | BMI: <25 (n=10) SAT >40 (n=16) VAT and SAT |
Number of CD40+ ATM was higher in obese vs. lean SAT, and higher in VAT vs. SAT in obese women. No difference in CD163+ and CD206+ ATM counts in obese vs. lean SAT. | (26) | |
| n=137 (133F) 42.6±1.2 yrs ethnicity: N/A |
Flow cytometry: CD3, CD4, CD8, CD14 | None | SAT: BMI range 19–43 (n=92) SAT and VAT paired samples: BMI 43±1 (n=45) |
Significant correlation between number of SAT CD3+, CD4+, and CD8+ cells and BMI; in paired tissue samples, the number of CD14+ ATM was 1.2-fold higher in VAT compared to SAT, while CD3+ cells were 3-fold higher in VAT compared to SAT. | (102) |
| n=66 (25F) 31±8 yrs Native Americans of Pima descent |
Gene expression: CSF1R, CD11B,
CD68,
IHC: CD68 |
Sex | None | Number of CD68+ ATM and mRNA expression of CSF1R, CD11B, and CD68 in SAT was positively associated with percent body fat and BMI. ATM number and mRNA was positively associated with age until years 31–33, then markers declined slightly with increasing age. | (357) |
| n=7 age: N/A ethnicity: N/A |
IHC: Tbet, FoxP3 | None | BMI: <25 (n=3) >30 (n=4) VAT from patients with colon cancer |
The ratio of IFNγ−secreting TH1 (Tbet+) cells to TH2 regulatory T-cells (FoxP3+) in VAT correlated with BMI and increased with increasing adiposity. | (512) |
| n=29F 20–61 yrs Caucasian |
IHC: CD11c, CD68, CD206 Flow cytometry: CD11c, CD14, CD206 |
None | BMI: formerly obese (n=5) obese (n=12) obese with metabolic syndrome (n=12), VAT and SAT paired samples |
CD11c+ ATM density was greater in SAT vs. VAT, CD206+CD11c+ ATMs form CLS and are higher in density in adipose tissue of women with metabolic syndrome vs. those without. | (508) |
| n=35F 50–60 yrs ethnicity: N/A |
Gene expression: 39-gene panel | Age | obese, T2DM (n=12) obese, non-diabetic (n=8) lean (n=15) |
Higher mRNA expression of CD68 and proinflammatory chemokines was found in SAT of obese vs. lean subjects, and this profile was further strengthened in obese subjects with T2DM | (330) |
| n=40 (32F) age: N/A Caucasian |
Gene expression: CD3E, CD8A, TBX21, FOXP3, GATA3, IFNG, IL4, TGFB | Age, sex | BMI: <30 (n=20) >40 (n=20) VAT and SAT paired samples |
All T cell markers were more highly expressed among obese vs. leaner controls in both VAT and SAT. Expression of T cell-produced cytokines (IFNG, IL4, and TGFB) was significantly increased with obesity in both depots. | (546) |
| n=24F 44.6±2.5 yrs Caucasian |
Flow cytometry: CD1c, CD11c, CD83 | None | None (mean BMI=28.3±1.2) | Dendritic cell populations (CD1c+, CD1c+CD11c+, and CD83+ cells) in SAT were positively associated with BMI. | (37) |
| n=24 (21F) age: N/A ethnicity: N/A |
Gene expression: CD1C, CD11C, CD83 | None | BMI<25 (n=4) BMI>40 (n=10) BMI>40, T2DM (n=10) |
mRNA expression of CD1C and CD83 in SAT was significantly greater in obese vs. lean subjects, and more so among obese with T2DM. | (37) |
| n=56 (48F) 21–61 yrs ethnicity: N/A |
Gene expression: IL6, TNFA | Age, sex | BMI: <25 (n=22) >40 (n=34) VAT and SAT paired samples |
Expression of IL6 and TNFA mRNA tended to be higher in VAT compared to SAT in obese subjects, and expression of IL6 and TNFA mRNA tended to be lower in VAT and SAT of lean compared to obese subjects, however the differences were not significant. | (368) |
| n=256 (162F) age: N/A ethnicity: N/A |
Gene expression: FAS, FASL | None | BMI: lean (n=56) overweight (n=32) obese (n=168) VAT and SAT paired samples |
Expression of FAS and FASL mRNA was significantly higher in VAT compared to SAT and was significantly higher in both depots in obese vs. lean subjects. FAS expression in VAT was significantly positively correlated with BMI and ATM infiltration. | (46) |
| n=30M 35–55 yrs ethnicity: N/A |
Gene expression: CD3, CD4, CD8, TBX21,
GATA3, FOXP3, CD68 Flow cytometry: CD14 |
None | WC: lean (<94 cm) (n=10)
overweight (94–102 cm) (n=10) obese (>102 cm) (n=10) |
Expression of CD4, CD68, and FOXP3 mRNA was significantly elevated in SAT of obese compared to lean men. Both CD4+ and CD8+ T-cell numbers in SAT, as well as CD14+ ATMs, were positively correlated with WC. | (471) |
| n=43 age: N/A ethnicity: N/A |
Gene expression: CD68, IL6, MCP1 | None | BMI: lean (n=12) obese (n=31) |
Expression of IL6, MCP1, and CD68 mRNA was significantly higher in SAT from obese vs. lean subjects | (286) |
| n=50 (37F) age: 37–46 yrs ethnicity: N/A |
Flow cytometry: CD4, CD39, IL-17, IL-23R,
Foxp3 Tissue stimulation: IL-1β, IL-6, IL-12, IL-23 |
Age, sex | BMI: <25 (n=20) >30, MHO (n=10) >30, MUO (n=20) |
VAT explants from MUO subjects secreted more pro-inflammatory cytokines compared to MHO and lean subjects, and CD4+ T-cells positive for CD39(Foxp3-), IL-17, and IL-23R, were more abundant in MUO compared to MHO and lean subjects. | (364) |
Abbreviations: ADIPOQ, adiponectin; ATM, adipose tissue macrophage; BMI, body mass index; CCL2 (MCP-1), C-C motif chemokine ligand 2; CCR5, C-C motif chemokine receptor type 5; CLS, crown-like structure; CSF(R), colony stimulating factor (receptor); ELISA, enzyme-linked immunosorbent assay; FoxP3, forkhead box P3; FAS (CD95); FASL, Fas ligand; GATA3, transcription factor GATA-3; HAM56, human alveolar macrophage-56; HIF1A, Hypoxia-inducible factor 1-alpha; IHC, immunohistochemistry; INFG/IFNγ, interferon gamma; IL, interleukin; IA, intra-abdominal; MAC2, (Galectin-3); MIP1A (CCL3), macrophage inflammatory protein 1-alpha; MHO, metabolically healthy obese; MUO, metabolically unhealthy obese; PLAUR, plasminogen activator urokinase receptor; RANTES (CCL5), regulated on activation, normal T cell expressed and secreted; SAT, subcutaneous (SC) adipose tissue; Tbet (TBX21), T-box transcription factor; TGFB, transforming growth factor beta; TNFA/TNFα, tumor necrosis factor alpha; T2DM, type 2 diabetes mellitus; VAT, visceral adipose tissue; WC, waist circumference
Challenges, controversies & knowledge gaps
A major challenge in the study of adipose tissue inflammation, in particular in humans, has been that no clear consensus exists how to assess or even quantify this complex biological process. It is also important to note that substantial differences exist between mice and humans in the cell surface markers used to identify and phenotype the cells, particularly in the case of myeloid cells such as macrophages. In mice, a combination of the markers CD11b, F4/80, CD11c, and CD206 (MMR) is commonly used to identify adipose macrophages (178, 295), even though it may also be debatable whether all of these markers have good specificity for macrophages. In humans, a standardized panel of ATM markers is lacking. Indeed, a wide panel of markers have been utilized by different groups to identify, classify, and quantify human ATM, including CD1c, CD11b, CD11c, CD14, CD31, CD40, CD68, CD163, CD206, MAC2, and HAM56 (26, 50, 56, 92, 93, 169, 504, 508, 545). In this regard, it is important to note that there are inherent limitations of employing a single marker to phenotype myeloid cells such as macrophages, as many of these markers lack lineage specificity or have traditionally been used to identify non-macrophage leukocytes (178, 219, 232, 252, 264, 547). In addition, distinguishing between true tissue macrophages and contaminating blood monocytes in SVC preparations of digested human adipose tissue requires at least two markers, CD14 and CD206 (545). Moreover, whereas rodent ATM are often characterized as pro- or anti-inflammatory based on their cell surface expression of CD11c and CD206, respectively (178, 280, 295), human ATM often exhibit both pro- and anti-inflammatory features and function simultaneously (253, 264, 545, 547). In our hands, we do neither observe a CD206-negative ATM population nor is CD206 specific to macrophages in adipose tissue, and is instead expressed on the cell surface of macrophages, dendritic cells and neutrophils, albeit to varying degrees (unpublished observation).
A consequence of the fact that the phenotyping of ATMs is not well standardized is that uncertainty exists as to whether the phenotype of ATMs in VAT vs. SAT differs. A major limitation of most studies that report ATM data from at least one visceral and one subcutaneous depot is that ATM numbers were almost always quantified using a single marker (MAC-2 or F4/80 in mice, HAM56, CD68, or CD206 in humans, using IHC) (16, 57, 168, 169, 194, 332, 355), which does not provide any information on phenotype, and – dependent on the marker used – may also lack specificity for macrophages. Of note, numerous published studies have attempted to obtain information on potential differences in the ATM phenotype between VAT and SAT by assessing “M1” macrophages by IHC or flow cytometric staining for markers such as CD11c or CD40, and “M2” macrophages by markers such as CD206. However, none of these markers have any specificity for classically activated M1 or alternatively activated M2 macrophages in mice or humans (34, 253). More importantly, as discussed further below, the initially proposed adipose tissue inflammation paradigm that focused on M1 vs. M2 macrophages could not be confirmed in mice and humans. Our data using multi-color flow cytometry, while probably still not sufficiently comprehensive, did not reveal any differences in the phenotype of ATM from VAT vs. SAT (253). Taken together, due to a lack of generally accepted staining approaches for differentially activated/polarized ATM, uncertainty exists about the phenotype of ATM in general, and potential phenotypic differences between VAT and SAT ATM in particular, in both mice and humans. In fact, the very concept of a homogenous ATM phenotype may be misleading. Instead, it seems possible that ATM in adipose tissue exhibit a wide spectrum of different phenotypes, with substantial between-subjects and even within-subject variability. This is in line with the concept that macrophages are highly plastic cells that can be activated within a very wide spectrum depending on their specific microenvironment (528).
Another challenge has to do with the phenotypic switch of ATM from non-activated or anti-inflammatory in lean adipose tissue to pro-inflammatory in obese adipose tissue, as nicely shown by Lumeng and colleagues (280). To summarize, in lean, insulin sensitive mice, macrophages present in adipose tissue exhibit a more anti-inflammatory phenotype, owing to the production of IL-10 and arginase 1. These macrophages were designated as M2-polarized, or alternatively activated, based on prior literature describing macrophages associated with tumors, parasitic infections, and the regulation of tissue repair and inflammation (149, 290). Importantly, these “M2” macrophages contribute to tissue remodeling and angiogenesis, two key functions that maintain normal tissue homeostasis (50, 61). When faced with nutrient excess, adipose tissue expands. The resulting adipocyte hypertrophy and concurrent release of chemokines, like MCP-1, by adipocytes, facilitates the recruitment of C-C motif chemokine receptor (CCR)2-positive monocytes to the expanding fat. Upon arrival, these monocytes differentiate into macrophages, and become activated cells that produce pro-inflammatory factors such as TNFα, IL-6, and in mice, iNOS (225, 484). Lumeng and colleagues proposed that these pro-inflammatory macrophages may be classically activated or M1 macrophages (280). While this was a plausible hypothesis at the time, it is now clear that the pro-inflammatory ATM in obese adipose tissue of mice and humans are not similar to M1 macrophages. First, it is important to note that the M1/M2 paradigm was established based on experiments in vitro. In vivo, the microenvironment acting upon macrophages is more complex, suggesting that ‘pure’ M1 or M2 macrophages are unlikely to be found (292). ATM have a complex phenotype, with numerous functional and phenotypic changes that are not similar to the pro-inflammatory activation seen during classical activation. For example, ATM show upregulation of pathways involved in lysosomal biogenesis, lipid metabolism, and autophagy, along with an activation of the NLRP3 inflammasome (272, 394, 486, 526). Still, as in classically activated macrophages, the expression of the classic pro-inflammatory cytokines such as TNFα is undoubtedly elevated in adipose tissue with increasing adiposity (29, 87, 188, 189, 229, 230), and macrophages in obese adipose tissue clearly have a more pro-inflammatory phenotype. Work by Xu et al. demonstrated that pro-inflammatory activation of macrophages isolated from the adipose tissue of obese mice was low-grade, as compared to a true M1 phenotype as was observed in the splenocytes of LPS injected mice (526). Further, the overall gene expression profile of ATM did not resemble that of classically activated M1 macrophages, leading these authors to conclude that ATMs do not display an obesity-driven switch towards a M1 phenotype (526).
Our own data support this conclusion. We recently demonstrated by a plasma membrane proteomics approach that the markers CD38, CD274, and CD319 are highly expressed on the cell surface of classically in vitro activated M1 macrophages, and on alveolar macrophages isolated from the airways of patients with cystic fibrosis (253). However, ATM isolated from both SAT and VAT of lean or obese humans did not express these surface markers (253). Thus, while we used a different experimental approach, we concluded similar to Xu et al. (526) that the pro-inflammatory macrophages present in the adipose tissue from obese mice or humans are not similar to classically activated M1 macrophages (253). Rather, ATM from obese humans express high levels of CD36 and ABCA1 on their cell surface, and the expression of these markers is upregulated in obese as compared to leaner individuals. This may be relevant because macrophages exposed to a combination of high physiologic concentrations of glucose, insulin, and palmitate in vitro similarly upregulate CD36 and ABCA1 expression on their cell surface, while also increasing their expression of TNFα, IL-1β, and IL-6, albeit at levels lower than that seen in in vitro activated M1 macrophages. While it is premature to conclude from these experiments that ATM may be pro-inflammatory cells due to what we called ‘metabolic activation’, we proposed that such metabolic activation is more reflective of the chronic, low intensity inflammation seen in obese persons with metabolic disease and may provide a better model than the M1/M2 paradigm (253). Still, the translatability of these in vitro experiments is limited by the fact that in vivo, macrophages will be exposed to a more complex microenvironment characterized by, for example, complex mixtures of different fatty acids and other lipids rather than isolated palmitate. In summary, there are clear changes in the composition, abundance, and function of ATM in response to increasing adiposity. While the M1/M2 paradigm is not consistent with the currently available data on the function and phenotype of ATM in mice and humans, questions remain as to the exact phenotype and function of macrophages and other immune cells in human adipose tissue, how adipose tissue inflammation can best be quantified, and which factors drive the inflammatory response in obese adipose tissue.
An important limitation of the literature, particularly in humans, is that the degree to which measures of adipose tissue inflammation in the different SAT depots (e.g., abdominal vs. peripheral) are correlated with those in the different VAT depots (e.g., omental vs. mesenteric). Another important limitation of the existing human literature in this regard is that the predominant source of VAT in most human studies is the omentum, because it is the most easily accessible VAT depot. The omentum is a distinct intra-abdominal fat depot that serves several immunological functions (309). Thus, immune cell infiltration and inflammatory signaling observed in omental VAT may be different from that occurring in other visceral depots, such as in mesenteric or peri-renal fat. It should also be pointed out that most of the human studies that have reported on VAT inflammation have relied predominantly on bariatric surgery patients. This is important to consider for two reasons. First, these patients are morbidly obese and the metabolic characteristics of VAT in these individuals may not be adequately representative of the inflammatory and/or metabolic state of VAT in individuals of lower BMI with T2DM or insulin resistance. Second, surgeons typically require patients to go on a low-calorie diet for several weeks prior to bariatric surgery; inflammation measured in adipose tissue samples collected during surgery may therefore be changed as a result of this active weight loss phase, and the relationship between adipose tissue inflammation and insulin resistance may be altered.
ADIPOSE TISSUE INFLAMMATION AND INSULIN RESISTANCE
Early data on role for inflammation in insulin resistance
A link between insulin resistance and inflammation was first suggested nearly 150 years ago when the administration of salicylates improved glucose homeostasis in patients with T2DM (428). Subsequently, there have been many other clinical studies that examined sepsis, viral infections, and rheumatoid arthritis, all of which revealed the presence of insulin resistance in concert with an active, ongoing inflammatory response (154). By the mid-1990’s, it was known that the canonical pro-inflammatory mediator TNFα is expressed in adipose tissue and induces insulin resistance in adipocytes (505). Additional pro-inflammatory factors secreted from adipose tissue that negatively impact insulin action have since been identified. In this section, we discuss the evidence generated from animal studies and human clinical studies on the association between adipose tissue inflammation and insulin resistance (Figure 11).
Figure 11. Insulin resistance and adipose tissue inflammation are strongly associated.
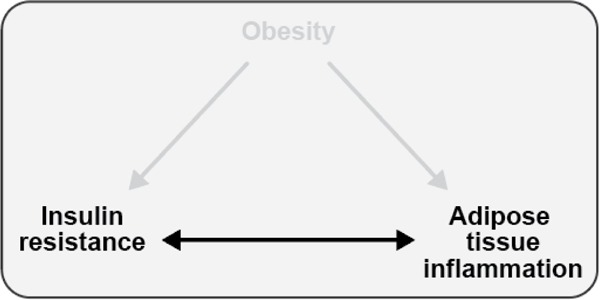
Although insulin resistance and adipose tissue inflammation generally characterize obese adipose tissue, the direction of causality has not been conclusively determined.
Teaching points: Although insulin resistance and adipose tissue inflammation generally characterize obese adipose tissue, the direction of causality has not been conclusively determined.
Adipose tissue inflammation and insulin resistance: rodent models
The etiology of insulin resistance and associated metabolic disease is complex, and rodent models have been among the most prominent tools utilized to understand pieces of this puzzle. Consistently, rodent studies have concluded that in the context of obesity, inflammation of adipose tissue is strongly implicated in the development of insulin resistance. The most common genetic strain of mice used in these studies, C57BL/6J, rapidly becomes obese, insulin resistant, and develops adipose tissue inflammation when fed a HFD (268, 350, 421). Genetic models of obesity and diabetes, such the Lepob/ob and Leprdb/db mice, also develop adipose tissue inflammation and insulin resistance concurrently with the increase in adiposity (221, 235, 458, 551).
To date, numerous genetic mouse models have been used to study the association between adipose tissue inflammation and insulin resistance in the context of obesity, typically employing the HFD regimen. The earliest such studies included alterations in the expression of some of the genes encoding key factors that had been implicated in adipose tissue inflammation, such as TNFα. A whole body KO of TNFα reduces adipose tissue inflammation and significantly improves insulin sensitivity (479, 490), while a KO of adiponectin significantly decreases insulin sensitivity (256). Adiponectin overexpression leads to improved insulin sensitivity associated with reduced adipose tissue inflammation (84, 235). However, all of these models also have an adiposity phenotype, i.e., the transgenic and wildtype mice differ with regard to adiposity. As a further limitation, the models used in the earlier studies were commonly not well phenotyped in terms of adipose tissue inflammation and other factors relevant for insulin sensitivity such as liver fat content or inflammation, which are likely to change along with adipose tissue inflammation as seen in Kim et al. (235), as discussed below. It is therefore hard to attribute any effect of the targeted genetic disruption on insulin sensitivity to changes in adipose tissue inflammation alone from these studies. What these early studies clearly established, however, is that key pro-inflammatory cytokines such as TNFα and anti-inflammatory factors such as adiponectin play central and antagonizing roles in energy and glucose homeostasis.
Several transgenic mouse models have shed light on the processes that contribute to adipose tissue inflammation, the specific cells and molecules involved, and the relative importance of adipose tissue inflammation for the development of insulin resistance. Here, we have identified those mouse models of obesity in which the aim was to influence inflammation, and in which adipose tissue inflammation and insulin sensitivity were assessed, and – in order to minimize confounding by fat mass and distribution – that do not have an adiposity phenotype (Table 3). The goal was to obtain a comprehensive overview of those mouse studies that are most likely to be informative about the importance of adipose tissue inflammation for the development of insulin resistance relative to other commonly associated factors. We identified a large number of studies that have consistently demonstrated that a KO of proteins involved in the initiation of inflammation, such as CCL2 (MCP-1), it’s receptor (CCR2), TLR4, or HIF-1α reduces the level of inflammation in adipose tissue when the animals are fed a HFD compared to wildtype (208, 221, 267, 380, 402, 503). Similar effects are seen with a KO of mediators of inflammation, including the leukotriene receptor-1 (BLT-1) or the cytotoxic T-cell surface marker CD8 (345, 435). In all of these cases, the reduction in measures of adipose tissue inflammation is associated with improved insulin sensitivity. In most cases in which a transgenic mouse does not display differential adipose tissue inflammation response with the onset of obesity, such as KO of PDCD4, IL-10, class A scavenger receptor, or the G protein-coupled receptor 120 (251, 354, 500, 552), this lack of a differential response in adipose tissue inflammation was associated with no change in insulin sensitivity. Cumulatively, these highly consistent associations of changes in adipose tissue inflammation and insulin sensitivity have been seen as supporting the hypothesis that adipose tissue inflammation is a key factor underlying the etiology of insulin resistance. It is important to note in this context, however, that many ‘adipocyte-specific’ transgenic mouse models use the aP2 (Fabp4) promoter to drive deletion/overexpression of the gene of interest. Results generated from such models should be interpreted cautiously as aP2 is also expressed in macrophages, and may therefore affect inflammation in tisues other than adipose. Indeed, in three such mouse models, Makowski et al. demonstrated increased expression of genes under control of the aP2 promoter in isolated peritoneal macrophages (287), although other studies have not found aP2 promoter-driven gene expression changes in macrophages in their transgenic models (267, 403). As illustrated in Table 3, one key limitation of almost all of these models, however, is that a reduction in adipose tissue inflammation is commonly also associated with a reduction in hepatic inflammation and/or liver fat content (for those studies that reported such data), making it impossible to confidently link the insulin sensitivity response (or lack thereof) to adipose tissue inflammation alone. At the same time, the very consistent association between adipose tissue inflammation and hepatic steatosis suggests a causal link, which may plausibly be mediated through adipose tissue inflammation-induced increased plasma concentrations of FFA and hypoadiponectinemia, as outlined earlier.
Table 3.
Targeted disruption of molecular targets involved in inflammation and its impact on insulin sensitivity, liver fat content, and inflammation in adipose tissue and liver in mice. Models were included if the transgenic mouse model did not have an adiposity phenotype.
| Target | Method | Diet (%Fat) Duration | Insulin Sensitivity | Adipose Inflammation | Liver Fat | Liver Inflammation | Ref |
|---|---|---|---|---|---|---|---|
| BLT-1 | WBKO | HFD (60%) 12 weeks |
↑ | ↓ | ↓ | ↓ | (435) |
| Cadherin-11 | WBKO | HFD (60%) 5–12 weeks |
↑ | ↓ | ↓ | n/a | (70) |
| Cap (Sorbs1) | WBKO | HFD (40% by wt) 6–8 weeks |
↑ | ↓ | ↓ | n/a | (271) |
| Catalase | WBKO | ND (18%) 21 weeks HFD (60%) 21 weeks |
↔ ↓ |
↑ (↑) |
n/a n/a |
n/a n/a |
(369) |
| CCL2 | AT Overexpression | ND (6% by wt) 8 weeks HFD (32% by wt) 8 weeks |
↓ ↓ |
↑
↔ ↑ |
↔ ↔ |
n/a n/a |
(218) |
| CCL2 | AT
Overexpression WBKO |
ND (n/a) 11 weeks HFD (56%) 12 weeks |
↓ ↑ |
↑ ↓ |
↑ ↓ |
n/a n/a |
(221) |
| CCR2 | WBKO | HFD (60%) 24 weeks |
↑ | ↓ | ↓ | n/a | (503) |
| CCR2 | Hematopoietic KO by BMT | HFD (60%) 20 weeks |
↔ | ↓ | n/a | (↓) | (159) |
| CD1d (iNKT) | WBKO | HFD (44%) 26 weeks |
↔ | ↔ | ↔ | ↔ | (289) |
| CD1d (iNKT) | WBKO | ND (10%), 19 weeks HFD (45%) 19 weeks |
↓ ↓ |
↔ ↑ ↔ |
↔ ↓ |
↔ ↑ |
(415) |
| CD1d (iNKT) | WBKO | HFD (60%) 4 days |
↓ | (↓) | n/a | ↔ | (207) |
| CD8 | Pharmacological inhibition | HFD (60%) 8 weeks |
↑ | ↓ | n/a | n/a | (345) |
| CD11c | BMT followed by selective depletion | HFD (60%) 16 weeks |
↑ | ↓ | ↓ | ↔ ↓ | (374) |
| CD18 | WBDNKI | HFD (45%) 20 weeks |
↓ | ↔ (↑) | n/a | ↑ | (303) |
| CD36 | Hematopoietic KO by BMT | HFD (42%) 20 weeks |
↔ | ↓ | n/a | n/a | (342) |
| CD95 (Fas) | Adipose KO | HFD (58%) 6 weeks |
↑ | ↓ | ↓ | (↓) | (520) |
| CXCR3 | WBKO | HFD (60%) 20 weeks |
↑ | ↓ | ↔ | n/a | (96) |
| CTRP7 | WBKO | HFD (60%) 12 weeks |
↔ | ↑ | ↔ | n/a | (376) |
| C5aR | WBKO | HFD (60%) 16–19 weeks |
(↑) | ↓ | ↔ | ↔ | (378) |
| Dab2 | Myeloid KO | HFD (60%) 12 weeks |
↑ | ↔ ↑ | n/a | n/a | (5) |
| FABP4 and FABP5 | WBKO BMT | HFD (60%) 15 weeks |
↑ | ↔ ↓ | (↔) | ↔ | (136) |
| Fetuin-A | WBKO | HFD (65%) 12 weeks |
↑ | ↓ | n/a | n/a | (361) |
| GPR120 | WBKO | ND (14%) 15–20 weeks HFD (60%) 15 weeks |
↓ ↔ |
↑ ↔ |
↔ ↔ |
↔ ↔ |
(354) |
| HIF-1α | Adipose KO | HFD (60%) 10 weeks |
↑ | ↓ | ↓ | ↓ | (267) |
| HIF-2α | WBKO (+/−) | HFD (60%) 16 weeks |
↓ | ↑ | n/a | n/a | (76) |
| Id3 | Lymphoid KO | HFD (60%) 12 weeks |
↔ | ↔ ↓ | n/a | n/a | (170) |
| Immunoglobulin μ heavy-chain | WBKO (B cell depletion) |
HFD (60%) 16 weeks |
↑ | ↔ ↓ | n/a | n/a | (511) |
| IR | Myeloid KO | HFD (55%) 12 weeks |
↑ | ↓ | n/a | ↔ | (297) |
| IFNγ | WBKO | HFD (60%) 13 weeks |
↑ | ↔ | n/a | ↓ | (350) |
| IL-10 | BMT WBKO | HFD (56%) 12 weeks |
↔ | ↔ | n/a | ↔ | (251) |
| Jα18 (iNKT) | WBKO | HFD (60%) 12–20 weeks |
↑ | ↓ | ↓ | ↓ | (519) |
| Jα18 (iNKT) | WBKO | HFD (32%) 18 weeks |
↔ | ↔ | ↔ | ↔ | (409) |
| JNK1 | Hematopoietic KO by BMT | HFD (60%) 20 weeks |
↑ | ↓ | ↔ | ↓ | (430) |
| JNK1 | Hematopoietic KO by BMT | HFD (60%) 26–36 weeks |
↑ | ↔ (↓) | ↔ | ↔ | (481) |
| JNK1 | Adipose KO | HFD (60%) 16 weeks |
↑ | ↔ ↓ | ↓ | ↔ | (403) |
| JNK1 | Muscle KO | HFD (60%) 16 weeks |
↑ | ↑ | ↑ | (↑) | (404) |
| JNK1 | Myeloid KO | HFD (60%) 4 weeks |
↑ | ↓ | n/a | ↓ | (165) |
| NAMPT | Adipose KO | ND (13%) 12–20 weeks |
↓ | ↑ | ↑ | ↔ | (446) |
| NLRP3 | WBKO | HFD (60%) 6 weeks |
↑ | ↓ | ↓ | n/a | (486) |
| Osteopontin | WBKO | HFD (60%) 25 weeks |
↑ | ↓ | n/a | n/a | (346) |
| Osteopontin | Mostly visceral ATM in ob/ob mice | ND (n/a) 5–7 weeks |
↑ | ↔ (↓ osteopontin in visceral ATM) |
n/a | ↔ | (22) |
| PDCD4 | WBKO | ND (n/a) 24 weeks |
↔ | ↔ | ↔ | ↔ | (500) |
| SR-A | WBKO | HFD (58%) 16 weeks |
↓ | ↔ | n/a | n/a | (552) |
| Sema3e | WBKO Overexpression |
HFHS (n/a) 8 weeks ND (n/a) 8 weeks |
↑ ↓ |
↓ ↑ |
n/a n/a |
n/a ↑ |
(425) |
| SOCS1 | Myeloid KO | ND (n/a) 12–15 weeks |
↓ | ↑ | n/a | ↑ | (405) |
| TNFα | Mostly visceral ATM in ob/ob mice | ND (n/a) 5–7 weeks |
↑ | ↔ (↓TNFα in visceral ATM) |
↔ | ↔ | (22) |
| TLR2 | WBKO | HFD (58%) 20 weeks |
↑ | ↓ | ↓ | ↓ | (104) |
| TLR4 | WBKO | HFD (60%) 16 weeks |
↑ | ↔ (↓) | n/a | n/a | (448) |
| TLR4 | WBKO | HFD (42%) 22 weeks |
↑ | ↓ | ↓ | (↓) | (380) |
| TLR4 | Hematopoietic KO by BMT | HFD (n/a) 16 weeks |
↑ | ↓ | ↓ | ↓ | (402) |
| TLR4 | WBKO | HFD (60%) 12 weeks |
↑ | ↔ | ↔ | n/a | (502) |
| TLR4 | WBKO | HFD (65%) 12 weeks |
↑ | ↓ | n/a | n/a | (361) |
| TLR4 | Liver KO | HFD (42%) 12 weeks |
↑ | ↓ | ↓ | ↓ | (208) |
| TLR9 | WBKO | HFD (60%) 12 weeks |
↑ | ↓ | n/a | n/a | (344) |
Abbreviations: AT, adipose tissue; ATM, adipose tissue macrophage; BLT-1, Leukotrin B4 receptor-1; BMT, bone marrow transfer; Cap, Cbl-associated protein (Sorbs1); C5aR, complement anaphylatoxin C5a receptor; CCL2, C-C motif chemokine ligand 2 (MCP1); CCR2, C-C motif chemokine receptor type 2; CD, cluster of differentiation; CXCR3, C-X-C motif chemokine receptor 3; CTRP7, C1q/TNF-related protein 7; Dab2, disabled homolog 2; FABP, fatty acid-binding proteins; GPR120, G protein-coupled receptor 120; GH-R, growth hormone-receptor; HFD, high-fat diet; HFHS, high-fat high-sucrose diet; HIF, hypoxia-inducible factor; iNKT, invariant natural killer T cell; IR, insulin receptor; IFNγ, interferon-γ; IL, interleukin; JNK1, c-Jun NH2-terminal kinase-1; KO, knockout; NAMPT, nicotinamide phosphoribosyl-transferase; ND, normal diet (chow); NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing 3; PDCD4, programmed cell death-4; Sema3e, Semaphorin E; SOCS1, suppressor of cytokine signaling-1; SR-A, class A scavenger receptor; TLR, toll-like receptor; WBKO, whole body knockout; WBDNKI, whole body dominant negative knock-in
Several notable exceptions, however, help disentangle the relative importance of adipose tissue inflammation vs. inflammation in other tissues in the development of insulin resistance and glucose intolerance. In one study, Aouadi and colleagues successfully employed small interfering RNA (siRNA) to selectively silence the expression of TNFα and osteopontin in ATMs (22), which led to improved insulin sensitivity and glucose tolerance. Another particularly informative study is based on a KO of fat-specific protein 27 (FSP27). Mice with this genotype exhibit reduced overall adiposity, reduced adipose tissue inflammation, and increased adiponectin concentrations when crossed into the Lepob/ob background or fed a HFD (551), a phenotype that one would expect to be associated with improved insulin sensitivity compared to wildtype. However, insulin sensitivity is reduced in this phenotype, possibly related to increased inflammation and fat content in the liver (551).This rare example of a dissociation between adipose tissue inflammation and insulin resistance illustrates the importance of inflammation and/or fat accumulation in the liver (and potentially other metabolically active tissues that were not considered in this paper) (551). Another study that provided insights into the importance of adipose tissue inflammation in the etiology of insulin restance included longitudinal analyses of adipose tissue inflammation and insulin resistance in wildtype and immunocompromised mice consuming a HFD (268). In wild-type mice, just three days of HFD feeding induced insulin resistance and upregulated pro-inflammatory mediators in adipose tissue. Over time, as the animals continued to gain weight, adipose tissue inflammation became more pronounced and insulin resistance worsened, consistent with a role for adipose tissue inflammation in the etiology of insulin resistance. Three models of immunocompromised models were used in this study, lymphocyte-deficient Rag (recombination activation gene)-1 knockout mice, mice treated with clodronate which depletes phagocytic cells, including macrophages and Kupffer cells, and mice treated with gadolinium, an inhibitor of Kupffer cells. With short-term (up to one week) HFD feeding, these immunocompromised models exhibited reduced glucose tolerance, as in the wildtype mice, indicating that inflammation may not play a major role in mediating the initial detrimental effects of a HFD on glucose tolerance and insulin sensitivity. Hepatic and skeletal muscle accumulation of FFA, diacylglycerol, and ceramide after three days of HFD feeding were not different between wildtype and clodronate treated mice, suggesting ectopic fat accumulation may be a potential mediator of early insulin resistance in response to short-term HFD feeding. As in the FSP27 KO mouse, the development of systemic insulin resistance in response to a HFD in the absence of the normal adipose tissue inflammation response supports the multi-factorial etiology of insulin resistance in the context of diet-induced obesity. However, these models did show some protection against impaired glucose tolerance under longer-term (14 weeks) HFD diet feeding conditions or in genetically obese Leprdb/db mice, consistent with the majority of the other transgenic mouse models shown in Table 3.
The relationship between lipid metabolism, immune cell function, and adipose tissue inflammation has been demonstrated through mouse models of genetic manipulation of lipid metabolism and transport enzymes. Adipose tissue-specific DGAT1 overexpression was associated with increased adiposity but improved metabolic phenotypes including improved insulin sensitivity and glucose tolerance and reduced adipose tissue inflammation and hepatic fat accumulation in HFD-fed mice (242). Macrophage-specific DGAT1 overexpression was sufficient to replicate the improvements in glucose tolerance, insulin sensitivity, and adipose tissue inflammation, possibly explained by the increase in lipid storage capacity of DGAT1 overexpressing macrophages. Surprisingly, these improvements occurred despite increased hepatic and skeletal muscle TG accumulation (242). Hematopoietic-specific loss of CD36 was associated with reduced adipose tissue inflammation and improved adipose tissue insulin signaling but without improvements in systemic insulin sensitivity (342). An adipose tissue-specific deletion of HSL was associated with lipodystrophic phenotypes including reduced adiposity, adipose tissue dysfunction, and impaired insulin sensitivity demonstrating that TG lipolysis is essential for maintenance of whole-body metabolic regulation (521). This is consistent with observations of humans with HSL deletion mutations, which are associated with impaired glucose homeostasis, with a redistribution of adipose tissue to visceral depots, adipose tissue inflammation, and an increased risk for T2DM (12).
Taken together, the data generated from these models are generally consistent with the hypothesis that adipose tissue inflammation is associated with insulin resistance, and that disruption of pro-inflammatory pathways results in reduced levels of adipose tissue inflammation and improved insulin sensitivity. Because other metabolic benefits such as reduced steatosis and inflammation in the liver are commonly associated with reduced levels of adipose tissue inflammation, it is difficult to isolate the contribution of adipose tissue inflammation to insulin resistance from these models. Models in which inflammation was selectively silenced in adipose tissue and exhibited improved insulin sensitivity demonstrate that adipose tissue inflammation is undoubtedly a major contributing factor in the etiology of insulin resistance. On the other hand, the few rare models in which adipose tissue inflammation is dissociated from insulin resistance make it clear that adipose tissue inflammation is almost certainly just one of several factors involved in the etiology of insulin resistance.
Adipose tissue inflammation and insulin resistance: human studies
How, then, should we apply the body of literature generated from rodent models to shape our understanding of the disease process in humans? Long-term overfeeding studies such as those carried out in rodents are obviously not feasible to the same extent in humans. Therefore, in humans, we must largely rely on observational evidence. In these observational studies, ‘adipose tissue inflammation’ is typically characterized by flow cytometry, immunohistological staining for markers of specific types or subtypes of leukocytes such as CD3 for T-cells or CD68 for macrophages, and/or quantification of transcript levels of genes encoding specific components of leukocytes or inflammatory mediators.
The positive association between plasma biomarkers of inflammation (CRP and IL-6) and insulin resistance was first observed in humans in the 1990’s. Yudkin et al. (542) noted that the secretion of CRP by the liver was regulated by circulating IL-6, which had been shown in vivo to be released from SAT (321). In a cross-sectional analysis, plasma CRP was significantly correlated with IL-6, and those subjects with CRP <1.35 μg/mL were significantly more insulin sensitive than those with elevated plasma CRP (542). While the authors did not adjust for confounders such as age or adiposity, these early observations suggested that inflammation was associated with insulin resistance. However, differences in the relationship between markers of inflammation and insulin sensitivity across different ethnic groups may occur. For example, in a study of healthy, overweight, premenopausal African American and Caucasian women matched for BMI, circulating CRP was independently and inversely associated with insulin sensitivity in the Caucasian women only, after adjustment for VAT (198). Insulin sensitivity was lower among the African American women independent of obesity, fat distribution, and inflammation (198).
Perhaps the strongest evidence of a relationship between adipose tissue inflammation and insulin resistance in humans comes from Kloeting et al. who have published using data from a repository of over 220 fat donors for whom hyperinsulinemic-euglycemic clamp data were also available (Table 4). In these morbidly obese individuals, the strongest predictor of insulin resistance was the extent of inflammation in VAT (238). In one of their studies, subjects were separated into insulin sensitive vs. insulin resistant groups and then age, sex, and BMI-matched to provide 30 pairs of subjects differing by insulin sensitivity (Figure 6). While there were no significant differences in SAT morphology between groups, VAT differed substantially. Tissue from insulin resistant subjects contained adipocytes of larger diameter and significantly lower insulin-stimulated glucose uptake as compared to that from insulin sensitive individuals. Independent of age, BMI, and whole body fat mass, VAT area was strongly, inversely associated with insulin sensitivity (238). Additionally, macrophage infiltration into omental adipose tissue was strongly inversely associated with insulin sensitivity, with no difference in macrophage infiltration into SAT between insulin sensitive and insulin resistant individuals (238). One of the most striking findings from this study was that a model including % macrophage infiltration into VAT and fasting plasma adiponectin concentrations almost perfectly predicted insulin sensitivity (GIR from hyperinsulinemic euglycemic clamp) (r2=0.98, p<0.0001) (238). Inclusion of liver fat content data (by magnetic resonance imaging) did not further improve the model (238).
Table 4.
Relationship between AT inflammation and insulin resistance in humans: evidence from observational studies.
| Subjects | Assessment method | Groups | Covariates | Outcome | Reference |
|---|---|---|---|---|---|
| n=77 (72F) 44±10 yrs 32–78 kg/m2 |
HOMA Gene expression: CD68, TNFA IHC: CD68 |
CLS- (n=27) CLS+ (n=50) |
None | ATM number was significantly higher in CLS+ group and correlated with HOMA and fasting insulin. CLS+ subjects had significantly higher HOMA and SAT expression of CD68 and TNFA mRNA compared to CLS- subjects. | (23) |
| n=33F 35–65 yrs 27–35 kg/m2 |
HOMA, insulin suppression test Gene expression: CD14, CD45, CD68, EMR1, IL6, IL8, INOS, CCL2, TNFA IHC (n=16): CD45, CD68 |
IS (n=19) IR (n=14) |
None | SAT expression of CD68, IL8, IL6, EMR1, and CCL2 mRNA and CD45+ cells was significantly greater in IR compared to IS tissue. | (300) |
| n=66 (25F) 31±8 yrs 34±7 kg/m2 |
Hyperinsulinemic euglycemic clamp Gene expression: CD68, CD11B, CSF1R, ICAM1, CCL2, PAI1, HIF1, VEGF, TNFA, CD11C IHC: CD68 |
None | Age, adiposity, sex | ATM number and SAT expression of CD68, CD11B, and CSF1R mRNA were not correlated with insulin sensitivity after adjustment for age, adiposity, and sex. SAT expression of CD11C and PAI1 mRNA were negatively correlated with insulin sensitivity after adjustment. | (357) |
| n=29F 20–61 yrs |
HOMA IHC: CD11c, CD68, CD206 Flow cytometry: CD11c, CD14, CD206 |
Formerly obese (n=5) obese (n=12) obese with metabolic syndrome (n=12) |
None | ATM (CD14+) in CLS were positive for CD11c, CD11c+ ATM density was positively correlated with insulin resistance. | (508) |
| n=13 (6F) 35–65 yrs 35.1–50.8 kg/m2 |
HOMA Flow cytometry: CD14, CD11b |
None | None | ATM (CD14+) number did not relate to measures of adiposity or insulin resistance in either SAT or VAT depot. | (492) |
| n=60 (40F) 41–49 yrs 42.8–47.8 kg/m2 |
Hyperinsulinemic euglycemic clamp Gene expression: CD68, ADIPOQ, IL6, IL8, CCL2, MIF, TRAP, CSF1 IHC: CD68 |
IS (n=30) IR (n=30) |
Age, BMI, adiposity, sex, liver fat content | IR subjects had a significantly greater number of ATM and expression of CD68 mRNA in VAT compared to age-, fat mass-, BMI-, and sex-matched IS subjects. SAT and VAT expression of ADIPOQ, SIRT1, and TRAP mRNA was significantly lower--while expression of HIF1, MIF, and CSF1 mRNA was significantly greater--in IR vs. IS subjects. | (238) |
| n=20 (14F) 31–57 yrs 39–60 kg/m2 |
HOMA Gene expression: microarray IHC: CD68 |
IS (n=10) IR (n=10) |
BMI | ATM infiltration into VAT was greater in IR compared to BMI-matched IS subjects and significantly correlated with HOMA, chemokines and chemokine receptor binding genes were upregulated in VAT of IR compared to IS subjects. | (167) |
| n=92 (79F) 42±11 yrs 44±10 kg/m2 |
HOMA IHC: CD68 |
CLS- (n=20) CLS+ (n=72) |
Age, BMI, WC, T2DM status | HOMA was positively associated with presence of CLS in SAT and VAT depots after adjustment for age, WC, BMI, and T2DM. | (40) |
| n=34 (29F) 26–57 yrs 20.8–52.8 kg/m2 |
HOMA, hyperinsulinemic euglycemic
clamp Gene expression: CD4, CCL5, IL7 |
Lean (n=9) IS-obese (n=12) IR-obese (n=13) |
None | SAT expression of CD4, CCL5, and IL7 mRNA increased progressively from lean, to IS-obese, to IR-obese. | (115) |
| n=171 (67F) 0–18 yrs |
HOMA Gene expression: CD68, TNFA, IL6 IHC: CD68 |
Lean children (n=106) obese children (n=65) |
None | HOMA was positively associated with ATM and the presence of CLS in SAT. There was no association between HOMA and SAT expression of TNFA or IL6 mRNA. | (259) |
Abbreviations: ADIPOQ, adiponectin; ATM, adipose tissue macrophage; BMI, body mass index; CCL2 (MCP-1), C-C motif chemokine ligand 2; CLS, crown-like structure; CSF(R), colony stimulating factor-1 (receptor); EMR1 (F4/80), EGF-like module-containing mucin-like hormone receptor-like 1; HOMA, homeostatic model assessment; IL, interleukin; IR/IS, insulin-resistant/-sensitive; MIF, macrophage migration inhibition factor; PAI1, plasminogen activator inhibitor-1; SAT, subcutaneous adipose tissue; SIRT1, sirtuin-1; TNFA, tumor necrosis factor alpha; TRAP, tartrate-resistant acid phosphatase; T2DM, type 2 diabetes mellitus; VAT, visceral adipose tissue; WC, waist circumference
Hardy et al. (167) observed a similar relationship in their study of obese insulin-resistant patients (HOMA2-IR > 2.4) vs. insulin-sensitive patients (HOMA2-IR < 2.4) of equivalent BMI prior to bariatric surgery. VAT of insulin resistant subjects contained adipoctyes of significantly larger diameter, with greater macrophage infiltration and greater expression of pro-inflammatory mediators compared to insulin sensitive patients, while there was no difference in these variables in SAT between the groups. Insulin resistance, but not BMI, was associated with adipocyte size and degree of macrophage infiltration into VAT. Evidence from these two studies suggest that inflammation of VAT specifically – independent of BMI and possibly independent of ectopic fat content – may play a key role in the etiology of insulin resistance.
In a population of healthy Pima Indians, SAT macrophage content and expression of macrophage-specific genes (CD68, CD11b, and CSF1R) was correlated with age and adiposity, but not with insulin sensitivity independent of adiposity (357). This is consistent with Kloeting et al.’s finding that inflammation of VAT as opposed to SAT is a more critical factor (238). These data suggest that the quantity of macrophages in SAT is not a direct contributor to the etiology of systemic insulin resistance. However, in contrast to the findings of Kloeting et al. (238) and Hardy et al. (167), Viardot et al. observed no correlation between SAT and VAT macrophage numbers and insulin resistance (HOMA-IR) in obese subjects undergoing bariatric surgery (492).
Based on the finding by Lumeng and colleagues that pro-inflammatory macrophages in obese mouse adipose tissue express CD11c (280) some investigators have used CD11c in an attempt to identify pro-inflammatory ATM in humans. For example, Wentworth et al. found that ATMs present in CLS stained positive for CD11c (508). In their study, CD11c+ ATM also expressed IL-1β, IL-6, IL-8, TNFα, and CCL-3, consistent with the idea that these macrophages may be a pro-inflammatory subtype. Interestingly, and contrary to findings by Kloeting et al. (238), the number of CD11c+ ATM was greater in SAT vs. VAT, and correlated with HOMA-IR (508). But overall, the evidence is not particularly strong that CD11c+ ATM in humans (or mice) constitute a phenotypically or functionally homogenous subtype of ATM that is fundamentally different from CD11c- ATM. One factor impeding progress in this area has been that no conclusive data exist as to which cell surface markers should be consistently used to specify pro-inflammatory ATM populations in humans.
Other than the study by Kloeting and colleagues (238), none of these studies measured ectopic fat content. While overall, the studies have relatively consistently reported associations between insulin resistance and different measures of adipose tissue inflammation, particularly the macrophage content of VAT, questions remain if adipose tissue inflammation would consistently remain a significant contributor to insulin resistance if adiposity, body fat distribution, and ectopic fat content were experimentally or statistically controlled for. The Kloeting et al. data showing that VAT macrophage infiltration, together with plasma adiponectin concentrations, was almost perfectly correlated with a gold-standard measure of insulin sensitivity, independent of liver fat content or any other measure of adiposity, is certainly intriguing (238). It is also not entirely clear why inflammation and macrophage accumulation in VAT tends to be more strongly associated with insulin resistance than inflammation in SAT. One hypothesis is that VAT has a more direct access to the liver through the portal vein, thereby more directly exposing the liver to inflammatory mediators or FFA from inflamed VAT (401).
One line of work that raises some doubts about the relative importance of adipose tissue inflammation in the etiology of insulin resistance and T2DM are genome-wide association studies (GWAS), which have not found associations between genetic variants in key mediators of inflammation and incidence T2DM [reviewed in (239)]. Instead, genetic analyses have suggested that limited storage capacity of peripheral subcutaneous adipose tissue is an important etiological contributor to insulin resistance (279). An intriguing hypothesis in this regard is the personal fat threshold hypothesis (461). This hypothesis proposes that interindividual differences in the storage capacity of adipose tissue, particularly SAT, determine the degree to which excess energy can be safely stored in adipose tissue, beyond which it would be deposited in ectopic depots, leading to metabolic dysfunction. The basis for the personal fat threshold hypothesis is the often-overlooked fact that T2DM affects numerous non-obese and even normal weight individuals, while many obese individuals do not develop T2DM. While to some degree genetic differences in pancreatic β-cell function may account for the fact that some individuals manifest glucose intolerance at a lowered BMI, this by itself is unlikely to explain the substantial heterogeneity in BMI and even body fat mass in people with T2DM. The work of Taylor and colleagues showing that weight loss through a very-low calorie diet can restore normal glucose tolerance in most individuals with T2DM (441, 442) is consistent with the hypothesis that absolute fat mass is less relevant than whether an individual personal fat threshold has been crossed. Once this threshold is passed, an individual is unable to safely store excess calories in SAT and storage of lipids in liver, muscle, and pancreas occurs, impairing the ability of these organs to maintain whole-body glucose homeostasis. At the same time, individual limitations in the storage capacity of SAT would be expected to trigger expansion and inflammation of VAT. Thus, another potential explanation for the strong association between macrophage accumulation and inflammation in VAT may be that the expansion and subsequent inflammation of VAT are a direct consequence of, and indicator for, limited storage capacity of peripheral SAT. It is possible that excessive ECM in the development of fibrosis in SAT, which may limit SAT expansion, or factors that inhibit adipogenesis itself, such as the accumulation of senescent preadipocytes, may at least partially mediate inter-individual differences in adipose tissue storage capacity.
Taken together, even though numerous lines of evidence link adipose tissue inflammation to insulin resistance and glucose intolerance, more data on the complex interrelationship between adiposity, fat distribution, adipose tissue inflammation, and ectopic fat deposition is needed to confidently conclude that adipose tissue inflammation is a major independent contributor in the development of insulin resistance. Doubts remain particularly because GWAS fail to link genetic variation in key mediators of inflammation to insulin resistance or T2DM, as outlined above, and because of conflicting data generated in the context of weight loss studies, as will be discussed in the next section.
Adipose tissue inflammation following bariatric surgery- or dietary caloric restriction-induced weight loss
Because obesity is strongly associated with insulin resistance, weight loss interventions offer an opportunity to determine whether adipose tissue inflammation and insulin resistance might be causally linked. If so, a caloric deficit should lead to reductions in inflammation with concomitant metabolic improvements. Dietary restriction-based weight loss studies have been inconsistent in the amount of weight loss and improvement in insulin sensitivity achieved. In contrast, surgical weight loss interventions have demonstrated unique efficacy in inducing rapid, massive weight loss, along with marked improvements in metabolic health and reductions in systemic inflammation (67, 181, 199–202, 325, 368, 468, 489, 491, 527) regardless of the type of bariatric surgery (gastric banding, vertical sleeve gastrectomy, or Roux-en-Y gastric bypass). Notably, metabolic improvements following surgery typically appear within days, and well before significant weight loss, which has prompted investigation into weight loss-independent mechanisms that might be driving the improvements in glucose homeostasis. A reduction in adipose tissue inflammation following surgery, but prior to weight loss, has emerged as a leading hypothesis that might explain this phenomenon. We have reviewed studies in this area that included surgery or diet-based interventions designed to reduce or increase body weight and that included assessments of both insulin sensitivity and adipose tissue inflammation (Table 5).
Table 5.
Effects of weight loss or gain on AT inflammation and insulin sensitivity in humans: evidence from surgical and dietary intervention studies.
| Baseline characteristics | Intervention | Assessment of AT inflammation | Assessment of insulin sensitivity | Measures of ectopic fat deposition | Outcome | Reference |
|---|---|---|---|---|---|---|
| n=17F 29–59 years 33–57 kg/m2 |
RYGB | SAT sample obtained at surgery and 3-months
post surgery. Gene expression: MCP1, CSF3, HIF1A, PLAUR IHC: HAM56, CD68 |
QUICKI | None | Three months following surgery subjects exhibited mean weight loss of 22.1 kg (17%), significant improvements in insulin sensitivity, and reductions in CD68+HAM56+ ATM in SAT. Expression of MCP1, CSF3, HIF1A, and PLAUR mRNA in SAT post-surgery was significantly lower compared to pre-surgery values. No correlation was observed between change in insulin sensitivity and change in ATM number in SAT. | (56) |
| n=50F 21–49 years 36.2±0.7 kg/m2 |
10-week energy restriction: ~70% of ETEE as HFLCD (n=25) vs. LFHCD (n=25) | SAT biopsy at baseline and 10
weeks. Gene expression: 38 genes (TNFA, ADIPOQ, IL6, PAI1) |
QUICKI | None | Subjects lost ~6.8% body weight on either diet and insulin sensitivity improved significantly. SAT expression of TNFA, ADIPOQ, IL6, and PAI1 mRNA did not change significantly with weight loss or by diet group. | (495) |
| n=22F premenopausal 35.3±1.0 kg/m2 |
Successive: 1-month VLCD (800 kcal/d), 2-month LCD (600 kcal/d deficit from ETEE), 3–4 month weight maintenance period | SAT biopsy at baseline and 1-, 2-, and 4-mths. Gene expression: 31 macrophage-specific markers | Hyperinsulinemic-euglycemic clamp | None | Insulin sensitivity increased significantly during the calorie restriction and weight maintenance phases compared to baseline concurrent with a mean weight loss of 10.2%. Expression of macrophage-specific genes was upregulated during the energy restriction phase and downregulated during weight stabilization. | (59) |
| n=16F 41.1±8.6 years 43.8±3.4 kg/m2 |
RYGB | SAT obtained at surgery and 3-mths post.
IHC: CD40, CD163, CD206, HAM56 |
QUICKI | None | At 3 months post-surgery, subjects had lost a mean 15% body weight and insulin sensitivity improved significantly. Number of HAM56+ and CD40+ cells decreased while CD206+ and CD163+ cells increased, and there was a 2-fold reduction of CD40 to CD206 ratio. | (26) |
| n=13 (6F) 35–65 years 35.1–50.8 kg/m2 |
24 week energy restriction (first 12 weeks VLCD, followed by gastric banding) | SAT sample and whole blood obtained at
baseline, 12 weeks, and 24 weeks. Flow cytometry: CD14, CD11b, CD66b, CD69, IFNγ, IL4 |
HOMA | None | At 12 weeks weight loss averaged 5%, and 13.5% by 24 weeks. HOMA showed a downward trend but changes were not significant at either time point. In SAT, macrophage number was significantly reduced after VLCD. No relationship was found between changes in immune cell activation, SAT ATM number, and improvements in HOMA. | (492) |
| n=27F 39±2 yrs 33.7±0.5 kg/m2 |
28-d VLCD (800 kcal/d), followed by 2 months of weight stabilization, then 3–4 months of weight maintenance | SAT biopsy at baseline, after calorie
restriction, and following weight stabilization. Gene expression: CD14, CD68, CD163, LYVE1 Flow cytometry: CD14, CD16, CD206 |
HOMA | None | VLCD led to significant weight loss (~10%), and significant improvements in HOMA. However, reductions in CD14+CD206+ ATM number and SAT expression of CD14, CD68, CD163 and LYVE1 mRNA were not detected until after the weight stabilization period that followed. | (250) |
| n=44M 18–55 years 18–30 kg/m2 |
56-d overfeeding (+760 kcal/d) | SAT biopsy at baseline, day 14, and day 56.
IHC (n=15): CD68 |
HOMA | None | Subjects consumed an additional 700 kcal daily and gained 3% of baseline body weight after the 56-d intervention. HOMA increased significantly. However, there was no significant change in ATM number in SAT. | (15) |
| n=34 (24F) 21–61 years 48.8±0.9 kg/m2 |
RYGB | SAT obtained at surgery, month 6, and month 12 post-surgery. Gene expression: CRP, IL6, TNFA | HOMA | None | Subjects lost 28% and 37% of body weight by months 6 and 12, respectively. HOMA decreased significantly compared to baseline by month 6 and was further reduced by month 12. No significant changes in SAT expression of IL6 or TNFA mRNA were detected at either 6 or 12 months post-surgery. In contrast, SAT expression of CRP mRNA decreased significantly 6 months after surgery and declined further at 12 months post-surgery. | (368) |
| n=23 (12F) 25–50 years 27.7±1.7 kg/m2 |
6-mo energy restriction (75% of ETEE) (n=12) vs. control (weight maintenance) (n=11) | SAT biopsy at baseline and 24 wks. Gene expression: TNFA, IL6, CD68, MIF, MCP1, PAI1, ADIPOQ |
FS-IVGTT | None | Subjects in CR group lost ~10% body weight and insulin sensitivity tended to improve. SAT expression of IL6, TNFA, CD68, MIF, MCP1, PAI1, and ADIPOQ mRNA did not change significantly with weight loss and did not differ by diet group. | (456) |
| n=13F 40.8±8.4 years 42.3±4.1 kg/m2 |
VSG | SAT biopsy at baseline (pre-surgery), month 6 (n=11), month 12 (n=11), and month 24 (n=8) post-surgery. Gene expression: 45 inflammation-related genes | HOMA | None | Subjects’ mean BMI decreased by 21% and 26% at months 6 and 12 post-surgery, respectively and tended to increase again by month 24. HOMA did not change to a significant degree at any time point compared to pre-surgery values. At 6 months post-surgery, SAT expression of inflammatory gene mRNAs were largely unchanged compared to baseline. By 12 and 24 months, the downregulation of some inflammation-related genes was more pronounced suggesting that VSG reduced the expression profile of inflammatory cytokines and chemokines in SAT. | (468) |
| n=16 (11F) 46±5.8 years 58.9±9.6 kg/m2 |
Two-step bariatric surgery: baseline VSG, RYGB at 12 months | SAT and VAT samples obtained at both
surgeries. Gene expression: FAS, FASL |
HOMA | None | Following VSG, BMI decreased by 25% and insulin sensitivity improved significantly. VAT and SAT expression of FAS mRNA decreased significantly by 40% and 33%, respectively. | (46) |
| n=29M 26.8±5.4 years 25.5±2.3 kg/m2 |
8-week overfeeding: 140% ETEE | SAT biopsy at baseline and 8
weeks. Gene expression: CD68, IL6, CCL2, ADIPOQ, NFKB, VCAM1 IHC: CD68 |
Hyperinsulinemic-euglycemic clamp | Intrahepatic and intramyocellular lipid: MRS | Subjects gained 10% body weight, and intrahepatic lipid content increased significantly while intramyocellular lipid content did not change. Insulin sensitivity decreased significantly. However, there was no change in SAT expression of CD68, IL6, CCL2, ADIPOQ, NFKB, or VCAM1 mRNA with overfeeding, nor was there a change in the numbers of ATM or CLS observed in SAT. | (212, 455) |
| n=7 (5F) 52.6±6.2 years 38.9±4.9 kg/m2 |
RYGB | SAT biopsy at baseline and at 7% weight loss
(13±2 days post-surgery). Gene expression: TNFA, IL1B, IL6, MCP1, ICAM1, ADIPOQ Flow Cytometry: ATM (CD14+CD206+CD11c+), neutrophils (CD15+CD16+), DC (CD1c+CD11c+), and T cells (CD3+CD4+CD8+) |
HOMA | None | Subjects lost 7% of initial body weight within the first 2 weeks following surgery and exhibited significant improvement in insulin sensitivity. SAT expression of TNFA, IL6, MCP1, and ICAM1 mRNA did not differ from baseline. In contrast, expression of IL1B was elevated, and ADIPOQ reduced, both significantly. Numbers of neutrophils in SAT exhibited significant increases over baseline, while ATM, DC, and T cell-numbers trended upward. | (254) |
| n=40 44±12 years 37.9±4.4 kg/m2 |
Energy restriction to achieve 5% weight loss (n=10) vs. 10% and 15% progressive weight loss (n=10) vs. weight maintenance (n=20) | SAT biopsy at baseline and 6-months (for
weight maintenance) or 3-weeks post weight stabilization. Gene expression: TNFA, IL6, CCL3, RANTES, CD68, EMR1, microarray |
Hyperinsulinemic-euglycemic clamp with stable isotope-labeled tracer infusion | Intrahepatic TG content: MRS | 5% weight loss decreased intrahepatic TG content, improved beta-cell function and insulin sensitivity in liver, SAT, and muscle while SAT inflammation remained unchanged. 11–16% weight loss was associated with a reduction in SAT inflammation while intrahepatic TG content decreased in a significant and linear manner with progressive weight loss along with further improvements in beta-cell function and insulin sensitivity in muscle. | (286) |
| n=17F 35±7 years 32.6±3.6 kg/m2 |
28-d energy restriction (800 kcal/d) | SAT biopsy at baseline, day 2, and day 28.
Gene expression: 14 macrophage and cytokine genes |
HOMA, QUICKI | None | Subjects lost ~9% body weight after 28-d intervention, and insulin sensitivity improved significantly. SAT expression of macrophage markers CD163, MSR1, IRF5, and CCR2 mRNA was significantly increased by day 28 and expression of IL8, MCP1, TNFA, IL6, and IL10 mRNA tended to increase but changes were not significant. | (436) |
| N=55 (38F) 44.5±11.0 years 53.6±7.3 kg/m2 |
VSG | SAT, VAT, and liver biopsy at baseline and after 1 year | HOMA (n=55) Hypersinsulinemic euglycemic clamp (n=11) |
None (But reduction in inflammation markers in liver) |
VSG similarly reduced body weight, body fat mass, and HOMA significantly after 1 year, independent of whether the weight loss was associated with a reduction in inflammation markers or ATM in VAT. | (417) |
| n=17 46.2±7.5 kg/m2 |
RYGB (n=7) VSG (n=10) |
SAT biopsy at baseline, 1 month, and
6–12 months after surgery. Gene expression: TNFA, IL1B, IL6, ADIPOQ Flow Cytometry: ATM (CD14+CD206+CD11c+), neutrophils (CD15+CD16+), DC (CD1c+CD11c+), and T cells (CD3+CD4+CD8+) |
HOMA Matsuda-ISI based on FS-OGTT |
None | In the first month, subjects lost ~10% of initial body weight, and HOMA was significantly reduced, with no change in the Matsuda-ISI. SAT expression of TNFA, IL1B, IL6, and ADIPOQ mRNA did not change from baseline, nor were there changes in the numbers of ATM, DC, or CD4+ T cells. The number of neutrophils and CD8+ T cells were elevated at 1-month post-surgery. By 12 months, subjects lost 26% of initial body weight, with an increase in Matsuda-ISI. SAT expression of TNFA, IL1B, and IL6 mRNA did not differ from baseline, while ADIPOQ declined. Neutrophils, DC, and CD8+ T cells all consistently exhibited increased numbers in AT relative to baseline, while ATM and CD4+ T cells also increased. | (162) |
Abbreviations: ADIPOQ, adiponectin; ATM, adipose tissue macrophage; BMI, body mass index; CCL2, C-C motif chemokine ligand 2 (MCP1); CCL3, C-C motif chemokine ligand 3; CCR2, C-C motif chemokine receptor type 2; CD, cluster of differentiation; CRP, C-reactive protein; CSF3, colony-stimulating factor-3; DC, dendritic cells; EMR1, EGF-like module-containing mucin-like hormone receptor-like 1; ETEE, estimated total energy expenditure; FAS, death receptor CD95; FASL, Fas-ligand; FS-OGTT: frequently sampled oral glucose tolerance test; FS-IVGT, frequently sampled-intravenous glucose tolerance test; T HFLCD, high-fat low-carbohydrate diet; HAM56, human alveolar macrophage 56; HIF1A, hypoxia-inducible factor-1α; HOMA, homeostasis model assessment; ICAM1, intracellular adhesion molecule 1; IHC, immunohistochemistry; IFNγ, interferon-γ; IL, interleukin; IRF5, interferon regulatory factor 5; ISI: Insulin sensitivity index; LCD, low-calorie diet; LFHCD, low-fat high-carbohydrate diet; LYVE1, lymphatic vessel endothelial hyaluronan receptor-1; MCP1, monocyte chemotactic protein-1; MIF, macrophage migration inhibitory factor; MRS, magnetic resonance spectroscopy; MSR1, macrophage scavenger receptor 1; NFKB, nuclear factor-κB; PAI1, plasminogen activator inhibitor 1; PLAUR, plasminogen activator urokinase receptor; QUICKI, quantitative insulin sensitivity check index; RANTES, regulated on activation normal T cell expressed and secreted; RYGB, Roux-en-Y gastric bypass; SAT, subcutaneous adipose tissue; TG, triglyceride; TNFα, tumor necrosis factor-α; VCAM1, vascular cell adhesion molecule-1; VLCD, very-low-calorie diet; VSG, vertical sleeve gastrectomy; VAT, visceral adipose tissue
Following bariatric surgery, systemic inflammation appears to persist at least through the first month, as indicated by circulating CRP, IL-6, and PAI-1 concentrations at or near pre-surgery levels (162, 254, 310, 368). Clear reductions in CRP are more apparent three to four months post-operatively, when presumably surgery-related inflammation has subsided (52, 202, 488), even though this is not seen in all studies (310, 483). Six to 12 months post-surgery, CRP, IL-6, PAI-1, and MCP-1 consistently are reduced relative to pre-surgery levels (6, 28, 108, 157, 162, 181, 199–202, 254, 310, 323, 327, 368, 468, 483, 489). There may also be changes in serum levels of the insulin sensitizing hormone adiponectin following bariatric surgery. While Sams et al. (407) observed significant increases in circulating adiponectin two weeks following surgery, our studies revealed no improvement one to 12 months post-surgery (162, 254).
With respect to adipose tissue, the data are less consistent (Table 5). Several studies reported that weight loss between 5% and ~10% in response to a dietary restriction-based intervention (59, 250, 436, 456, 495) led to improvements in insulin sensitivity that were not associated with concurrent reductions in adipose tissue inflammation when follow-up measurements were taken immediately at the end of the weight loss period (i.e., when participants were still in a state of caloric deficit). This dissociation between insulin sensitivity and adipose tissue inflammation was even apparent in one study in which participants were kept weight stable for three weeks following 5% weight loss (286). In that study, insulin sensitivity (by clamp) was improved in response to weight loss, leading the authors to conclude that reductions in adipose tissue inflammation are not necessary for an increase in insulin sensitivity (286). Two other studies found that a weight stabilization period following active weight loss eventually led to reductions in measures of adipose tissue inflammation (59, 250), indicating that – aside from the active phase of caloric deficit and weight loss – adipose tissue inflammation will be reduced from a reduction in body fat mass. However, insulin sensitivity improved only during the period of active weight loss when adipose tissue inflammation was unchanged, while insulin sensitivity did not improve further when measures of adipose tissue inflammation were reduced after the weight stabilization period (59, 250). Greater weight loss of 11%−16%, however, was associated with a reduction in the expression of inflammation-related genes in adipose tissue, concurrent with improvements in insulin sensitivity and reduction in liver fat content (286).
In some studies of weight loss of 15% - 17% following bariatric surgery, improvements in insulin sensitivity tended to be concurrent with reductions in measures of adipose tissue inflammation (26, 56, 492), even though these measures showed no correlation with one another in two of these studies (56, 492). Other investigators found either no or only very minor changes in measures of adipose tissue inflammation following weight loss of 7% to 37% following bariatric surgery (162, 254, 368), in spite of sometimes substantial improvements in insulin sensitivity, again illustrating a dissociation between adipose tissue inflammation and insulin resistance.
It is a potential important limitation in all of these longitudinal weight loss studies that they were forced to rely on SAT, because VAT cannot feasibly be collected twice in an intervention study for before-and-after comparison. Because some authors, most notably Kloeting and colleagues as discussed above find stronger associations between insulin resistance and measures of adipose tissue inflammation in VAT vs. SAT, it is possible that potentially relevant effects of weight loss on VAT inflammation that may (partly) mediate improvements in insulin sensitivity were not detectable. The one exception in this regard is a particularly informative study by Schmitz and colleagues (417). These authors were able to obtain SAT, VAT, and liver biopsies during a vertical sleeve gastrectomy (VSG) surgery (baseline) from 55 morbidly obese individuals, and again one year later, following substantial weight loss, when subjects were undergoing Roux-en-Y gastric bypass surgery. They found that while VSG-induced weight loss was associated with a reduction in measures of adipose tissue inflammation and the number of ATM in VAT in most individuals, no changes in the number of ATM or inflammation markers in VAT were seen in 23 participants. Nevertheless, improvements in systemic insulin sensitivity (assessed by HOMA in all subjects, and confirmed by clamp in a subgroup) were seen in all participants, independent of whether VAT adipose tissue inflammation was reduced.
Another potential limitation of studies in this area is the persisting uncertainty about the specific functional and phenotypical characteristics of adipose tissue macrophages and other leukocyte populations. This impairs our ability to assess whether weight loss-related changes in the phenotype of adipose tissue leukocytes may contribute to improvements in insulin sensitivity.
Taken together, improvements in insulin sensitivity during periods of weight loss from either dietary restriction or bariatric surgery are commonly not or not consistently associated with reductions in measures of adipose tissue inflammation, thus illustrating that active weight loss is an example of dissociation between adipose tissue inflammation and insulin resistance.
Adipose tissue inflammation following overfeeding
To understand whether chronic, low-grade inflammation plays a causal role in insulin resistance in humans, Tam and colleagues overfed 36 healthy normal to overweight men and women by 1,250 kcal/day for four weeks to observe the relationship between obesity, inflammation, and insulin resistance (457) (Table 5). Despite a gain in body weight and fat mass of approximately 3.5% of baseline levels, and a reduction in insulin sensitivity that corresponded with elevated circulating CRP and MCP-1 concentrations, there were no measurable increases in the expression of pro-inflammatory gene expression (CD68, IL-6, CCL2, ADIPOQ, NF-κB, and VCAM) or evidence of increased CD68-positive macrophage infiltration into SAT (457). Similar results were seen by Johannsen et al., who overfed 29 men over eight weeks by ~40%, for an average weight gain of ~10% (212). While the weight gain was associated with a decline in insulin sensitivity, no change in a variety of measures of SAT inflammation was seen. Both of these overfeeding studies exposed relatively young and healthy individuals to a substantial caloric overfeeding paradigm, and saw body weight gain associated with reductions in insulin sensitivity, in spite of no change in measures of adipose tissue inflammation. Thus, these overfeeding studies constitute another example of dissociation between adipose tissue inflammation and insulin resistance.
Anti-inflammatory drugs: impact on adipose tissue inflammation and insulin resistance
Another potential way to obtain insight into the relative contribution of low-grade chronic inflammation to insulin resistance is to consider the response to anti-inflammatory drugs. Several naturally occurring and bioengineered compounds that reduce inflammation have been tested in clinical trials to determine their effect on insulin resistance. Salicylates are naturally occurring compounds that have been used since antiquity to treat pain, inflammation, and fever. Functionally, salicylates target cyclooxygenase (COX) 1 and 2, which regulate the production of pro-inflammatory prostaglandins (145, 277). Prostaglandins are powerful vasodilators that also contribute to inflammatory processes by prolonging and enhancing the effects of other pro-inflammatory mediators (261). At high doses, salicylates also interfere with IKKβ-dependent activation of the NF-κB pathway (539), which activates the inflammasome and the maturation and release of IL-1β (113, 264, 444). Acetylsalicylic acid (aspirin) was introduced in the late 19th century. Aspirin reduces blood glucose in patients with T2DM (390) but not in individuals with normal glucose tolerance (142, 337). More recently, several studies, including six randomized, blinded and placebo controlled trials, demonstrated marked improvements in glucose tolerance and reduced HbA1c with salsalate treatment among obese patients with and without T2DM (13, 117, 130, 143–145, 196). In addition, in five of the six trials, salsalate treatment reduced markers of systemic inflammation (CRP, IL-6, or CD40L) or increased adiponectin (13, 130, 143, 145, 196). However, none of these trials directly measured adipose tissue inflammation.
Another approach used to target inflammatory pathways utilizes bioengineered compounds. Studies in rodents using such compounds to selectively inhibit TNFα and IL-1β significantly reduced obesity-associated inflammation (139, 189, 299, 478, 479, 506). Based on the compelling results from the rodent-based studies, several anti-inflammatory drug trials have since been conducted in humans, the results of which have been less promising. For example, a four-week intervention using a single dose of either a TNFα-neutralizing antibody, CDP571, or the antagonist Ro 45–2081, a fusion protein of the soluble TNF-receptor linked to the Fc portion of human IgG1, did not improve insulin sensitivity (351, 366). Similarly, a four-week course of the TNFα antagonist etanercept, given twice weekly at 25 mg by subcutaneous injection, failed to improve insulin sensitivity relative to non-treated controls despite clear reductions in systemic CRP concentrations (99). Longer treatment of six months with 50 mg of etanercept in obese individuals without autoimmune or inflammatory conditions improved fasting glucose and circulating adiponectin compared to placebo, although insulin sensitivity did not change (437). However, in individuals with inflammatory joint disease, long-term use of the TNFα-inhibitor infliximab improved insulin sensitivity (353, 535, 536).
Selective targeting of IL-1β has produced more promising results from randomized controlled trials. In patients with T2DM, daily subcutaneous injections of 100 mg of the recombinant IL-1 receptor antagonist anakinra for 13 weeks did not alter insulin sensitivity but improved HbA1c and β-cell function, and reduced systemic inflammation when compared to placebo (260). Results from other studies that used human monoclonal neutralizing antibodies against IL-1β were inconsistent. For example, intravenous injections of gevokizumab reduced HbA1c, improved insulin sensitivity, and reduced circulating CRP (62). In contrast, in a large trial of adults with controlled T2DM, four different doses of canakinumab did not significantly alter fasting glucose, insulin, or HbA1c relative to placebo (392).
Statins are commonly prescribed drugs that lower cholesterol levels by inhibiting the rate-limiting enzyme of endogenous cholesterol synthesis. However, this is a pleiotropic class of drugs that also exerts several anti-inflammatory effects (43, 544). Despite these anti-inflammatory effects, several studies have now shown statin use to be associated with an increased risk for T2DM (89, 91, 381, 410). Furthermore, results from Cederberg et al. suggest that statins reduce insulin sensitivity and insulin secretion (63). On the other hand, thiazolidinediones (TZDs) are commonly prescribed antidiabetic insulin-sensitizing drugs that also exert anti-inflammatory effects (66). TZDs are potent PPARγ ligands and work in mice demonstrated that macrophage PPARγ activity is required to elicit their full capacity to increase insulin sensitivity and glucose tolerance (177). Adipocyte PPARγ and adipogenesis/lipid metabolism as one alternative pathway through which TZDs may affect insulin sensitivity (269). At the same time, the degree to which the insulin sensitizing effects of TZDs are due to their anti-inflammatory actions as compared to other pathways is not fully understood.
Collectively, results from human clinical trials that selectively targeted key components of pro-inflammatory pathways have yielded mixed results on insulin sensitivity and glycemic control. It is important to emphasize, however, that despite any metabolic improvements, there is no evidence that such improvements arise directly from reductions in adipose tissue inflammation. These studies only provide indirect evidence of a relationship between inflammation and insulin resistance, and these studies therefore provide limited insight to what extent inflammation of adipose tissue, relative to inflammation of other tissues, contributes to systemic insulin resistance.
Summary and conclusion
Hotamisligil and colleagues pioneered work on adipose tissue inflammation and its importance for insulin sensitivity, with their seminal paper showing that in rodents, TNFα interferes with insulin signaling, and that by blocking inflammation, obese mice were protected from developing insulin resistance (189, 479). Extensive data from both rodent models and humans have since largely confirmed that accumulation of excess adipose tissue is strongly associated with adipose tissue inflammation, and that adipose tissue inflammation is associated with insulin resistance. A key limitation in studies in this area is that adipose tissue inflammation is strongly associated with other factors known or suspected to affect insulin sensitivity, most importantly liver inflammation and steatosis. Most studies in this field have been unable to experimentally or statistically separate the relative contribution of adipose tissue inflammation (in various depots) from that of liver fat content, for example, making it impossible to confidently conclude at this time to which degree both are contributors in the etiology of insulin resistance. Some limited evidence suggests that insulin resistance and adipose tissue inflammation can be dissociated. These include FSP27 KO mice that are insulin resistant in spite of reduced adipose tissue inflammation when made obese (551), or human studies following acute weight loss or gain in which insulin sensitivity improved and decreased, respectively, in spite of unchanged measures of adipose tissue inflammation (Table 5). All of these pieces of evidence suggest that insulin resistance is affected to a substantial degree by factors other than adipose tissue inflammation. At the same time, one of the largest and strongest studies, by Kloeting and colleagues, suggests that adipose tissue inflammation (as assessed by quantifying macrophages) in VAT, together with fasting plasma adiponectin concentrations (which are also affected by adipose tissue inflammation), can perfectly predict systemic insulin sensitivity (238). Thus, taken together, the data collectively demonstrate that while adipose tissue inflammation is clearly a key factor, insulin resistance is affected by other factors as well. These include most likely inflammation and fat deposition in other metabolically active tissues such as the liver.
Adipose tissue inflammation: a physiologic adaptive response to chronic caloric excess
Obesity, adipose tissue inflammation, and insulin resistance – a short history
It was nearly 25 years ago that the first reports were published that obesity is associated with an increase in the adipose tissue expression of inflammatory cytokines, such as TNFα (188, 189, 479), and that this inflammation in adipose tissue may be a key mediator between increased adiposity and insulin resistance (479). Two landmark papers published in 2003 then solidified these initial findings of a key role of adipose tissue inflammation in insulin resistance. Weisberg and colleagues demonstrated that macrophages infiltrate adipose tissue in large numbers in obesity, and that these cells are the major source of key inflammatory cytokines (504). Xu and colleagues showed that low-grade inflammation in obesity was to a large extent specific to adipose tissue, and that the timing of development of adipose tissue inflammation and insulin resistance is quite strongly associated in a diet-induced obesity mouse model (523). In quick succession, as discussed in detail previously (Table 3), numerous reports, mostly from KO mouse models, were published showing that impairing the ability of a mouse’s immune system to mount an inflammatory response made them more insulin sensitive and glucose tolerant when obese. Cumulatively, the mouse models led to a clear and straightforward paradigm: an increase in adiposity triggers low-grade chronic inflammation in adipose tissue, which suggests that adipose tissue inflammation may be a major contributor to insulin resistance in obesity. As will be discussed in the next section, this paradigm would benefit from some minor and major additions or corrections to more adequately reflect new data illustrating that the relationship between adiposity, adipose tissue inflammation, and insulin resistance is more complex than previously appreciated. Specifically, it is now clear that the immune cells infiltrating adipose tissue in obesity, and their pro-inflammatory activation, play an important role in the ability of the tissue to expand in response to chronic caloric excess. Further, the complexity of the interrelationship between adipose tissue inflammation and other factors impairing insulin sensitivity was, and still may be, underappreciated. And lastly, it is now clear that rodent models of obesity differ in some ways from human obesity, even though insights into the specific series of events leading to adipose tissue inflammation and its relationship to insulin resistance in humans are more scarce, and constitute a major knowledge gap that needs to be addressed in future studies.
A newly emerging paradigm
The immune system plays an underappreciated role in the normal physiology of adipose tissue, particularly in the adaptive processes that become necessary in the context of long-term excessive caloric intake (Figure 12). Specifically, macrophages break down and secrete extracellular matrix proteins (213, 418), an essential continuous process that allows the tissue to expand through hyperplasia or hypertrophy. Similarly, new blood vessels are needed to provide oxygen and nutrients to expanded adipose tissue, and macrophages play a critical role in this angiogenesis (75), through mechanisms that are not entirely clear. The activation of pro-inflammatory pathways within immune cells such as macrophages, resulting in the production of pro-inflammatory cytokines including TNFα, IL-1β, and IL-6 that induce insulin resistance in adipocytes (and potentially other cell types) is an important mechanism limiting further growth of hypertrophic adipocytes that would otherwise increase their risk of cell death (211, 324). Macrophages are also able to store TG from the lipid droplets of dead adipocytes (79, 382), or buffer FFA that adipocytes are unable to store (526).
Figure 12. Key functions of macrophages in adipose tissue physiology.
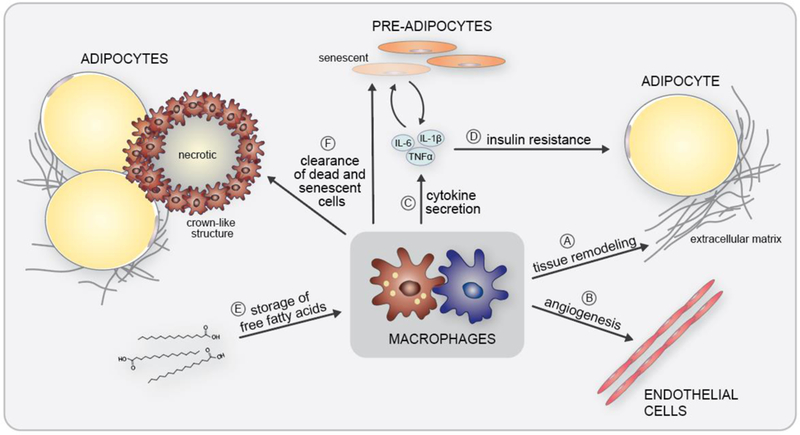
Leukocytes infiltrating adipose tissue, particularly macrophages, play key roles in processes that allow growth of adipose tissue in the context of chronic caloric excess. These include the breakdown of extracellular matrix proteins as cells expand and to create room for new adipocytes, and laying down new extracellular matrix, processes that are collectively known as ‘tissue remodeling’ (A). Macrophages also play a critical role in the formation of new blood vessels as the tissue expands, i.e., angiogenesis (B). The activation of inflammatory pathways in adipocytes and immune cells (C) also may play an important physiological role, because the induction of insulin resistance in adipocytes (D) may serve to limit excessive hypertrophy of these cells, which is known to trigger cell death. Macrophages are also able to intermittently store lipid (E), which could become important whenever the lipid-storage capacity of adipocytes is (temporarily) restricted. And lastly, macrophages play a key role in the removal of cellular debris from necrotic or apoptotic adipocytes or senescent cells (F).
Teaching points: Although obesity is associated with the development of chronic, low-grade adipose tissue inflammation, the immune cells that infiltrate the adipose tissue perform several different functions that allow for healthy expansion of adipose tissue during chronic caloric excess. Examples of functions of macrophages in adipose tissue include the breakdown of extracellular matrix proteins as cells expand and to create room for new adipocytes, and laying down new extracellular matrix, processes that are collectively known as ‘tissue remodeling’ (A). Macrophages also play a critical role in the formation of new blood vessels as the tissue expands, i.e., angiogenesis (B). The activation of inflammatory pathways in adipocytes and immune cells (C) also may play an important physiological role because the induction of insulin resistance in adipocytes (D) may serve to limit excessive hypertrophy of these cells, which is known to trigger cell death. Macrophages are also able to intermittently store lipid (E), which could become important whenever the lipid-storage capacity of adipocytes is (temporarily) restricted. And lastly, macrophages play a key role in the removal of cellular debris from necrotic or apoptotic adipocytes or senescent cells (F).
Considering these essential adaptive physiologic functions of immune cells and particularly macrophages in adipose tissue, the ‘old’ paradigm that regarded adipose tissue inflammation as a purely pathophysiological event, to be treated with anti-inflammatory drugs, is inadequate. Instead, currently available evidence suggests that immune cells and particularly macrophages play important physiological and adaptive roles in adipose tissue. While numerous details have yet to be worked out, a new paradigm has begun to emerge, which we would like to summarize and outline here as a working concept:
Compensated expansion of adipose tissue: Excessive caloric intake over extended periods of time will initially be compensated by adequate and healthy expansion of adipose tissue, particularly SAT. This will occur initially largely by hyperplasia, i.e., the recruitment of pre-adipocytes that differentiate into mature adipocytes (153). This expanded tissue will be characterized by still mostly small adipocytes, with little to no increased immune cell infiltration and very few, if any, dead adipocytes. Evidence from time-series studies in mice fed an obesogenic HFD suggests that the caloric excess induced by the highly palatable HFD promotes a mild increase in the expression of genes encoding several key mediators of inflammation, such as TNFα, MCP-1, or IL-6 within as few as three days of switching to the HFD (268). However, the development of more substantial adipose tissue inflammation is typically not seen until week 16.
Reaching adipocyte storage capacity triggers adipose tissue inflammation: One current hypothesis posits that the main storage organ for excess fuel, SAT, can initially expand sufficiently to store excess nutrients as TG in enlarging existing adipocytes (hypertrophy) or newly-differentiated adipocytes (hyperplasia). As the ability of the tissue to induce adipogenesis and differentiate preadipocytes becomes limited, which may occur for a number of reasons, the burden of continued exposure to excess nutrients is increased for existing enlarged adipocytes causing them to upregulate pro-inflammatory pathways, which increases the expression of key chemokines including MCP-1 (209). The resulting influx of monocytes differentiating to macrophages may have a number of different functions, as outlined above, from intermittent lipid storage (buffering) (526) to induction of insulin resistance in adipocytes to prevent their death to removal of adipocytes once they have undergone apoptosis or necrosis (79, 382).
When exactly SAT becomes unable to expand to compensate for the chronic nutrient overexposure may differ substantially between individuals, as has been suggested in the personal fat threshold hypothesis (461). Factors that may affect when the individual storage capacity has been reached may include fibrotic adipose tissue, age-related senescence of preadipocytes, or genetic variability in genes involved in adipogenesis, as discussed in previous sections. Once an individual is unable to safely store excess calories in SAT, low-grade chronic inflammation in SAT will develop, storage of lipids will shift to VAT, eventually triggering inflammation in VAT, and ultimately leading to gradual shifts in lipid storage to ectopic depots such as liver, muscle, and pancreas.
Negative metabolic consequences of persistent long-term adipose tissue inflammation: During short-term exposure to caloric excess, modest ATM activation and associated induction of inflammatory signaling pathways may provide more time for the tissue to appropriately expand to ultimately accommodate storage of the excess nutrients (or the insult, caloric excess, to subside), thereby preventing flux of lipid to ectopic depots such as the liver or muscle. However, if the caloric excess and the resulting adipose tissue inflammation persists over extended periods of time, then the inflammation itself may contribute to ectopic fat storage, partly through the continued adipocyte insulin resistance, which attenuates the ability of insulin to inhibit lipolysis in the postprandial state, thereby leading to increased flux of FFA to ectopic depots, partly through inhibited adiponectin expression leading to hypoadiponectinemia (440). Thus, while the basic macrophage functions and inflammatory signaling pathways may be very similar under short- and long-term caloric excess, the effect on whole-body metabolic regulation may differ due to the longer-term consequences of continued elevated flux of FFA to ectopic depots and chronic hypoadiponectinemia. Available evidence suggests that inflammation of VAT is particularly strongly associated with metabolic dysfunction, possibly due to the more direct delivery of inflammatory mediators and FFA to the liver (401).
Adipose tissue inflammation and ectopic fat deposition as key components of metabolic dysfunction: Persistent adipose tissue inflammation, together with systemic inflammation, hypoadiponectinemia, and ectopic fat accumulation, are the principal players in the etiology of systemic insulin resistance. It is important to emphasize the strong interrelationship of these factors with each other, making it hard in many cases to disentangle to relative contribution of one factor over the others in the development of insulin resistance. As illustrated in Figure 13, it is clear that adipocyte size remains smaller, with less inflammatory activity in adipose tissue and less ectopic fat content in obese individuals who remain relatively insulin sensitive. It is also clear that the average size of adipocytes tends to be larger in obese, insulin resistant subjects, associated with more substantial leukocyte infiltration and pro-inflammatory cytokine secretion in adipose tissue, increased plasma FFA concentrations, hypoadiponectinemia, and increased TG content in liver and muscle. The key differentiating factor between these two scenarios is likely the ability vs. inability of adipose tissue, particularly SAT, to expand in a healthy fashion in response to chronic caloric excess.
Human studies conducted in the context of bariatric surgery- or lifestyle change-induced weight loss suggest that while adipose tissue inflammation is a major contributor to systemic insulin resistance, it is by itself not sufficient, and substantial improvements in insulin sensitivity are associated with a reduction in ectopic fat mass despite persistent inflammation in SAT and VAT (286, 417).
Figure 13. Adipose tissue inflammation, ectopic fat deposition, and insulin resistance in obesity.
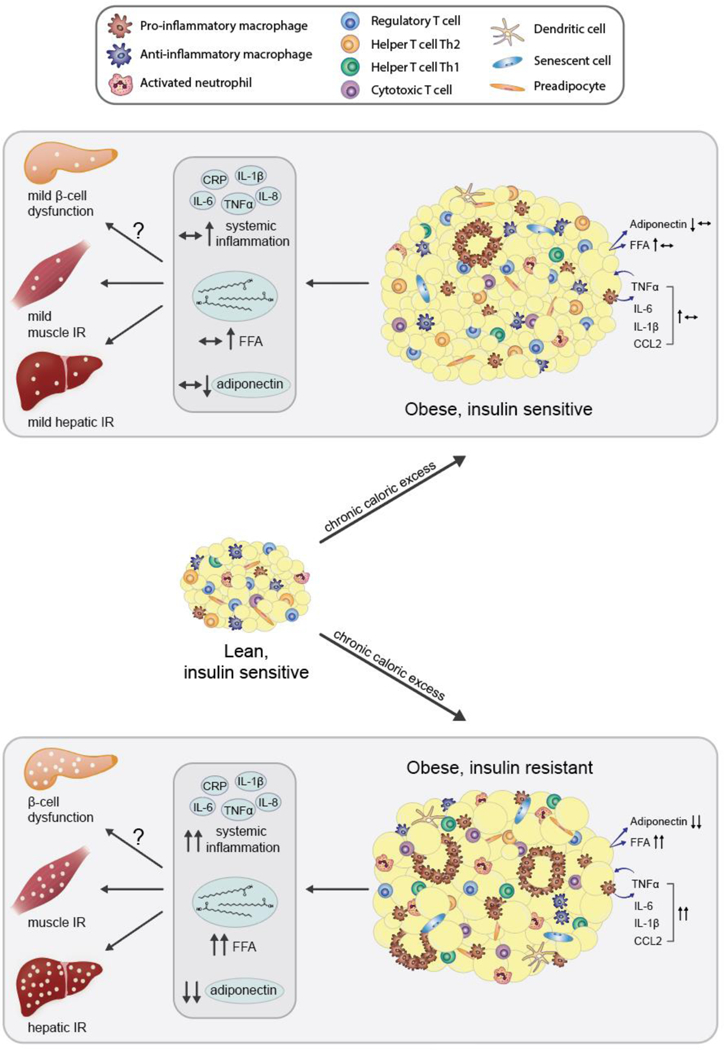
In the context of chronic caloric excess, adipose tissue is challenged to store excess calories in the form of TG. This requires either the differentiation of preadipocytes to mature adipocytes (hyperplasia) or hypertrophy of existing adipocytes. Currently available evidence suggests that the ability of adipose tissue, particularly subcutaneous adipose tissue, to expand in a healthy fashion is reliant primarily upon hyperplasia, which prevents excessive adipocyte hypertrophy. This is one key differentiating factor between obese individuals that remain relatively insulin sensitive (upper panel) from those that become insulin resistant (lower panel). Larger adipocytes are more susceptible to cell death and are more strongly associated with immune cell infiltration and activation of pro-inflammatory pathways within adipocytes and infiltrating leukocytes. Together these processes promote insulin resistance within the expanded adipose tissue. Inflammation and insulin resistance in adipose tissue are major contributors to low-grade chronic systemic inflammation, hypoadiponectinemia, and an elevated flux of free fatty acids (FFA) to the liver, muscle, and pancreas, eventually contributing to excessive ectopic fat storage in these organs. Elevated concentrations of TG in liver and muscle are considered a major contributor to insulin resistance in these organs, and TG stored in the pancreas may contribute to pancreatic β-cell dysfunction, i.e., the inability of the β-cell to fully compensate for insulin resistance. The importance of adipose tissue inflammation in driving systemic insulin resistance relative to the other, interlinked factors outlined here is currently unclear, particularly in humans.
Teaching points: During chronic caloric excess, a major challenge for adipose tissue is to store the excess calories as triglycerides. This requires the adipose tissue to expand, either by increasing the number of adipocytes (hyperplasia) or by increasing the size of existing adipocytes (hypertrophy). Currently available evidence suggests that the ability of adipose tissue, particularly subcutaneous adipose tissue, to expand in a healthy fashion is reliant primarily upon hyperplasia. An increase in the number of functional lipid-storing adipocytes prevents excessive adipocyte hypertrophy. This is one key differentiating factor between obese individuals that remain relatively insulin sensitive (upper panel) from those that become insulin resistant (lower panel). Larger adipocytes are more susceptible to cell death and are more strongly associated with immune cell infiltration, activation of pro-inflammatory pathways, and a state of insulin resistance. Inflammation and insulin resistance in adipose tissue are major contributors to low-grade chronic systemic inflammation, low adiponectin concentrations in the circulation, and an elevated flux of adipose tissue-derived free fatty acids (FFA) to organs such as liver, muscle, and pancreas. Accumulation of triglycerides in liver and muscle are considered a major contributor to insulin resistance in these organs, and triglycerides stored in the pancreas may impair the inability of the β-cell to fully compensate for insulin resistance. The importance of adipose tissue inflammation in driving systemic insulin resistance relative to the other, interlinked factors outlined here is currently unclear, particularly in humans.
Conclusion
Adipose tissue inflammation, particularly in visceral fat depots, is clearly a contributor in the etiology of systemic insulin resistance, glucose intolerance, and T2DM. However, it is critical to recognize that the immune system and inflammation play important roles in the adaptive adipose tissue response to chronic caloric excess, rather than regarding adipose tissue inflammation as a purely pathophysiological process. There is some uncertainty about the relative impact of adipose tissue inflammation on insulin resistance relative to ectopic fat content and inflammation in other metabolically active tissues. Several pieces of evidence, such as rare transgenic mouse models in which adipose tissue inflammation and insulin resistance are dissociated, as well as the often substantially improved insulin sensitivity in the context of human weight loss in spite of persistent adipose tissue inflammation, suggest that adipose tissue inflammation is neither necessary nor sufficient to induce systemic insulin resistance, and that ectopic fat accumulation and inflammation in organs such as liver and muscle are almost certainly critical contributors. It cannot be ruled out, however, that the sequence of events that ultimately induce insulin resistance in obesity is distinct from the sequence of events that ameliorate insulin resistance upon weight loss. That is, while the evidence suggests that reduced adipose tissue inflammation is not a major mediator of improved insulin sensitivity in individuals with significantly improved insulin sensitivity and glucose tolerance upon moderate or even substantial weight loss, it may play a more important role in the gradual development of insulin resistance that is associated with weight gain. It is also worth emphasizing that in almost all models discussed in this paper, both rodent and human, adipose tissue inflammation is associated with, and may well be an important contributor to, ectopic fat deposition. Thus, taken together, the currently available evidence suggests that adipose tissue inflammation is an important factor in the development of insulin resistance and glucose intolerance in obesity, along with other factors that likely include ectopic fat deposition and inflammation in other metabolically active tissues.
Didactic Synopsis.
Major teaching points:
Low-grade chronic inflammation includes the accumulation of pro-inflammatory macrophages and other immune cells in adipose tissue, and their secretion of mediators of inflammation such as tumor necrosis factor α and interleukins 1β and 6.
Low-grade chronic adipose tissue inflammation is strongly and consistently associated with excess body fat mass in mouse models of obesity as well as overweight and obese humans.
Adipose tissue inflammation is also consistently associated with systemic insulin resistance, a major determinant of glucose intolerance, and genetic alterations that reduce adipose tissue inflammation are very consistently associated with improved insulin sensitivity.
Rare exceptions in which adipose tissue inflammation and insulin resistance are not associated with one another illustrate that other factors contribute to the etiology of insulin resistance as well. These include lipid accumulation and inflammation in other metabolically active tissues such as the liver.
Acknowledegements
We would like to acknowledge funding support for M.B. from the National Cancer Institute (R25CA094880) and the National Institute of Diabetes and Digestive and Kidney Diseases (T32DK007247), for K.S. from the National Cancer Institute (R25CA094880, T32CA094880), and for M.K. from National Institute of Diabetes and Digestive and Kidney Diseases (R01DK102960, P30DK017047).
References
- 1.Abate N, Garg A, Peshock RM, Stray-Gundersen J and Grundy SM. Relationships of generalized and regional adiposity to insulin sensitivity in men. J Clin Invest 96: 88–98, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abate N, Sallam HS, Rizzo M, Nikolic D, Obradovic M, Bjelogrlic P and Isenovic ER. Resistin: an inflammatory cytokine. Role in cardiovascular diseases, diabetes and the metabolic syndrome. Curr Pharm Des 20: 4961–9, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Abdullah A, Peeters A, de Courten M and Stoelwinder J. The magnitude of association between overweight and obesity and the risk of diabetes: a meta-analysis of prospective cohort studies. Diabetes Res Clin Pract 89: 309–19, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Adams LA, Angulo P and Lindor KD. Nonalcoholic fatty liver disease. CMAJ 172: 899–905, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adamson SE, Griffiths R, Moravec R, Senthivinayagam S, Montgomery G, Chen W, Han J, Sharma PR, Mullins GR, Gorski SA, Cooper JA, Kadl A, Enfield K, Braciale TJ, Harris TE and Leitinger N. Disabled homolog 2 controls macrophage phenotypic polarization and adipose tissue inflammation. J Clin Invest 126: 1311–22, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aghamohammadzadeh R, Greenstein AS, Yadav R, Jeziorska M, Hama S, Soltani F, Pemberton PW, Ammori B, Malik RA, Soran H and Heagerty AM. Effects of bariatric surgery on human small artery function: evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity. J Am Coll Cardiol 62: 128–35, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aguilar-Salinas CA, Garcia EG, Robles L, Riano D, Ruiz-Gomez DG, Garcia-Ulloa AC, Melgarejo MA, Zamora M, Guillen-Pineda LE, Mehta R, Canizales-Quinteros S, Tusie Luna MT and Gomez-Perez FJ. High adiponectin concentrations are associated with the metabolically healthy obese phenotype. J Clin Endocrinol Metab 93: 4075–9, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Ahima RS and Lazar MA. Adipokines and the peripheral and neural control of energy balance. Mol Endocrinol (Baltimore, Md 22: 1023–31, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahl S, Guenther M, Zhao S, James R, Marks J, Szabo A and Kidambi S. Adiponectin Levels Differentiate Metabolically Healthy vs Unhealthy Among Obese and Nonobese White Individuals. J Clin Endocrinol Metab 100: 4172–80, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahren B and Pacini G. Age-related reduction in glucose elimination is accompanied by reduced glucose effectiveness and increased hepatic insulin extraction in man. J Clin Endocrinol Metab 83: 3350–6, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Ahren B and Pacini G. Insufficient islet compensation to insulin resistance vs. reduced glucose effectiveness in glucose-intolerant mice. Am J Physiol Endocrinol Metab 283: E738–44, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Albert JS, Yerges-Armstrong LM, Horenstein RB, Pollin TI, Sreenivasan UT, Chai S, Blaner WS, Snitker S, O’Connell JR, Gong DW, Breyer RJ 3rd, Ryan AS, McLenithan JC, Shuldiner AR, Sztalryd C and Damcott CM. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. New Engl J Med 370: 2307–2315, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alderete TL, Sattler FR, Richey JM, Allayee H, Mittelman SD, Sheng X, Tucci J, Gyllenhammer LE, Grant EG and Goran MI. Salsalate treatment improves glycemia without altering adipose tissue in nondiabetic obese hispanics. Obesity (Silver Spring) 23: 543–51, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aleman JO, Iyengar NM, Walker JM, Milne GL, Da Rosa JC, Liang Y, Giri DD, Zhou XK, Pollak MN, Hudis CA, Breslow JL, Holt PR and Dannenberg AJ. Effects of Rapid Weight Loss on Systemic and Adipose Tissue Inflammation and Metabolism in Obese Postmenopausal Women. J Endocr Soc 1: 625–637, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alligier M, Meugnier E, Debard C, Lambert-Porcheron S, Chanseaume E, Sothier M, Loizon E, Hssain AA, Brozek J, Scoazec JY, Morio B, Vidal H and Laville M. Subcutaneous adipose tissue remodeling during the initial phase of weight gain induced by overfeeding in humans. J Clin Endocrinol Metab 97: E183–92, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Altintas MM, Azad A, Nayer B, Contreras G, Zaias J, Faul C, Reiser J and Nayer A. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J Lipid Res 52: 480–8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermudez-Humaran LG, Smirnova N, Berge M, Sulpice T, Lahtinen S, Ouwehand A, Langella P, Rautonen N, Sansonetti PJ and Burcelin R. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3: 559–72, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amati F, Dube JJ, Coen PM, Stefanovic-Racic M, Toledo FG and Goodpaster BH. Physical inactivity and obesity underlie the insulin resistance of aging. Diabetes Care 32: 1547–9, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.American Diabetes Association. The cost of diabetes. http://www.diabetes.org/advocacy/news-events/cost-of-diabetes.html. Accessed on August 31, 2017.
- 20.Anderson EK, Gutierrez DA, Kennedy A and Hasty AH. Weight cycling increases T-cell accumulation in adipose tissue and impairs systemic glucose tolerance. Diabetes 62: 3180–8, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anstee QM and Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 87: 1–16, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aouadi M, Tencerova M, Vangala P, Yawe JC, Nicoloro SM, Amano SU, Cohen JL and Czech MP. Gene silencing in adipose tissue macrophages regulates whole-body metabolism in obese mice. Proc Natl Acad Sci U S A 110: 8278–83, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apovian CM, Bigornia S, Mott M, Meyers MR, Ulloor J, Gagua M, McDonnell M, Hess D, Joseph L and Gokce N. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscler Thromb Vasc Biol 28: 1654–9, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Araneta MR and Barrett-Connor E. Ethnic differences in visceral adipose tissue and type 2 diabetes: Filipino, African-American, and white women. Obesity Res 13: 1458–65, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T and Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Aron-Wisnewsky J, Tordjman J, Poitou C, Darakhshan F, Hugol D, Basdevant A, Aissat A, Guerre-Millo M and Clement K. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab 94: 4619–23, 2009. [DOI] [PubMed] [Google Scholar]
- 27.Auerbach P, Nordby P, Bendtsen LQ, Mehlsen JL, Basnet SK, Vestergaard H, Ploug T and Stallknecht B. Differential effects of endurance training and weight loss on plasma adiponectin multimers and adipose tissue macrophages in younger, moderately overweight men. Am J Physiol Regul Integr Comp Physiol 305: R490–8, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Baena-Fustegueras JA, Pardina E, Balada E, Ferrer R, Catalan R, Rivero J, Casals I, Lecube A, Fort JM, Vargas V and Peinado-Onsurbe J. Soluble CD40 ligand in morbidly obese patients: effect of body mass index on recovery to normal levels after gastric bypass surgery. JAMA Surg 148: 151–6, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J and Feve B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw 17: 4–12, 2006. [PubMed] [Google Scholar]
- 30.Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, Robert JJ, Capeau J and Hainque B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab 87: 2084–9, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Basu R, Breda E, Oberg AL, Powell CC, Dalla Man C, Basu A, Vittone JL, Klee GG, Arora P, Jensen MD, Toffolo G, Cobelli C and Rizza RA. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes 52: 1738–48, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Bates SH, Kulkarni RN, Seifert M and Myers MG Jr., Roles for leptin receptor/STAT3-dependent and -independent signals in the regulation of glucose homeostasis. Cell Metab 1: 169–78, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Batt R and Mialhe P. Insulin resistance of the inherently obese mouse--obob. Nature 212: 289–90, 1966. [DOI] [PubMed] [Google Scholar]
- 34.Becker L, Liu NC, Averill MM, Yuan W, Pamir N, Peng Y, Irwin AD, Fu X, Bornfeldt KE and Heinecke JW. Unique proteomic signatures distinguish macrophages and dendritic cells. PLoS One 7: e33297, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benoit B, Plaisancie P, Awada M, Geloen A, Estienne M, Capel F, Malpuech-Brugere C, Debard C, Pesenti S, Morio B, Vidal H, Rieusset J and Michalski MC. High-fat diet action on adiposity, inflammation, and insulin sensitivity depends on the control low-fat diet. Nutr Res 33: 952–60, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Bergman RN, Stefanovski D, Buchanan TA, Sumner AE, Reynolds JC, Sebring NG, Xiang AH and Watanabe RM. A better index of body adiposity. Obesity (Silver Spring) 19: 1083–9, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, Blin-Wakkach C, Anty R, Iannelli A, Gugenheim J, Tran A, Bouloumie A, Gual P and Wakkach A. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 61: 2238–47, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Best JD, Kahn SE, Ader M, Watanabe RM, Ni TC and Bergman RN. Role of glucose effectiveness in the determination of glucose tolerance. Diabetes Care 19: 1018–30, 1996. [DOI] [PubMed] [Google Scholar]
- 39.Bierman EL, Bagdade JD and Porte D Jr., Obesity and diabetes: the odd couple. Am J Clin Nutr 21: 1434–7, 1968. [DOI] [PubMed] [Google Scholar]
- 40.Bigornia SJ, Farb MG, Mott MM, Hess DT, Carmine B, Fiscale A, Joseph L, Apovian CM and Gokce N. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes 2: e30, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bjorntorp P, Bergman H and Varnauskas E. Plasma free fatty acid turnover rate in obesity. Acta Med Scand 185: 351–6, 1969. [DOI] [PubMed] [Google Scholar]
- 42.Blachnio-Zabielska AU, Baranowski M, Hirnle T, Zabielski P, Lewczuk A, Dmitruk I and Gorski J. Increased bioactive lipids content in human subcutaneous and epicardial fat tissue correlates with insulin resistance. Lipids 47: 1131–41, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanco-Colio LM, Tunon J, Martin-Ventura JL and Egido J. Anti-inflammatory and immunomodulatory effects of statins. Kidney Int 63: 12–23, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Bluher M Are metabolically healthy obese individuals really healthy? Eur J Endocrinol 171: R209–19, 2014. [DOI] [PubMed] [Google Scholar]
- 45.Bluher M The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr Opin Lipidol 21: 38–43, 2010. [DOI] [PubMed] [Google Scholar]
- 46.Bluher M, Kloting N, Wueest S, Schoenle EJ, Schon MR, Dietrich A, Fasshauer M, Stumvoll M and Konrad D. Fas and FasL expression in human adipose tissue is related to obesity, insulin resistance, and type 2 diabetes. J Clin Endocrinol Metab 99: E36–44, 2014. [DOI] [PubMed] [Google Scholar]
- 47.Boden G, Chen X, Ruiz J, White JV and Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest 93: 2438–46, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogardus C, Lillioja S, Mott D, Reaven GR, Kashiwagi A and Foley JE. Relationship between obesity and maximal insulin-stimulated glucose uptake in vivo and in vitro in man. Int J Obes 9 Suppl 1: 149, 1985. [PubMed] [Google Scholar]
- 49.Bonora E Relationship between regional fat distribution and insulin resistance. Int J Obes Relat Metab Disord 24 Suppl 2: S32–5, 2000. [DOI] [PubMed] [Google Scholar]
- 50.Bourlier V, Zakaroff-Girard A, Miranville A, De Barros S, Maumus M, Sengenes C, Galitzky J, Lafontan M, Karpe F, Frayn KN and Bouloumie A. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation 117: 806–15, 2008. [DOI] [PubMed] [Google Scholar]
- 51.Breslin WL, Strohacker K, Carpenter KC, Esposito L and McFarlin BK. Weight gain in response to high-fat feeding in CD-1 male mice. Lab Anim 44: 231–7, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Brethauer SA, Heneghan HM, Eldar S, Gatmaitan P, Huang H, Kashyap S, Gornik HL, Kirwan JP and Schauer PR. Early effects of gastric bypass on endothelial function, inflammation, and cardiovascular risk in obese patients. Surg Endosc 25: 2650–9, 2011. [DOI] [PubMed] [Google Scholar]
- 53.Browning LM, Krebs JD, Magee EC, Fruhbeck G and Jebb SA. Circulating markers of inflammation and their link to indices of adiposity. Obes Facts 1: 259–65, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buettner R, Parhofer KG, Woenckhaus M, Wrede CE, Kunz-Schughart LA, Scholmerich J and Bollheimer LC. Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J Mol Endocrinol 36: 485–501, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Bultman SJ, Michaud EJ and Woychik RP. Molecular characterization of the mouse agouti locus. Cell 71: 1195–204, 1992. [DOI] [PubMed] [Google Scholar]
- 56.Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, Coupaye M, Pelloux V, Hugol D, Bouillot JL, Bouloumie A, Barbatelli G, Cinti S, Svensson PA, Barsh GS, Zucker JD, Basdevant A, Langin D and Clement K. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 54: 2277–2286, 2005. [DOI] [PubMed] [Google Scholar]
- 57.Cancello R, Tordjman J, Poitou C, Guilhem G, Bouillot JL, Hugol D, Coussieu C, Basdevant A, Bar Hen A, Bedossa P, Guerre-Millo M and Clement K. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 55: 1554–61, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM and Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57: 1470–81, 2008. [DOI] [PubMed] [Google Scholar]
- 59.Capel F, Klimcakova E, Viguerie N, Roussel B, Vitkova M, Kovacikova M, Polak J, Kovacova Z, Galitzky J, Maoret JJ, Hanacek J, Pers TH, Bouloumie A, Stich V and Langin D. Macrophages and adipocytes in human obesity: adipose tissue gene expression and insulin sensitivity during calorie restriction and weight stabilization. Diabetes 58: 1558–67, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carneiro IP, Elliott SA, Siervo M, Padwal R, Bertoli S, Battezzati A and Prado CM. Is Obesity Associated with Altered Energy Expenditure? Adv Nutr 7: 476–87, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Castoldi A, de Souza Naffah C, Camara NO and Moraes-Vieira PM. The Macrophage Switch in Obesity Development. Front Immunol 6: 637, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, Solinger AM, Mandrup-Poulsen T, Dinarello CA and Donath MY. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 35: 1654–62, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cederberg H, Stancakova A, Yaluri N, Modi S, Kuusisto J and Laakso M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: a 6 year follow-up study of the METSIM cohort. Diabetologia 58: 1109–17, 2015. [DOI] [PubMed] [Google Scholar]
- 64.Cefalu WT, Wang ZQ, Werbel S, Bell-Farrow A, Crouse JR 3rd, Hinson WH, Terry JG and Anderson R. Contribution of visceral fat mass to the insulin resistance of aging. Metabolism 44: 954–9, 1995. [DOI] [PubMed] [Google Scholar]
- 65.Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2017. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf. Accessed on August 31, 2017.
- 66.Ceriello A Thiazolidinediones as anti-inflammatory and anti-atherogenic agents. Diabetes/Metab Res Rev 24: 14–26, 2008. [DOI] [PubMed] [Google Scholar]
- 67.Chakaroun R, Raschpichler M, Kloting N, Oberbach A, Flehmig G, Kern M, Schon MR, Shang E, Lohmann T, Dressler M, Fasshauer M, Stumvoll M and Bluher M. Effects of weight loss and exercise on chemerin serum concentrations and adipose tissue expression in human obesity. Metabolism 61: 706–14, 2012. [DOI] [PubMed] [Google Scholar]
- 68.Chan JC, Malik V, Jia W, Kadowaki T, Yajnik CS, Yoon KH and Hu FB. Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA 301: 2129–40, 2009. [DOI] [PubMed] [Google Scholar]
- 69.Chandrasekharan JA and Sharma-Walia N. Lipoxins: nature’s way to resolve inflammation. J Inflamm Res 8: 181–92, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chang SK, Kohlgruber AC, Mizoguchi F, Michelet X, Wolf BJ, Wei K, Lee PY, Lynch L, Duquette D, Ceperuelo-Mallafre V, Banks AS and Brenner MB. Stromal cell cadherin-11 regulates adipose tissue inflammation and diabetes. J Clin Invest 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DL, Gopalan V, Prakash KN, Velan SS, Bulchand S, Tsong TJ, Wang M, Siddique MM, Yuguang G, Sigmundsson K, Mellet NA, Weir JM, Meikle PJ, Bin MYMS, Shabbir A, Shayman JA, Hirabayashi Y, Shiow ST, Sugii S and Summers SA. Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metab 24: 820–834, 2016. [DOI] [PubMed] [Google Scholar]
- 72.Chavez JA and Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 15: 585–94, 2012. [DOI] [PubMed] [Google Scholar]
- 73.Chawla A, Nguyen KD and Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol 11: 738–49, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI and Morgenstern JP. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84: 491–5, 1996. [DOI] [PubMed] [Google Scholar]
- 75.Cho CH, Koh YJ, Han J, Sung HK, Lee Jong H, Morisada T, Schwendener RA, Brekken RA, Kang G, Oike Y, Choi TS, Suda T, Yoo OJ and Koh GY. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ Res 100: e47–57, 2007. [DOI] [PubMed] [Google Scholar]
- 76.Choe SS, Shin KC, Ka S, Lee YK, Chun JS and Kim JB. Macrophage HIF-2alpha ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes 63: 3359–71, 2014. [DOI] [PubMed] [Google Scholar]
- 77.Chun TH, Inoue M, Morisaki H, Yamanaka I, Miyamoto Y, Okamura T, Sato-Kusubata K and Weiss SJ. Genetic link between obesity and MMP14-dependent adipogenic collagen turnover. Diabetes 59: 2484–94, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cildir G, Akincilar SC and Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med 19: 487–500, 2013. [DOI] [PubMed] [Google Scholar]
- 79.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS and Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46: 2347–55, 2005. [DOI] [PubMed] [Google Scholar]
- 80.Cnop M, Havel PJ, Utzschneider KM, Carr DB, Sinha MK, Boyko EJ, Retzlaff BM, Knopp RH, Brunzell JD and Kahn SE. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia 46: 459–69, 2003. [DOI] [PubMed] [Google Scholar]
- 81.Coleman DL. Diabetes-obesity syndromes in mice. Diabetes 31: 1–6, 1982. [DOI] [PubMed] [Google Scholar]
- 82.Collaboration NCDRF. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 387: 1377–1396, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Collaboration NCDRF. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 387: 1513–30, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, Nawrocki AR, Rajala MW, Parlow AF, Cheeseboro L, Ding YY, Russell RG, Lindemann D, Hartley A, Baker GR, Obici S, Deshaies Y, Ludgate M, Rossetti L and Scherer PE. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 145: 367–83, 2004. [DOI] [PubMed] [Google Scholar]
- 85.Conte C, Fabbrini E, Kars M, Mittendorfer B, Patterson BW and Klein S. Multiorgan insulin sensitivity in lean and obese subjects. Diabetes Care 35: 1316–21, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Coon PJ, Rogus EM, Drinkwater D, Muller DC and Goldberg AP. Role of body fat distribution in the decline in insulin sensitivity and glucose tolerance with age. J Clin Endocrinol Metab 75: 1125–32, 1992. [DOI] [PubMed] [Google Scholar]
- 87.Cottam DR, Mattar SG, Barinas-Mitchell E, Eid G, Kuller L, Kelley DE and Schauer PR. The chronic inflammatory hypothesis for the morbidity associated with morbid obesity: implications and effects of weight loss. Obes Surg 14: 589–600, 2004. [DOI] [PubMed] [Google Scholar]
- 88.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N and Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112: 645–57, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crandall JP, Mather K, Rajpathak SN, Goldberg RB, Watson K, Foo S, Ratner R, Barrett-Connor E and Temprosa M. Statin use and risk of developing diabetes: results from the Diabetes Prevention Program. BMJ Open Diabetes Res Care 5: e000438, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crewe C, An YA and Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest 127: 74–82, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Culver AL, Ockene IS, Balasubramanian R, Olendzki BC, Sepavich DM, Wactawski-Wende J, Manson JE, Qiao Y, Liu S, Merriam PA, Rahilly-Tierny C, Thomas F, Berger JS, Ockene JK, Curb JD and Ma Y. Statin use and risk of diabetes mellitus in postmenopausal women in the Women’s Health Initiative. Arch Intern Med 172: 144–52, 2012. [DOI] [PubMed] [Google Scholar]
- 92.Curat CA, Miranville A, Sengenes C, Diehl M, Tonus C, Busse R and Bouloumie A. From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes 53: 1285–92, 2004. [DOI] [PubMed] [Google Scholar]
- 93.Curat CA, Wegner V, Sengenes C, Miranville A, Tonus C, Busse R and Bouloumie A. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 49: 744–747, 2006. [DOI] [PubMed] [Google Scholar]
- 94.Davies LC, Rosas M, Jenkins SJ, Liao CT, Scurr MJ, Brombacher F, Fraser DJ, Allen JE, Jones SA and Taylor PR. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat Commun 4: 1886, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davis JE, Gabler NK, Walker-Daniels J and Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 16: 1248–55, 2008. [DOI] [PubMed] [Google Scholar]
- 96.Deiuliis JA, Oghumu S, Duggineni D, Zhong J, Rutsky J, Banerjee A, Needleman B, Mikami D, Narula V, Hazey J, Satoskar AR and Rajagopalan S. CXCR3 modulates obesity-induced visceral adipose inflammation and systemic insulin resistance. Obesity (Silver Spring) 22: 1264–74, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dietze-Schroeder D, Sell H, Uhlig M, Koenen M and Eckel J. Autocrine action of adiponectin on human fat cells prevents the release of insulin resistance-inducing factors. Diabetes 54: 2003–11, 2005. [DOI] [PubMed] [Google Scholar]
- 98.Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre-Millo M, Poitou C, Zucker JD, Bedossa P and Clement K. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 59: 2817–25, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, Spohr C, Kober L, Vaag A and Torp-Pedersen C. Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res 42: 517–25, 2005. [DOI] [PubMed] [Google Scholar]
- 100.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD and Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115: 1343–51, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dube S, Errazuriz-Cruzat I, Basu A and Basu R. The forgotten role of glucose effectiveness in the regulation of glucose tolerance. Curr Diab Rep 15: 605, 2015. [DOI] [PubMed] [Google Scholar]
- 102.Duffaut C, Zakaroff-Girard A, Bourlier V, Decaunes P, Maumus M, Chiotasso P, Sengenes C, Lafontan M, Galitzky J and Bouloumie A. Interplay between human adipocytes and T lymphocytes in obesity: CCL20 as an adipochemokine and T lymphocytes as lipogenic modulators. Arterioscler Thromb Vasc Biol 29: 1608–14, 2009. [DOI] [PubMed] [Google Scholar]
- 103.Eglit T, Ringmets I and Lember M. Obesity, high-molecular-weight (HMW) adiponectin, and metabolic risk factors: prevalence and gender-specific associations in Estonia. PLoS One 8: e73273, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ehses JA, Meier DT, Wueest S, Rytka J, Boller S, Wielinga PY, Schraenen A, Lemaire K, Debray S, Van Lommel L, Pospisilik JA, Tschopp O, Schultze SM, Malipiero U, Esterbauer H, Ellingsgaard H, Rutti S, Schuit FC, Lutz TA, Boni-Schnetzler M, Konrad D and Donath MY. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 53: 1795–806, 2010. [DOI] [PubMed] [Google Scholar]
- 105.Eissing L, Scherer T, Todter K, Knippschild U, Greve JW, Buurman WA, Pinnschmidt HO, Rensen SS, Wolf AM, Bartelt A, Heeren J, Buettner C and Scheja L. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat Commun 4: 1528, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Elgazar-Carmon V, Rudich A, Hadad N and Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 49: 1894–903, 2008. [DOI] [PubMed] [Google Scholar]
- 107.Elisha B, Karelis AD, Imbeault P and Rabasa-Lhoret R. Effects of acute hyperinsulinaemia on total and high-molecular-weight adiponectin concentration in metabolically healthy but obese postmenopausal women: a Montreal-Ottawa New Emerging Team (MONET) study. Diabetes Metab 36: 319–21, 2010. [DOI] [PubMed] [Google Scholar]
- 108.Emery CF, Fondow MD, Schneider CM, Christofi FL, Hunt C, Busby AK, Needleman BJ, Melvin WS and Elsayed-Awad HM. Gastric bypass surgery is associated with reduced inflammation and less depression: a preliminary investigation. Obes Surg 17: 759–63, 2007. [DOI] [PubMed] [Google Scholar]
- 109.Engeli S, Feldpausch M, Gorzelniak K, Hartwig F, Heintze U, Janke J, Mohlig M, Pfeiffer AF, Luft FC and Sharma AM. Association between adiponectin and mediators of inflammation in obese women. Diabetes 52: 942–7, 2003. [DOI] [PubMed] [Google Scholar]
- 110.Erridge C and Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arteriosclerosis, thrombosis, and vascular biology 29: 1944–9, 2009. [DOI] [PubMed] [Google Scholar]
- 111.Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985) 106: 326–32, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Esser N, L’Homme L, De Roover A, Kohnen L, Scheen AJ, Moutschen M, Piette J, Legrand-Poels S and Paquot N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 56: 2487–97, 2013. [DOI] [PubMed] [Google Scholar]
- 113.Esser N, Legrand-Poels S, Piette J, Scheen AJ and Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract 105: 141–50, 2014. [DOI] [PubMed] [Google Scholar]
- 114.Evans DJ, Hoffmann RG, Kalkhoff RK and Kissebah AH. Relationship of body fat topography to insulin sensitivity and metabolic profiles in premenopausal women. Metabolism 33: 68–75, 1984. [DOI] [PubMed] [Google Scholar]
- 115.Fabbrini E, Cella M, McCartney SA, Fuchs A, Abumrad NA, Pietka TA, Chen Z, Finck BN, Han DH, Magkos F, Conte C, Bradley D, Fraterrigo G, Eagon JC, Patterson BW, Colonna M and Klein S. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 145: 366–74 e1–3, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, Okunade A and Klein S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A 106: 15430–5, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Faghihimani E, Aminorroaya A, Rezvanian H, Adibi P, Ismail-Beigi F and Amini M. Salsalate improves glycemic control in patients with newly diagnosed type 2 diabetes. Acta Diabetol 50: 537–43, 2013. [DOI] [PubMed] [Google Scholar]
- 118.Fain JN. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. VitamHorm 74: 443–477, 2006. [DOI] [PubMed] [Google Scholar]
- 119.Fantuzzi G Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 115: 911–9; quiz 920, 2005. [DOI] [PubMed] [Google Scholar]
- 120.Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J and Paschke R. Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3T3-L1 adipocytes. Biochem Biophys Res Commun 301: 1045–50, 2003. [DOI] [PubMed] [Google Scholar]
- 121.Ferrannini E, Camastra S, Coppack SW, Fliser D, Golay A and Mitrakou A. Insulin action and non-esterified fatty acids. The European Group for the Study of Insulin Resistance (EGIR). Proc Nutr Soc 56: 753–61, 1997. [DOI] [PubMed] [Google Scholar]
- 122.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S and Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15: 930–9, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Filippin-Monteiro FB, de Oliveira EM, Sandri S, Knebel FH, Albuquerque RC and Campa A. Serum amyloid A is a growth factor for 3T3-L1 adipocytes, inhibits differentiation and promotes insulin resistance. Int J Obes (Lond) 36: 1032–9, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fink RI, Kolterman OG, Griffin J and Olefsky JM. Mechanisms of insulin resistance in aging. J Clin Invest 71: 1523–35, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Finkelstein EA, DiBonaventura M, Burgess SM and Hale BC. The costs of obesity in the workplace. J Occup Environ Med 52: 971–6, 2010. [DOI] [PubMed] [Google Scholar]
- 126.Finkelstein EA, Trogdon JG, Cohen JW and Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff (Millwood) 28: w822–31, 2009. [DOI] [PubMed] [Google Scholar]
- 127.Fisher FM, Trujillo ME, Hanif W, Barnett AH, McTernan PG, Scherer PE and Kumar S. Serum high molecular weight complex of adiponectin correlates better with glucose tolerance than total serum adiponectin in Indo-Asian males. Diabetologia 48: 1084–7, 2005. [DOI] [PubMed] [Google Scholar]
- 128.Flegal KM, Carroll MD, Kuczmarski RJ and Johnson CL. Overweight and obesity in the United States: prevalence and trends, 1960–1994. Int J Obes Relat Metab Disord 22: 39–47, 1998. [DOI] [PubMed] [Google Scholar]
- 129.Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD and Ogden CL. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 315: 2284–91, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fleischman A, Shoelson SE, Bernier R and Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31: 289–94, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fontana L Modulating human aging and age-associated diseases. Biochim Biophys Acta 1790: 1133–8, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fraulob JC, Ogg-Diamantino R, Fernandes-Santos C, Aguila MB and Mandarim-de-Lacerda CA. A Mouse Model of Metabolic Syndrome: Insulin Resistance, Fatty Liver and Non-Alcoholic Fatty Pancreas Disease (NAFPD) in C57BL/6 Mice Fed a High Fat Diet. J Clin Biochem Nutr 46: 212–23, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fresno M, Alvarez R and Cuesta N. Toll-like receptors, inflammation, metabolism and obesity. Arch Physiol Biochem 117: 151–64, 2011. [DOI] [PubMed] [Google Scholar]
- 134.Fried SK, Bunkin DA and Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 83: 847–50, 1998. [DOI] [PubMed] [Google Scholar]
- 135.Fujisaka S, Usui I, Ikutani M, Aminuddin A, Takikawa A, Tsuneyama K, Mahmood A, Goda N, Nagai Y, Takatsu K and Tobe K. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1alpha-dependent and HIF-1alpha-independent manner in obese mice. Diabetologia 56: 1403–12, 2013. [DOI] [PubMed] [Google Scholar]
- 136.Furuhashi M, Fucho R, Gorgun CZ, Tuncman G, Cao H and Hotamisligil GS. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest 118: 2640–50, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M and Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–61, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gabriely I, Ma XH, Yang XM, Atzmon G, Rajala MW, Berg AH, Scherer P, Rossetti L and Barzilai N. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes 51: 2951–8, 2002. [DOI] [PubMed] [Google Scholar]
- 139.Garcia MC, Wernstedt I, Berndtsson A, Enge M, Bell M, Hultgren O, Horn M, Ahren B, Enerback S, Ohlsson C, Wallenius V and Jansson JO. Mature-onset obesity in interleukin-1 receptor I knockout mice. Diabetes 55: 1205–13, 2006. [DOI] [PubMed] [Google Scholar]
- 140.Garg A. Acquired and inherited lipodystrophies. New Engl J Med 350: 1220–34, 2004. [DOI] [PubMed] [Google Scholar]
- 141.Gavi S, Qurashi S, Melendez MM, Mynarcik DC, McNurlan MA and Gelato MC. Plasma retinol-binding protein-4 concentrations are elevated in human subjects with impaired glucose tolerance and type 2 diabetes: response to Cho et al. Diabetes Care 30: e7; author reply e8, 2007. [DOI] [PubMed] [Google Scholar]
- 142.Giugliano D, Sacca L, Scognamiglio G, Ungaro B and Torella R. Influence of acetylsalicylic acid on glucose turnover in normal man. Diabete Metab 8: 279–82, 1982. [PubMed] [Google Scholar]
- 143.Goldfine AB, Fonseca V, Jablonski KA, Chen YD, Tipton L, Staten MA, Shoelson SE and Targeting Inflammation Using Salsalate in Type 2 Diabetes Study T. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med 159: 1–12, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE and Team T-TDS. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 152: 346–57, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J and Shoelson SE. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 1: 36–43, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Goodpaster BH, Thaete FL, Simoneau JA and Kelley DE. Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes 46: 1579–85, 1997. [DOI] [PubMed] [Google Scholar]
- 147.Goossens GH, Bizzarri A, Venteclef N, Essers Y, Cleutjens JP, Konings E, Jocken JW, Cajlakovic M, Ribitsch V, Clement K and Blaak EE. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation 124: 67–76, 2011. [DOI] [PubMed] [Google Scholar]
- 148.Gordon RS Jr., and Cherkes A Unesterified fatty acid in human blood plasma. J Clin Invest 35: 206–12, 1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gordon S Alternative activation of macrophages. Nat Rev Immunol 3: 23–35, 2003. [DOI] [PubMed] [Google Scholar]
- 150.Gosejacob D, Jager PS, Vom Dorp K, Frejno M, Carstensen AC, Kohnke M, Degen J, Dormann P and Hoch M. Ceramide Synthase 5 Is Essential to Maintain C16:0-Ceramide Pools and Contributes to the Development of Diet-induced Obesity. J Biol Chem 291: 6989–7003, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Goyal R, Faizy AF, Siddiqui SS and Singhai M. Evaluation of TNF-alpha and IL-6 Levels in Obese and Non-obese Diabetics: Pre- and Postinsulin Effects. N Am J Med Sci 4: 180–4, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Graham TE, Yang Q, Bluher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson PA, Smith U and Kahn BB. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. New Engl J Med 354: 2552–63, 2006. [DOI] [PubMed] [Google Scholar]
- 153.Gregoire FM, Smas CM and Sul HS. Understanding adipocyte differentiation. Physiol Rev 78: 783–809, 1998. [DOI] [PubMed] [Google Scholar]
- 154.Gregor MF and Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415–45, 2011. [DOI] [PubMed] [Google Scholar]
- 155.Gregor MF and Hotamisligil GS. Thematic review series: Adipocyte Biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res 48: 1905–14, 2007. [DOI] [PubMed] [Google Scholar]
- 156.Grigsby RJ and Dobrowsky RT. Inhibition of ceramide production reverses TNF-induced insulin resistance. Biochem Biophys Res Commun 287: 1121–4, 2001. [DOI] [PubMed] [Google Scholar]
- 157.Gumbau V, Bruna M, Canelles E, Guaita M, Mulas C, Bases C, Celma I, Puche J, Marcaida G, Oviedo M and Vazquez A. A prospective study on inflammatory parameters in obese patients after sleeve gastrectomy. Obes Surg 24: 903–8, 2014. [DOI] [PubMed] [Google Scholar]
- 158.Gupta D, Krueger CB and Lastra G. Over-nutrition, obesity and insulin resistance in the development of beta-cell dysfunction. Curr Diabetes Rev 8: 76–83, 2012. [DOI] [PubMed] [Google Scholar]
- 159.Gutierrez DA, Kennedy A, Orr JS, Anderson EK, Webb CD, Gerrald WK and Hasty AH. Aberrant accumulation of undifferentiated myeloid cells in the adipose tissue of CCR2-deficient mice delays improvements in insulin sensitivity. Diabetes 60: 2820–9, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Haase J, Weyer U, Immig K, Kloting N, Bluher M, Eilers J, Bechmann I and Gericke M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 57: 562–71, 2014. [DOI] [PubMed] [Google Scholar]
- 161.Hadad N, Burgazliev O, Elgazar-Carmon V, Solomonov Y, Wueest S, Item F, Konrad D, Rudich A and Levy R. Induction of cytosolic phospholipase a2alpha is required for adipose neutrophil infiltration and hepatic insulin resistance early in the course of high-fat feeding. Diabetes 62: 3053–63, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hagman DK, Larson I, Kuzma JN, Cromer G, Makar K, Rubinow KB, Foster-Schubert KE, van Yserloo B, Billing PS, Landerholm RW, Crouthamel M, Flum DR, Cummings DE and Kratz M. The short-term and long-term effects of bariatric/metabolic surgery on subcutaneous adipose tissue inflammation in humans. Metabolism 70: 12–22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA and Scherer PE. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29: 4467–83, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB and Gudkov AV. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 8: 1294–315, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA and Davis RJ. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339: 218–22, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Haque WA, Shimomura I, Matsuzawa Y and Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab 87: 2395, 2002. [DOI] [PubMed] [Google Scholar]
- 167.Hardy OT, Perugini RA, Nicoloro SM, Gallagher-Dorval K, Puri V, Straubhaar J and Czech MP. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg Obes Relat Dis 7: 60–7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Harlev A, Aricha-Tamir B, Shaco-Levy R, Tarnovscki T, Bashan N, Rudich A, Sheiner E, Press F and Wiznitzer A. Macrophage infiltration and stress-signaling in omental and subcutaneous adipose tissue in diabetic pregnancies. J Matern Fetal Neonatal Med 27: 1189–94, 2014. [DOI] [PubMed] [Google Scholar]
- 169.Harman-Boehm I, Bluher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, Shai I, Kloting N, Stumvoll M, Bashan N and Rudich A. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab 92: 2240–7, 2007. [DOI] [PubMed] [Google Scholar]
- 170.Harmon DB, Srikakulapu P, Kaplan JL, Oldham SN, McSkimming C, Garmey JC, Perry HM, Kirby JL, Prohaska TA, Gonen A, Hallowell P, Schirmer B, Tsimikas S, Taylor AM, Witztum JL and McNamara CA. Protective Role for B-1b B Cells and IgM in Obesity-Associated Inflammation, Glucose Intolerance, and Insulin Resistance. Arterioscler Thromb Vasc Biol 36: 682–91, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA and Kirwan JP. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58: 337–43, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Henegar C, Tordjman J, Achard V, Lacasa D, Cremer I, Guerre-Millo M, Poitou C, Basdevant A, Stich V, Viguerie N, Langin D, Bedossa P, Zucker JD and Clement K. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol 9: R14, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Heni M, Machann J, Staiger H, Schwenzer NF, Peter A, Schick F, Claussen CD, Stefan N, Haring HU and Fritsche A. Pancreatic fat is negatively associated with insulin secretion in individuals with impaired fasting glucose and/or impaired glucose tolerance: a nuclear magnetic resonance study. Diabetes Metab Res Rev 26: 200–5, 2010. [DOI] [PubMed] [Google Scholar]
- 174. Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, Klein S and Kahn BB. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484: 333–8, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hernandez-Corbacho MJ, Canals D, Adada MM, Liu M, Senkal CE, Yi JK, Mao C, Luberto C, Hannun YA and Obeid LM. Tumor Necrosis Factor-alpha (TNFalpha)-induced Ceramide Generation via Ceramide Synthases Regulates Loss of Focal Adhesion Kinase (FAK) and Programmed Cell Death. J Biol Chem 290: 25356–73, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Herrero L, Shapiro H, Nayer A, Lee J and Shoelson SE. Inflammation and adipose tissue macrophages in lipodystrophic mice. Proc Natl Acad Sci U S A 107: 240–5, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, Watt MJ, Benner C, Febbraio MA, Nguyen AK, Folian B, Subramaniam S, Gonzalez FJ, Glass CK and Ricote M. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest 117: 1658–69, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hill AA, Bolus Reid W and Hasty AH. A decade of progress in adipose tissue macrophage biology. Immunol Rev 262: 134–52, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hodson L, Humphreys SM, Karpe F and Frayn KN. Metabolic signatures of human adipose tissue hypoxia in obesity. Diabetes 62: 1417–25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hoenicke L and Zender L. Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis 33: 1123–6, 2012. [DOI] [PubMed] [Google Scholar]
- 181.Holdstock C, Lind L, Engstrom BE, Ohrvall M, Sundbom M, Larsson A and Karlsson FA. CRP reduction following gastric bypass surgery is most pronounced in insulin-sensitive subjects. Int J Obes (Lond) 29: 1275–80, 2005. [DOI] [PubMed] [Google Scholar]
- 182.Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE and Summers SA. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121: 1858–70, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ and Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5: 167–79, 2007. [DOI] [PubMed] [Google Scholar]
- 184.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA and Scherer PE. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17: 55–63, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, Quittner-Strom E, Tippetts TS, Gordillo R and Scherer PE. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab 6: 267–275, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M and Shimomura I. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56: 901–11, 2007. [DOI] [PubMed] [Google Scholar]
- 187. Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860–7, 2006. [DOI] [PubMed] [Google Scholar]
- 188.Hotamisligil GS, Arner P, Caro JF, Atkinson RL and Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95: 2409–15, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hotamisligil GS, Shargill NS and Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87–91, 1993. [DOI] [PubMed] [Google Scholar]
- 190.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, Nishida M, Kihara S, Sakai N, Nakajima T, Hasegawa K, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Hanafusa T and Matsuzawa Y. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 20: 1595–9, 2000. [DOI] [PubMed] [Google Scholar]
- 191.Hsu WC, Araneta MR, Kanaya AM, Chiang JL and Fujimoto W. BMI cut points to identify at-risk Asian Americans for type 2 diabetes screening. Diabetes Care 38: 150–8, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hu E, Liang P and Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem 271: 10697–703, 1996. [DOI] [PubMed] [Google Scholar]
- 193.Huang-Doran I, Sleigh A, Rochford JJ, O’Rahilly S and Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol 207: 245–55, 2010. [DOI] [PubMed] [Google Scholar]
- 194.Huber J, Kiefer FW, Zeyda M, Ludvik B, Silberhumer GR, Prager G, Zlabinger GJ and Stulnig TM. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J Clin Endocrinol Metab 93: 3215–21, 2008. [DOI] [PubMed] [Google Scholar]
- 195.Hummel KP, Dickie MM and Coleman DL. Diabetes, a new mutation in the mouse. Science 153: 1127–8, 1966. [DOI] [PubMed] [Google Scholar]
- 196.Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE and Shulman GI. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 109: 1321–6, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hunter GR, Gower BA and Kane BL. Age Related Shift in Visceral Fat. Int J Body Compos Res 8: 103–108, 2010. [PMC free article] [PubMed] [Google Scholar]
- 198.Hyatt TC, Phadke RP, Hunter GR, Bush NC, Munoz AJ and Gower BA. Insulin sensitivity in African-American and white women: association with inflammation. Obesity (Silver Spring) 17: 276–82, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Iannelli A, Anty R, Piche T, Dahman M, Gual P, Tran A and Gugenheim J. Impact of laparoscopic Roux-en-Y gastric bypass on metabolic syndrome, inflammation, and insulin resistance in super versus morbidly obese women. Obes Surg 19: 577–82, 2009. [DOI] [PubMed] [Google Scholar]
- 200.Iannelli A, Anty R, Schneck AS, Tran A and Gugenheim J. Inflammation, insulin resistance, lipid disturbances, anthropometrics, and metabolic syndrome in morbidly obese patients: a case control study comparing laparoscopic Roux-en-Y gastric bypass and laparoscopic sleeve gastrectomy. Surgery 149: 364–70, 2011. [DOI] [PubMed] [Google Scholar]
- 201.Iannelli A, Martini F, Rodolphe A, Schneck AS, Gual P, Tran A, Hebuterne X and Gugenheim J. Body composition, anthropometrics, energy expenditure, systemic inflammation, in premenopausal women 1 year after laparoscopic Roux-en-Y gastric bypass. Surg Endosc 28: 500–7, 2014. [DOI] [PubMed] [Google Scholar]
- 202.Illan-Gomez F, Gonzalvez-Ortega M, Orea-Soler I, Alcaraz-Tafalla MS, Aragon-Alonso A, Pascual-Diaz M, Perez-Paredes M and Lozano-Almela ML. Obesity and inflammation: change in adiponectin, C-reactive protein, tumour necrosis factor-alpha and interleukin-6 after bariatric surgery. Obes Surg 22: 950–5, 2012. [DOI] [PubMed] [Google Scholar]
- 203.Ingalls AM, Dickie MM and Snell GD. Obese, a new mutation in the house mouse. J Hered 41: 317–8, 1950. [DOI] [PubMed] [Google Scholar]
- 204.Jaworski K, Sarkadi-Nagy E, Duncan RE, Ahmadian M and Sul HS. Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am J Physiol Gastrointest Liver Physiol 293: G1–4, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS and Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332: 1284–8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Jensen MD, Haymond MW, Rizza RA, Cryer PE and Miles JM. Influence of body fat distribution on free fatty acid metabolism in obesity. J Clin Invest 83: 1168–73, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ji Y, Sun S, Xia S, Yang L, Li X and Qi L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem 287: 24378–86, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, Lee S, Scherer PE and Elmquist JK. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun 5: 3878, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jiao P, Chen Q, Shah S, Du J, Tao B, Tzameli I, Yan W and Xu H. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes 58: 104–15, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jin C and Flavell RA. Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol 132: 287–94, 2013. [DOI] [PubMed] [Google Scholar]
- 211.Jo J, Guo J, Liu T, Mullen S, Hall KD, Cushman SW and Periwal V. Hypertrophy-driven adipocyte death overwhelms recruitment under prolonged weight gain. Biophys J 99: 3535–44, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Johannsen DL, Tchoukalova Y, Tam CS, Covington JD, Xie W, Schwarz JM, Bajpeyi S and Ravussin E. Effect of 8 weeks of overfeeding on ectopic fat deposition and insulin sensitivity: testing the “adipose tissue expandability” hypothesis. Diabetes Care 37: 2789–97, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Jones PA and Scott-Burden T. Activated macrophages digest the extracellular matrix proteins produced by cultured cells. Biochem Biophys Res Commun 86: 71–7, 1979. [DOI] [PubMed] [Google Scholar]
- 214.Kadowaki T and Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev 26: 439–51, 2005. [DOI] [PubMed] [Google Scholar]
- 215.Kahn SE, Cooper ME and Prato Del S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383: 1068–83, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kahn SE, Hull RL and Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840–6, 2006. [DOI] [PubMed] [Google Scholar]
- 217.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC and Palmer JP. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42: 1663–1672, 1993. [DOI] [PubMed] [Google Scholar]
- 218.Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, Yamauchi T, Ueki K, Oishi Y, Nishimura S, Manabe I, Hashimoto H, Ohnishi Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Nagai R and Kadowaki T. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 281: 26602–14, 2006. [DOI] [PubMed] [Google Scholar]
- 219.Kammoun HL, Kraakman MJ and Febbraio MA. Adipose tissue inflammation in glucose metabolism. Rev Endocr Metab Disord 15: 31–44, 2014. [DOI] [PubMed] [Google Scholar]
- 220.Kanasaki K and Koya D. Biology of obesity: lessons from animal models of obesity. J Biomed Biotechnol 2011: 197636, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K and Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116: 1494–1505, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S and Zender L. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–51, 2011. [DOI] [PubMed] [Google Scholar]
- 223.Kappes A and Loffler G. Influences of ionomycin, dibutyryl-cycloAMP and tumour necrosis factor-alpha on intracellular amount and secretion of apM1 in differentiating primary human preadipocytes. Horm Metab Res 32: 548–54, 2000. [DOI] [PubMed] [Google Scholar]
- 224.Karpe F, Dickmann JR and Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes 60: 2441–9, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Katakura T, Miyazaki M, Kobayashi M, Herndon DN and Suzuki F. CCL17 and IL-10 as effectors that enable alternatively activated macrophages to inhibit the generation of classically activated macrophages. J Immunol 172: 1407–13, 2004. [DOI] [PubMed] [Google Scholar]
- 226.Kaufman RJ, Scheuner D, Schroder M, Shen X, Lee K, Liu CY and Arnold SM. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol 3: 411–21, 2002. [DOI] [PubMed] [Google Scholar]
- 227.Kawasaki N, Asada R, Saito A, Kanemoto S and Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep 2: 799, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kern PA, Di Gregorio GB, Lu T, Rassouli N and Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 52: 1779–85, 2003. [DOI] [PubMed] [Google Scholar]
- 229.Kern PA, Ranganathan S, Li C, Wood L and Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol 280: E745–51, 2001. [DOI] [PubMed] [Google Scholar]
- 230.Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R and Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 95: 2111–2119, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S and Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29: 1575–91, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Khazen W, M’Bika JP, Tomkiewicz C, Benelli C, Chany C, Achour A and Forest C. Expression of macrophage-selective markers in human and rodent adipocytes. FEBS Lett 579: 5631–4, 2005. [DOI] [PubMed] [Google Scholar]
- 233.Kim CS, Park HS, Kawada T, Kim JH, Lim D, Hubbard NE, Kwon BS, Erickson KL and Yu R. Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. Int J Obes (Lond) 30: 1347–55, 2006. [DOI] [PubMed] [Google Scholar]
- 234.Kim DD and Basu A. Estimating the Medical Care Costs of Obesity in the United States: Systematic Review, Meta-Analysis, and Empirical Analysis. Value Health 19: 602–13, 2016. [DOI] [PubMed] [Google Scholar]
- 235.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ and Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621–37, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, Fischer-Posovszky P, Barth TF, Dragun D, Skurk T, Hauner H, Bluher M, Unger T, Wolf AM, Knippschild U, Hombach V and Marx N. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 28: 1304–10, 2008. [DOI] [PubMed] [Google Scholar]
- 237.Kishida K, Kim KK, Funahashi T, Matsuzawa Y, Kang HC and Shimomura I. Relationships between circulating adiponectin levels and fat distribution in obese subjects. J Atheroscler Thromb 18: 592–5, 2011. [DOI] [PubMed] [Google Scholar]
- 238.Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M, Stumvoll M and Bluher M. Insulin-sensitive obesity. Am J Physiol 299: E506–15, [DOI] [PubMed] [Google Scholar]
- 239.Kodama S, Fujihara K, Ishiguro H, Horikawa C, Ohara N, Yachi Y, Tanaka S, Shimano H, Kato K, Hanyu O and Sone H. Quantitative Relationship Between Cumulative Risk Alleles Based on Genome-Wide Association Studies and Type 2 Diabetes Mellitus: A Systematic Review and Meta-analysis. J Epidemiol 28: 3–18, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kodama S, Horikawa C, Fujihara K, Heianza Y, Hirasawa R, Yachi Y, Sugawara A, Tanaka S, Shimano H, Iida KT, Saito K and Sone H. Comparisons of the strength of associations with future type 2 diabetes risk among anthropometric obesity indicators, including waist-to-height ratio: a meta-analysis. Am J Epidemiol 176: 959–69, 2012. [DOI] [PubMed] [Google Scholar]
- 241.Kolaczkowska E and Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13: 159–75, 2013. [DOI] [PubMed] [Google Scholar]
- 242.Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K, Naylor S, Rao M, Hubbard B and Farese RV Jr., DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J Clin Invest 120: 756–67, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Konner AC and Bruning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab 22: 16–23, 2011. [DOI] [PubMed] [Google Scholar]
- 244.Korenblat KM, Fabbrini E, Mohammed BS and Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 134: 1369–75, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Korkmaz B, Horwitz MS, Jenne DE and Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62: 726–59, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Korou LM, Doulamis IP, Tzanetakou IP, Mikhailidis DP and Perrea DN. The effect of biological age on the metabolic responsiveness of mice fed a high-fat diet. Lab Anim 47: 241–4, 2013. [DOI] [PubMed] [Google Scholar]
- 247.Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R and Ferrante AW Jr., Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest 120: 3466–79, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Koster A, Ding J, Stenholm S, Caserotti P, Houston DK, Nicklas BJ, You T, Lee JS, Visser M, Newman AB, Schwartz AV, Cauley JA, Tylavsky FA, Goodpaster BH, Kritchevsky SB, Harris TB and Health ABCs. Does the amount of fat mass predict age-related loss of lean mass, muscle strength, and muscle quality in older adults? J Gerontol A Biol Sci Med Sci 66: 888–95, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kotronen A, Yki-Jarvinen H, Sevastianova K, Bergholm R, Hakkarainen A, Pietilainen KH, Juurinen L, Lundbom N and Sorensen TI. Comparison of the relative contributions of intra-abdominal and liver fat to components of the metabolic syndrome. Obesity (Silver Spring) 19: 23–8, 2011. [DOI] [PubMed] [Google Scholar]
- 250.Kovacikova M, Sengenes C, Kovacova Z, Siklova-Vitkova M, Klimcakova E, Polak J, Rossmeislova L, Bajzova M, Hejnova J, Hnevkovska Z, Bouloumie A, Langin D and Stich V. Dietary intervention-induced weight loss decreases macrophage content in adipose tissue of obese women. Int J Obes (Lond) 35: 91–8, 2011. [DOI] [PubMed] [Google Scholar]
- 251.Kowalski GM, Nicholls HT, Risis S, Watson NK, Kanellakis P, Bruce CR, Bobik A, Lancaster GI and Febbraio MA. Deficiency of haematopoietic-cell-derived IL-10 does not exacerbate high-fat-diet-induced inflammation or insulin resistance in mice. Diabetologia 54: 888–99, 2011. [DOI] [PubMed] [Google Scholar]
- 252.Kraakman MJ, Murphy AJ, Jandeleit-Dahm K and Kammoun HL. Macrophage polarization in obesity and type 2 diabetes: weighing down our understanding of macrophage function? Front Immunol 5: 470, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK and Becker L. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20: 614–25, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kratz M, Hagman DK, Kuzma JN, Foster-Schubert KE, Chan CP, Stewart S, van Yserloo B, Westbrook EO, Arterburn DE, Flum DR and Cummings DE. Improvements in glycemic control after gastric bypass occur despite persistent adipose tissue inflammation. Obesity (Silver Spring) 24: 1438–45, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M and Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42: 113–6, 1999. [DOI] [PubMed] [Google Scholar]
- 256.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T and Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem 277: 25863–6, 2002. [DOI] [PubMed] [Google Scholar]
- 257.Lackey DE, Burk DH, Ali MR, Mostaedi R, Smith WH, Park J, Scherer PE, Seay SA, McCoin CS, Bonaldo P and Adams SH. Contributions of adipose tissue architectural and tensile properties toward defining healthy and unhealthy obesity. Am J Physiol 306: E233–46, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, Weir J, Mellett NA, Pernes G, Conway JRW, Lee MKS, Timpson P, Murphy AJ, Masters SL, Gerondakis S, Bartonicek N, Kaczorowski DC, Dinger ME, Meikle PJ, Bond PJ and Febbraio MA. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab 27: 1096–1110 e5, 2018. [DOI] [PubMed] [Google Scholar]
- 259.Landgraf K, Rockstroh D, Wagner IV, Weise S, Tauscher R, Schwartze JT, Loffler D, Buhligen U, Wojan M, Till H, Kratzsch J, Kiess W, Bluher M and Korner A. Evidence of early alterations in adipose tissue biology and function and its association with obesity-related inflammation and insulin resistance in children. Diabetes 64: 1249–61, 2015. [DOI] [PubMed] [Google Scholar]
- 260.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T and Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. New Engl J Med 356: 1517–26, 2007. [DOI] [PubMed] [Google Scholar]
- 261.Larsen GL and Henson PM. Mediators of inflammation. Annu Rev Immunol 1: 335–59, 1983. [DOI] [PubMed] [Google Scholar]
- 262.Lawler HM, Underkofler CM, Kern PA, Erickson C, Bredbeck B and Rasouli N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J Clin Endocrinol Metab 101: 1422–8, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Le KA, Ventura EE, Fisher JQ, Davis JN, Weigensberg MJ, Punyanitya M, Hu HH, Nayak KS and Goran MI. Ethnic differences in pancreatic fat accumulation and its relationship with other fat depots and inflammatory markers. Diabetes Care 34: 485–90, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lee BC and Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta 1842: 446–62, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lee JW, Im JA, Lee HR, Shim JY, Youn BS and Lee DC. Visceral adiposity is associated with serum retinol binding protein-4 levels in healthy women. Obesity (Silver Spring) 15: 2225–32, 2007. [DOI] [PubMed] [Google Scholar]
- 266.Lee MJ, Wu Y and Fried SK. Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Aspects Med 34: 1–11, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, Quehenberger O, Johnson RS and Olefsky JM. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 157: 1339–52, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, Bandyopadhyay GK, Schwendener R, Olefsky J and Kim JB. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60: 2474–83, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lefterova MI, Haakonsson AK, Lazar MA and Mandrup S. PPARgamma and the global map of adipogenesis and beyond. Trends Endocrinol Metab 25: 293–302, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Leibel RL, Edens NK and Fried SK. Physiologic basis for the control of body fat distribution in humans. Annu Rev Nutr 9: 417–43, 1989. [DOI] [PubMed] [Google Scholar]
- 271.Lesniewski LA, Hosch SE, Neels JG, de Luca C, Pashmforoush M, Lumeng CN, Chiang SH, Scadeng M, Saltiel AR and Olefsky JM. Bone marrow-specific Cap gene deletion protects against high-fat diet-induced insulin resistance. Nat Med 13: 455–62, 2007. [DOI] [PubMed] [Google Scholar]
- 272.Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, Ryu D, Koo SH, Kim HL, Kim J, Komatsu M and Lee MS. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun 5: 4934, 2014. [DOI] [PubMed] [Google Scholar]
- 273.Liska D, Dufour S, Zern TL, Taksali S, Cali AM, Dziura J, Shulman GI, Pierpont BM and Caprio S. Interethnic differences in muscle, liver and abdominal fat partitioning in obese adolescents. PLoS One 2: e569, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, Doria A, Libby P, Blumberg RS, Kahn BB, Hotamisligil GS and Shi GP. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 15: 940–5, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Liu Q, Yuan B, Lo KA, Patterson HC, Sun Y and Lodish HF. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc Natl Acad Sci U S A 109: 14568–73, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Liu X, Miyazaki M, Flowers MT, Sampath H, Zhao M, Chu K, Paton CM, Joo DS and Ntambi JM. Loss of Stearoyl-CoA desaturase-1 attenuates adipocyte inflammation: effects of adipocyte-derived oleate. Arterioscler Thromb Vasc Biol 30: 31–8, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Loll PJ, Picot D and Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol 2: 637–43, 1995. [DOI] [PubMed] [Google Scholar]
- 278.Lopez-Otin C, Blasco MA, Partridge L, Serrano M and Kroemer G. The hallmarks of aging. Cell 153: 1194–217, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, De Lucia Rolfe E, Stewart ID, Wheeler E, Willems SM, Adams C, Yaghootkar H, Consortium EP-I, Cambridge FC, Forouhi NG, Khaw KT, Johnson AD, Semple RK, Frayling T, Perry JR, Dermitzakis E, McCarthy MI, Barroso I, Wareham NJ, Savage DB, Langenberg C, O’Rahilly S and Scott RA. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 49: 17–26, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Lumeng CN, Bodzin JL and Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of clinical investigation 117: 175–84, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lutz TA and Woods SC. Overview of animal models of obesity. Curr Protoc Pharmacol Chapter 5: Unit5 61, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Lynch L, O’Shea D, Winter DC, Geoghegan J, Doherty DG and O’Farrelly C. Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol 39: 1893–901, 2009. [DOI] [PubMed] [Google Scholar]
- 283.Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y and Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem Biophys Res Commun 221: 286–9, 1996. [DOI] [PubMed] [Google Scholar]
- 284.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I and Matsuzawa Y. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 50: 2094–9, 2001. [DOI] [PubMed] [Google Scholar]
- 285.Magkos F, Fabbrini E, Mohammed BS, Patterson BW and Klein S. Increased whole-body adiposity without a concomitant increase in liver fat is not associated with augmented metabolic dysfunction. Obesity (Silver Spring) 18: 1510–5, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, de Las Fuentes L, He S, Okunade AL, Patterson BW and Klein S. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab 23: 591–601, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, Parker RA, Suttles J, Fazio S, Hotamisligil GS and Linton MF. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med 7: 699–705, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Manolopoulos KN, Karpe F and Frayn KN. Gluteofemoral body fat as a determinant of metabolic health. Int J Obes (Lond) 34: 949–59, 2010. [DOI] [PubMed] [Google Scholar]
- 289.Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ and O’Doherty RM. Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS One 6: e19831, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Mantovani A, Sozzani S, Locati M, Allavena P and Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–55, 2002. [DOI] [PubMed] [Google Scholar]
- 291.Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS and Kahn CR. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 340: 925–9, 1992. [DOI] [PubMed] [Google Scholar]
- 292.Martinez FO and Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000 Prime Rep 6: 13, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Martinez JA, Milagro FI, Claycombe KJ and Schalinske KL. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr 5: 71–81, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Maskarinec G, Grandinetti A, Matsuura G, Sharma S, Mau M, Henderson BE and Kolonel LN. Diabetes prevalence and body mass index differ by ethnicity: the Multiethnic Cohort. Ethn Dis 19: 49–55, 2009. [PMC free article] [PubMed] [Google Scholar]
- 295.Mathis D Immunological goings-on in visceral adipose tissue. Cell Metab 17: 851–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Matos LN, Giorelli Gde V and Dias CB. Correlation of anthropometric indicators for identifying insulin sensitivity and resistance. Sao Paulo Med J 129: 30–5, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Bluher M, Kolanus W, Kahn CR and Bruning JC. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet 6: e1000938, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Mayer J, Bates MW and Dickie MM. Hereditary diabetes in genetically obese mice. Science 113: 746–7, 1951. [DOI] [PubMed] [Google Scholar]
- 299.McGillicuddy FC, Harford KA, Reynolds CM, Oliver E, Claessens M, Mills KH and Roche HM. Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes 60: 1688–98, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.McLaughlin T, Deng A, Gonzales O, Aillaud M, Yee G, Lamendola C, Abbasi F, Connolly AJ, Sherman A, Cushman SW, Reaven G and Tsao PS. Insulin resistance is associated with a modest increase in inflammation in subcutaneous adipose tissue of moderately obese women. Diabetologia 51: 2303–8, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.McLaughlin T, Lamendola C, Liu A and Abbasi F. Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J Clin Endocrinol Metab 96: E1756–60, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.McNelis JC and Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity 41: 36–48, 2014. [DOI] [PubMed] [Google Scholar]
- 303.Meakin PJ, Morrison VL, Sneddon CC, Savinko T, Uotila L, Jalicy SM, Gabriel JL, Kang L, Ashford ML and Fagerholm SC. Mice Lacking beta2-Integrin Function Remain Glucose Tolerant in Spite of Insulin Resistance, Neutrophil Infiltration and Inflammation. PLoS One 10: e0138872, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Medzhitov R Origin and physiological roles of inflammation. Nature 454: 428–35, 2008. [DOI] [PubMed] [Google Scholar]
- 305.Meikle PJ and Summers SA. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat Rev Endocrinol 13: 79–91, 2017. [DOI] [PubMed] [Google Scholar]
- 306.Menke A, Casagrande S, Geiss L and Cowie CC. Prevalence of and Trends in Diabetes Among Adults in the United States, 1988–2012. JAMA 314: 1021–9, 2015. [DOI] [PubMed] [Google Scholar]
- 307.Menke A, Orchard TJ, Imperatore G, Bullard KM, Mayer-Davis E and Cowie CC. The prevalence of type 1 diabetes in the United States. Epidemiology 24: 773–4, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Metter EJ, Conwit R, Tobin J and Fozard JL. Age-associated loss of power and strength in the upper extremities in women and men. J Gerontol A Biol Sci Med Sci 52: B267–76, 1997. [DOI] [PubMed] [Google Scholar]
- 309.Meza-Perez S and Randall TD. Immunological Functions of the Omentum. Trends Immunol 38: 526–536, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Miller GD, Nicklas BJ and Fernandez A. Serial changes in inflammatory biomarkers after Roux-en-Y gastric bypass surgery. Surg Obes Relat Dis 7: 618–24, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Miller KN, Burhans MS, Clark JP, Howell PR, Polewski MA, DeMuth TM, Eliceiri KW, Lindstrom MJ, Ntambi JM and Anderson RM. Aging and caloric restriction impact adipose tissue, adiponectin, and circulating lipids. Aging Cell 16: 497–507, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H and Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105: 1541–4, 2002. [DOI] [PubMed] [Google Scholar]
- 313.Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F and Komuro I. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 15: 1082–7, 2009. [DOI] [PubMed] [Google Scholar]
- 314.Minster RL, Hawley NL, Su CT, Sun G, Kershaw EE, Cheng H, Buhule OD, Lin J, Reupena MS, Viali S, Tuitele J, Naseri T, Urban Z, Deka R, Weeks DE and McGarvey ST. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat Genet 48: 1049–1054, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Mittendorfer B, Magkos F, Fabbrini E, Mohammed BS and Klein S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity (Silver Spring) 17: 1872–7, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Mitterberger MC, Lechner S, Mattesich M and Zwerschke W. Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose-derived stromal/progenitor cells. J Gerontol A Biol Sci Med Sci 69: 13–24, 2014. [DOI] [PubMed] [Google Scholar]
- 317.Miyazaki M, Bruggink SM and Ntambi JM. Identification of mouse palmitoyl-coenzyme A Delta9-desaturase. J Lipid Res 47: 700–4, 2006. [DOI] [PubMed] [Google Scholar]
- 318.Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X and Ntambi JM. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab 6: 484–96, 2007. [DOI] [PubMed] [Google Scholar]
- 319.Miyazaki M, Man WC and Ntambi JM. Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J Nutr 131: 2260–8, 2001. [DOI] [PubMed] [Google Scholar]
- 320.Miyazaki M, Sampath H, Liu X, Flowers MT, Chu K, Dobrzyn A and Ntambi JM. Stearoyl-CoA desaturase-1 deficiency attenuates obesity and insulin resistance in leptin-resistant obese mice. Biochem Biophys Res Commun 380: 818–22, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, Klein S and Coppack SW. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab 82: 4196–200, 1997. [DOI] [PubMed] [Google Scholar]
- 322.Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, Feigenbaum L, Lee E, Aoyama T, Eckhaus M, Reitman ML and Vinson C. Life without white fat: a transgenic mouse. Genes Dev 12: 3168–81, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Monte SV, Caruana JA, Ghanim H, Sia CL, Korzeniewski K, Schentag JJ and Dandona P. Reduction in endotoxemia, oxidative and inflammatory stress, and insulin resistance after Roux-en-Y gastric bypass surgery in patients with morbid obesity and type 2 diabetes mellitus. Surgery 151: 587–93, 2012. [DOI] [PubMed] [Google Scholar]
- 324.Monteiro R, de Castro PM, Calhau C and Azevedo I. Adipocyte size and liability to cell death. Obes Surg 16: 804–6, 2006. [DOI] [PubMed] [Google Scholar]
- 325.Morinigo R, Casamitjana R, Delgado S, Lacy A, Deulofeu R, Conget I, Barcelo-Batllori S, Gomis R and Vidal J. Insulin resistance, inflammation, and the metabolic syndrome following Roux-en-Y gastric bypass surgery in severely obese subjects. Diabetes Care 30: 1906–8, 2007. [DOI] [PubMed] [Google Scholar]
- 326.Morton GJ, Cummings DE, Baskin DG, Barsh GS and Schwartz MW. Central nervous system control of food intake and body weight. Nature 443: 289–95, 2006. [DOI] [PubMed] [Google Scholar]
- 327.Moschen AR, Molnar C, Geiger S, Graziadei I, Ebenbichler CF, Weiss H, Kaser S, Kaser A and Tilg H. Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut 59: 1259–64, 2010. [DOI] [PubMed] [Google Scholar]
- 328.Moussa NM and Claycombe KJ. The yellow mouse obesity syndrome and mechanisms of agouti-induced obesity. Obesity Res 7: 506–14, 1999. [DOI] [PubMed] [Google Scholar]
- 329.Mraz M and Haluzik M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol 222: R113–27, 2014. [DOI] [PubMed] [Google Scholar]
- 330.Mraz M, Lacinova Z, Drapalova J, Haluzikova D, Horinek A, Matoulek M, Trachta P, Kavalkova P, Svacina S and Haluzik M. The effect of very-low-calorie diet on mRNA expression of inflammation-related genes in subcutaneous adipose tissue and peripheral monocytes of obese patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 96: E606–13, 2011. [DOI] [PubMed] [Google Scholar]
- 331.Muir LA, Neeley CK, Meyer KA, Baker NA, Brosius AM, Washabaugh AR, Varban OA, Finks JF, Zamarron BF, Flesher CG, Chang JS, DelProposto JB, Geletka L, Martinez-Santibanez G, Kaciroti N, Lumeng CN and O’Rourke RW. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity (Silver Spring) 24: 597–605, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M and Cinti S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res 49: 1562–8, 2008. [DOI] [PubMed] [Google Scholar]
- 333.Murphy K, Travers P and Walport M. Janeway’s Immunobiology. Garland Science, Taylor & Francis Group, LLC, New York, 2008. [Google Scholar]
- 334.Nadler ST, Stoehr JP, Schueler KL, Tanimoto G, Yandell BS and Attie AD. The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc Natl Acad Sci U S A 97: 11371–6, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Nakano Y, Tobe T, Choi-Miura NH, Mazda T and Tomita M. Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J Biochem 120: 803–12, 1996. [DOI] [PubMed] [Google Scholar]
- 336.Nathan C and Neutrophils immunity: challenges and opportunities. Nat Rev Immunol 6: 173–82, 2006. [DOI] [PubMed] [Google Scholar]
- 337.Newman WP and Brodows RG. Aspirin causes tissue insensitivity to insulin in normal man. J Clin Endocrinol Metab 57: 1102–6, 1983. [DOI] [PubMed] [Google Scholar]
- 338.Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM and Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480: 104–8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG and Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 282: 35279–92, 2007. [DOI] [PubMed] [Google Scholar]
- 340.Ni Y, Zhao L, Yu H, Ma X, Bao Y, Rajani C, Loo LW, Shvetsov YB, Yu H, Chen T, Zhang Y, Wang C, Hu C, Su M, Xie G, Zhao A, Jia W and Jia W. Circulating Unsaturated Fatty Acids Delineate the Metabolic Status of Obese Individuals. EBioMedicine 2: 1513–22, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Niccoli T and Partridge L. Ageing as a risk factor for disease. Curr Biol 22: R741–52, 2012. [DOI] [PubMed] [Google Scholar]
- 342.Nicholls HT, Kowalski G, Kennedy DJ, Risis S, Zaffino LA, Watson N, Kanellakis P, Watt MJ, Bobik A, Bonen A, Febbraio M, Lancaster GI and Febbraio MA. Hematopoietic cell-restricted deletion of CD36 reduces high-fat diet-induced macrophage infiltration and improves insulin signaling in adipose tissue. Diabetes 60: 1100–10, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA and Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) 17: 2014–8, 2009. [DOI] [PubMed] [Google Scholar]
- 344.Nishimoto S, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Murata C, Kim-Kaneyama JR, Sato F, Bando M, Yagi S, Soeki T, Hayashi T, Imoto I, Sakaue H, Shimabukuro M and Sata M. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci Adv 2: e1501332, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T and Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15: 914–20, 2009. [DOI] [PubMed] [Google Scholar]
- 346.Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, Jones KL, Kawamori R, Cassis LA, Tschop MH and Bruemmer D. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest 117: 2877–88, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM and Attie AD. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci U S A 99: 11482–6, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.O’Connell J, Lynch L, Cawood TJ, Kwasnik A, Nolan N, Geoghegan J, McCormick A, O’Farrelly C and O’Shea D. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PLoS One 5: e9997, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.O’Rourke RW, White AE, Metcalf MD, Olivas AS, Mitra P, Larison WG, Cheang EC, Varlamov O, Corless CL, Roberts CT Jr., and Marks DL. Hypoxia-induced inflammatory cytokine secretion in human adipose tissue stromovascular cells. Diabetologia 54: 1480–90, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.O’Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, Zhu X and Marks DL. Systemic inflammation and insulin sensitivity in obese IFN-gamma knockout mice. Metabolism 61: 1152–61, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Ofei F, Hurel S, Newkirk J, Sopwith M and Taylor R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45: 881–5, 1996. [DOI] [PubMed] [Google Scholar]
- 352.Ogden CL, Carroll MD, Kit BK and Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 311: 806–14, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Oguz FM, Oguz A and Uzunlulu M. The effect of infliximab treatment on insulin resistance in patients with rheumatoid arthritis. Acta Clin Belg 62: 218–22, 2007. [DOI] [PubMed] [Google Scholar]
- 354.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM and Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687–98, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Okamoto Y, Higashiyama H, Rong JX, McVey MJ, Kinoshita M, Asano S and Hansen MK. Comparison of mitochondrial and macrophage content between subcutaneous and visceral fat in db/db mice. Exp Mol Pathol 83: 73–83, 2007. [DOI] [PubMed] [Google Scholar]
- 356.Olefsky JM and Glass CK. Macrophages, inflammation, and insulin resistance. Ann Rev Physiol 72: 219–46, 2010. [DOI] [PubMed] [Google Scholar]
- 357.de Victoria Ortega Martinez E, Xu X, Koska J, Francisco AM, Scalise M, Ferrante AW Jr., and Krakoff J. Macrophage content in subcutaneous adipose tissue: associations with adiposity, age, inflammatory markers, and whole-body insulin action in healthy Pima Indians. Diabetes 58: 385–93, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH and Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–61, 2004. [DOI] [PubMed] [Google Scholar]
- 359.Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M and Scherer PE. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem 278: 9073–85, 2003. [DOI] [PubMed] [Google Scholar]
- 360.Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, Utzschneider KM, Kahn SE, Olefsky JM, Buchanan TA and Scherer PE. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J Biol Chem 279: 12152–62, 2004. [DOI] [PubMed] [Google Scholar]
- 361.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS and Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med 18: 1279–85, 2012. [DOI] [PubMed] [Google Scholar]
- 362.Palmer AK and Kirkland JL. Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp Gerontol 86: 97–105, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB and Storlien LH. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46: 983–8, 1997. [DOI] [PubMed] [Google Scholar]
- 364.Pandolfi JB, Ferraro AA, Sananez I, Gancedo MC, Baz P, Billordo LA, Fainboim L and Arruvito L. ATP-Induced Inflammation Drives Tissue-Resident Th17 Cells in Metabolically Unhealthy Obesity. J Immunol 196: 3287–96, 2016. [DOI] [PubMed] [Google Scholar]
- 365.Paolisso G, Tataranni PA, Foley JE, Bogardus C, Howard BV and Ravussin E. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia 38: 1213–7, 1995. [DOI] [PubMed] [Google Scholar]
- 366.Paquot N, Castillo MJ, Lefebvre PJ and Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 85: 1316–9, 2000. [DOI] [PubMed] [Google Scholar]
- 367.Paradisi G, Smith L, Burtner C, Leaming R, Garvey WT, Hook G, Johnson A, Cronin J, Steinberg HO and Baron AD. Dual energy X-ray absorptiometry assessment of fat mass distribution and its association with the insulin resistance syndrome. Diabetes Care 22: 1310–7, 1999. [DOI] [PubMed] [Google Scholar]
- 368.Pardina E, Ferrer R, Baena-Fustegueras JA, Rivero J, Lecube A, Fort JM, Vargas V, Catalan R and Peinado-Onsurbe J. Only C-reactive protein, but not TNF-alpha or IL6, reflects the improvement in inflammation after bariatric surgery. Obes Surg 22: 131–9, 2012. [DOI] [PubMed] [Google Scholar]
- 369.Park YS, Uddin MJ, Piao L, Hwang I, Lee JH and Ha H. Novel Role of Endogenous Catalase in Macrophage Polarization in Adipose Tissue. Mediators Inflamm 2016: 8675905, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, Mehrabian M, Ursell LK, He A, Castellani LW, Zinker B, Kirby M, Drake TA, Drevon CA, Knight R, Gargalovic P, Kirchgessner T, Eskin E and Lusis AJ. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17: 141–52, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, Castellani LW, Rau CD, Pan C, Phun J, Zhou Z, Yang WP, Neuhaus I, Gargalovic PS, Kirchgessner TG, Graham M, Lee R, Tontonoz P, Gerszten RE, Hevener AL and Lusis AJ. Genetic architecture of insulin resistance in the mouse. Cell Metab 21: 334–46, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, Rood JC, Burk DH and Smith SR. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58: 718–25, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Patel P and Abate N. Role of subcutaneous adipose tissue in the pathogenesis of insulin resistance. J Obes 2013: 489187, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8: 301–309, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P and Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 109: 1345–50, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Petersen PS, Lei X, Wolf RM, Rodriguez S, Tan SY, Little HC, Schweitzer MA, Magnuson TH, Steele KE and Wong GW. CTRP7 deletion attenuates obesity-linked glucose intolerance, adipose tissue inflammation, and hepatic stress. Am J Physiol 312: E309–E325, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6: 541–50, 2006. [DOI] [PubMed] [Google Scholar]
- 378.Phieler J, Chung KJ, Chatzigeorgiou A, Klotzsche-von Ameln A, Garcia-Martin R, Sprott D, Moisidou M, Tzanavari T, Ludwig B, Baraban E, Ehrhart-Bornstein M, Bornstein SR, Mziaut H, Solimena M, Karalis KP, Economopoulou M, Lambris JD and Chavakis T. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol 191: 4367–74, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Piya MK, McTernan PG and Kumar S. Adipokine inflammation and insulin resistance: the role of glucose, lipids and endotoxin. J Endocrinol 216: T1–T15, 2013. [DOI] [PubMed] [Google Scholar]
- 380.Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, Peiretti F, Verdier M, Juhan-Vague I, Tanti JF, Burcelin R and Alessi MC. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50: 1267–76, 2007. [DOI] [PubMed] [Google Scholar]
- 381.Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, Braunwald E, Kastelein JJ, de Lemos JA, Blazing MA, Pedersen TR, Tikkanen MJ, Sattar N and Ray KK. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA 305: 2556–64, 2011. [DOI] [PubMed] [Google Scholar]
- 382.Prieur X, Mok CY, Velagapudi VR, Nunez V, Fuentes L, Montaner D, Ishikawa K, Camacho A, Barbarroja N, O’Rahilly S, Sethi JK, Dopazo J, Oresic M, Ricote M and Vidal-Puig A. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes 60: 797–809, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Qatanani M and Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev 21: 1443–55, 2007. [DOI] [PubMed] [Google Scholar]
- 384.Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, Dogra S, Ohman MK, Takeda K, Sugii S, Pewzner-Jung Y, Futerman AH and Summers SA. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 20: 687–95, 2014. [DOI] [PubMed] [Google Scholar]
- 385.Raji A, Seely EW, Arky RA and Simonson DC. Body fat distribution and insulin resistance in healthy Asian Indians and Caucasians. J Clin Endocrinol Metab 86: 5366–71, 2001. [DOI] [PubMed] [Google Scholar]
- 386.Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14: 263–83, 1998. [DOI] [PubMed] [Google Scholar]
- 387.Randle PJ, Garland PB, Newsholme EA and Hales CN. The glucose fatty acid cycle in obesity and maturity onset diabetes mellitus. Ann N Y Acad Sci 131: 324–33, 1965. [DOI] [PubMed] [Google Scholar]
- 388.Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B, Perusse L and Bouchard C. The human obesity gene map: the 2005 update. Obesity (Silver Spring) 14: 529–644, 2006. [DOI] [PubMed] [Google Scholar]
- 389.Reaven GM, Hollenbeck C, Jeng CY, Wu MS and Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 37: 1020–4, 1988. [DOI] [PubMed] [Google Scholar]
- 390.Reid J, Macdougall AI and Andrews MM. Aspirin and diabetes mellitus. Br Med J 2: 1071–4, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC and Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 74: 1–9, 2017. [DOI] [PubMed] [Google Scholar]
- 392.Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, Thuren T and Group CPI. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation 126: 2739–48, 2012. [DOI] [PubMed] [Google Scholar]
- 393.Robbins AL and Savage DB. The genetics of lipid storage and human lipodystrophies. Trends Mol Med 21: 433–8, 2015. [DOI] [PubMed] [Google Scholar]
- 394.Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KL, Shenoy MK, Volmer R, Iwawaki T and Koliwad SK. Saturated Fatty Acids Engage an IRE1alpha-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep 14: 2611–23, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW and Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97: 2859–65, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Rong JX, Qiu Y, Hansen MK, Zhu L, Zhang V, Xie M, Okamoto Y, Mattie MD, Higashiyama H, Asano S, Strum JC and Ryan TE. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 56: 1751–60, 2007. [DOI] [PubMed] [Google Scholar]
- 397.Rosen ED and Spiegelman BM. What we talk about when we talk about fat. Cell 156: 20–44, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Rowe JW, Minaker KL, Pallotta JA and Flier JS. Characterization of the insulin resistance of aging. J Clin Invest 71: 1581–7, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Ruan H and Dong LQ. Adiponectin signaling and function in insulin target tissues. J Mol Cell Biol 8: 101–9, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Rutkowski JM, Stern JH and Scherer PE. The cell biology of fat expansion. J Cell Biol 208: 501–12, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Rytka JM, Wueest S, Schoenle EJ and Konrad D. The portal theory supported by venous drainage-selective fat transplantation. Diabetes 60: 56–63, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM and Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab 10: 419–29, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK and Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322: 1539–43, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK and Davis RJ. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol 30: 106–15, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Sachithanandan N, Graham KL, Galic S, Honeyman JE, Fynch SL, Hewitt KA, Steinberg GR and Kay TW. Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes 60: 2023–31, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM and Whitington PF. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol 287: G1035–43, 2004. [DOI] [PubMed] [Google Scholar]
- 407.Sams VG, Blackledge C, Wijayatunga N, Barlow P, Mancini M, Mancini G and Moustaid-Moussa N. Effect of bariatric surgery on systemic and adipose tissue inflammation. Surg Endosc 2015. [DOI] [PubMed] [Google Scholar]
- 408.Santomauro AT, Boden G, Silva ME, Rocha DM, Santos RF, Ursich MJ, Strassmann PG and Wajchenberg BL. Overnight lowering of free fatty acids with Acipimox improves insulin resistance and glucose tolerance in obese diabetic and nondiabetic subjects. Diabetes 48: 1836–41, 1999. [DOI] [PubMed] [Google Scholar]
- 409.Satoh M, Andoh Y, Clingan CS, Ogura H, Fujii S, Eshima K, Nakayama T, Taniguchi M, Hirata N, Ishimori N, Tsutsui H, Onoe K and Iwabuchi K. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS One 7: e30568, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK and Ford I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 375: 735–42, 2010. [DOI] [PubMed] [Google Scholar]
- 411.Sawada M, Kiyono T, Nakashima S, Shinoda J, Naganawa T, Hara S, Iwama T and Sakai N. Molecular mechanisms of TNF-alpha-induced ceramide formation in human glioma cells: P53-mediated oxidant stress-dependent and -independent pathways. Cell Death Differ 11: 997–1008, 2004. [DOI] [PubMed] [Google Scholar]
- 412.Schafer MJ, White TA, Evans G, Tonne JM, Verzosa GC, Stout MB, Mazula DL, Palmer AK, Baker DJ, Jensen MD, Torbenson MS, Miller JD, Ikeda Y, Tchkonia T, van Deursen JM, Kirkland JL and LeBrasseur NK. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 65: 1606–15, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Scheja L, Heese B, Zitzer H, Michael MD, Siesky AM, Pospisil H, Beisiegel U and Seedorf K. Acute-phase serum amyloid A as a marker of insulin resistance in mice. Exp Diabetes Res 2008: 230837, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Scherer PE, Williams S, Fogliano M, Baldini G and Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270: 26746–9, 1995. [DOI] [PubMed] [Google Scholar]
- 415.Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, Prop S, Meerding J, Hamers N, Besra G, Boon L, Nieuwenhuis EE, Elewaut D, Prakken B, Kersten S, Boes M and Kalkhoven E. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest 122: 3343–54, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Schmidt-Arras D and Rose-John S. IL-6 pathway in the liver: From physiopathology to therapy. J Hepatol 64: 1403–15, 2016. [DOI] [PubMed] [Google Scholar]
- 417.Schmitz J, Evers N, Awazawa M, Nicholls HT, Bronneke HS, Dietrich A, Mauer J, Bluher M and Bruning JC. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol Metab 5: 328–39, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, Rauterberg J and Lorkowski S. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol 180: 5707–19, 2008. [DOI] [PubMed] [Google Scholar]
- 419.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ and Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336: 86–90, 2012. [DOI] [PubMed] [Google Scholar]
- 420.Schwartz MW, Seeley RJ, Tschop MH, Woods SC, Morton GJ, Myers MG and D’Alessio D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 503: 59–66, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, Shao M, Zhao F, He S, Yang L, Zhang M, Nan F, Li J, Liu J, Liu J, Jia W, Qiu Y, Song B, Han JJ, Rui L, Duan SZ and Liu Y. The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol 18: 519–529, 2017. [DOI] [PubMed] [Google Scholar]
- 422.Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, Rasouli N, Spencer HJ, Yao-Borengasser A and Elbein SC. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab 93: 4532–41, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H and Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015–25, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Shi Y and Cheng D. Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am J Physiol 297: E10–8, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Shimizu I, Yoshida Y, Moriya J, Nojima A, Uemura A, Kobayashi Y and Minamino T. Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab 18: 491–504, 2013. [DOI] [PubMed] [Google Scholar]
- 426.Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL and Brown MS. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev 12: 3182–94, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, Yamamoto T, Anzai A, Isobe S, Yoshida N, Itoh H, Manabe I, Sekai M, Hamazaki Y, Fukuda K, Minato N and Sano M. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest 126: 4626–4639, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Shoelson SE, Lee J and Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–801, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Snodgrass RG, Boss M, Zezina E, Weigert A, Dehne N, Fleming I, Brune B and Namgaladze D. Hypoxia Potentiates Palmitate-induced Pro-inflammatory Activation of Primary Human Macrophages. J Biol Chem 291: 413–24, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris A, Scadeng M, Olefsky JM and Karin M. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab 6: 386–97, 2007. [DOI] [PubMed] [Google Scholar]
- 431.Soukas A, Cohen P, Socci ND and Friedman JM. Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev 14: 963–80, 2000. [PMC free article] [PubMed] [Google Scholar]
- 432.Speakman J, Hambly C, Mitchell S and Krol E. Animal models of obesity. Obesity Rev 8 Suppl 1: 55–61, 2007. [DOI] [PubMed] [Google Scholar]
- 433.Spencer M, Unal R, Zhu B, Rasouli N, McGehee RE Jr., Peterson CA and Kern PA. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J Clin Endocrinol Metab 96: E1990–8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Spieker EA and Pyzocha N. Economic Impact of Obesity. Prim Care 43: 83–95, viii-ix, 2016. [DOI] [PubMed] [Google Scholar]
- 435.Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, Bhatnagar A, Jala VR and Haribabu B. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J Immunol 187: 1942–9, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Sramkova V, Rossmeislova L, Krauzova E, Kracmerova J, Koc M, Langin D, Stich V and Siklova M. Comparison of Early (2 Days) and Later (28 Days) Response of Adipose Tissue to Very Low-Calorie Diet in Obese Women. J Clin Endocrinol Metab 101: 5021–5029, 2016. [DOI] [PubMed] [Google Scholar]
- 437.Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS and Grinspoon SK. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab 96: E146–50, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Stefan N, Haring HU, Hu FB and Schulze MB. Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol 1: 152–62, 2013. [DOI] [PubMed] [Google Scholar]
- 439.Stefan N, Kantartzis K, Machann J, Schick F, Thamer C, Rittig K, Balletshofer B, Machicao F, Fritsche A and Haring HU. Identification and characterization of metabolically benign obesity in humans. Arch Intern Med 168: 1609–16, 2008. [DOI] [PubMed] [Google Scholar]
- 440.Stern JH, Rutkowski JM and Scherer PE. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab 23: 770–84, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Steven S, Hollingsworth KG, Al-Mrabeh A, Avery L, Aribisala B, Caslake M and Taylor R. Very Low-Calorie Diet and 6 Months of Weight Stability in Type 2 Diabetes: Pathophysiological Changes in Responders and Nonresponders. Diabetes Care 39: 808–15, 2016. [DOI] [PubMed] [Google Scholar]
- 442.Steven S, Hollingsworth KG, Small PK, Woodcock SA, Pucci A, Aribisala B, Al-Mrabeh A, Daly AK, Batterham RL and Taylor R. Weight Loss Decreases Excess Pancreatic Triacylglycerol Specifically in Type 2 Diabetes. Diabetes Care 39: 158–65, 2016. [DOI] [PubMed] [Google Scholar]
- 443.Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, Kersten S, Muller M, van den Berg WB, van Rooijen N, Wabitsch M, Kullberg BJ, van der Meer JW, Kanneganti T, Tack CJ and Netea MG. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 12: 593–605, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Stienstra R, Tack CJ, Kanneganti TD, Joosten LA and Netea MG. The inflammasome puts obesity in the danger zone. Cell Metab 15: 10–8, 2012. [DOI] [PubMed] [Google Scholar]
- 445.Strable MS and Ntambi JM. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit Rev Biochem Mol Biol 45: 199–214, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC, Qi N, Imai S and Yoshino J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep 16: 1851–60, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Succurro E, Marini MA, Frontoni S, Hribal ML, Andreozzi F, Lauro R, Perticone F and Sesti G. Insulin secretion in metabolically obese, but normal weight, and in metabolically healthy but obese individuals. Obesity (Silver Spring) 16: 1881–6, 2008. [DOI] [PubMed] [Google Scholar]
- 448.Suganami T, Mieda T, Itoh M, Shimoda Y, Kamei Y and Ogawa Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem Biophys Res Commun 354: 45–9, 2007. [DOI] [PubMed] [Google Scholar]
- 449.Summers SA, Garza LA, Zhou H and Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol 18: 5457–64, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA and Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37: 1163–7, 1988. [DOI] [PubMed] [Google Scholar]
- 451.Swarbrick MM and Havel PJ. Physiological, pharmacological, and nutritional regulation of circulating adiponectin concentrations in humans. Metab Syndr Relat Disord 6: 87–102, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Takaya K, Ogawa Y, Isse N, Okazaki T, Satoh N, Masuzaki H, Mori K, Tamura N, Hosoda K and Nakao K. Molecular cloning of rat leptin receptor isoform complementary DNAs--identification of a missense mutation in Zucker fatty (fa/fa) rats. Biochem Biophys Res Commun 225: 75–83, 1996. [DOI] [PubMed] [Google Scholar]
- 453.Takeuchi K and Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol 296: E1195–209, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB and Olefsky JM. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18: 1407–12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Tam CS, Covington JD, Bajpeyi S, Tchoukalova Y, Burk D, Johannsen DL, Zingaretti CM, Cinti S and Ravussin E. Weight gain reveals dramatic increases in skeletal muscle extracellular matrix remodeling. J Clin Endocrinol Metab 99: 1749–57, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Tam CS, Covington JD, Ravussin E, Redman LM and Pennington CT. Little evidence of systemic and adipose tissue inflammation in overweight individuals(dagger). Front Genet 3: 58, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Tam CS, Viardot A, Clement K, Tordjman J, Tonks K, Greenfield JR, Campbell LV, Samocha-Bonet D and Heilbronn LK. Short-term overfeeding may induce peripheral insulin resistance without altering subcutaneous adipose tissue macrophages in humans. Diabetes 59: 2164–70, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Tamura Y, Sugimoto M, Murayama T, Ueda Y, Kanamori H, Ono K, Ariyasu H, Akamizu T, Kita T, Yokode M and Arai H. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol 28: 2195–201, 2008. [DOI] [PubMed] [Google Scholar]
- 459.Tataranni PA and Ortega E. A burning question: does an adipokine-induced activation of the immune system mediate the effect of overnutrition on type 2 diabetes? Diabetes 54: 917–27, 2005. [DOI] [PubMed] [Google Scholar]
- 460.Tateya S, Kim F and Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Front Endocrinol (Lausanne) 4: 93, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Taylor R and Holman RR. Normal weight individuals who develop type 2 diabetes: the personal fat threshold. Clin Sci (Lond) 128: 405–10, 2015. [DOI] [PubMed] [Google Scholar]
- 462.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD and Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell 9: 667–84, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD and Kirkland JL. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab 17: 644–56, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tschop MH and Schwartz MW. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122: 153–62, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Thorpe KE, Florence CS, Howard DH and Joski P. The impact of obesity on rising medical spending. Health Aff (Millwood) Suppl Web Exclusives: W4–480-6, 2004. [DOI] [PubMed] [Google Scholar]
- 466.Thorpe LE, Upadhyay UD, Chamany S, Garg R, Mandel-Ricci J, Kellerman S, Berger DK, Frieden TR and Gwynn C. Prevalence and control of diabetes and impaired fasting glucose in New York City. Diabetes Care 32: 57–62, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Tilg H and Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6: 772–83, 2006. [DOI] [PubMed] [Google Scholar]
- 468.Trachta P, Dostalova I, Haluzikova D, Kasalicky M, Kavalkova P, Drapalova J, Urbanova M, Lacinova Z, Mraz M and Haluzik M. Laparoscopic sleeve gastrectomy ameliorates mRNA expression of inflammation-related genes in subcutaneous adipose tissue but not in peripheral monocytes of obese patients. Mol Cell Endocrinol 383: 96–102, 2014. [DOI] [PubMed] [Google Scholar]
- 469.Trak-Smayra V, Paradis V, Massart J, Nasser S, Jebara V and Fromenty B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int J Exp Pathol 92: 413–21, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Tran TT, Yamamoto Y, Gesta S and Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab 7: 410–20, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Travers RL, Motta AC, Betts JA, Bouloumie A and Thompson D. The impact of adiposity on adipose tissue-resident lymphocyte activation in humans. Int J Obes (Lond) 39: 762–9, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Trayhurn P, Wang B and Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr 100: 227–35, 2008. [DOI] [PubMed] [Google Scholar]
- 473.Trayhurn P and Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 92: 347–55, 2004. [DOI] [PubMed] [Google Scholar]
- 474.Tsigos C, Kyrou I, Chala E, Tsapogas P, Stavridis JC, Raptis SA and Katsilambros N. Circulating tumor necrosis factor alpha concentrations are higher in abdominal versus peripheral obesity. Metabolism 48: 1332–5, 1999. [DOI] [PubMed] [Google Scholar]
- 475.Turer AT and Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia 55: 2319–26, 2012. [DOI] [PubMed] [Google Scholar]
- 476.Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, Mauer J, Xu E, Hammerschmidt P, Bronneke HS, Trifunovic A, LoSasso G, Wunderlich FT, Kornfeld JW, Bluher M, Kronke M and Bruning JC. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 20: 678–86, 2014. [DOI] [PubMed] [Google Scholar]
- 477.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP and Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–6, 2000. [DOI] [PubMed] [Google Scholar]
- 478.Uysal KT, Wiesbrock SM and Hotamisligil GS. Functional analysis of tumor necrosis factor (TNF) receptors in TNF-alpha-mediated insulin resistance in genetic obesity. Endocrinology 139: 4832–8, 1998. [DOI] [PubMed] [Google Scholar]
- 479.Uysal KT, Wiesbrock SM, Marino MW and Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389: 610–4, 1997. [DOI] [PubMed] [Google Scholar]
- 480.Vague J The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr 4: 20–34, 1956. [DOI] [PubMed] [Google Scholar]
- 481.Vallerie SN, Furuhashi M, Fucho R and Hotamisligil GS. A predominant role for parenchymal c-Jun amino terminal kinase (JNK) in the regulation of systemic insulin sensitivity. PLoS One 3: e3151, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.van Beek L, Lips MA, Visser A, Pijl H, Ioan-Facsinay A, Toes R, Berends FJ, Willems van Dijk K, Koning F and van Harmelen V. Increased systemic and adipose tissue inflammation differentiates obese women with T2DM from obese women with normal glucose tolerance. Metabolism 63: 492–501, 2014. [DOI] [PubMed] [Google Scholar]
- 483.van Dielen FM, Buurman WA, Hadfoune M, Nijhuis J and Greve JW. Macrophage inhibitory factor, plasminogen activator inhibitor-1, other acute phase proteins, and inflammatory mediators normalize as a result of weight loss in morbidly obese subjects treated with gastric restrictive surgery. J Clin Endocrinol Metab 89: 4062–8, 2004. [DOI] [PubMed] [Google Scholar]
- 484.Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Hassanzadeh Ghassabeh G, Raes G and De Baetselier P. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood 108: 525–35, 2006. [DOI] [PubMed] [Google Scholar]
- 485.van Vliet-Ostaptchouk JV, Nuotio ML, Slagter SN, Doiron D, Fischer K, Foco L, Gaye A, Gogele M, Heier M, Hiekkalinna T, Joensuu A, Newby C, Pang C, Partinen E, Reischl E, Schwienbacher C, Tammesoo ML, Swertz MA, Burton P, Ferretti V, Fortier I, Giepmans L, Harris JR, Hillege HL, Holmen J, Jula A, Kootstra-Ros JE, Kvaloy K, Holmen TL, Mannisto S, Metspalu A, Midthjell K, Murtagh MJ, Peters A, Pramstaller PP, Saaristo T, Salomaa V, Stolk RP, Uusitupa M, van der Harst P, van der Klauw MM, Waldenberger M, Perola M and Wolffenbuttel BH. The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: a collaborative analysis of ten large cohort studies. BMC Endocr Disord 14: 9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM and Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17: 179–88, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Vasiliauskaite-Brooks I, Sounier R, Rochaix P, Bellot G, Fortier M, Hoh F, De Colibus L, Bechara C, Saied EM, Arenz C, Leyrat C and Granier S. Structural insights into adiponectin receptors suggest ceramidase activity. Nature 544: 120–123, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Vazquez LA, Pazos F, Berrazueta JR, Fernandez-Escalante C, Garcia-Unzueta MT, Freijanes J and Amado JA. Effects of changes in body weight and insulin resistance on inflammation and endothelial function in morbid obesity after bariatric surgery. J Clin Endocrinol Metab 90: 316–22, 2005. [DOI] [PubMed] [Google Scholar]
- 489.Vendrell J, Broch M, Vilarrasa N, Molina A, Gomez JM, Gutierrez C, Simon I, Soler J and Richart C. Resistin, adiponectin, ghrelin, leptin, and proinflammatory cytokines: relationships in obesity. Obesity Res 12: 962–71, 2004. [DOI] [PubMed] [Google Scholar]
- 490.Ventre J, Doebber T, Wu M, MacNaul K, Stevens K, Pasparakis M, Kollias G and Moller DE. Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes 46: 1526–31, 1997. [DOI] [PubMed] [Google Scholar]
- 491.Viana EC, Araujo-Dasilio KL, Miguel GP, Bressan J, Lemos EM, Moyses MR, de Abreu GR, de Azevedo JL, Carvalho PS, Passos-Bueno MR, Errera FI and Bissoli NS. Gastric Bypass and Sleeve Gastrectomy: the Same Impact on IL-6 and TNF-alpha. Prospective Clinical Trial. Obes Surg 23: 1252–61, 2013. [DOI] [PubMed] [Google Scholar]
- 492.Viardot A, Lord RV and Samaras K. The effects of weight loss and gastric banding on the innate and adaptive immune system in type 2 diabetes and prediabetes. J Clin Endocrinol Metab 95: 2845–50, 2010. [DOI] [PubMed] [Google Scholar]
- 493.Vicennati V, Vottero A, Friedman C and Papanicolaou DA. Hormonal regulation of interleukin-6 production in human adipocytes. Int J Obes Relat Metab Disord 26: 905–11, 2002. [DOI] [PubMed] [Google Scholar]
- 494.Vieira-Potter VJ. Inflammation and macrophage modulation in adipose tissues. Cell Microbiol 16: 1484–92, 2014. [DOI] [PubMed] [Google Scholar]
- 495.Viguerie N, Vidal H, Arner P, Holst C, Verdich C, Avizou S, Astrup A, Saris WH, Macdonald IA, Klimcakova E, Clement K, Martinez A, Hoffstedt J, Sorensen TI and Langin D. Adipose tissue gene expression in obese subjects during low-fat and high-fat hypocaloric diets. Diabetologia 48: 123–131, 2005. [DOI] [PubMed] [Google Scholar]
- 496.Villaret A, Galitzky J, Decaunes P, Esteve D, Marques MA, Sengenes C, Chiotasso P, Tchkonia T, Lafontan M, Kirkland JL and Bouloumie A. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 59: 2755–63, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Visser M, Bouter LM, McQuillan GM, Wener MH and Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA 282: 2131–5, 1999. [DOI] [PubMed] [Google Scholar]
- 498.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev 21: 697–738, 2000. [DOI] [PubMed] [Google Scholar]
- 499.Wang M and Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529: 326–35, 2016. [DOI] [PubMed] [Google Scholar]
- 500.Wang Q, Dong Z, Liu X, Song X, Song Q, Shang Q, Jiang Y, Guo C and Zhang L. Programmed cell death-4 deficiency prevents diet-induced obesity, adipose tissue inflammation, and insulin resistance. Diabetes 62: 4132–43, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Warden CH and Fisler JS. Comparisons of diets used in animal models of high-fat feeding. Cell Metab 7: 277, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Watanabe Y, Nakamura T, Ishikawa S, Fujisaka S, Usui I, Tsuneyama K, Ichihara Y, Wada T, Hirata Y, Suganami T, Izaki H, Akira S, Miyake K, Kanayama HO, Shimabukuro M, Sata M, Sasaoka T, Ogawa Y, Tobe K, Takatsu K and Nagai Y. The radioprotective 105/MD-1 complex contributes to diet-induced obesity and adipose tissue inflammation. Diabetes 61: 1199–209, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL and Ferrante AW, Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115–24, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL and Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Wellen KE and Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 115: 1111–9, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ and Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12: 408–15, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Wensveen FM, Valentic S, Sestan M, Wensveen Turk T and Polic B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur J Immunol 45: 2446–56, 2015. [DOI] [PubMed] [Google Scholar]
- 508.Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, Wabitsch M, O’Brien PE and Harrison LC. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 59: 1648–56, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.West DB, Boozer CN, Moody DL and Atkinson RL. Dietary obesity in nine inbred mouse strains. The American journal of physiology 262: R1025–32, 1992. [DOI] [PubMed] [Google Scholar]
- 510.Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE and Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86: 1930–5, 2001. [DOI] [PubMed] [Google Scholar]
- 511.Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, Leong HX, Glassford A, Caimol M, Kenkel JA, Tedder TF, McLaughlin T, Miklos DB, Dosch HM and Engleman EG. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17: 610–7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, Maezawa Y, Drucker DJ, Engleman E, Winer D and Dosch HM. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15: 921–9, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Withrow D and Alter DA. The economic burden of obesity worldwide: a systematic review of the direct costs of obesity. Obesity Rev 12: 131–41, 2011. [DOI] [PubMed] [Google Scholar]
- 514.Wolf AM, Wolf D, Rumpold H, Enrich B and Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun 323: 630–5, 2004. [DOI] [PubMed] [Google Scholar]
- 515.Wong VW, Wong GL, Yeung DK, Abrigo JM, Kong AP, Chan RS, Chim AM, Shen J, Ho CS, Woo J, Chu WC and Chan HL. Fatty pancreas, insulin resistance, and beta-cell function: a population study using fat-water magnetic resonance imaging. Am J Gastroenterol 109: 589–97, 2014. [DOI] [PubMed] [Google Scholar]
- 516.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A and Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332: 243–7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Wu D, Ren Z, Pae M, Han SN and Meydani SN. Diet-induced obesity has a differential effect on adipose tissue and macrophage inflammatory responses of young and old mice. Biofactors 39: 326–33, 2013. [DOI] [PubMed] [Google Scholar]
- 518.Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL, Sweeney JF, Peterson LE, Chan L, Smith CW and Ballantyne CM. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 115: 1029–38, 2007. [DOI] [PubMed] [Google Scholar]
- 519.Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, Kim S, Mendez-Fernandez YV, Besra GS, Lomenick JP, Williams B, Wasserman DH and Van Kaer L. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A 109: E1143–52, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Wueest S, Rapold RA, Schumann DM, Rytka JM, Schildknecht A, Nov O, Chervonsky AV, Rudich A, Schoenle EJ, Donath MY and Konrad D. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest 120: 191–202, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Xia B, Cai GH, Yang H, Wang SP, Mitchell GA and Wu JW. Adipose tissue deficiency of hormone-sensitive lipase causes fatty liver in mice. PLoS Genet 13: e1007110, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, Sifuentes AJ, McDonald JG, Gordillo R and Scherer PE. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab 22: 266–278, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA and Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–30, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Xu J, Yeh CH, Chen S, He L, Sensi SL, Canzoniero LM, Choi DW and Hsu CY. Involvement of de novo ceramide biosynthesis in tumor necrosis factor-alpha/cycloheximide-induced cerebral endothelial cell death. J Biol Chem 273: 16521–6, 1998. [DOI] [PubMed] [Google Scholar]
- 525.Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, Sepe A, Johnson KO, Stout MB, Giorgadze N, Jensen MD, LeBrasseur NK, Tchkonia T and Kirkland JL. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 4: e12997, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ and Ferrante AW, Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18: 816–30, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Xu XJ, Apovian C, Hess D, Carmine B, Saha A and Ruderman N. Improved Insulin Sensitivity 3 Months After RYGB Surgery Is Associated With Increased Subcutaneous Adipose Tissue AMPK Activity and Decreased Oxidative Stress. Diabetes 64: 3155–9, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T and Schultze JL. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40: 274–88, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C and Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–60, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Yamauchi T, Iwabu M, Okada-Iwabu M and Kadowaki T. Adiponectin receptors: a review of their structure, function and how they work. Best Pract Res Clin Endocrinol Metab 28: 15–23, 2014. [DOI] [PubMed] [Google Scholar]
- 531. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB and Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8: 1288–95, 2002. [DOI] [PubMed] [Google Scholar]
- 532.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P and Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7: 941–946, 2001. [DOI] [PubMed] [Google Scholar]
- 533.Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, Ito Y, Kamon J, Tsuchida A, Kumagai K, Kozono H, Hada Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Awazawa M, Takamoto I, Froguel P, Hara K, Tobe K, Nagai R, Ueki K and Kadowaki T. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med 13: 332–9, 2007. [DOI] [PubMed] [Google Scholar]
- 534.Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, Snitker S, Horenstein RB, Hull K, Goldberg NH, Goldberg AP, Shuldiner AR, Fried SK and Gong DW. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med 3: e287, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Yazdani-Biuki B, Mueller T, Brezinschek HP, Hermann J, Graninger W and Wascher TC. Relapse of diabetes after interruption of chronic administration of anti-tumor necrosis factor-alpha antibody infliximab: a case observation. Diabetes Care 29: 1712–3, 2006. [DOI] [PubMed] [Google Scholar]
- 536.Yazdani-Biuki B, Stelzl H, Brezinschek HP, Hermann J, Mueller T, Krippl P, Graninger W and Wascher TC. Improvement of insulin sensitivity in insulin resistant subjects during prolonged treatment with the anti-TNF-alpha antibody infliximab. Eur J Clin Invest 34: 641–2, 2004. [DOI] [PubMed] [Google Scholar]
- 537.Yazdi FT, Clee SM and Meyre D. Obesity genetics in mouse and human: back and forth, and back again. Peer J 3: e856, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Ye J, Gao Z, Yin J and He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol 293: E1118–28, 2007. [DOI] [PubMed] [Google Scholar]
- 539.Yin MJ, Yamamoto Y and Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396: 77–80, 1998. [DOI] [PubMed] [Google Scholar]
- 540.Yin X, Lanza IR, Swain JM, Sarr MG, Nair KS and Jensen MD. Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J Clin Endocrinol Metab 99: E209–16, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Yki-Jarvinen H, Sammalkorpi K, Koivisto VA and Nikkila EA. Severity, duration, and mechanisms of insulin resistance during acute infections. J Clin Endocrinol Metab 69: 317–23, 1989. [DOI] [PubMed] [Google Scholar]
- 542.Yudkin JS, Stehouwer CD, Emeis JJ and Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 19: 972–8, 1999. [DOI] [PubMed] [Google Scholar]
- 543.Zamarron BF, Mergian TA, Cho KW, Martinez-Santibanez G, Luan D, Singer K, DelProposto JL, Geletka LM, Muir LA and Lumeng CN. Macrophage Proliferation Sustains Adipose Tissue Inflammation in Formerly Obese Mice. Diabetes 66: 392–406, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Zeiser R Immune modulatory effects of statins. Immunology 154: 69–75, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Zeyda M, Farmer D, Todoric J, Aszmann O, Speiser M, Gyori G, Zlabinger GJ and Stulnig TM. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int J Obes (Lond) 31: 1420–8, 2007. [DOI] [PubMed] [Google Scholar]
- 546.Zeyda M, Huber J, Prager G and Stulnig TM. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity (Silver Spring) 19: 743–8, 2011. [DOI] [PubMed] [Google Scholar]
- 547.Zeyda M and Stulnig TM. Adipose tissue macrophages. Immunol Lett 112: 61–7, 2007. [DOI] [PubMed] [Google Scholar]
- 548.Zhang J and Liu F. Tissue-specific insulin signaling in the regulation of metabolism and aging. IUBMB Life 66: 485–95, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L and Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 372: 425–32, 1994. [DOI] [PubMed] [Google Scholar]
- 550.Zheng Y, Eilertsen KJ, Ge L, Zhang L, Sundberg JP, Prouty SM, Stenn KS and Parimoo S. Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat Genet 23: 268–70, 1999. [DOI] [PubMed] [Google Scholar]
- 551.Zhou L, Park SY, Xu L, Xia X, Ye J, Su L, Jeong KH, Hur JH, Oh H, Tamori Y, Zingaretti CM, Cinti S, Argente J, Yu M, Wu L, Ju S, Guan F, Yang H, Choi CS, Savage DB and Li P. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27-deficient mice. Nat Commun 6: 5949, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Zhu X, Zong G, Zhu L, Jiang Y, Ma K, Zhang H, Zhang Y, Bai H, Yang Q, Ben J, Li X, Xu Y and Chen Q. Deletion of class A scavenger receptor deteriorates obesity-induced insulin resistance in adipose tissue. Diabetes 63: 562–77, 2014. [DOI] [PubMed] [Google Scholar]
- 553.Zoico E, Garbin U, Olioso D, Mazzali G, Pasini Fratta AM, Di Francesco V, Sepe A, Cominacini L and Zamboni M. The effects of adiponectin on interleukin-6 and MCP-1 secretion in lipopolysaccharide-treated 3T3-L1 adipocytes: role of the NF-kappaB pathway. Int J Mol Med 24: 847–51, 2009. [DOI] [PubMed] [Google Scholar]
- 554.Zucker LM and Antoniades HN. Insulin and obesity in the Zucker genetically obese rat “fatty”. Endocrinology 90: 1320–30, 1972. [DOI] [PubMed] [Google Scholar]
- 555.Zucker TF and Zucker LM. Fat accretion and growth in the rat. J Nutr 80: 6–19, 1963. [DOI] [PubMed] [Google Scholar]