

ABSTRACT
Hormone therapy (HT) is associated with increased risk of breast cancer, strongly dependent on type, duration, and recency of use. HT use could affect cancer risk by changing breast tissue transcriptional programs. We hypothesize that these changes are preceded by changes in DNA methylation. To explore this hypothesis we used histologically normal-appearing breast tissue from the Normal Breast Study (NBS). DNA methylation β-values were obtained using the Illumina HumanMethylation 450 BeadChips for 90 samples including all NBS-participants who used HT within 5 y before surgery. Data were analyzed using the reference-free cell mixture method. Cancer Genome Atlas (TCGA) mRNA-Seq data were used to assess correlation between DNA methylation and gene expression. We identified 527 CpG sites in 403 genes that were associated with ever using HT at genome wide significance (FDR q < 0.05), of these, 68 sites were also significantly associated with duration of use or recency of use. Twelve sites reached significance in all analyses one of which was cg01382688 in ARHGEF4 (p < 1.2x10−7). Mutations in ARHGEF4 have been reported in breast tumors, but this is the first report of possible breast cancer-related DNA methylation changes. In addition, 22 genes included more than one significant CpG site and a majority of these sites were significantly correlated with gene expression. Although based on small numbers, these findings support the hypothesis that HT is associated with epigenetic alterations in breast tissue, and identifies genes with altered DNA methylation states which could be linked to breast cancer development.
KEYWORDS: Hormone therapy, normal breast tissue, DNA methylation, epigenetics, epigenome wide association
Introduction
Hormone therapy (HT), either with estrogen alone or combined with progesterone, has been used to alleviate menopausal symptoms since the 1940s [1]. Although health benefits such as reduced risk of cardiovascular disease, osteoporosis and decreased mortality have been observed [2–4], the balance between risk and benefit remains an area of clinical concern [2,5].
Concern about the safety of HT was raised after the Women’s Health Initiative clinical trials in 2002 indicated that combined (estrogen plus progestin) HT use increased breast cancer risk [6]. Since then, epidemiological efforts have focused on timing and duration of exposure and elucidating the mechanisms underlying the risk [7,8]. The current consensus is that estrogen alone therapy or short-term combined HT use (initiated around the time of menopause) does not appear to increase breast cancer risk. However, long-term combined HT-use, starting at menopause, is associated with breast cancer with larger risks associated with longer duration of use [8,9].
HT leads to transcriptional changes in breast tissue and estrogen exposure has been connected to epigenetic alterations, including changes to DNA methylation in different target tissues [10–12]. Methylation changes are known to be extensive in breast tumor tissue [13–15] and are likely to represent some of the early events in cancer development [16,17]. Identifying differential DNA methylation patterns resulting from HT use could therefore serve as an intermediate endpoint for studying the role of hormonal exposures in relation to breast cancer risk.
We hypothesized that exposure to HT would lead to differential DNA methylation detectable in DNA from breast tissue. To investigate this, we used DNA extracted from fresh frozen histologically normal-appearing breast tissue and compared women who reported using HT therapy vs. women who reported never using HT.
Results
Participant characteristics
Participant characteristics are depicted in Table 1 and Supplementary Table S1. All women were participants in the Normal Breast Study (NBS). NBS inclusion criteria resulted in some heterogeneity regarding reasons for surgery as both women undergoing surgery for invasive or in situ breast cancer (76 women, 84%) as well as women with benign tumors, prophylactic or reduction surgeries (14 women, 16%) were included. Ages ranged from 42 to 85 y with a median age of 60, all women were reportedly postmenopausal at the time of surgery. There were no statistically significant differences between HT-users and non-users among common characteristics related to breast cancer risk, including age, BMI, and parity.
Table 1.
Participant characteristics (All).
| 36 Never users |
17 Former users |
37 Current users |
||
|---|---|---|---|---|
| Characteristic | N (%) | N (%) | N (%) | p-valuea |
| Age at surgery | ||||
| >55 | 13 (36) | 6 (35) | 9 (24) | 0.58 |
| 55–65 | 11 (31) | 7 (41) | 18 (49) | |
| <65 | 12 (33) | 4 (24) | 10 (27) | |
| BMI | ||||
| <19 | 12 (33) | 4 (24) | 13 (35) | 0.87 |
| 19–25 | 12 (33) | 5 (29) | 11 (30) | |
| >25 | 12 (33) | 8 (47) | 13 (35) | |
| Race | ||||
| White | 29 (81) | 12 (80) | 30 (81) | 0.58 |
| Black | 6 (17) | 1 (7) | 5 (14) | |
| Other | 1 (3) | 2 (13) | 2 (5) | |
| Smoking Status | ||||
| Never | 20 (56) | 7 (41) | 19 (51) | 0.83 |
| Former | 13 (36) | 9 (53) | 16 (43) | |
| Current | 3 (8) | 1 (6) | 2 (5) | |
| Parity | ||||
| 0 | 10 (28) | 0 (0) | 6 (16) | 0.24 |
| 1 | 7 (19) | 2 (12) | 7 (19) | |
| 2 | 9 (25) | 11 (65) | 15 (41) | |
| >2 | 10 (28) | 4 (23) | 9 (24) | |
| Lactation | ||||
| No | 11 (42) | 3 (18) | 12 (38) | 0.22 |
| Yes | 15 (58) | 14 (82) | 19 (61) | |
| Reason for surgery | ||||
| Invasive | 25 (69) | 9 (53) | 22 (59) | 0.46 |
| In Situ | 7 (19) | 3 (18) | 10 (27) | |
| Benign | 1 (3) | 2 (12) | 3 (8) | |
| Prophylactic | 3 (8) | 2 (12) | 2 (5) | |
| Reduction | 0 | 1 (6) | 0 |
aP-values are from chi-square tests
Of 54 women classified as current or former users, 26 (48%) reported using combined HT (estrogen + progesterone), 12 (22%) reported using estrogen only, and 2 (4%) reported using progesterone only (data on precise type of progesterone was not available). Fourteen women (26%) did not specify which type of HT they had used. Duration of use was 0–5 y for 19 (35%), 6–10 y for 9 (17%) and >10 y for 26 women (48%).
EWAS analysis
In our initial analysis we compared ever users of HT to never users. Results were adjusted for potential confounders and for multiple comparisons using an FDR cutoff of q < 0.05 and are presented as a Manhattan plot in Figure 1. We identified 527 CpG sites with q < 0.05 that, based on Illumina 450K annotation, are located in 403 genes and 97 intergenic regions (Supplementary Table S2). Compared to never-users, DNA methylation was higher in HT-users for 59% (311) of the significant CpG sites and lower in 41% (216) of the CpGs. At the 527 significant CpGs sites the average percentage DNA methylation difference between HT-users and never users (mean βHTusers – mean βnever users) ranged from −10.1% to 8.7%. Differences were larger among the 211 CpGs that exhibited lower DNA methylation in HT-users (3.6% mean difference compared to 1.7% mean difference among the 311 CpG sites where DNA methylation was higher).
Figure 1.
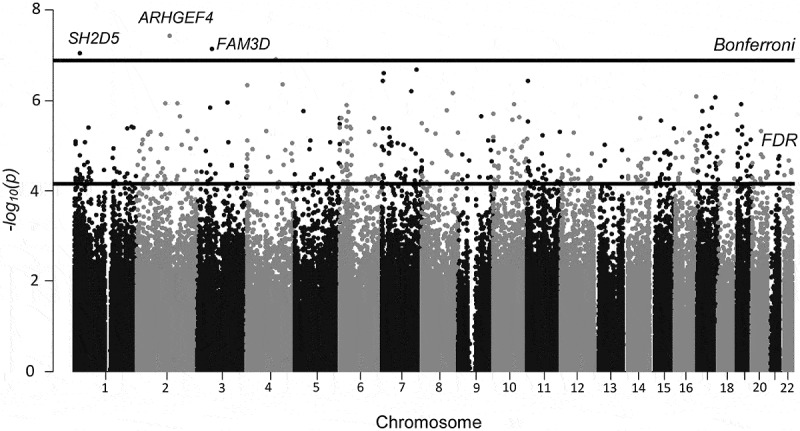
Manhattan plot depicting differentially methylated CpG sites (by location and – log10 p-value) from the analysis comparing ever vs. never HT-users (n = 90). Five hundred twenty-seven CpGs passed the FDR cutoff (q < 0.05), represented by the lower black line. CpGs from 3 genes (cg01382688 in ARHGEF4, cg26334888 in FAM3D and cg03472655 in SH2D5), on chromosomes 2, 3, and 1 passed a strict Bonferroni correction of p < 1.2x10−7 (higher black line).
Three CpG sites (cg03472655 in SH2D5, cg01382688 in ARHGEF4, and cg26334888 in FAM3D) passed a Bonferroni corrected cutoff of p < 1.2x10−7. All three CpG sites displayed increased DNA methylation in HT-users vs nonusers (mean % increase ranging from 0.7% (cg03472655) to 1.9% (cg26334888). Both cg01382688 in ARHGEF4 and cg26334888 in FAM3D were hyper-methylated (mean β-values >0.75) whereas cg03472655 in SH2D5 was hypo-methylated with a mean β-value of 0.05. We verified array methylation β-values by pyrosequencing cg26334888 in FAM3D using the same samples evaluated in the EWAS (n = 83). There was high correlation between the raw β-values and % methylation derived by pyrosequencing (Pearson’s correlation = 0.71, Supplementary Fig. S1). Mean methylation values obtained by pyrosequencing were consistently larger than those from the array, but showed the same relative relationship for methylation values between HT users and never users (Supplementary Table S3).
We further examined if duration of use (1–5, 6–10, >10 y) and recency of use (never user, former user, current user) influenced DNA methylation among the 527 identified CpG sites. Sixty-eight CpG sites exhibited DNA methylation changes that were significantly associated with either duration of HT-use (49 sites) or recency of use (31 sites) with 12 sites significant in both analyses (Table 2, Supplementary Table S2). Nine (75%) of these 12 sites displayed increased DNA methylation with both longer duration of use and more recent use; this direction was consistent with the direction observed when comparing ever and never users. One intergenic CpG site (cg02245004 on 15q24) displayed consistent decreased DNA methylation related to duration, past use, and ever/never use, and two sites (cg16416165 in RGS12 and cg26705583 in SGTB) displayed inconsistent results with increasing DNA methylation observed with more recent HT-use but decreasing DNA methylation observed with a longer duration of use (Supplementary Table S4).
Table 2.
CpGs significant in ever/nevera analysis and at least one trend test.
| Probe | CHR | Gene/Location | Ever/Never |
Duration of use |
Recency of use |
|||
|---|---|---|---|---|---|---|---|---|
| p-value | q-value | p-value | q-value | p-value | q-value | |||
| cg01382688 | 2 | ARHGEF4 | 3.7E-08 | 0.01 | 2.7E-06 | 0.03 | 4.3E-06 | 0.04 |
| cg26334888 | 3 | FAM3D | 7.3E-08 | 0.01 | 2.7E-04 | 0.09 | 1.2E-08 | 0.00 |
| cg03472655 | 1 | SH2D5 | 9.1E-08 | 0.01 | 1.9E-04 | 0.08 | 2.4E-06 | 0.04 |
| cg08460153 | 7 | TNRC18 | 2.5E-07 | 0.01 | 1.4E-05 | 0.04 | 4.8E-07 | 0.02 |
| cg11034191 | 4 | INPP4B | 4.3E-07 | 0.01 | 3.1E-09 | 0.00 | 6.3E-06 | 0.05 |
| cg16416165 | 4 | RGS12 | 4.6E-07 | 0.01 | 2.3E-05 | 0.05 | 6.9E-06 | 0.05 |
| cg09180239 | 16 | GSE1 | 8.2E-07 | 0.02 | 1.1E-05 | 0.04 | 3.3E-05 | 0.07 |
| cg11151929 | 3 | LRRC58 | 1.1E-06 | 0.02 | 2.0E-07 | 0.01 | 6.5E-05 | 0.08 |
| cg09423413 | 6 | TRIM40 | 1.3E-06 | 0.02 | 5.1E-06 | 0.03 | 1.5E-04 | 0.10 |
| cg19049616 | 17 | RNFT1 | 1.4E-06 | 0.02 | 1.0E-04 | 0.07 | 1.9E-06 | 0.03 |
| cg15028339 | 5 | RAI14 | 1.7E-06 | 0.02 | 7.3E-06 | 0.03 | 5.1E-07 | 0.02 |
| cg01571735 | 2 | SP5 | 2.2E-06 | 0.02 | 1.9E-05 | 0.04 | 6.1E-06 | 0.05 |
| cg06133876 | 6 | PBOV1 | 2.4E-06 | 0.02 | 2.2E-05 | 0.05 | 2.3E-05 | 0.07 |
| cg18291238 | 14 | 14q24 | 2.5E-06 | 0.02 | 3.4E-04 | 0.09 | 3.2E-06 | 0.04 |
| cg04763519 | 6 | 6p25 | 4.0E-06 | 0.02 | 1.5E-04 | 0.07 | 5.3E-07 | 0.02 |
| cg17771569 | 19 | ATP8B3 | 4.6E-06 | 0.02 | 1.5E-05 | 0.04 | 2.7E-04 | 0.11 |
| cg01953797 | 8 | GPIHBP1 | 5.2E-06 | 0.02 | 1.9E-05 | 0.05 | 4.0E-05 | 0.07 |
| cg17472832 | 2 | PROM2 | 5.6E-06 | 0.03 | 1.4E-05 | 0.04 | 1.3E-05 | 0.06 |
| cg12542656 | 2 | NA | 5.6E-06 | 0.03 | 2.2E-08 | 0.00 | 8.6E-05 | 0.09 |
| cg07273415 | 11 | CYB561A3 | 6.1E-06 | 0.03 | 2.7E-06 | 0.03 | 7.5E-06 | 0.05 |
| cg08030662 | 17 | KIF2B | 6.3E-06 | 0.03 | 1.1E-04 | 0.07 | 5.7E-08 | 0.01 |
| cg26705583 | 5 | SGTB | 7.6E-06 | 0.03 | 3.1E-05 | 0.05 | 6.7E-08 | 0.01 |
| cg00099017 | 16 | RBFOX1 | 8.5E-06 | 0.03 | 5.0E-03 | 0.21 | 1.0E-06 | 0.03 |
| cg08274097 | 19 | ZNF607 | 9.1E-06 | 0.03 | 7.0E-03 | 0.22 | 1.7E-06 | 0.03 |
| cg04671476 | 5 | MGAT1 | 9.3E-06 | 0.03 | 1.4E-05 | 0.04 | 3.1E-05 | 0.07 |
| cg25640096 | 6 | NA | 1.2E-05 | 0.03 | 6.5E-07 | 0.02 | 3.8E-05 | 0.07 |
| cg08255481 | 16 | BANP | 1.3E-05 | 0.03 | 9.2E-06 | 0.04 | 4.4E-04 | 0.12 |
| cg13030786 | 7 | TNRC18 | 1.3E-05 | 0.03 | 4.7E-04 | 0.10 | 5.9E-06 | 0.05 |
| cg15437133 | 10 | ZNF503-AS2 | 1.3E-05 | 0.03 | 2.5E-07 | 0.01 | 3.1E-05 | 0.07 |
| cg06194738 | 6 | GPSM3 | 1.4E-05 | 0.03 | 1.6E-04 | 0.08 | 6.3E-06 | 0.05 |
| cg24290251 | 7 | 7p15 | 1.5E-05 | 0.03 | 1.0E-03 | 0.14 | 9.0E-07 | 0.03 |
| cg26791665 | 1 | ALDH4A1 | 1.7E-05 | 0.03 | 5.0E-06 | 0.03 | 8.6E-05 | 0.09 |
| cg00352027 | 10 | AIFM2 | 1.8E-05 | 0.03 | 9.0E-04 | 0.12 | 2.3E-06 | 0.04 |
| cg27269561 | 16 | THOC6 | 1.8E-05 | 0.03 | 3.3E-05 | 0.05 | 2.4E-04 | 0.11 |
| cg14415844 | 14 | CCDC88C | 1.9E-05 | 0.03 | 6.8E-06 | 0.03 | 2.1E-04 | 0.10 |
| cg14662522 | 4 | NA | 2.1E-05 | 0.04 | 4.3E-07 | 0.01 | 2.0E-03 | 0.19 |
| cg26185079 | 18 | SLMO1 | 2.3E-05 | 0.04 | 1.2E-05 | 0.04 | 1.0E-06 | 0.03 |
| cg08048620 | 17 | TTYH2 | 2.5E-05 | 0.04 | 1.0E-06 | 0.02 | 0.0E+00 | 0.07 |
| cg06538238 | 6 | GABBR1 | 2.6E-05 | 0.04 | 2.3E-04 | 0.08 | 5.0E-07 | 0.02 |
| cg09413013 | 17 | TMC8 | 2.7E-05 | 0.04 | 6.4E-06 | 0.03 | 2.6E-05 | 0.07 |
| cg09071838 | 13 | CENPJ | 2.8E-05 | 0.04 | 2.2E-05 | 0.05 | 2.0E-04 | 0.10 |
| cg03606898 | 19 | NA | 3.0E-05 | 0.04 | 2.9E-05 | 0.05 | 2.5E-04 | 0.11 |
| cg25140751 | 15 | CIB2 | 3.3E-05 | 0.04 | 4.8E-06 | 0.03 | 8.8E-05 | 0.09 |
| cg26279783 | 6 | COL11A2 | 3.5E-05 | 0.04 | 1.8E-05 | 0.04 | 5.1E-04 | 0.13 |
| cg09742643 | 7 | PTPRN2 | 3.5E-05 | 0.04 | 1.1E-05 | 0.04 | 1.6E-06 | 0.03 |
| cg08975834 | 10 | TCERG1L | 3.7E-05 | 0.04 | 9.5E-06 | 0.04 | 8.8E-07 | 0.03 |
| cg26365623 | 11 | 11p15 | 3.8E-05 | 0.04 | 5.6E-02 | 0.40 | 3.8E-06 | 0.04 |
| cg19793640 | 21 | SH3BGR | 4.0E-05 | 0.04 | 2.0E-03 | 0.14 | 5.9E-06 | 0.05 |
| cg25044701 | 12 | GDF3 | 4.3E-05 | 0.05 | 1.1E-05 | 0.04 | 8.0E-05 | 0.08 |
| cg02601249 | 2 | 2p12 | 4.6E-05 | 0.05 | 4.0E-03 | 0.19 | 4.7E-06 | 0.04 |
| cg06362313 | 12 | GAPDH | 5.0E-05 | 0.05 | 5.8E-08 | 0.01 | 1.4E-04 | 0.10 |
| cg01360281 | 5 | NA | 5.0E-05 | 0.05 | 1.0E-05 | 0.04 | 4.0E-03 | 0.22 |
| cg16518142 | 20 | CDH26 | 5.1E-05 | 0.05 | 2.0E-03 | 0.15 | 3.0E-06 | 0.04 |
| cg03902160 | 1 | PTPRU | 5.2E-05 | 0.05 | 3.0E-07 | 0.01 | 1.9E-05 | 0.07 |
| cg02245004 | 15 | 15q24 | 5.4E-05 | 0.05 | 2.8E-06 | 0.03 | 1.9E-06 | 0.03 |
| cg06894891 | 14 | TPPP2 | 5.4E-05 | 0.05 | 5.6E-05 | 0.06 | 4.4E-06 | 0.04 |
| cg20677939 | 6 | ARID1B | 5.4E-05 | 0.05 | 2.8E-06 | 0.03 | 1.1E-05 | 0.06 |
| cg11847964 | 12 | NA | 5.5E-05 | 0.05 | 3.3E-06 | 0.03 | 3.7E-04 | 0.12 |
| cg23697855 | 7 | NA | 5.6E-05 | 0.05 | 2.8E-06 | 0.03 | 1.0E-03 | 0.16 |
| cg18072095 | 5 | C5orf30 | 6.1E-05 | 0.05 | 2.0E-03 | 0.15 | 6.9E-06 | 0.05 |
| cg00274357 | 8 | PUF60 | 6.6E-05 | 0.05 | 7.4E-06 | 0.03 | 1.0E-02 | 0.29 |
| cg02861082 | 2 | MCM6 | 6.8E-05 | 0.05 | 9.5E-06 | 0.04 | 1.4E-04 | 0.10 |
| cg09100343 | 16 | CPNE2 | 7.0E-05 | 0.05 | 6.4E-06 | 0.03 | 2.3E-04 | 0.11 |
| cg19216162 | 20 | C20orf24 | 7.3E-05 | 0.05 | 3.4E-06 | 0.03 | 6.3E-04 | 0.14 |
| cg26098117 | 6 | 6q22 | 7.6E-05 | 0.05 | 4.4E-06 | 0.03 | 3.9E-06 | 0.04 |
| cg17045804 | 5 | NPR3 | 7.8E-05 | 0.05 | 1.0E-05 | 0.04 | 2.5E-04 | 0.11 |
| cg03836747 | 7 | NA | 7.9E-05 | 0.05 | 7.8E-06 | 0.03 | 5.0E-03 | 0.23 |
| cg24141382 | 1 | CTPS1 | 8.0E-05 | 0.05 | 4.5E-06 | 0.03 | 1.0E-03 | 0.16 |
a Adjusted for: age at surgery, race (White, Black, Other), reason for surgery (invasive cancer, in situ cancer, benign, prophylactic, reduction) and BMI (treated as a continuous variable)
Twenty-two genes (Table 3) included multiple CpGs that reached genome-wide significance in the ever/never analysis; and three (CTBP1-AS1, PTPRN2 and GRB7) included three or more significant CpG sites. The significant CpG-sites in CTBP1-AS2 (3 sites, mean difference −0.03 β-values in HT-users) and GRB7 (4 sites, mean difference −0.06 β-values in HT-users) all showed decreased DNA methylation in HT-users compared to never users and all sites were located in the 5ʹ end of the gene, either upstream in the promoter or in close proximity to exon 1 (Figure 2). PTPRN2, a large gene on chromosome 7, had three significant CpG sites located in the gene body, which all showed increased DNA methylation in HT-users.
Table 3.
Genes with multiple significant CpGs.
| Ever/Never |
Correlation with gene expression |
|||||
|---|---|---|---|---|---|---|
| Gene/Location | CHR | Probe | p-value | q-value | cor. | p-value |
| LPGAT1 | 1 | cg07097417 | 4.3E-06 | 0.02 | −0.18 | 1.6E-07 |
| 1 | cg07569918 | 4.2E-05 | 0.04 | −0.17 | 9.3E-07 | |
| CTBP1-AS2 | 4 | cg20743744 | 7.8E-06 | 0.03 | NA | NA |
| 4 | cg23835219 | 4.7E-05 | 0.05 | NA | NA | |
| 4 | cg25897951 | 1.6E-05 | 0.03 | NA | NA | |
| MGAT1 | 5 | cg15847249 | 2.6E-05 | 0.04 | NA | NA |
| 5 | cg04671476 | 9.3E-06 | 0.03 | NA | NA | |
| GPR116 | 6 | cg00629395 | 2.7E-05 | 0.04 | 0.07 | 0.03 |
| 6 | cg13975362 | 7.0E-06 | 0.03 | −0.17 | 7.7E-07 | |
| TNRC18 | 7 | cg13030786 | 1.3E-05 | 0.03 | −0.03 | 0.43 |
| 7 | cg08460153 | 2.5E-07 | 0.01 | 0.01 | 0.67 | |
| 7p15 | 7 | cg10083572 | 1.5E-05 | 0.03 | NA | NA |
| 7 | cg14826425 | 4.1E-06 | 0.02 | NA | NA | |
| EEPD1 | 7 | cg17328052 | 8.4E-06 | 0.03 | −0.47 | < 1E-30 |
| 7 | cg26556065 | 7.8E-05 | 0.05 | −0.47 | < 1E-30 | |
| TFPI2 | 7 | cg07380959 | 4.7E-05 | 0.05 | −0.28 | 3.2E-17 |
| 7 | cg03333330 | 3.4E-05 | 0.04 | −0.37 | 8.3E-29 | |
| PTPRN2 | 7 | cg02161503 | 2.2E-05 | 0.04 | 0.63 | < 1E-30 |
| 7 | cg09742643 | 3.5E-05 | 0.04 | 0.75 | < 1E-30 | |
| 7 | cg03899215 | 7.8E-05 | 0.05 | 0.70 | < 1E-30 | |
| ERICH1-AS1 | 8 | cg00602245 | 3.3E-06 | 0.02 | NA | NA |
| 8 | cg14041855 | 4.5E-05 | 0.05 | NA | NA | |
| ARHGEF10 | 8 | cg09126794 | 7.9E-05 | 0.05 | 0.33 | < 1E-30 |
| 8 | cg14390047 | 6.8E-05 | 0.05 | −0.12 | 6.2E-04 | |
| COMMD3-BMI1 | 10 | cg14014799 | 4.9E-05 | 0.05 | −0.04 | 0.19 |
| 10 | cg19378631 | 3.2E-05 | 0.04 | −0.07 | 0.03 | |
| AIFM2 | 10 | cg04859918 | 3.2E-05 | 0.04 | −0.15 | 5.5E-06 |
| 10 | cg00352027 | 1.8E-05 | 0.03 | NA | NA | |
| LOC100188947 | 10 | cg19192065 | 6.5E-05 | 0.05 | 0.20 | 5.9E-09 |
| 10 | cg00526835 | 4.1E-05 | 0.04 | 0.16 | 5.2E-06 | |
| TCERG1L | 10 | cg08975834 | 3.7E-05 | 0.04 | NA | NA |
| 10 | cg10371050 | 2.0E-05 | 0.03 | 0.27 | 5.3E-12 | |
| MICAL2 | 11 | cg23044178 | 8.1E-05 | 0.05 | −0.37 | < 1E-30 |
| 11 | cg04468741 | 4.5E-05 | 0.05 | −0.39 | < 1E-30 | |
| ENOX1 | 13 | cg26872968 | 9.6E-06 | 0.03 | 0.13 | 1.1E-04 |
| 13 | cg10448831 | 2.2E-05 | 0.04 | 0.11 | 1.2E-03 | |
| PNMA1 | 14 | cg10523105 | 3.1E-05 | 0.04 | −0.27 | 4.4E-16 |
| 14 | cg09238801 | 2.4E-05 | 0.04 | −0.30 | 5.4E-20 | |
| ADAMTSL3 | 15 | cg01152302 | 1.5E-05 | 0.03 | −0.07 | 0.05 |
| 15 | cg21377071 | 2.6E-05 | 0.04 | −0.08 | 0.02 | |
| GRB7 | 17 | cg14263391 | 1.1E-05 | 0.03 | −0.34 | < 1E-30 |
| 17 | cg08284496 | 4.6E-05 | 0.05 | −0.30 | 2.5E-19 | |
| 17 | cg11183072 | 9.4E-06 | 0.03 | −0.20 | 6.1E-09 | |
| 17 | cg17740645 | 6.1E-06 | 0.03 | −0.28 | 7.3E-17 | |
| GIPC3 | 19 | cg14202338 | 6.2E-05 | 0.05 | 0.10 | 4.7E-03 |
| 19 | cg07679230 | 2.6E-05 | 0.04 | 0.15 | 4.5E-06 | |
| DDA1 | 19 | cg08142263 | 1.9E-05 | 0.03 | −0.26 | 1.4E-14 |
| 19 | cg10664184 | 7.9E-05 | 0.05 | 0.01 | 0.80 | |
NA = Not Available
Figure 2.
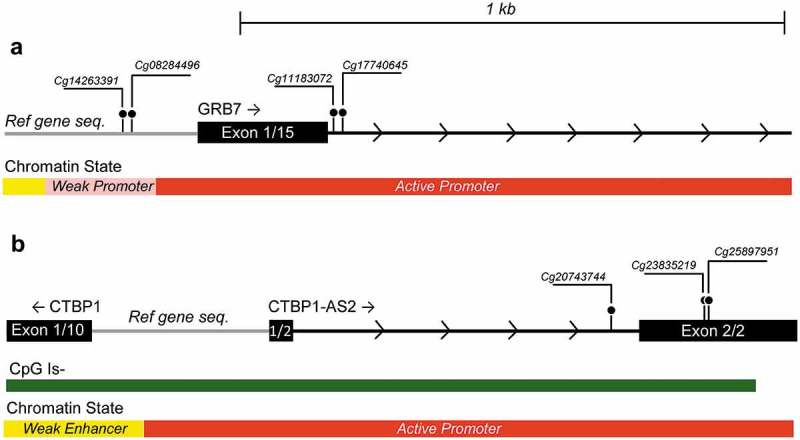
Genetic location of significant CpG sites in (a) GRB7 (from left to right: cg14263391, cg08284496, cg11183072 and cg17740645) and (b) CTBP1-AS2 (from left to right: cg20743744, cg23835219 and cg25897951. CpG sites are depicted as ball-and-stick representations. Gene sequence, CpG island location and Chromatin State are adapted from http://genome.ucsc.edu/using the human GRCh73/hg 19 build. Scale is the same in both figures.
Relationship between DNA methylation and gene expression
In order to assess the degree of correlation between DNA methylation levels and expression of nearby genes we obtained gene expression data from the Cancer Genome Atlas (TCGA) database. TCGA data were available for 275 out of the 526 CpG sites that reached genome wide significance in the never/ever analysis. Two hundred and seven of these 275 CpG sites (75%) showed significant correlation between expression and DNA methylation (p < 0.05) (Supplementary Table S2). Furthermore, TCGA data were available for 18 of the 22 genes (37 out of 48 CpG sites) that contained multiple significant CpG sites. CpG sites in 11 of these 18 genes were both concordant for direction of methylation and displayed significant correlations between DNA methylation and gene expression (Table 3). Using the data from TCGA we also assessed the correlation between ARHGEF4 expression and breast cancer stage (I-IV) and lobular or ductal subtype, but these were not correlated (Supplementary Fig. S2a and S2b).
Discussion
The use of HT and its impact on health has been the focus of several decades of research. There is growing consensus that short term use of HT for peri-menopausal symptoms provides considerable benefit without undue cancer risk, whereas long term postmenopausal use has cancer risks that outweigh other benefits [5]. HT-use has a wide variety of biological effects within breast tissue that may affect breast cancer risk [7]. These include changes to transcriptional programs that may result in epigenetic changes that persist long after HT has been discontinued. In this study, we investigated the hypothesis that HT is contributing to epigenetic changes in breast tissue.
In the primary analysis of ever vs never users, three CpG sites (cg01382688 in ARHGEF4, cg26334888 in FAM3D and cg03472655 in SH2D5) passed the strict Bonferroni correction and remained significant in one or more of the sensitivity analyses examining duration of use and recency of use analyses. Of these, the CpG that reached the highest overall significance was cg01382688 in ARHGEF4 located in an enhancer region just upstream of the ARHGEF4 transcriptional start site (TSS). Cg01382688 displayed hyper-methylation in all tissue samples with an increase observed in HT users compared to never users. The ARHGEF4 gene codes for a guanine nucleotide exchange factor, commonly known as ASEF [18], which is activated by the well-known tumor suppressor APC (adenomatous polyposis coli) [19]. APC is known for its role in colorectal cancer development [20]: silencing mutations in APC are responsible for the autosomal dominant disorder familial adenomatous polyposis [21] and are often found in sporadic colorectal cancer [22]. Although mutations in the APC gene are not widely seen in breast cancer, APC promoter hyper-methylation is found in primary breast tumors [23] and may be associated with specific breast cancer phenotypes [24]. Interestingly, ARHGEF4 mutations (although absent from colorectal tumors) have been reported in a variety of tumors, but appear in less than 1% of breast cancer [25,26]. APC activated ASEF may function as a tumor suppressor with loss of ASEF function, through either mutations or other mechanisms, promoting tumor progression [27].
The second CpG to pass Bonferroni correction was cg26334888 in FAM3D (Family with Sequence Similarity 3, Member D) which is located approximately 700bp from the transcriptional start site. FAM3D is part of a family of cytokines containing four genes (FAM3A, FAM3B, FAM3C and FAM3D) that is mainly expressed in highly proliferative tissue such as placenta and the gastrointestinal tract [28]. It has been suggested to play a role in cell proliferation, and functions as an activator of the ERK1/2 and p38MAPK signaling pathways [29]. Finally, the third highly significant CpG (cg03472655) was located in the gene SH2D5 (SH2 Domain Containing 5 protein) which is an adaptor-like protein expressed mainly in the brain [30]. Cg03472655 is situated in a CpG island in the first exon of the gene – a region that appears to be actively regulated. The CpG was hypo-methylated in all our samples but DNA methylation increased further in HT-users compared to never users.
Twelve CpGs were significant in all three analyses (never vs. ever, duration of use, and recency of use). Except for cg01382688 in ARHGEF4 (described above) one of the most interesting of these is cg11034191 in INPP4B (Inositol Polyphosphate-4-Phosphatase Type II B). The INPP4B CpG is in a CpG island approximately 200bp upstream of the INPP4B TSS and showed increased DNA methylation in current and former users compared to never users. Increased methylation of this CpG island is associated with transcriptional silencing of INPP4B [31]. INPP4B is a tumor suppressor gene whose expression is frequently lost in primary breast tumors and is associated with higher grade and size [32]. INPP4B expression is induced by estrogen receptor alpha binding, with INPP4B providing inhibition of AKT phosphorylation and inhibition of cell growth [33].
We also identified several genes with multiple CpGs that were significantly differentially methylated in HT-users. Interestingly DNA methylation at the majority of these genes was highly correlated with expression of the genes. One of the genes, GRB7 had four CpG sites in potential regulatory regions that all reached genome-wide significance: two within 100pb 5'; of the TSS (cg14263391 and cg08284496) and two in the first exon (cg11183072 and cg17740645). All four CpGs exhibited decreased DNA methylation in HT users and were significantly negatively correlated with GRB7 expression. GRB7 codes for an adapter protein that can interact with different downstream signaling molecules, including the HER2 receptor. Increased mRNA expression of GRB7 has been suggested as a prognostic marker for recurrence in triple-negative breast cancer [34] and may be related to metastatic potential [35]. In addition, the GRB7 protein has been implicated as conferring estrogen independence in breast cancer cell lines [36].
In this study we used fresh frozen histologically normal breast tissue enabling us to extract high quality DNA for methylation analysis. However breast tissue samples contain a mixture of different cell types with different methylation profiles which could confound the statistical association tests. Because methylation profiles from individual cell types found in breast tissue are unavailable, we applied the reference-free method [37] to adjust for the cell mixture effect. The reference-free method is reported to be similar to reference-based methods and to produce more biologically meaningful results than surrogate variable analysis (SVA) [37,38]. McGregor et al. [39] used simulation to compare alternative methods and showed that although the reference-free method could increase false positives, it had improved power and evaluations of several real datasets using both the reference-free method and SVA produced similar results.
Additional study strengths were detailed information on HT use, including time since last use and duration of use. A high proportion of women selected for this analysis also actively used HT up until the date of surgery. Weaknesses include the small sample size, which limited our ability to explore effects of ER status, type of hormone therapy, years since menopause, and initiation of HT (gap years). The inclusion of participants with different reasons for surgery may be regarded as a weakness. However, the majority of women underwent surgery for invasive or in situ breast cancer and tumor adjacent tissue was always used. Another potential weakness of the study are the small magnitude of effects that we identified which would need to be replicated in order to confirm that they are true findings, but we did not have access to additional datasets with the necessary information on HT use in order to attempt this. To account for potential confounding effects in the association analyses, we adjusted all analyses for age at surgery, reason for surgery, BMI, and race as well as the top 10 surrogate variables derived from internal control variables. Despite this, the results should be interpreted with caution and mainly viewed as exploratory examples of the potential impact of HT-use on breast tissue methylation.
In conclusion, despite a small sample set, we identify a number of genes whose methylation state is associated with HT use, and that have plausible links to breast cancer development. Replication of these results in independent samples remain, together with testing the hypothesis that methylation of these genes is altered in HT-associated breast tumors.
Materials and methods
Study population
Normal breast tissue samples used in this study come from women enrolled in the NBS [40]. NBS contacted 526 women above 18 y of age who were undergoing breast surgery at the University of North Carolina Hospitals. Eligible surgery types included total mastectomy, partial mastectomy, and excisional biopsy for women with breast tumors; prophylactic mastectomy for women at high risk of breast cancer; and elective surgery (reduction mammoplasty or mastopexy). Of the patients contacted for participation, 23 declined and 29 lacked available breast tissue. The final study population was therefore set at 474 patients. Participants donated snap frozen normal breast tissue, two tubes of blood, and completed a telephone interview to assess demographics and risk factors. After surgery, medical record abstraction was conducted to obtain anthropometric data.
One hundred and six women reported using HT at some point prior to surgery; we selected for inclusion in our study all 54 women who had used HT within the past 5 y (excluding 52 women that reported using HT but were either missing information on time since last use or had stopped using HT >5 y prior to surgery). The 54 HT-users consisted of 37 ‘current users’ who reported that they had used HT within one year of surgery and 17 ‘former users’ who reported stopping HT use 1–5 y prior to surgery (Supplementary Table S1). Of the women who reported never using HT (n = 368) we used frequency matching by age to select 36 women from different age categories similar to the ages of our current and former users (Supplementary Fig. S3). All HT users except one former user had used HT for a minimum of 12 months.
Sample preparation
Samples of fresh-frozen normal-appearing breast tissue were sectioned front and back and stained with hematoxylin and eosin (H&E). For women with breast cancer, normal breast tissue was obtained at a distance of at least two centimeters from the tumor margin. The H&E slides were scanned into high-resolution digital images using the Aperio Scan-Scope XT Slide Scanner at the Translational Pathology Lab (TPL) at UNC. Image analysis was used to segment images and calculate proportions of epithelial, stromal and fat tissue as described in Sandhu et al [41]. Frozen tissue adjacent to the H&E sections were used for DNA extraction.
DNA extraction and bisulfite conversion
20–25mg tissue samples were digested with 20 µl proteinase K and 180 µl buffer ATL at 56°C for 2 hours or until liquid was clear. DNA was extracted using DNeasy (Qiagen, Cat No: 69,506), according to the manufacturer’s instructions. Extracted DNA was eluted in 100 µL buffer AE and quantified using the Qubit fluorometer (Thermo Fisher Scientific), all samples were adjusted to a final volume of 45 µl containing 1000 ng of DNA. Extracted DNA samples were bisulfite converted simultaneously using the Zymo EZ DNA methylation kit (Zymo Research, Cat No: D5002) according to the manufacturer’s instructions. Briefly, 5 µl of M-dilution buffer was added to each sample yielding a final volume of 50 µl. Samples were incubated at 37°C for 15 min after which 100 µl of prepared CT-conversion reagent was added. The samples were then incubated for 16 cycles of 95°C for 30 sec followed by 50°C for 60 min, using the manufacturer’s recommendation for samples intended for the Illumina HumanMethylation450 BeadChip. After this, samples were kept at – 80°C until epigenome-wide analysis.
Epigenome-wide analysis
Genome wide DNA methylation was assessed using the Illumina HumanMethylation450 BeadChip (Illumina, Cat No: WG-314–1003) which provides information on 485,577 CpG sites with 99% coverage of RefSeq genes and an average of 17 CpG sites per gene including sites in the promoter, 5ʹUTR, first exon, gene body, and 3ʹUTR. Four µl of bisulfite-converted DNA from 90 samples, 4 duplicate samples, and 2 DNA methylation laboratory controls consisting of human methylated (100%) and non-methylated DNA (Zymo Research, Cat No: D5014) were randomly assigned to eight 450K chips with 12 samples on each chip. DNA was hybridized to the array following the manufacturer’s protocol and then scanned with an Illumina iScan. DNA methylation array data has been deposited into the NCBI Gene Expression Omnibus (GSE108213).
Pyrosequencing
Pyrosequencing primers were constructed for one of the top three CpGs associated with HT use that also passed a strict Bonferroni correction (cg26334888 in FAM3D – Supplementary Table S5). Pyrosequencing was performed as previously described [42]. Briefly, bisulfite converted DNA from all 96 samples was subjected to a PCR containing 5 pmol of each primer (forward and reverse) 10xPCR buffer (Thermo Fisher Scientific, Cat No: 10,342,020), 3 mM MgCl2, 1 mM dNTP, and 0.8 units of Taq polymerase (Thermo Fisher Scientific, Cat No: 10,342,020), heated to 95°C for 15 minutes, followed by 45 PCR cycles (95°C for 20 seconds, 58°C for 20 seconds and 72°C for 20 seconds) with a final extension at 72°C for 5 minutes. The pyrosequencing was carried out using PyroMark Q96 MD System (Qiagen) according to the manufacturer’s instructions. Percentage methylation was quantified using the Pyro Q-CpG Software (Qiagen).
Data analysis
The ENmix R package was used to preprocess raw DNA methylation data in order to improve data quality (https://bioconductor.org/packages/release/bioc/html/ENmix.html). Briefly, we used the ENmix method to reduce background noise [43]; the RELIC method to correct fluorescent dye-bias [44]; and quantile inter-array normalization on the methylation intensity values and RCP method to reduce Infinium I and II probe design bias [45]. We excluded 5 samples due to data quality issue: 3 samples with >0.05% low quality methylation values (detection p > 1 x 10−6 or number of beads <3) or with an average bisulfite intensity of <4000, and 2 samples with missing phenotype data. A total of 77,941 CpG probes were excluded: 11,796 CpG probes with >5% low quality data; 66,145 probes with: 1) common SNPs (MAF>0.05 in HapMap CEU dataset) within 2 base pairs of the probe target region, 2) non-specific CpGs mapping to multiple genomic locations, 3) Illumina designed SNP probes, 4) located on the Y chromosome or 5) multiple mode CpGs identified by ENmix R package.
We examined the association between ever use of HT (n = 51) and never use of HT (n = 34) adjusting for age at surgery, race (White, Black, Other), reason for surgery (invasive cancer, in situ cancer, benign, prophylactic, reduction) and BMI (treated as a continuous variable). To account for the possibility of different cell type composition in the tissue samples we used a reference-free method [37] to test the association between HT use-status and methylation level at each CpG site. Experimental batch effects and other unknown confounders were accounted for by adjusting for the top ten surrogate variables derived using the singular value decomposition (SVD) analysis of the 235 non-negative internal control probes on the array [46]. We then used the same reference-free cell mixture method to perform trend tests between methylation level and categorical phenotype variables for the effects of duration of use (HT-use 1: 0–5 y (n = 19), 2: 6–10 y (n = 8) and 3: >10 y (n = 24)) and recency of use (1: never users (n = 34), 2: former users (n = 17), 3: current users (n = 34)). We used the false discovery rate (FDR q < 0.05) to adjust for multiple comparisons.
Associations between DNA methylation and gene expression evaluated using Spearman correlation in breast tissue datasets from The Cancer Genome Atlas (TCGA). DNA methylation and mRNA-Seq data were downloaded from the Broad Institute website (http://gdac.broadinstitute.org). A total of 868 TCGA samples had both DNA methylation and gene expression data available for breast tissue. CpGs were first mapped to genes according to the NCBI build 37 gene annotation, and then correlation analyses were performed for each CpG and gene expression pairs.
Funding Statement
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. This research was also funded by the North Carolina University Cancer Research Fund, and several grants from the NCI (P30-CA016086, U54 CA156733, U01 CA 179715).
Acknowledgments
We thank the women who volunteered to participate in the Normal Breast Study and Kevin Gerrish and Laura Wharey at the NIEHS microarray core for processing and running the Illumina 450K bead arrays.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethical permissions
The study was approved by the Institutional Review Board (IRB) at the University of North Carolina (UNC) at Chapel Hill and all participants signed an informed written consent at the time of enrolment.
Supplemental data
Supplemental data for this article can be accessed here.
References
- [1].Chlebowski R, Anderson G.. Changing concepts: menopausal hormone therapy and breast cancer. J Natl Cancer Inst. 2012;104(7):517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lobo RA. Hormone-replacement therapy: current thinking. Nat Rev Endocrinol. 2016. DOI: 10.1038/nrendo.2016.164 [DOI] [PubMed] [Google Scholar]
- [3].Salpeter SR, Walsh JME, Greyber E, et al. Brief Report: coronary heart disease events associated with hormone therapy in younger and older women. J Gen Intern Med. 2006;21(4):363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhu L, Jiang X, Sun Y, et al. Effect of hormone therapy on the risk of bone fractures: a systematic review and meta-analysis of randomized controlled trials. Menopause. 2016;23(4):461. [DOI] [PubMed] [Google Scholar]
- [5].Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the women’s health initiative randomized trials. JAMA. 2013;310(13):1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women’s health initiative randomized controlled trial. JAMA. 2002. July 17;288(3):321–333. joc21036 [pii]. PubMed PMID: 12117397; eng [DOI] [PubMed] [Google Scholar]
- [7].Flores VA, Taylor HS. The effect of menopausal hormone therapies on breast cancer: avoiding the risk. Endocrinol Metab Clin North Am. 2015. September;44(3):587–602. PubMed PMID: 26316245; PubMed Central PMCID: PMCPMC4555991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Narod SA. Hormone replacement therapy and the risk of breast cancer. Nat Rev Clin Oncol. 2011. November;8(11):669–676. PubMed PMID: 21808267; eng [DOI] [PubMed] [Google Scholar]
- [9].Chlebowski RT, Rohan TE, Manson JE, et al. Breast cancer after use of estrogen plus progestin and estrogen alone: analyses of data from 2 women’s health initiative randomized clinical trials. Breast cancer after use of estrogen plus progestin and estrogen alone: analyses of data from 2 women’s health initiative randomized clinical trials. JAMA Oncol. 2015;1(3). DOI: 10.1001/jamaoncol.2015.0494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hall P, Ploner A, Bjöhle J, et al. Hormone-replacement therapy influences gene expression profiles and is associated with breast-cancer prognosis: a cohort study. BMC Med. 2006;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li S, Hursting SD, Davis BJ, et al. Environmental exposure, DNA methylation, and gene regulation: lessons from diethylstilbesterol-induced cancers [Review]. Ann N Y Acad Sci. 2003. March;983:161–169. PubMed PMID: 12724221; eng [DOI] [PubMed] [Google Scholar]
- [12].Sieuwerts A, De Napoli G, van Galen A, et al. Hormone replacement therapy dependent changes in breast cancer-related gene expression in breast tissue of healthy postmenopausal women. Mol Oncol. 2011;5(6):504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hartmann O, Spyratos F, Harbeck N, et al. DNA methylation markers predict outcome in node-positive, estrogen receptor-positive breast cancer with adjuvant anthracycline-based chemotherapy. Clin Cancer Res. 2009. January 1;15(1):315–323. PubMed PMID: 19118060. [DOI] [PubMed] [Google Scholar]
- [14].Novak P, Jensen T, Oshiro MM, et al. Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res. 2008. October 15;68(20):8616–8625. PubMed PMID: 18922938; PubMed Central PMCID: PMCPMC2680223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Soares J, Pinto AE, Cunha CV, et al. Global DNA hypomethylation in breast carcinoma: correlation with prognostic factors and tumor progression. Cancer. 1999. January 1;85(1):112–118. PubMed PMID: 9921982 [PubMed] [Google Scholar]
- [16].Rauscher GH, Kresovich JK, Poulin M, et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer. 2015;15:816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wajed SA, Laird PW, DeMeester TR. DNA methylation: an alternative pathway to cancer. Ann Surg. 2001;234(1):10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thiesen S, Kubart S, Ropers HH, et al. Isolation of two novel human RhoGEFs, ARHGEF3 and ARHGEF4, in 3p13–21 and 2q22. Biochem Biophys Res Commun. 2000. June 24;273(1):364–369. PubMed PMID: 10873612. [DOI] [PubMed] [Google Scholar]
- [19].Kawasaki Y, Senda T, Ishidate T, et al. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289(5482):1194–1197. [DOI] [PubMed] [Google Scholar]
- [20].Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120(19). DOI: 10.1242/jcs.03485 [DOI] [PubMed] [Google Scholar]
- [21].Waller A, Findeis S, Lee MJ. Familial adenomatous polyposis. J Pediatr Genet. 2016;5(2):78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Christie M, Jorissen RN, Mouradov D, et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/β-catenin signalling thresholds for tumourigenesis. Oncogene. 2012;32:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jin Z, Tamura G, Tsuchiya T, et al. Adenomatous polyposis coli (APC) gene promoter hypermethylation in primary breast cancers. Br J Cancer. 2001;85(1):69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Van der Auwera I, Van Laere SJ, Van Den Bosch SM, et al. Aberrant methylation of the Adenomatous Polyposis Coli (APC) gene promoter is associated with the inflammatory breast cancer phenotype. Br J Cancer. 2008;99(10):1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science (New York, NY). 2006;314(5797):268–274. [DOI] [PubMed] [Google Scholar]
- [26].Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2018. DOI: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mitin N, Betts L, Yohe ME, et al. Release of autoinhibition of ASEF by APC leads to CDC42 activation and tumor suppression. Nat Struct Mol Biol. 2007;14(9):814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhu Y, Xu G, Patel A, et al. Cloning, expression, and initial characterization of a novel cytokine-like gene family. Genomics. 2002. August;80(2):144–150. PubMed PMID: 12160727 [DOI] [PubMed] [Google Scholar]
- [29].Peng X, Xu E, Liang W, et al. Identification of FAM3D as a new endogenous chemotaxis agonist for the formyl peptide receptors. J Cell Sci. 2016;129(9):1831–1842. [DOI] [PubMed] [Google Scholar]
- [30].Gray EJ, Petsalaki E, James AD, et al. Src Homology 2 Domain Containing Protein 5 (SH2D5) binds the breakpoint cluster region protein, BCR, and regulates levels of Rac1-GTP. J Biol Chem. 2014;289(51):35397–35408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yuen JW, Chung GT, Lun SW, et al. Epigenetic inactivation of inositol polyphosphate 4-phosphatase B (INPP4B), a regulator of PI3K/AKT signaling pathway in EBV-associated nasopharyngeal carcinoma. PLoS One. 2014;9(8):e105163 PubMed PMID: 25126743; PubMed Central PMCID: PMC4134277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fedele CG, Ooms LM, Ho M, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010. December 21;107(51):22231–22236. PubMed PMID: 21127264; PubMed Central PMCID: PMCPMC3009830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hsu I, Yeh CR, Slavin S, et al. Estrogen receptor alpha prevents bladder cancer via INPP4B inhibited akt pathway in vitro and in vivo. Oncotarget. 2014. September 15;5(17):7917–7935. PubMed PMID: 25277204; PubMed Central PMCID: PMCPMC4202170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sparano JA, Goldstein LJ, Childs BH, et al. Relationship between quantitative GRB7 RNA expression and recurrence after adjuvant anthracycline chemotherapy in triple-negative breast cancer. Clin Cancer Res. 2011. November 15;17(22):7194–7203. 10.1158/1078-0432.CCR-10-3357. PubMed PMID: 21933890; PubMed Central PMCID: PMCPMC3570203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Giricz O, Calvo V, Pero SC, et al. GRB7 is required for triple-negative breast cancer cell invasion and survival. Breast Cancer Res Treat. 2012. June;133(2):607–615. PubMed PMID: 22005836. [DOI] [PubMed] [Google Scholar]
- [36].van Agthoven T, Veldscholte J, Smid M, et al. Functional identification of genes causing estrogen independence of human breast cancer cells. Breast Cancer Res Treat. 2009. March;114(1):23–30. PubMed PMID: 18351453. [DOI] [PubMed] [Google Scholar]
- [37].Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014. May 15;30(10):1431–1439. PubMed PMID: 24451622; PubMed Central PMCID: PMC4016702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Leek J, Storey J. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McGregor K, Bernatsky S, Colmegna I, et al. An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol. 2016. May 3;17:84 10.1186/s13059-016-0935-y. PubMed PMID: 27142380; PubMed Central PMCID: PMCPMC4855979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hair BY, Xu Z, Kirk EL, et al. Body mass index associated with genome-wide methylation in breast tissue. Breast Cancer Res Treat. 2015. June;151(2):453–463. PubMed PMID: 25953686; PubMed Central PMCID: PMC4474159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sandhu R, Chollet-Hinton L, Kirk EL, et al. Digital histologic analysis reveals morphometric patterns of age-related involution in breast epithelium and stroma. Hum Pathol. 2016. February;48:60–68. PubMed PMID: 26772400; PubMed Central PMCID: PMCPMC4727830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Harlid S, Xu Z, Panduri V, et al. CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the sister study. Environ Health Perspect. 2014. July;122(7):673–678. PubMed PMID: 24704585; PubMed Central PMCID: PMC4080519. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xu Z, Niu L, Li L, et al. ENmix: a novel background correction method for Illumina HumanMethylation450 Beadchip. Nucleic Acids Res. 2016. February 18;44(3):e20 PubMed PMID: 26384415; PubMed Central PMCID: PMCPMC4756845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu Z, Langie SA, De Boever P, et al. RELIC: a novel dye-bias correction method for Illumina methylation Beadchip. BMC Genomics. 2017. January 03;18(1):4 10.1186/s12864-016-3426-3. PubMed PMID: 28049437; PubMed Central PMCID: PMCPMC5209853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Niu L, Xu Z, Taylor JA. RCP: a novel probe design bias correction method for Illumina methylation Beadchip. Bioinformatics. 2016. September 01;32(17):2659–2663. PubMed PMID: 27153672; PubMed Central PMCID: PMCPMC5013906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wall ME, Rechtsteiner A, Rocha LM. Singular value decomposition and principal component analysis In: Berrar DP, Dubitzky W, Granzow M, editors. A practical approach to microarray data analysis. Kluwer, Norwell, MA; 2003. Chapter 5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.