

ABSTRACT
Several miRNAs are dysregulated in gastrointestinal stromal tumours (GIST), and miR-221/222 appear to have a prominent role in GIST biology. Therefore, we investigated the role of DNA variants located in miR-221/222 precursor sequences and their target KIT 3'UTR. Ninety-five polymorphisms were analysed in 115 GIST cases and 88 healthy controls. KIT 3'UTR rs17084733 and pri-miR-222 rs75246947 were found significantly associated with GIST susceptibility. Specifically, KIT rs17084733 A allele was more common in GIST, particularly in KIT wild-type (WT) patients (Padj = 0.017). rs17084733 variant is located within one of the three miR-221/222 binding sites in the KIT 3'UTR, resulting in a mismatch in this seed region. Conversely, KIT mRNA levels were lower in patients carrying the variant allele, except for KIT mutant GIST. Luciferase assay data in GIST cells, generated using a construct containing all the three miR-221/222 binding sites, are consistent with KIT mRNA levels in GIST patients. Reporter assay data, generated using a construct containing only the site encompassing rs17084733, confirmed that this is a functional variant disrupting the miR-221/222 binding site. In conclusion, this is the first study investigating the role of SNPs on miR-221/222 precursor sequences and their binding region on KIT 3'UTR in GIST. We identified the KIT variant rs17084733 as a possible novel genetic biomarker for risk of developing KIT-WT GIST. Moreover, our findings suggest the role of one of the three miR-221/222 binding sites on KIT 3'UTR as endogenous sponge, soaking up and subtracting miR-221/222 to the other two sites characterized by a higher affinity.
KEYWORDS: GIST, miRSNP, SNP, miRNA sponge, epigenetics, imatinib, gastrointestinal stromal tumor, KIT, miR-221/222
Introduction
Gastrointestinal stromal tumour (GIST) emerged as a distinct disease entity after the identification of KIT and PDGFRA oncogenic mutations in GIST tumorigenesis in about 85–90% of cases [1–5]. The discovery of KIT/PDGFRA mutations led to the introduction of tyrosine kinase inhibitors (TKIs) with KIT inhibitory activity, such as imatinib, sunitinib, and regorafenib, which effectively bind to and inhibit KIT and PDGFRA oncogenic signalling, thereby impacting favourably in GIST patients survival [6–10]. In addition, approximately 10–15% of the GIST are wild-type (WT) for KIT/PDGFRA mutations. This group has distinctive molecular hallmarks, including defects in SDH complex, or oncogenic activation of RAS/MAPK pathway. KIT/PDGFRA WT GIST are considered therapeutic orphans, given that no treatment has demonstrated any clinical benefit [11].
For a long time, research has focused on genetic traits associated with susceptibility to develop GIST and/or to predict treatment response [12–19]. In recent years, a wealth of evidence supports a relevant role for microRNA (miRNA) in GIST oncogenesis and tumour progression [20]. miRNAs, endogenous non-coding RNAs, negatively regulate gene expression by binding to the 3ʹuntranslated regions (3ʹUTR) of target genes [21,22]. miRNAs derive from a two-step process: generation of pre-miRNA (60–70 nt long) from pri-miRNA (500–3000 nt long) in the nucleus, followed by generation of mature miRNA from pre-miRNA in the cytoplasm [23]. miRNA-mRNA base pairing, and therefore gene expression modulation, may be influenced by different factors, including polymorphisms in both miRNAs and miRNA targets [23–25]. Indeed, genetic variants within the miRNA binding site (miR-SNPs) can affect miRNA-mRNA interactions, influencing several cellular processes, including susceptibility, prognosis, and clinical outcome of complex diseases, such as cancer [26–29].
A limited number of studies in GIST have analysed the role of miRNAs in tumour development, classification, diagnosis, and prognosis [20,30–34]. miR-221/222 down-regulation correlates with high KIT expression [30]. However, it is still controversial miR-221/222 function across GIST genotypes [30–32]. Therefore, we first analysed miR-221/222 expression in GIST patients, considering GIST genotype. Second, we evaluated the influence of genetic variants in pri-miR-221/222 and KIT 3ʹUTR on GIST prognosis and clinical outcome with first-line imatinib. Finally, we explored the role of KIT 3ʹUTR rs17084733 in regulating KIT expression in GIST cell lines.
Results
miR-221/222 and KIT expression levels in GIST patients according to tumour genotype
We analysed a cohort of 34 patients, 19 KIT and 7 PDGFRA mutants, and 8 KIT/PDGFRA WT GIST. As shown in Figure 1, miR-221-3p and miR-222-3p expression levels did not differ significantly in the three GIST genotypes (KIT mutant vs PDGFRA mutant vs KIT/PDGFRA WT: 0.17 ± 0.21 vs 0.10 ± 0.06 vs 0.16 ± 0.16 (miR-221), P= 0.438; 0.10 ± 0.16 vs 0.020 ± 0.010 vs 0.070 ± 0.080 (miR-222), P= 0.365). Inverse correlation between miR-221/222 and KIT expression levels was observed (Supplementary Figure S1 and S2). Nevertheless, miR-221/222 expression was significantly lower compared to papillary thyroid carcinoma (PTC) (miR-221/222 positive control) regardless of GIST genotype (Figure 1).
Figure 1.
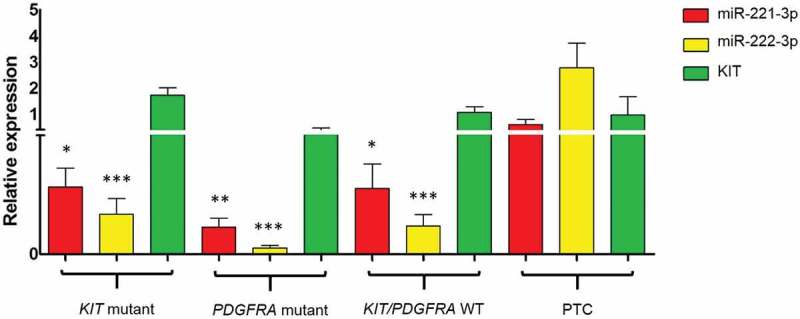
Expression level of miR-221, miR-222 and KIT in KIT mutant, PDGFRA mutant and KIT/PDGFRA WT GIST patients and in papillary thyroid carcinoma (PTC), used as positive controls. (Significantly lower compared to PTC, * P≤ 0.05, ** P≤ 0.01, *** P≤ 0.001).
pri-miR-221/222 and KIT 3ʹUTR genotype distribution in GIST patients and controls
A total of 95 germinal variants in pri-miR-221/222 and KIT 3ʹUTR were genotyped in 115 GIST and 88 healthy controls (Supplementary Table S1). Eighty-seven polymorphisms were homozygous WT and consequently excluded from further analysis. The remaining eight polymorphisms were consistent with the Hardy–Weinberg equilibrium in both cases and controls and thus were tested for association with risk of GIST development (Supplementary Table S2). rs17084733 (G>A) on KIT and rs75246947 (T>A) on pri-miR-222 polymorphisms were found significantly associated with GIST susceptibility. GG genotype in KIT rs17084733 was present in 88.6% of controls, compared to 74.8% of GIST patients. Interestingly, it was less frequent in patients with no mutations in KIT (KIT-WT GIST) compared to KIT mutant GIST (60.7% vs 79.8%). In particular, GG genotype was associated with a significantly lower risk to develop GIST in KIT WT compared to the controls (age- and gender-adjusted OR= 0.25, 95% CI 0.08–0.81, P= 0.021). Regarding pri-miR-222 rs75246947 variant, carriers of heterozygous genotype (TA) were more common among controls compared with GIST (TA: 39.2% and 21.1%, respectively; P= 0.020). The presence of the minor allele – A – was correlated with a lower risk to develop GIST (age-, gender-adjusted OR= 0.22, 95% CI 0.090–0.53, P= 0.001). None of the other investigated polymorphisms showed significant association with GIST susceptibility (Supplementary Table S2).
KIT 3'UTR and pri-miR-221/222 genotype predict outcomes in metastatic GIST patients treated with imatinib
Our cohort of GIST patients treated with imatinib for metastatic disease had a median time to progression (TTP) of 20.3 months, which is in line with reported data [35]. We first focused on KIT-mutant GIST patients, as miR-221 and miR-222 are modulators of KIT expression through binding to its 3ʹUTR [20]. KIT polymorphism rs17084733 G > A was significantly associated with shorter TTP, and was further confirmed in the multivariate analysis (age- and gender-adjusted HR= 15.5, 95% CI 1.70–141.3, P= 0.015; recessive model). A significant association between rs75246947, located in pri-miR-222 (T>A), and TTP was also observed in the analysis. Specifically, patients homozygous for the major allele (TT) had longer TTP (TA vs TT: age- and gender-adjusted HR= 6.05, 95% CI 1.92–19.07, P= 0.0020). This result was also confirmed correlating alleles with TTP based on the Kaplan–Meier method (median TTP TT vs TA: 23.9 vs 16.9, P= 0.009; Figure 2). Notably, this variant maintained the significance in the overall GIST population, which includes PDGFRA-mutant and KIT/PDGFRA WT GIST (TA vs TT: age- and gender-adjusted HR= 5.13, 95% CI 1.91–13.82, P= 0.001). None of the other investigated polymorphisms was associated with TTP (Supplementary Table S3 and S4).
Figure 2.
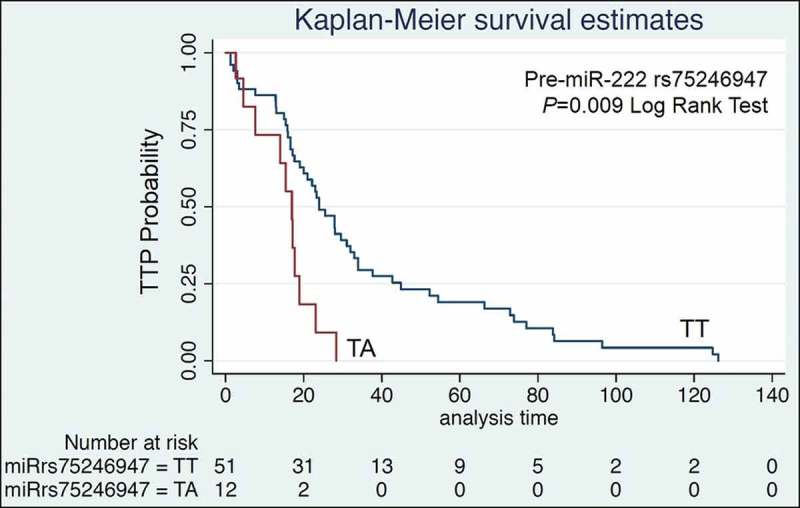
Kaplan-Meier TTP estimates for pri-miR-222 rs75246947. Patients harbouring the TT genotype showed longer PFS compared to TA.
Impact of rs17084733 variant on KIT mRNA levels in GIST patients
All GIST cases were KIT positive by immunohistochemistry (IHC), and data on KIT expression were retrieved from whole transcriptome paired-end RNA sequencing analysis (WTS) [34]. Lower levels of KIT mRNA were detected in patients carrying at least one variant allele in all molecular GIST subtypes, except for KIT-mutant GIST. With regard to the KIT-WT subgroup we observed a significantly lower KIT expression in patients carrying the rs17084733 variant allele compared to rs17084733 WT patients (9.70 ± 1.21 vs 11.22 ± 0.80, P= 0.046; Figure 3), suggesting that rs17084733 variant might represent a specific biomarker for this GIST subgroup.
Figure 3.
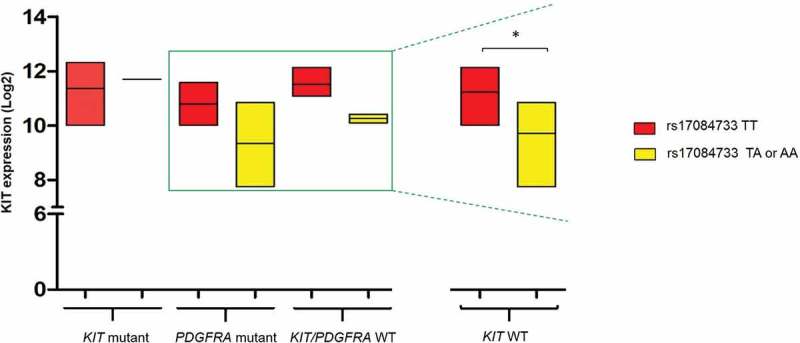
KIT expression in patients WT or carrying the rs17084733 variant allele according to tumour mutational status. KIT expression was significantly lower in KIT WT GIST patients harbouring the rs17084733 variant allele (TA or AA) compared to rs17084733 WT locus (TT) (* P≤ 0.05).
Functional analysis of rs17084733 variant in GIST cell lines
The KIT 3ʹUTR has been validated as a target of the cluster miR-221/222 [36,37] and three binding sites have been computationally predicted [38] being the rs17084733 locus located within one of these miR-221/222 interaction domains. Presence of the variant allele (A) leads to a conformational change responsible for an increased free energy (Supplementary Figure S3), indicative of base pairing disruption within the seed sequence of the miR-221/222 complementary site. However, our data show that patients harbouring rs17084733 variant allele present down-expression of the KIT oncogene, whereas disruption of the miRNA binding site should result in an increased expression. In order to elucidate the putative role of this locus on miRNA binding efficiency, we generated two constructs, herein referred to as maxiKIT constructs, containing all the three binding sites, with and without the rs17084733 KIT variant. The two constructs were cloned into a Renilla luciferase reporter vector; presence or absence of the variant in the constructs was confirmed by Sanger sequencing (Figure 4a). The presence of the variant allele led to 2.4 times and 1.5 times decrease in luciferase activity compared to the WT constructs in GIST-48 (P= 0.02) and GIST-882 (P= 0.017), respectively (Figure 4b). Thus, the rs17084733 SNP causes enhanced binding of the cluster miR-221/222 to the KIT 3ʹUTR in vitro. This result agrees with the observed lower KIT mRNA levels in GIST carrying the rs17084733 variant compared to WT patients (Figure 3). We next investigated how rs17084733 variant affected the miR-221/222 binding domain in vitro. To this purpose, we designed two constructs (herein referred to as miniKIT) in which the KIT 3ʹUTR – containing only the binding site encompassing the rs17084733 locus – was cloned into a Renilla luciferase reporter, with and without the rs17084733 variant. In both GIST-48 and GIST-882 cell lines, the presence of the variant allele led to a higher normalized luciferase activity compared to the WT locus (Figure 4c), indicating a potential disruption of the miRNA seed sequence. In particular, in GIST-48 transfected cells, the single mismatch caused by the variant allele resulted in a significant 1.7 times increased luciferase activity (P= 0.030). GIST-882 transfected cells showed 1.4 times increased luciferase level, reaching a borderline significance (P= 0.050).
Figure 4.
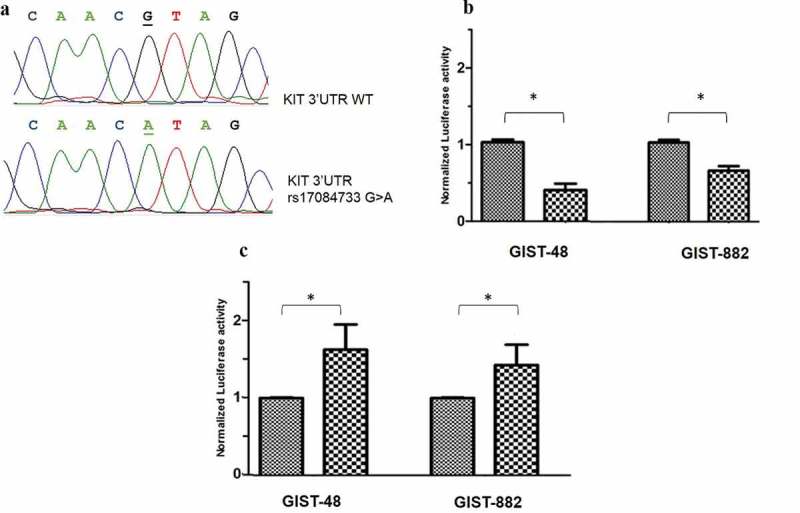
Functional analysis: luciferase assay. (a): Sanger sequence analysis carried out for luciferase construct verification of the KIT 3'UTR. Upper panel: KIT 3'UTR WT construct (underlined the rs17084733 wild-type allele – G); lower panel: KIT 3'UTR variant construct, (underlined the rs17084733 variant allele - A). (b and c): Putative role of the KIT 3'UTR rs17084733 on miRNA binding by Luciferase assay. (b): Transfection with the maxiKIT constructs – containing all the three miR-221/222 binding sites – in GIST-48 and GIST-882 lines: the presence of the minor allele A led to a significant decrease in luciferase activity in both GIST cell lines (* P< 0.05). (c): Transient transfection performed in GIST-48 and GIST-882 cells using the miniKIT construct: in both cell lines, presence of the minor allele A led to a significantly higher normalized luciferase activity (* P< 0.05).
Together, these results suggest that rs17084733 locus buffers the activity of miR-221/222 on the KIT target. In particular, rs17084733 WT sequence soaks up miR-221/222 and competitively sequester them from the other two sites on KIT 3ʹUTR, thus abrogating their repressive activity (Figure 5).
Figure 5.
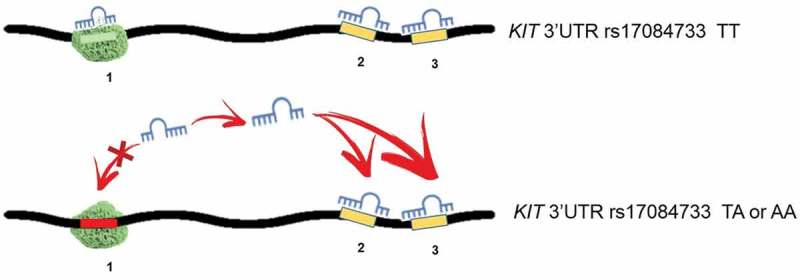
Proposed mechanism of a miRNA binding site acting as a sponge for miR-221/222. The seed sequence 1 encompasses the rs17084733. (Upper panel) The sequence with WT (TT) allele acts like a sponge, soaking up miR-221/222 and therefore, reducing their availability for sites 2 and 3. (Lower panel) Presence of the rs17085733 variant allele (TA or AA) disrupts the sponge site, increasing miR-221/222 availability for sites 2 and 3. Overall, disruption of the sponge contributes to repress KIT expression.
Discussion
GIST are driven by a variety of molecular events, including the well-known KIT/PDGFRA mutations, and other genetic events, such as SDH deficiency or BRAF/RAS/NF1 alterations [5,39,40]. However, in recent years specific epigenetic mechanisms, including miRNA deregulation, have emerged as responsible for the gradual malignant transformation of GIST [41]. miRNAs have an established role in cancer biology by controlling the expression of their targets’ mRNAs to promote tumour growth, invasion, angiogenesis, and immune evasion [23,25]. In particular, miRNAs post-transcriptionally repress the expression of target genes by binding to 3ʹUTR of the target mRNAs, causing inhibition of translation or degradation of the mRNA target [42]. The highly conserved miR-221 and miR-222, sharing the same seed sequence and encoded in tandem by the X chromosome, have been reported either as oncomiRs or as tumour suppressor-miRs [43]. In recent years, it has been ascribed an important role to miR-221/222 cluster as a regulator of KIT expression in GIST [20]. Indeed, the 3ʹUTR region of KIT harbours three putative binding sites for these two miRNAs. In our study, we evaluated for the first time miR-221/222 expression levels, taking into account tumour mutational status (KIT mutant vs PDGFRA mutant vs KIT/PDGFRA WT). Interestingly, we did not observe any significant difference regarding tumor genotype, leading us to speculate that miR-221/222 might play a role in the pathogenesis of GIST. This prompted us to investigate DNA alterations on pri-miR-221 and 222. A small number of studies have so far investigated the impact of SNPs located in pri-miR-221 and pri-miR-222 [38,44–46], but none of them have been performed in GIST. Mature miRNAs are composed of 18 to 24 nts in length, but only 2 to 7 nts form the seed region, which specifically identifies target mRNAs binding to miRNAs [47]. The small size of the seed sequence implies that a change as small as a SNP may deeply affect the miRNA binding capacity. Therefore, we analysed 95 polymorphisms within the pri-miR-221 and pri-miR-222 regions, and in the KIT 3ʹUTR. The majority of them did not show the minor allele (MA), as we also evaluated variants with low MA frequency (though > 0.01). We observed a significant difference in the distribution of the KIT 3ʹUTR variant rs17084733 (*217 G→A transition) between cases and controls. rs17084733 minor allele (A) was more common in GIST, particularly in KIT-WT GIST, potentially representing a disease susceptibility factor for this group of patients. Our data agree with those reported by Godshalk and collaborators, showing that rs17084733 A allele was more common among acral melanoma patients – KIT-mutant tumours – compared to the healthy population [38]. We also observed a significant correlation between rs75246947 variant and TTP in GIST patients under imatinib treatment. rs75246947, a −619 T>A transversion, is located within an intronic region of the X chromosome (45746731–45747263, GRCh38), encompassing the pri-miR-222. The underlying mechanism behind the correlation between rs75246947 and TTP remains unclear; however, given the regulatory role of miR-222, the finding is worthy of further comprehensive exploration. Notably, chromosome X also contains the tumour suppressor DMD gene, which is deleted in more than 60% of myogenic cancers, including GIST [48]. The deletion occurs in a chromosome region that does not include miR-222 and its rs75246947 variant, which therefore may continue to exert its function. Using the HaploReg database [49], rs75246947 is predicted to alter the Foxp1 and RREB-1 regulatory motifs, which are located 107 bp downstream of miR-222. These two regulatory motifs function as oncogenes in different cancer types [50], and their gene products exert some role in altered immune response [51,52].
Considering the impact of rs17084733 on KIT expression, our studies, as well as a prior report in PTC [44] showed lower KIT expression in KIT-WT GIST harbouring the rs17084733 variant compared to rs17084733 WT patients. By contrast, two independent microarray studies performed in melanoma displayed increased levels of KIT mRNA associated with rs17084733 WT due to disruption of miR-221/222 binding sequence on the KIT 3ʹUTR region [38]. Therefore, this study suggests a mechanism of KIT de-repression likely due to a decreased base pairing of the miRNA: target duplex in the miRNAs seed region. To further clarify these controvert results, we performed functional studies to elucidate the effect of the KIT 3ʹUTR rs17084733 variant on miRNA interaction in GIST cell lines. Luciferase reporter assay using maxiKIT constructs, covering the three binding sites, demonstrated a reduced luciferase activity, consistent with decreased KIT mRNA levels, which in turn disagrees with the hypothesis that rs17084733 variant breaks miR221/222 binding site. However, rs17084733 disrupts a site poorly predicted and therefore considered of minor importance. Indeed, all the main tools used for binding prediction (including MiRanda, TargetScan and DIANA-microT) identified all the binding sites except the one encompassing the rs17084733 variant. Only the RNAhybrid tool, based on two RNA hybridization energy calculations, correctly identified all the three sites. The hybridization energy computed for the site encompassing rs17084733 is affected by the minor allele, changing from −23.5 to −19.3 Kcal/mol for miR-221 and from 23.9 to −20.8 Kcal/mol for miR-222, highlighting a lower binding affinity (Supplementary Figure S3). We confirmed this binding prediction in GIST models using miniKIT constructs only covering the rs17084733 locus. Indeed, we reported an increased luciferase activity consistent with rs17084733 behaving as a functional variant disrupting the miR-221/222 binding site. Specifically, the binding disadvantage caused by the variant allele (A) drives the miR-221/222 to bind to the other two sites (Figure 5). Therefore, these two sites, predicted to be the most important miR-221/222 targets, represent the main players in KIT expression regulation, while the site including the rs17084733 locus acts as a natural sponge and competes for miR-221/222 binding.
In conclusion, to the best of our knowledge, this is the first study (i) showing comparable miR-221/222 expression levels between different GIST subtypes and (ii) investigating the role of polymorphisms in pri-miR-221/222 and on their binding regions in the KIT 3ʹUTR region. Specifically, this work identifies the rs17084733 variant in the KIT 3ʹUTR as a possible novel genetic biomarker for the risk of KIT-WT GIST development. Lastly, the most intriguing finding is the identification of an endogenous DNA sequence likely acting as a sponge to sequester miR-221/222, adding a new intricate facet to the miRNA regulatory dimension. Regulating the regulators: similarly to protein-coding mRNAs, miRNAs are subjected to regulatory mechanisms at different stages, and miRNA sponges constitute a recently recognized layer of this mechanism [53,54]. Given the preliminary nature of the data, we can only speculate on the existence of endogenous DNA sequences acting as miRNA sponges in GIST. Therefore, additional insights are needed to address this hypothesis and understand the complexity of miRNA regulatory machinery in GIST.
Materials and methods
Study population
A total of 149 unresectable/metastatic GIST patients and 88 healthy controls were retrospectively included in this study, at the Sant’Orsola-Malpighi Hospital, Bologna. With regard to the 149 GIST patients, 34 had only RNA, 98 had only germinal DNA and 17 had both germinal DNA and RNA available; the workflow of the analysis performed according to the available biological samples is reported in Figure 6.
Figure 6.
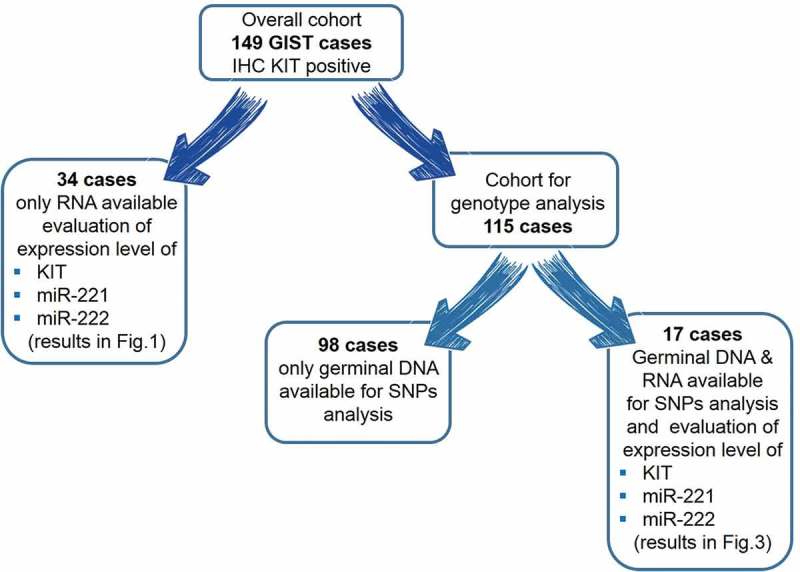
Workflow of the analysis performed according to the available biological samples.
Clinical information of the 115 GIST patients used in SNPs analysis was collected from patients’ medical records and are summarized in Table 1. KIT protein (CD117) level was evaluated at diagnosis by IHC; all patients included in the present study were IHC CD117 positive. KIT/PDGFRA mutational status was investigated through Sanger sequencing. Among the 115 patients with available DNA, 79 (68.7%) harbored a KIT mutation (68 in exon 11 and 11 in exon 9), 9 (7.8%) a PDGFRA mutation, 19 (16.5%) were KIT/PDGFRA WT, and 8 (7%) were missing. Data on clinical outcome were available for 71 GIST patients. These patients underwent standard first-line imatinib (400 mg or 600 mg daily); TTP was calculated from the beginning of imatinib therapy to the date of disease progression, documented by the CT scan performed approximately every 3–4 months. For patients who did not progress at the last follow-up, TTP evaluation was censored at that time. All patients were followed up until death; none of them has dropped out during surveillance. In order to evaluate disease susceptibility, we also genotyped 88 controls, anonymous blood donors from the Centro Trasfusionale, Sant’Orsola-Malpighi Hospital, Bologna (Table 1). The analysis, approved by the hospital Ethics Committee, was performed after written informed consent for study participation and anonymous data publication in accordance with national legislation and the Helsinki Declaration.
Table 1.
Demographic and disease characteristics of GIST patients and controls.
| Total PB cases (n= 115) | Controls (n= 88) | |
|---|---|---|
| Gender, n (%)a | ||
| Female | 48 (41.4) | 30 (34.1) |
| Male | 67 (58.3) | 58 (65.9) |
| Age, yearsb | ||
| Mean ± SD (range) | 57.5 ± 13.9 (21–88) | 46.6 ± 13.4 (21–79) |
| Tumor site, n (%) | ||
| Small intestine | 49 (42.6) | |
| Stomach | 55 (47.8) | |
| Peritoneum | 4 (3,5) | |
| Rectum | 1 (0,9) | |
| Missing | 6 (5.2) | |
| Tumor size, n (%) | ||
| ≤5 cm | 22 (19.1) | |
| 6–10 cm | 35 (30.5) | |
| ≥10 cm | 46 (40.0) | |
| Missing | 12 (10.4) | |
| Status at onset, n (%) | ||
| Localized | 59 (51.3) | |
| Metastatic | 56 (48.7) |
aNo difference in gender distribution between cases and controls (P= 0.68).
bNo difference in age distribution between cases and controls (P= 0.21); for cases, age is intended at diagnosis.
Cell cultures
GIST-48 and GIST-882 cell lines were kindly provided by Dr Fletcher (Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA). GIST-48 was established from a patient that had progressed – after an initial clinical response – during imatinib therapy; this cell line is characterized by a primary, homozygous KIT exon 11 mutation (p.V560D) and a secondary, heterozygous KIT exon 17 mutation (p.D820A). Cells were cultured in IMDM (Gibco, Thermo Fisher Scientific Inc brand, Waltham, MA, USA) supplemented with 15% fetal bovine serum (FBS) and 1 mM L-Glutamine (Invitrogen, Thermo Fisher Scientific Inc brand). GIST-882 was established from an untreated, human primary tumour harbouring a homozygous imatinib sensitive mutation in KIT exon 13 (p.K642E). Cells were maintained in RPMI-1640 (Gibco), supplemented with 15% FBS and 1 mM L-Glu (Invitrogen). All experiment were performed in newly thawed cells. GIST lines were routinely monitored by Sanger sequencing to confirm their KIT mutational status and to exclude additional secondary mutations in KIT.
miR-221/222 and KIT expression level in GIST patients and cell lines
Total RNA was isolated from GIST tissue samples, GIST cells and papillary thyroid carcinoma (PTC) FFPE samples using Trizol reagent (Invitrogen), according to the manufacturer’s instructions. PTC samples were included as a positive control for miR-221/222 expression. Ten nanograms of total RNA was reverse transcribed using the TaqMan microRNA Reverse Transcription kit (Applied Biosystems, Thermo Fisher Scientific Inc brand). For detection of miR-221-3p and miR-222-3p, expression levels were evaluated using Taqman microRNA single assays (Applied Biosystems), in accordance with the instructions supplied by the manufacturer. RT-PCR reactions were performed in triplicate in a 7900 HT Real-Time PCR (Applied Biosystems); negative controls and RNU48 and RNU44, both used as endogenous controls, were included in each run.
To evaluate KIT mRNA expression, total RNA was reverse transcribed through High-Capacity RNA-to-cDNA Kit (Applied Biosystems) and expression level was evaluated using Taqman gene expression assays (assay ID Hs00174029, Thermo Fisher). Relative expression was estimated by the ΔΔCt method, using GAPDH (assay ID Hs99999905, Thermo Fisher) as housekeeping control.
Polymorphisms selection and genotyping
miRNA binding sites in the KIT sequence were predicted with different tools as described by Krek et al. [49]. On the basis of the retrieved sequences, we selected a total of 95 polymorphisms, 77 miR-SNPs (28 in pri-miR-221 and 49 in pri-miR-222) and 18 polymorphisms in the KIT 3ʹUTR (Supplementary Table S1). Genomic DNA was extracted from fresh or frozen whole blood using a DNA isolation kit from Qiagen (QIAamp® DNA Mini Kit, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Genotyping of the 77 miR-SNPs on pri-miR-221 and pri-miR-222 was conducted by Sanger sequencing into an ABI PRISM® 310 Genetic Analyzer (Applied Biosystems). In particular, primers amplified two regions of 517 bp and 533 bp for miR-221 and miR-222, respectively (Hs00509011_CE and Hs00509012_CE, Thermo Fisher). Amplicons were purified using the QIAquick PCR Purification Kit (Qiagen) and sequenced using the BigDye® Terminator v1.1 Cycle Sequencing kit (Applied Biosystems). Sequence data were analysed by BLAST and manual review of chromatograms; all variants were confirmed by bidirectional (forward and reverse) sequencing. Concerning the 18 variants in KIT 3ʹUTR, genotyping was performed either by real-time PCR using Taqman® Assay (Applied Biosystems) as recommended by manufacturer or by Sanger sequencing. With regard to the variants analysed by sequencing, we amplified the region harbouring two binding domains, one for miR-221 and one for miR-222. This region also contained variants analysed by Taqman® Assays, and both the results of the overlapping variants were 100% concordant. Genotyping performed by real-time PCR included positive and negative controls in each reaction as quality control. For further quality control, all genotypes were performed blind to control or case status. Moreover, 10% of the collected samples were repeated for genotyping assay (variants performed by Taqman® Assay not overlapping with Sanger sequencing were selected), and the results were 100% concordant.
KIT 3′UTR plasmid constructs and luciferase assay
The pMirNanoGlo2 dual-Luciferase vector, containing both the Renilla and the Firefly luciferase genes, was purchased from Promega (Promega, Madison, WI, USA). pMirNanoGlo2 constructs containing a 1977 bp portion of KIT 3ʹUTR, WT (G) or with the variant allele (A) rs17084733, were generated (maxiKIT constructs) as follows. The KIT 3′UTR regions (G or A allele) were amplified from human genomic DNA, introducing the NheI and XhoI restriction sites. The two fragments (WT or with the variant) were cloned into a pGEM® vector using the pGEM-T Easy Vector Systems (Promega) according to the manufacturer’s instructions. The amplified DNA fragments were subcloned into the pMirNanoGlo2 vector, downstream of the Renilla luciferase stop coding region. This amplicon included all three binding sites (at the position 214–222, 1030–1037, 1959–1966 of KIT 3ʹUTR; Figure 7). Similarly, two constructs containing a 616 bp KIT 3ʹUTR region (miniKIT constructs) WT or with the variant allele, were generated. The 616 bp amplicon enclosed only the binding site at position 214–222, encompassing the rs17084733 locus. All constructs were sequence-verified prior to use. GIST-882 and GIST-48 cells were transfected using Amaxa Nucleofector II device (Lonza AG, Basel, Switzerland). In brief, 1 × 106 cells were resuspended in 100 μl of Ingenio Electroporation solution (Mirus Bio, LLC; Madison, WI, USA) and mixed with 50 pmol of miRNA mimics (Mirvana miRNA mimic, Ambion, Thermo Fisher Scientific Inc brand). Cells were then electroporated using the program T-20, and seeded in triplicate in a 96 wells plate. After 24 h, Firefly and Renilla Luciferase activities were quantified using the Dual-Luciferase system (Promega), through an EnSpire Multimode Plate Reader (Perkin Elmer, Inc, Waltham, MA USA). Renilla Luciferase expression was normalized on the Firefly Luciferase expression. Independent triplicate experiments were performed for each plasmid construct.
Figure 7.
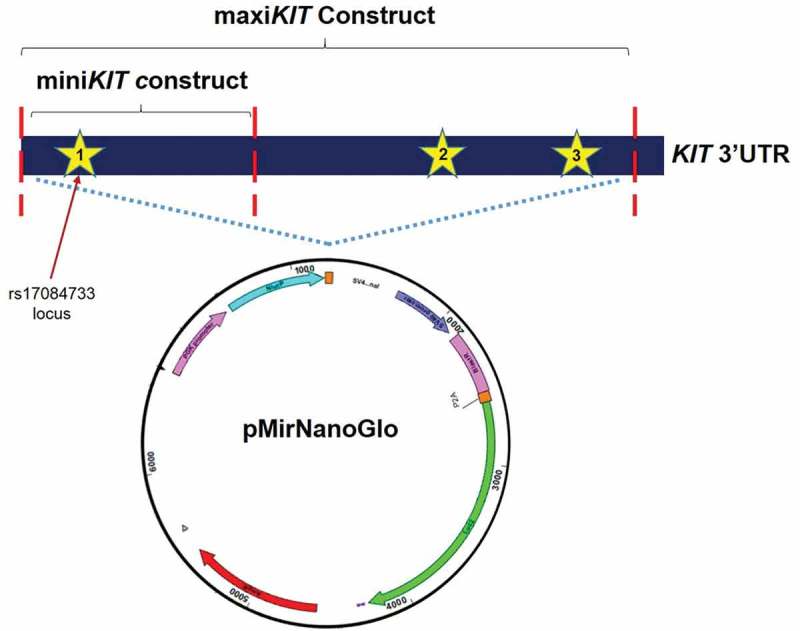
pMirNanoGlo vector. Stars indicate the three miRNA binding sites. MaxiKIT construct includes a 1977 bp amplicon carrying the three binding sites (at the positions 214–222, 1030–1037, 1959–1966 of KIT 3'UTR). MiniKIT construct includes a 616 bp amplicon including only the first binding site (position 214–222 of KIT 3'UTR).
MiRNA target site and hybridization prediction
Four popular tools for miR-221 and miR-222 binding prediction – MiRanda, TargetScan, DIANA-microT and RNAhybrid – were used [55]. The 3ʹUTR of the KIT transcript (ENSEMBL identifier ENST00000288135) was used as target. MiRanda and RNAhybrid were run locally using the stand-alone software version, DIANA-microT using its web interface, and TargetScan using both the stand-alone and the web interface. All tools were run using default parameters. Hybridization predictions for the rs17084733 WT and variant allele transcripts were evaluated with the following criteria: total score for MiRanda; context score and conservation for TargetScan; score and conservation for DIANA-microT; hybridization minimum free energy for RNAhybrid.
Statistical analysis
The distribution of genotypes was tested for departures from the Hardy–Weinberg equilibrium using the χ2 test. The frequency distributions of categorical variables were compared using Pearson’s chi-square and Fisher’s exact test as appropriate. Survival analysis methods were used to examine the relationship between genotypes (homozygous WT, heterozygous and homozygous for the variant allele) and TTP. Results were analysed using both the recessive (AA + Aa vs. aa) and dominant (AA vs. Aa + aa) models. In univariate analysis, the survival curves were estimated and plotted with the Kaplan–Meier method. The curves were compared with the log-rank test of equality of survivor functions (statistical significance defined as P< 0.05). In multivariate analysis, hazard ratios (HR) and 95% confidence interval (95% CI) were estimated by Cox proportional hazards models, using gender and age at diagnosis, as covariates in addition to the genotype. The proportional hazards assumption was tested using Schoenfeld residuals. Multiple logistic regression was used to assess the relation between individual polymorphisms and primary resistance. Given the limited small size of the present study, probability values and additional parameter estimates were not adjusted for multiplicity. Results should be interpreted as exploratory. Significant changes in mRNA and miR-221/222 expression and in reporter assay activity were assessed via Student’s t-test. All the analysis, except where otherwise specified, were conducted using Stata Intercooled version 12.0 [56].
Funding Statement
This work was supported by the Ministry of Education, University and Research of Italy [MIUR, grant number 2015Y3C5KP_002 to SA].
Acknowledgments
Gloria Ravegnini was supported by an MSD Italia fellowship granted by and on behalf of Merck Sharp & Dohme Corporation and is now supported by the L’Oréal-UNESCO for Women in Science. Giulia Sammarini is supported by Fondazione Famiglia Parmiani.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics approval and consent to participate
The analysis, approved by the hospital Ethics Committee, was performed after written informed consent for study participation and anonymous data publication in accordance with national legislation and the Helsinki Declaration.
Consent for publication
Not applicable.
Availability of data and material
All data generated or analysed during this study are included in this published article and its supplementary information files.
Author contribution statement
GR and SA conceived and designed the project; GR, GS, ER, and MU performed the analysis; MN and MAP acquired and managed patients; GR, VS, FF, CS, RR, MN, MAP, and SA analysed and interpreted the data; GR, CS, PH, and SA wrote the manuscript; all authors approved the final version of the manuscript.
Supplementary material:
Supplementary data for this article can be accessed here.
References
- [1].Biasco G, Velo D, Angriman I, et al. Gastrointestinal stromal tumors: report of an audit and review of the literature. Eur J Cancer Prev. 2009;18:106–116. [DOI] [PubMed] [Google Scholar]
- [2].Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998. [cited 2016 November23]; 279:577–580. [DOI] [PubMed] [Google Scholar]
- [3].Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. [DOI] [PubMed] [Google Scholar]
- [4].Corless CL, Barnett CM, Heinrich MC.. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–878. [DOI] [PubMed] [Google Scholar]
- [5].Angelini S, Ravegnini G, Fletcher JA, et al. Clinical relevance of pharmacogenetics in gastrointestinal stromal tumor treatment in the era of personalized therapy. Pharmacogenomics. 2013;14:941–956. [DOI] [PubMed] [Google Scholar]
- [6].Ravegnini G, Nannini M, Sammarini G, et al. Personalized medicine in gastrointestinal stromal tumor (GIST): clinical implications of the somatic and germline DNA analysis. Int J Mol Sci. 2015;16:15592–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Casali PG, Zalcberg J, Le Cesne A, et al. Ten-year progression-free and overall survival in patients with unresectable or metastatic gi stromal tumors: long-term analysis of the european organisation for research and treatment of cancer, Italian sarcoma group, and Australasian gastrointestinal Tr. J Clin Oncol. 2017;35:1713–1720. [DOI] [PubMed] [Google Scholar]
- [8].Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results. JAMA Oncol. 2017;3:944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Serrano C, Wang Y, Mariño-Enríquez A, et al. KRAS and KIT gatekeeper mutations confer polyclonal primary imatinib resistance in GI stromal tumors: relevance of concomitant phosphatidylinositol 3-kinase/AKT dysregulation. J Clin Oncol. 2015;33:e93–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ravegnini G, Sammarini G, Angelini S, et al. Pharmacogenetics of tyrosine kinase inhibitors in gastrointestinal stromal tumor and chronic myeloid leukemia. Expert Opin Drug Metab Toxicol. 2016;12:733–742. [DOI] [PubMed] [Google Scholar]
- [11].Pantaleo MA, Biasco G.. Management of GIST—go beyond imatinib: treat resistant subtypes. Nat Rev Clin Oncol. 2015;12:440–442. [DOI] [PubMed] [Google Scholar]
- [12].Ravegnini G, Urbini M, Simeon V, et al. An exploratory study by DMET array identifies a germline signature associated with imatinib response in gastrointestinal stromal tumor. Pharmacogenomics J. 2018. DOI: 10.1038/s41397-018-0050-4. [DOI] [PubMed] [Google Scholar]
- [13].O’Brien KM, Orlow I, Antonescu CR, et al. Gastrointestinal stromal tumors, somatic mutations and candidate genetic risk variants. PLoS One. 2013;8:e62119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Angelini S, Pantaleo MAMA, Ravegnini G, et al. Polymorphisms in OCTN1 and OCTN2 transporters genes are associated with prolonged time to progression in unresectable gastrointestinal stromal tumours treated with imatinib therapy. Pharmacol Res. 2013;68:1–6. [DOI] [PubMed] [Google Scholar]
- [15].Kang BW, Kim JG, Chae YS, et al. Clinical significance of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 gene polymorphisms in patients with gastrointestinal stromal tumors. Asia Pac J Clin Oncol. 2014;10:e40–e45. [DOI] [PubMed] [Google Scholar]
- [16].Angelini S, Ravegnini G, Nannini M, et al. Folate-related polymorphisms in gastrointestinal stromal tumours: susceptibility and correlation with tumour characteristics and clinical outcome. Eur J Hum Genet. 2015;23:817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang R, Zhao J, Xu J, et al. Genetic variations in the TERT and CLPTM1L gene region and gastrointestinal stromal tumors risk. Oncotarget. 2015;6:31360–31367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ravegnini G, Nannini M, Simeon V, et al. Polymorphisms in DNA repair genes in gastrointestinal stromal tumours: susceptibility and correlation with tumour characteristics and clinical outcome. Tumour Biol. 2016;37:13413–13423. [DOI] [PubMed] [Google Scholar]
- [19].Ravegnini G, Nannini M, Zenesini C, et al. An exploratory association of polymorphisms in angiogenesis-related genes with susceptibility, clinical response and toxicity in gastrointestinal stromal tumors receiving sunitinib after imatinib failure. Angiogenesis 20 DOI: 10.1007/s10456-016-9534-5 [DOI] [PubMed] [Google Scholar]
- [20].Nannini M, Ravegnini G, Angelini S, et al. miRNA profiling in gastrointestinal stromal tumors: implication as diagnostic and prognostic markers. Epigenomics. 2015;7:1033–1049. [DOI] [PubMed] [Google Scholar]
- [21].Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annu Rev Med. 2009;60:167–179. [DOI] [PubMed] [Google Scholar]
- [22].Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. [DOI] [PubMed] [Google Scholar]
- [23].Kasinski AL, Slack FJ. Epigenetics and genetics. microRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol Mech Dis. 2014;9:287–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Stahlhut C, Slack FJ. MicroRNAs and the cancer phenotype: profiling, signatures and clinical implications. Genome Med. 2013;5:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shah MY, Ferracin M, Pileczki V, et al. Cancer-associated rs6983267 SNP and its accompanying long noncoding RNA CCAT2 induce myeloid malignancies via unique SNP-specific RNA mutations. Genome Res. 2018;28:432–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Iorio MV, Visone R, Di Leva G, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–8707. [DOI] [PubMed] [Google Scholar]
- [28].Yousef GM. miRSNP-based approach identifies a miRNA that regulates prostate-specific antigen in an allele-specific manner. Cancer Discov. 2015;5:351–352. [DOI] [PubMed] [Google Scholar]
- [29].Peckham-Gregory EC, Thapa DR, Martinson J, et al. MicroRNA-related polymorphisms and non-Hodgkin lymphoma susceptibility in the multicenter AIDS cohort study. Cancer Epidemiol. 2016;45:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Koelz M. Down-regulation of miR-221 and miR-222 correlates with pronounced Kit expression in gastrointestinal stromal tumors. Int J Oncol. 2011;38 DOI: 10.3892/ijo.2010.857. [DOI] [PubMed] [Google Scholar]
- [31].Gits CMM, van Kuijk PF, Jonkers MBE, et al. MiR-17-92 and miR-221/222 cluster members target KIT and ETV1 in human gastrointestinal stromal tumours. Br J Cancer. 2013;109:1625–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ihle MA, Trautmann M, Kuenstlinger H, et al. miRNA-221 and miRNA-222 induce apoptosis via the KIT/AKT signalling pathway in gastrointestinal stromal tumours. Mol Oncol. 2015;9:1421–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pantaleo MA, Ravegnini G, Astolfi A, et al. Integrating miRNA and gene expression profiling analysis revealed regulatory networks in gastrointestinal stromal tumors. Epigenomics. 2016;8:1347–1366. [DOI] [PubMed] [Google Scholar]
- [34].Pantaleo MA, Urbini M, Indio V, et al. Genome-wide analysis identifies MEN1 and MAX mutations and a neuroendocrine-like molecular heterogeneity in quadruple WT GIST. Mol Cancer Res. 2017;15:553–562. [DOI] [PubMed] [Google Scholar]
- [35].Balachandran VP, Dematteo RP. Targeted therapy for cancer: the gastrointestinal stromal tumor model. Surg Oncol Clin N Am. 2013;22:805–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Felicetti F, Errico MC, Bottero L, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68:2745–2754. [DOI] [PubMed] [Google Scholar]
- [37].Igoucheva O, Alexeev V. MicroRNA-dependent regulation of cKit in cutaneous melanoma. Biochem Biophys Res Commun. 2009;379:790–794. [DOI] [PubMed] [Google Scholar]
- [38].Godshalk SE, Paranjape T, Nallur S, et al. A variant in a microRNA complementary site in the 3' UTR of the KIT oncogene increases risk of acral melanoma. Oncogene. 2011;30:1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang J, Du X, Lazar AJF, et al. Genetic aberrations of gastrointestinal stromal tumors. Cancer. 2008;113:1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nannini M, Biasco G, Astolfi A, et al. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J Med Genet. 2013;50:653–661. [DOI] [PubMed] [Google Scholar]
- [41].Sioulas AD, Vasilatou D, Pappa V, et al. Epigenetics in gastrointestinal stromal tumors: clinical implications and potential therapeutic perspectives. Dig Dis Sci. 2013;58:3094–3102. [DOI] [PubMed] [Google Scholar]
- [42].Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. [DOI] [PubMed] [Google Scholar]
- [43].Garofalo M, Quintavalle C, Romano G, et al. miR221/222 in cancer: their role in tumor progression and response to therapy. Curr Mol Med. 2012. [cited 2017 April13]; 12:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].He H, Jazdzewski K, Li W, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci. 2005;102:19075–19080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shaker O, Alhelf M, Morcos G, et al. miRNA-101-1 and miRNA-221 expressions and their polymorphisms as biomarkers for early diagnosis of hepatocellular carcinoma. Infect Genet Evol. 2017;51:173–181. [DOI] [PubMed] [Google Scholar]
- [46].Abelson JF, Kwan KY, O’Roak BJ, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–320. [DOI] [PubMed] [Google Scholar]
- [47].Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222. [DOI] [PubMed] [Google Scholar]
- [48].Wang Y, Marino-Enriquez A, Bennett RR, et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat Genet. 2014;46(6):601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Katoh MM, Igarashi M, Fukuda H, et al. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 2013;328:198–206. [DOI] [PubMed] [Google Scholar]
- [51].Jonsson H, Peng SL. Forkhead transcription factors in immunology. Cell Mol Life Sci. 2005;62:397–409. [DOI] [PubMed] [Google Scholar]
- [52].Flajollet S, Poras I, Carosella ED, et al. RREB-1 is a transcriptional repressor of HLA-G. J Immunol. 2009;183:6948–6959. [DOI] [PubMed] [Google Scholar]
- [53].Bak RO, Mikkelsen JG. miRNA sponges: soaking up miRNAs for regulation of gene expression. Wiley Interdiscip Rev RNA. 2014;5:317–333. [DOI] [PubMed] [Google Scholar]
- [54].Pan X, Wenzel A, Jensen LJ, et al. Genome-wide identification of clusters of predicted microRNA binding sites as microRNA sponge candidates. PLoS One. 2018;13:e0202369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].STATA Corporation Stata statistical software, release 11. College Station, TX, 2011 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.