

Abstract
Inflammation in the synovium is known to mediate joint destruction in several forms of arthritis. Fibroblast-like synoviocytes (FLS) are cells that reside in the synovial lining of joints and are known to be key contributors to inflammation associated with arthritis. FLS are a major source of inflammatory cytokines and catabolic enzymes that promote joint degeneration. We now know that there exists a direct correlation between the signaling pathways that are activated by the pro-inflammatory molecules produced by the FLS, and the severity of joint degeneration in arthritis. Research focused on understanding the signaling pathways that are activated by these pro-inflammatory molecules has led to major advancements in the understanding of the joint pathology in arthritis. Transcription factors (TFs) that act as downstream mediators of the pro-inflammatory signaling cascades in various cell types have been reported to play an important role in inducing the deleterious transformation of the FLS. Interestingly, recent studies have started uncovering that several TFs that were previously reported to play role in embryonic development and cancer, but not known to have pronounced roles in tissue inflammation, can actually play crucial roles in the regulation of the pathological properties of the FLS. In this review, we will discuss reports that have been able to impart novel arthritogenic roles to TFs that are specialized in embryonic development. We also discuss the therapeutic potential of targeting these newly identified regulators of FLS transformation in the treatment of arthritis.
Keywords: joint disease, transcription factors, arthritis, inflammation, fibroblasts
Graphical Abstract
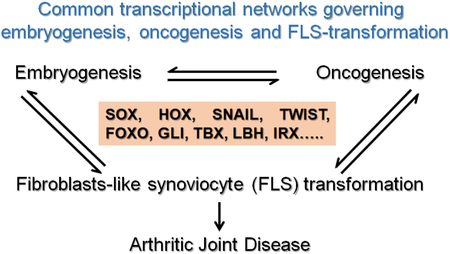
1. Introduction.
Regulation of gene expression dictates normal development and adult life of an organism and de-regulation of gene expression leads to disease. Changes in the expression of genes under physiological and pathological conditions is regulated at multiple levels. At the top of the level are external stimuli, such as growth factors, hormones and cytokines as well as mechanical and chemical stimuli. They in turn activate intracellular signaling cascades that transduce the extracellular messages into the cell nuclei. Gene expression is then achieved by transcription, where the genetic information in DNA is copied into an RNA transcript (1). Transcription factors (TFs) are a class of proteins that directly bind to DNA and provide a surface for the assembly of large multi-protein transcriptional complexes whose cooperative actions, alter gene expression and influence properties such as cell growth, differentiation, and homeostasis. Aberrations at any level of these biological processes can lead to a variety of developmental disorders and pathologies ranging from cancers to musculoskeletal and joint diseases (1, 2). Over the past years there has been a lot of emphasis on developing targeted therapeutics for correcting biological stimuli and intermediates of the signaling cascades. Although hugely successful in treating various diseases, vast amount of redundancy and overlap between the signaling cascades, has made the task very difficult. More so because these pathologies can be triggered by multiple environmental and biological factors and they are likely to be influenced by patient-specific factors. Transcription factors, until recently were not considered as attractive drug targets, because of their relatively low abundance and the lack of availability of small molecules that can block their actions. However, recent research has made available, critical information about the molecular architecture and mode of action transcription factors in multiple contexts (3, 4). Latest technologies such as, nanoparticles, small RNA’s, genome editing coupled with improved means of drug delivery and availability are creating previously unavailable opportunities for targeting transcription factors that are pivotal for the regulation of gene expression (5–7). Attempts to target transcription factors that exhibit aberrant activities are already underway in the cancer field (4, 8). We predict that similar strategies could be employed for targeting of fibroblast-like synoviocytes (FLS) in joint diseases, since the pathological properties of the FLS in arthritic disease are comparable to that of cancer-associated fibroblasts (CAF) in various cancers (9).
Fibroblasts are a ubiquitous cell type that exist in multiple organs and play vital roles in embryonic development and adult tissue homeostasis. Multiple lines of evidence points to the existence of specialized fibroblasts that are mainly involved in the production of extracellular matrix components of connective tissues and are needed for homeostasis (10). Fibroblasts also act as a source of adult stem cells that participate in reparative responses (10, 11). The specialized fibroblasts located in the synovial lining of joints are commonly called synovial fibroblasts or fibroblast-like synoviocytes (FLS) (9, 12). They specialize in the production of joint lubricants. In pathological conditions (CAF in cancers, and FLS in arthritis) acquire an aggressive and transformed phenotype, which is characterized by the production of inflammatory cytokines and catabolic enzymes, increased proliferation, evasion of apoptosis and the host immune system (13). For a long time, FLS transformation was considered a classical feature present only in inflammatory forms of arthritis, which are often associated auto-immune diseases, such as rheumatoid arthritis (RA). Recent studies suggest that FLS transformation and synovitis can contribute to osteoarthritis (OA), where the underlying cause for joint degeneration is age or trauma-induced wear and tear of the articular cartilage (14). Transformed FLS, are known to produce catabolic molecules that degrade the adjacent bone and cartilage in the joints (9, 12, 15). In addition to above mentioned similarities between CAF and transformed-FLS, de-regulation of the activities of several TF’s is evident in both types of fibroblasts (Fig 1). TF’s and pathways that are involved in FLS-transformation, includes the ones that work down stream of pro-inflammatory stimuli with proven arthritogenic roles (reviewed elsewhere(12)), as well as TF’s that are well-known for their roles in embryonic development and cancer and currently being investigated for their potential roles in FLS transformation during inflammatory arthritic diseases. In this review we will primarily focus on the TF’s with roles in embryonic development and discuss the possibility of developing new and effective TF-targeting strategies that could prove to be valuable in the treatment of arthritic and other inflammatory diseases.
Figure 1. Comparison between the properties of transformed FLS and CAF:

Aberrant activation of transcription factors occurs when FLS and CAF are exposed to pro-inflammatory ligands, including cytokines, chemokines and growth factors. Binding of the ligands to the cell surface receptors results in the activation of signaling cascades that eventually results in the binding of the TFs to their respective binding sites in the enhancers/promoters of their target genes. The activated target genes function as mediators of pro-inflammatory processes, extra-cellular matrix (ECM) degradation, epithelial-to-mesenchymal transition (EMT) and promoters of cell survival and proliferation. Together these molecular changes lead to the proliferative and invasive behavior of FLS and CAF.
2. SOX transcription factors:
The mammalian SOX family consists of 20 proteins, defined by the presence of a SOX-HMG signature DNA binding domain. They are divided in eight groups (A-H) depending on their homology (16). The canonical functions of SOX factors affect diverse processes and tissue systems, including: preimplantation development, germ cell differentiation, pluripotency, primitive and definitive endoderm induction, hematopoiesis, as well as development and regulation of the skeletal, pituitary, cardiac, neural crest, and nervous systems(16, 17). Reflecting this potential, SOX factors are highly implicated in developmental disorders and cancer (18). The following sections provide evidence suggesting that the SOX family members, SOX5, SOX4 and SOX11 play a role in FLS transformation.
2.1. SOX5:
SOX5 belongs to the D group of SOX family transcription factor and is expressed in multiple cell linages including the spermatids, neurons, fetal brain, striated muscles, chondrocytes and B cells(19). Sox5 primarily plays important roles in the regulation of embryonic development of the tissues it is expressed, in particular, nervous system and skeleton (20–22). Because of these critical developmental roles, haploinsufficiency of SOX5 induced by genomic deletions has been linked to developmental delay, intellectual disability including motor disturbances (23). Although the role of SOX5 in adult tissue homeostasis are not completely understood, it is known that SOX5 is a marker for poor prognosis of prostrate, adenocarcinoma and non-small cell lung cancer among others (24–26). The link between SOX5 and FLS-activation was recently identified by Dr. Tan’s research group (27, 28). Their studies showed that SOX5 mRNA and protein can be detected at higher level in the synovium and synovial fluid of RA patient synovium vs OA synovium. Mechanistically, these studies showed that SOX5 increases lamellipodia formation, migration and invasive properties of human RA-FLS. Further, localized shRNA-mediated silencing of Sox5 in the joints of collagen-induced arthritis mouse model showed that downregulation of Sox5 diminished the percentage of RANKL positive bone eroding cells and pannus formation. In support of these studies another group reported that SOX5 is a direct target of miR-212–3p in human RA-FLS and that over-expression of SOX5 reversed the effects of miR-212–3p (29). At the mechanistic level, it was proposed that miR-212–3p could reduce cell proliferation, but promoted cell apoptosis of RA-FLS, via repressing SOX5.
2.2. SOX4 and SOX11:
SOX4, SOX11, and SOX12 form group C of the transcription factors containing a SOX DNA-binding domain. SOXC proteins share more identity with one another than with other SOX proteins (16). Studies from mouse development, showed that Sox4 and Sox11 are primarily co-expressed in various types of multipotent progenitor cells, and that they act largely in redundancy to determine the behavior and survival of these mesenchymal and neural progenitor cells. Sox4-null, Sox11-null, and Sox4/11–double- conditional- null mutant mice therefore exhibit major defects in the development of organs such as the skeleton, heart, brain, and eyes (30). SOX4 and SOX11 have been shown to be highly expressed in prostate, breast, leukemia, colorectal, and other forms of cancer in humans (31). SOX4 has been recognized as a master regulator of cell proliferation and metastasis in several cancer types, with SOX11 recognized as a poor prognosis marker in lymphoma and breast cancer subtypes (32). Our laboratory recently identified that SOXC proteins, 4 and 11 play a critical role in the pathological behavior of the both OA-FLS and RA-FLS(33). By performing conditional deletion of SOXC genes in the cells that express the joint lubricant, Lubricin (Prg4), we showed that SOXC proteins are dispensable for joint integrity in juvenile and young adult mice, but are required for synovial pannus formation and articular cartilage degeneration in TNF-induced arthritic disease. At the molecular level, we were able to demonstrate that SOXC proteins are very unstable in FLS under basal conditions but robustly stabilized upon stimulation with TNF and other proinflammatory cytokines. Our data suggest that similar to that of SOX5, SOX4/11, reduce apoptosis, increase proliferation and support the invasiveness of FLS. Thus, SOX5 and SOX4/11 TFs are likely to amplify the expression of genes that promote FLS survival and migration, which are critical events in inflammation-induced FLS transformation.
2.3. SOX2:
SOX2 belongs to SOXB1 group. It is one of the Yamanaka factors famous for its ability to transform various somatic cells into pluripotent stem cells and is considered to be master regulator of embryonic and cancer stem cell fates(34). SOX2 has not been directly linked to FLS-transformation, but is known to be expressed by synovial sarcoma cells (35). We speculate that SOX2 may have a role in the stem cell-like fibroblasts in the synovial lining that are suggested to participate in joint repair. Whether or not SOX2 has a role in transforming the stem cell-like synovial fibroblasts needs to be determined.
3. HOX family.
The HOX’s are an evolutionarily conserved group of genes that encode for Homeobox transcription factors (36, 37). In humans there are 39 HOX family members that are located in four clusters (A-D) on different chromosomes. Their coordinated expression in both space and time defines cellular identities along the body axes and therefore they are critical for embryonic patterning (36). HOX genes continue to be expressed in postnatal life (37). They are associated with cancer development, including prostate, renal, breast, ovarian and lung cancer and hematological malignancies (38). Seminal work from several labs is beginning to provide evidence that, beyond their roles in embryogenesis and cancer HOX genes have the ability to confer location-specific disease susceptibility to adult cell-types in the skeleton, including bone marrow MSCs, articular cartilage and synovium (39–42). For instance, it was found that OA cartilage from hip and knee joints could be differentiated from each other by distinct patterns of HOX gene expression and DNA methylation (40). Another recent large scale transcriptome analysis study showed that the HOX signature of synovial fibroblasts of proximal joints (hands) and the distal joints (shoulder and knee) are distinct and they recapitulate key features of the embryonic positional HOX gene expression along the proximal–distal developmental axes (42). Transcripts encoded in the 5′ end of the HOXA (HOXA11-AS, HOXA13, HOTTIP) and HOXD genomic loci (HOXD10, HOXD11, HOXD13) reflected the positional identity to distal joint fibroblasts, while the shoulder-specific HOX transcripts encoded in the 3′ end of the HOXA, HOXB and HOXD clusters. These studies speculated that HOX code could describe the intrinsic variability in the FLS and articular cartilage from various joints. These studies also suggested that HOX code may also explain differences in susceptibility, progression and degeneration of various joints.
4. SNAIL family:
This family genes encode zinc finger transcriptional repressors. SNAIL family genes are best known for their role in the induction of epithelial to mesenchymal transition (EMT) (43). Snail-induced EMT converts epithelial cells into mesenchymal cells with migratory properties that contribute to the formation of many tissues during embryonic development and to the acquisition of invasive properties in epithelial tumors (43, 44). With respect to RA, a study utilizing RA-FLS and rodent collagen-induced arthritis model, demonstrated that Snai1 (Snail) regulates TNFα-mediated activation of synovial fibroblasts in the rheumatoid joint (45). They showed that loss of Snail expression ameliorated arthritis, with reduced Cadherin11 expression and reduced levels of extracellular matrix deposition in the joints of rats with collagen-induced arthritis, whereas overexpression of Snail exacerbated arthritis, with increased Cadherin11 expression and increased levels of extracellular matrix deposition. Mechanistically, Snail is required for the formation of invadosomes, induces extracellular matrix degradation in synovial cells by repressing PTEN, resulting in increased phosphorylation of platelet-derived growth factor receptor and activation of the phosphatidylinositol 3-kinase/AKT pathway (46–48). Other studies demonstrated that SNAI2 (SLUG) is overexpressed in human RA synovium and that suppression of SNAI2 gene facilitates apoptosis of FLS by increasing Puma transactivation and platelet derived growth factor receptor-alpha activation (49).
5. TWIST family.
This family consists of two members TWIST1 and 2 which are basic helix–loop–helix transcription factors that is essential for the development of mesodermally derived tissues, including the skeleton, muscle and heart (50). Mutations in the human TWIST1 gene are associated with Saethre-Chotzen syndrome, which is characterized by craniosynostosis (premature fusion of cranial sutures) and limb deformities (51). On the other hand, TWIST1 is known to be an important inducer of epithelial–mesenchymal transition (EMT) in a variety of epithelial cancer cells and increased TWIST1 expression is associated with poor prognosis (52). The association of TWIST1 with FLS-transformation was suggested from a systems biology base approach to identify gene networks that regulate interleukin1 beta -induced activation of RA-FLS (53). TWIST1 and TWIST2 were among the selected 13 key RA-FLS regulators in this study. In vitro assays confirmed that TWIST1 expression was elevated in RA-FLSs and is crucial for migration and invasion of FLSs stimulated with interleukin1 beta. Further studies are required to confirm the roles of TWIST family proteins in FLS transformation.
6. FOXO transcription factors:
Forkhead proteins, are a family of transcriptional regulators characterized by a conserved DNA-binding domain termed the ‘forkhead box’ (54). FOXO group is comprised of 4 members, FOXO1, FOXO3, FOXO4, and FOXO6. The genes exhibit both redundant and unique roles in development and disease and control cell fate during embryonic development, tissue homeostasis, aging and disease. They are critically required for cardiovascular, pancreatic beta cell, liver and T-cell development.. Some of the biological processes regulated by FOXO proteins include modulation of apoptosis, cell cycle transitions, DNA repair, oxidative stress and glucose metabolism. High expression or activation of FOXOs is often associated with cell-cycle arrest and apoptosis enabling them to function as tumor suppressors. Murine studies showed that Foxo1/Foxo3/Foxo4 are redundant with respect to tumor suppression (55). FOXO1 and FOXO3a, are expressed and phosphorylated in synovial tissue from both RA patients and OA patients and in RA-FLS (56). Downregulation of FOXO1 expression is required to promote FLS survival in RA (57). Similarly, up-regulation of FOXO3a signaling in synovial fibroblasts via simvastatin treatment played a beneficial role in inflammatory arthritis (58). It is currently not known whether FOXO factors act in redundancy during FLS transformation. Continued study of the pathways linking FOXO proteins, and the inflammatory responses of RA-FLS may provide new insights into their functions.
7. GLI1:
GLI family proteins are transcription factors that share five highly conserved tandem zinc fingers and a consensus histidine-cysteine linker sequence between zinc fingers. GLI proteins are the main downstream effectors of the Hedgehog signaling pathway that plays an essential role in the growth, development, and homeostasis of many tissues in vertebrates (59). Binding of hedgehog ligands to their receptor Patched1 (PTCH1) induces conformational changes in PTCH1, finally resulting in the stabilization of GLI family zinc finger transcription factors, which stimulate the transcription of hedgehog target genes (60). Altered activation of the hedgehog pathway has also been implicated in the pathogenesis of malignancies such as basal cell carcinoma, pancreatic carcinoma, and gioblastoma and medulloblastoma (61). Of the four GLI members, GLI1 has been implicated in the transformation of FLS. It was initially reported that GLI1 and other hedgehog pathway components including, Sonic hedgehog, Smoothened and PTCH1 are expressed at higher levels in RA synovial tissue than in healthy synovial tissue (62). In a rat model of collagen-induced arthritis, inhibition of Gli1 activation, by a small molecule antagonist, GANT61 resulted in reduced proliferation of RA-FLS, which was accompanied by increase in their apoptosis rate (63). These data were also confirmed by another study which showed that inhibiting hedgehog signaling in human RA-FLS by cyclopamine, resulted in a G1 phase arrest in the cell cycle (64). In cancer cells GLI1 was shown to regulate the AKT/mTOR pathway, suggesting that a similar mechanism may also be employed during FLS transformation (65) (66).
8. TBX5:
The T-box gene family refers to a group of transcription factors that share a highly conserved, sequence-specific DNA-binding domain (T-box) (67). TBX5 is the most studied member of the T-box transcription factor family because of its role in cardiac and forelimb development. Patients with dominant mutations in TBX5 develop Holt-Oram syndrome, which is characterized by defects of the cardiac septa, cardiac conduction system, and the anterior forelimb (68). Postnatally, expression of TBX5 is linked to cancer, where it plays context dependent roles. Higher levels of TBX5 expression are associated with unfavorable survival rates in patients with stage I and II gastric adenocarcinoma, but TBX5 overexpression markedly suppressed in vitro non-small cell lung cancer cell proliferation, colony formation, and invasion and induced apoptosis (68) (69). An unexpected role for TBX5 was uncovered by a study, which showed that TBX5 gene was less methylated in RA synovium and RA-FLS than in OA samples (70). In RA synovium, TBX5 expression was primarily localized to the synovial lining. This study also revealed that the chemokines, interleukin 8, CXCL12, and CCL20 were common targets of TBX5 in FLS. Thus, they concluded that RA-FLS contribute to the inflammatory processes operating in the pathogenesis of RA via epigenetic control of TBX5. In addition to DNA methylation, the levels of TBX5 in FLS is also likely to be regulated by miR-10a-5p that targets TBX5.
9. LBH:
Limb bud and heart (LBH) is a highly conserved, tissue-specific transcription cofactor in vertebrates and is a target of the canonical WNT signaling pathway. During mouse embryonic development, Lbh is expressed in the limb buds, heart, gut, kidney, gonads and nervous system (71) (72). Deletion of Lbh does not affect mouse development, but overexpression is detrimental to mouse cardiac development and Chick limb morphogenesis (72, 73). Similarly, aberrant gain-of function of LBH in humans, results in a rare genetic disorder characterized by multiple congenital anomalies including cardiovascular, skeletal, and limb malformations(74) (73). In cancer, LBH plays a context depend role. It is negatively associated with the poor prognosis of lung adenocarcinoma and nasopharyngeal carcinoma, but positively with poor prognosis of aggressive basal subtype human breast cancers and hepatocellular carcinoma. After identifying that the LBH locus has risk alleles associated with RA/celiac disease and lupus Firestein’s group extensively investigated the link between LBH and inflammation-induced FLS transformation(75) (76). They then showed that the RA-associated single-nucleotide polymorphism (SNP) decreases LBH gene transcription and supported this observation by showing that Lbh knockout exacerbated the disease severity in the K/BxN serum transfer arthritis mouse model. In another study this group reported that LBH deficiency increases interleukin1 beta secretion, causes S phase arrest by decreasing expression of the catalytic subunit of DNA polymerase alpha, increasing DNA damage, and activating the S phase checkpoint, thus enhancing synovial inflammation (77).
10. IRX1:
Iroquois homeobox (IRX), containing TFs that are widely in various embryonic tissues. They play a crucial role in the patterning of tissues and organs during development and mostly function as transcriptional repressors (78). Irx1 is highly expressed in the developing brain, lung, limbs, kidney, testis and developing teeth(79, 80) (81). Irx1 null mice die at birth due to pulmonary immaturity (82). In relation to cancer, Irx1 was shown to be a tumor suppressor in gastric cancer and head and neck squamous cell carcinoma (83) (84). Inactivation of Irx1 through epigenetic mechanism was shown to promote metastasis of gastric cancer cells (85), while overexpression was shown to induce growth arrest and prevented pulmonary metastasis of gastric cancer cells in murine models (86). On the other hand, a gain of chromosome 5p15.33 (chromosomal location of IRX1) has been frequently detected in osteosarcoma cell lines (87). Further investigations revealed that IRX1 is epigenetically activated in highly metastatic osteosarcoma cell lines. Downregulation of IRX1 in osteosarcoma cell lines resulted in the inhibition of NFκB activity, and suppression of metastasis (88). A genome-wide association study (GWAS) using RF-positive and RF-negative patients identified single nucleotide polymorphism in the IRX1 gene locus and predicted it to be a risk locus for rheumatoid factor positivity in rheumatoid arthritis (89). Another study analyzed genome-wide DNA methylation and CpG analysis from FLS isolated from RA, OA or healthy subjects and reported that IRX1 and IRX6 was among the 13 gene loci that were hypermethylated in RA-FLS(90). They speculated that down regulation of Irx1 may play a role in RA pathogenesis. Further studies are needed to understand the positive and negative effects of Irx genes on RA pathogenesis.
12. Targeting TF’s:
Indirect targeting of transcription factors through the inhibition of upstream signaling effectors (e.g., activating ligand or kinase) is a proven therapeutic strategy in case of cancer and other auto-immune diseases. It is also becoming increasingly evident that direct targeting of TF’s, which integrate inputs from various signaling pathways, is likely to be a more specific approach, with reduced off-target effects(3, 4). Some of the approaches that are being developed, as illustrated in Figure 2 include, blocking TF expression by small RNA’s (siRNA, shRNA, miRNAs) or by regulation of epigenetics, blocking TF-co-factor interactions, blocking TF-DNA interactions, altering the protein stability of TF’s and finally CRISPR mediated genome editing to correct the genetic mutations in TF functions(3, 4, 91, 92). The following findings, show that some of the TF’s discussed above can indeed be targeted under different cellular contexts, providing a proof-of-concept for attempting similar strategies for targeting TF’s to treat arthritic diseases.
Figure 2. Strategies for targeting TFs:

Several currently available strategies that could be utilized to block the pathological functions of TFs are listed here. lightning bolt, small molecule/drug; red circle, post-translational modification of protein; me, methylation; acacetylation.
12.1. Targeting the SOX’s:
SOX18 inhibitors that interfere with DNA binding and disrupt protein-protein interaction with its transcriptional partner RBPJ (Notch signaling transcription factor) have already been developed and proven to delay progression of breast cancer(93, 94), suggesting that it is feasible to develop drugs that target other SOX family TF’s. SOX4 and 11 could additionally be targeted by altering their protein stability as suggested by our previous work (33). SOX5 could be targeted by modulating the expression of miR-212–3p in RA-FLS (29).
12.2. Targeting the HOX’s:
In case of HOX proteins inhibition of TF-co-factor binding is being developed as a potential approach to treat cancers. Elegant work has proven that HOX proteins interact with co-factors that enhances their DNA binding specificity (95). Pre-B-cell Leukemia (PBX) proteins are HOX co-factors that form strong complexes with HOX1–11. This interaction is mediated by a highly conserved hexapeptide sequence in HOX proteins. Short peptides that block this interaction are being considered for therapeutic purposes (96).
12.3. Targeting the FOX’s:
Elegant studies have shown that inhibition of the FOXO1 has beneficial effects on diabetic hyperglycaemia, but also promotes lipogenesis. Langlet et al. identified SIN3a as co-repressor that is required for FOXO mediated regulation of glucose levels, but not for other FOXO1 functions. This study also identified a FOXO1 inhibitor that targeted the activity of the co-repressor resulting in the inhibition of glucose production without activating lipogenesis (97). It remains to be investigated whether SIN3a or another co-factor is involved in FOXO mediated FLS transformation.
12.4. Targeting GLI’s:
Specific inhibitors for blocking GLI transcription have been developed and found to be effective in limiting cancer cell proliferation and metastasis in the laboratory setting, but clinical trials are yet to be carried out. GANT61, an inhibitor of GLI transcriptional activity has been extensively studied at the preclinical level. This molecule was shown to bind to the 5-zinc finger GLI1 protein between zinc fingers 2 and 3 at without affecting the GLI-DNA binding region, but severely compromising its transactivation function (98, 99). The safety of this compound is yet to be determined, as this molecule may influence other pathways in addition to canonical GLI functions on AKT/mTOR axis (100). Plant based compounds that inhibit GLI1 are also reported, but further evaluation is required.
12.5. Targeting SNAIL:
There are two classes of inhibitors that have been proven to be successful in preventing SNAIL mediated transcription in cancers at the pre-clinical level. The first class of inhibitors exploit the interaction between p53 and SNAIL. It was shown that one of the mechanisms through which p53 is eliminated from cancer cells is through its interaction with SNAIL. GN25 and GN29 are small molecules that inhibit SNAIL binding to p53 and thereby prevent p53 loss in cancer cells (101). Another group developed a Co(III) Schiff base complex modified with a 17-bp DNA sequence that can selectively inhibit DNS binding of SNAIL family TF’s (Snail, Slug and Smuc) (102). Further evaluations are still needed for determining the safety and efficacy of these products.
12.6. Targeting TWIST 1:
TWIST 1 is known to play a role in tumor initiation, stemness, angiogenesis, invasion, metastasis, and chemo-resistance in a variety of carcinomas, sarcomas, and hematological malignances and is an attractive candidate therapeutic target for cancer treatment in the clinic. Harmine, is an alkaloid compound identified as a TWIST 1 inhibitor through a chemical–bioinformatic approach using connectivity mapping analysis (103). This compound could effectively block TWIST 1 functions and cancer progression in patient-derived xenograft mouse model of non–small cell lung cancer. At the mechanistic level, harmine promoted TWIST1 protein degradation.
12. Conclusions and Future directions:
In this review we complied and discussed recent reports that have started to identify that several TF’s with key roles in embryonic development are also responsible for the transformed behavior of FLS in inflammatory arthritic diseases. In doing so, we revealed that these TF’s function also have key roles in cancer progression either by acting as tumor suppressors or oncogenes that regulate tumor growth and metastasis. Although, data regarding the roles of these TF’s in embryogenesis and tumorigenesis are very strong, and supported by observations in vitro and in vivo from both mice and humans, the findings related to FLS transformation and arthritic disease are currently in the emerging phase. Nonetheless these new findings represent significant advancement in our knowledge about the molecular mechanisms, governing FLS transformation in arthritic disease. Further research will be need to understand the reasons for the aberrant activation of these TF’s in the FLS that are exposed to chronic inflammation and to determine, targeting which of these TF’s will yield therapeutic success. We also believe that inflammatory diseases with complex pathophysiology, involving multiple cell types and biological processes require drugs that can target more than one target protein or pathway. It is likely that combinatorial approaches coupled with effective drug delivery methods will greatly improve current treatment strategies.
Acknowledgements:
PB is supported by the National Institute of Arthritis and Musculoskeletal and Skin diseases grant (AR070736) and start-up support from the Department of Orthopaedics, Emory University School of Medicine.
List of Abbreviations:
- FLS
Fibroblast-like synoviocytes
- CAF
Cancer-associated fibroblast
- TF
Transcription factor
- RA
Rheumatoid arthritis
- OA
Osteoarthritis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lelli KM, Slattery M, Mann RS. Disentangling the many layers of eukaryotic transcriptional regulation. Annu Rev Genet. 2012;46:43–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagenbuchner J, Ausserlechner MJ. Targeting transcription factors by small compounds--Current strategies and future implications. Biochem Pharmacol. 2016;107:1–13. [DOI] [PubMed] [Google Scholar]
- 4.Lambert M, Jambon S, Depauw S, David-Cordonnier MH. Targeting Transcription Factors for Cancer Treatment. Molecules. 2018;23(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35(3):222–9. [DOI] [PubMed] [Google Scholar]
- 6.Tran S, DeGiovanni PJ, Piel B, Rai P. Cancer nanomedicine: a review of recent success in drug delivery. Clin Transl Med. 2017;6(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adli M The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Staalduinen J, Baker D, Ten Dijke P, van Dam H. Epithelial-mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene. 2018. [DOI] [PubMed] [Google Scholar]
- 9.Bhattaram P, Chandrasekharan U. The joint synovium: A critical determinant of articular cartilage fate in inflammatory joint diseases. Semin Cell Dev Biol. 2017;62:86–93. [DOI] [PubMed] [Google Scholar]
- 10.Furtado MB. Tissue fibroblasts: From bystanders to proactive modulators of homeostasis and disease. Differentiation. 2016;92(3):65. [DOI] [PubMed] [Google Scholar]
- 11.Ichim TE, O’Heeron P, Kesari S. Fibroblasts as a practical alternative to mesenchymal stem cells. J Transl Med. 2018;16(1):212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2018. [DOI] [PubMed] [Google Scholar]
- 14.Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bustamante MF, Garcia-Carbonell R, Whisenant KD, Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefebvre V, Dumitriu B, Penzo-Mendez A, Han Y, Pallavi B. Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int J Biochem Cell Biol. 2007;39(12):2195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Julian LM, McDonald AC, Stanford WL. Direct reprogramming with SOX factors: masters of cell fate. Curr Opin Genet Dev. 2017;46:24–36. [DOI] [PubMed] [Google Scholar]
- 18.Xu YR, Yang WX. SOX-mediated molecular crosstalk during the progression of tumorigenesis. Semin Cell Dev Biol. 2017;63:23–34. [DOI] [PubMed] [Google Scholar]
- 19.Lefebvre V The SoxD transcription factors--Sox5, Sox6, and Sox13--are key cell fate modulators. Int J Biochem Cell Biol. 2010;42(3):429–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai T, Jabaudon D, Molyneaux BJ, Azim E, Arlotta P, Menezes JR, et al. SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron. 2008;57(2):232–47. [DOI] [PubMed] [Google Scholar]
- 21.Kwan KY, Lam MM, Krsnik Z, Kawasawa YI, Lefebvre V, Sestan N. SOX5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. Proc Natl Acad Sci USA. 2008;105(41):16021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu CF, Lefebvre V. The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 2015;43(17):8183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nesbitt A, Bhoj EJ, McDonald Gibson K, Yu Z, Denenberg E, Sarmady M, et al. Exome sequencing expands the mechanism of SOX5-associated intellectual disability: A case presentation with review of sox-related disorders. Am J Med Genet A. 2015;167A(11):2548–54. [DOI] [PubMed] [Google Scholar]
- 24.Zou H, Wang S, Wang S, Wu H, Yu J, Chen Q, et al. SOX5 interacts with YAP1 to drive malignant potential of non-small cell lung cancer cells. Am J Cancer Res. 2018;8(5):866–78. [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Fu Y, Xu H, Teng P, Xie Q, Zhang Y, et al. SOX5 predicts poor prognosis in lung adenocarcinoma and promotes tumor metastasis through epithelial-mesenchymal transition. Oncotarget. 2018;9(13):10891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma S, Chan YP, Woolcock B, Hu L, Wong KY, Ling MT, et al. DNA fingerprinting tags novel altered chromosomal regions and identifies the involvement of SOX5 in the progression of prostate cancer. Int J Cancer. 2009;124(10):2323–32. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y, Wu Q, Xuan W, Feng X, Wang F, Tsao BP, et al. Transcription Factor SOX5 Promotes the Migration and Invasion of Fibroblast-Like Synoviocytes in Part by Regulating MMP-9 Expression in Collagen-Induced Arthritis. Front Immunol. 2018;9:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng X, Shi Y, Xu L, Peng Q, Wang F, Wang X, et al. Modulation of IL-6 induced RANKL expression in arthritic synovium by a transcription factor SOX5. Sci Rep. 2016;6:32001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Zhang XL, Li XF, Tang YC, Zhao X. miR-212–3p reduced proliferation, and promoted apoptosis of fibroblast-like synoviocytes via down-regulating SOX5 in rheumatoid arthritis. Eur Rev Med Pharmacol Sci. 2018;22(2):461–71. [DOI] [PubMed] [Google Scholar]
- 30.Bhattaram P, Penzo-Mendez A, Kato K, Bandyopadhyay K, Gadi A, Taketo MM, et al. SOXC proteins amplify canonical WNT signaling to secure nonchondrocytic fates in skeletogenesis. J Cell Biol. 2014;207(5):657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhattaram P, Penzo-Mendez A, Sock E, Colmenares C, Kaneko KJ, Vassilev A, et al. Organogenesis relies on SoxC transcription factors for the survival of neural and mesenchymal progenitors. Nat Commun. 2010;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhattaram P, Kato K, Lefebvre V. Progenitor cell fate, SOXC and WNT. Oncotarget. 2015;6(28):24596–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattaram P, Muschler G, Wixler V, Lefebvre V. Inflammatory Cytokines Stabilize SOXC Transcription Factors to Mediate the Transformation of Fibroblast-Like Synoviocytes in Arthritic Disease. Arthritis Rheumatol. 2018;70(3):371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wuebben EL, Rizzino A. The dark side of SOX2: cancer - a comprehensive overview. Oncotarget. 2017;8(27):44917–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naka N, Takenaka S, Araki N, Miwa T, Hashimoto N, Yoshioka K, et al. Synovial sarcoma is a stem cell malignancy. Stem Cells. 2010;28(7):1119–31. [DOI] [PubMed] [Google Scholar]
- 36.Mallo M Reassessing the Role of Hox Genes during Vertebrate Development and Evolution. Trends Genet. 2018;34(3):209–17. [DOI] [PubMed] [Google Scholar]
- 37.Rux DR, Wellik DM. Hox genes in the adult skeleton: Novel functions beyond embryonic development. Dev Dyn. 2017;246(4):310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan R, El-Tanani M, Hunter KD, Harrington KJ, Pandha HS. Targeting HOX/PBX dimers in cancer. Oncotarget. 2017;8(19):32322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelttari K, Barbero A, Martin I. A potential role of homeobox transcription factors in osteoarthritis. Ann Transl Med. 2015;3(17):254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.den Hollander W, Ramos YF, Bos SD, Bomer N, van der Breggen R, Lakenberg N, et al. Knee and hip articular cartilage have distinct epigenomic landscapes: implications for future cartilage regeneration approaches. Ann Rheum Dis. 2014;73(12):2208–12. [DOI] [PubMed] [Google Scholar]
- 41.Rux DR, Song JY, Swinehart IT, Pineault KM, Schlientz AJ, Trulik KG, et al. Regionally Restricted Hox Function in Adult Bone Marrow Multipotent Mesenchymal Stem/Stromal Cells. Dev Cell. 2016;39(6):653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frank-Bertoncelj M, Trenkmann M, Klein K, Karouzakis E, Rehrauer H, Bratus A, et al. Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat Commun. 2017;8:14852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132(14):3151–61. [DOI] [PubMed] [Google Scholar]
- 44.Baulida J Epithelial-to-mesenchymal transition transcription factors in cancer-associated fibroblasts. Mol Oncol. 2017;11(7):847–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen SY, Shiau AL, Li YT, Lin CC, Jou IM, Liu MF, et al. Transcription factor snail regulates tumor necrosis factor alpha-mediated synovial fibroblast activation in the rheumatoid joint. Arthritis Rheumatol. 2015;67(1):39–50. [DOI] [PubMed] [Google Scholar]
- 46.Lauzier A, Lavoie RR, Charbonneau M, Gouin-Boisvert B, Harper K, Dubois CM. Snail Is a Critical Mediator of Invadosome Formation and Joint Degradation in Arthritis. Am J Pathol. 2016;186(2):359–74. [DOI] [PubMed] [Google Scholar]
- 47.Lauzier A, Charbonneau M, Harper K, Jilaveanu-Pelmus M, Dubois CM. Formation of invadopodia-like structures by synovial cells promotes cartilage breakdown in collagen-induced arthritis: involvement of the protein tyrosine kinase Src. Arthritis Rheum. 2011;63(6):1591–602. [DOI] [PubMed] [Google Scholar]
- 48.Charbonneau M, Lavoie RR, Lauzier A, Harper K, McDonald PP, Dubois CM. Platelet-Derived Growth Factor Receptor Activation Promotes the Prodestructive Invadosome-Forming Phenotype of Synoviocytes from Patients with Rheumatoid Arthritis. J Immunol. 2016. [DOI] [PubMed] [Google Scholar]
- 49.Cha HS, Bae EK, Ahn JK, Lee J, Ahn KS, Koh EM. Slug suppression induces apoptosis via Puma transactivation in rheumatoid arthritis fibroblast-like synoviocytes treated with hydrogen peroxide. Exp Mol Med. 2010;42(6):428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang AT, Liu Y, Ayyanathan K, Benner C, Jiang Y, Prokop JW, et al. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev. 2015;29(6):603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15(1):36–41. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Z, Rahman MA, Chen ZG, Shin DM. Multiple biological functions of Twist1 in various cancers. Oncotarget. 2017;8(12):20380–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.You S, Yoo SA, Choi S, Kim JY, Park SJ, Ji JD, et al. Identification of key regulators for the migration and invasion of rheumatoid synoviocytes through a systems approach. Proc Natl Acad Sci USA. 2014;111(1):550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39(4):159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128(2):309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ludikhuize J, de Launay D, Groot D, Smeets TJ, Vinkenoog M, Sanders ME, et al. Inhibition of forkhead box class O family member transcription factors in rheumatoid synovial tissue. Arthritis Rheum. 2007;56(7):2180–91. [DOI] [PubMed] [Google Scholar]
- 57.Grabiec AM, Angiolilli C, Hartkamp LM, van Baarsen LG, Tak PP, Reedquist KA. JNK-dependent downregulation of FoxO1 is required to promote the survival of fibroblast-like synoviocytes in rheumatoid arthritis. Ann Rheum Dis. 2015;74(9):1763–71. [DOI] [PubMed] [Google Scholar]
- 58.Kok SH, Lin LD, Hou KL, Hong CY, Chang CC, Hsiao M, et al. Simvastatin inhibits cysteine-rich protein 61 expression in rheumatoid arthritis synovial fibroblasts through the regulation of sirtuin-1/FoxO3a signaling. Arthritis Rheum. 2013;65(3):639–49. [DOI] [PubMed] [Google Scholar]
- 59.Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–37. [DOI] [PubMed] [Google Scholar]
- 60.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–6. [DOI] [PubMed] [Google Scholar]
- 61.Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn J Basic Med Sci. 2018;18(1):8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang M, Zhu S, Peng W, Li Q, Li Z, Luo M, et al. Sonic hedgehog signaling drives proliferation of synoviocytes in rheumatoid arthritis: a possible novel therapeutic target. J Immunol Res. 2014;2014:401903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin S, Sun D, Li X, Kong F, Yu Q, Hua H, et al. GANT61 alleviates arthritic symptoms by targeting fibroblast-like synoviocytes in CIA rats. J Orthop Sci. 2018. [DOI] [PubMed] [Google Scholar]
- 64.Peng WX, Zhu SL, Zhang BY, Shi YM, Feng XX, Liu F, et al. Smoothened Regulates Migration of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis via Activation of Rho GTPase Signaling. Front Immunol. 2017;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh R, Dhanyamraju PK, Lauth M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget. 2017;8(1):833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP, Shankar S, et al. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget. 2015;6(31):32039–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu T, Qiao L, Wang Q, Mi R, Chen J, Lu Y, et al. T-box family of transcription factor-TBX5, insights in development and disease. Am J Transl Res. 2017;9(2):442–53. [PMC free article] [PubMed] [Google Scholar]
- 68.Mori AD, Bruneau BG. TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr Opin Cardiol. 2004;19(3):211–5. [DOI] [PubMed] [Google Scholar]
- 69.Ma R, Yang Y, Tu Q, Hu K. Overexpression of T-box Transcription Factor 5 (TBX5) Inhibits Proliferation and Invasion in Non-Small Cell Lung Carcinoma Cells. Oncol Res. 2017;25(9):1495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karouzakis E, Trenkmann M, Gay RE, Michel BA, Gay S, Neidhart M. Epigenome analysis reveals TBX5 as a novel transcription factor involved in the activation of rheumatoid arthritis synovial fibroblasts. J Immunol. 2014;193(10):4945–51. [DOI] [PubMed] [Google Scholar]
- 71.Briegel KJ, Joyner AL. Identification and characterization of Lbh, a novel conserved nuclear protein expressed during early limb and heart development. Dev Biol. 2001;233(2):291–304. [DOI] [PubMed] [Google Scholar]
- 72.Conen KL, Nishimori S, Provot S, Kronenberg HM. The transcriptional cofactor Lbh regulates angiogenesis and endochondral bone formation during fetal bone development. Dev Biol. 2009;333(2):348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Briegel KJ, Baldwin HS, Epstein JA, Joyner AL. Congenital heart disease reminiscent of partial trisomy 2p syndrome in mice transgenic for the transcription factor Lbh. Development. 2005;132(14):3305–16. [DOI] [PubMed] [Google Scholar]
- 74.Lurie IW, Ilyina HG, Gurevich DB, Rumyantseva NV, Naumchik IV, Castellan C, et al. Trisomy 2p: analysis of unusual phenotypic findings. Am J Med Genet. 1995;55(2):229–36. [DOI] [PubMed] [Google Scholar]
- 75.Hammaker D, Whitaker JW, Maeshima K, Boyle DL, Ekwall AH, Wang W, et al. LBH Gene Transcription Regulation by the Interplay of an Enhancer Risk Allele and DNA Methylation in Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68(11):2637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ekwall AK, Whitaker JW, Hammaker D, Bugbee WD, Wang W, Firestein GS. The Rheumatoid Arthritis Risk Gene LBH Regulates Growth in Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2015;67(5):1193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matsuda S, Hammaker D, Topolewski K, Briegel KJ, Boyle DL, Dowdy S, et al. Regulation of the Cell Cycle and Inflammatory Arthritis by the Transcription Cofactor LBH Gene. J Immunol. 2017;199(7):2316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cavodeassi F, Modolell J, Gomez-Skarmeta JL. The Iroquois family of genes: from body building to neural patterning. Development. 2001;128(15):2847–55. [DOI] [PubMed] [Google Scholar]
- 79.Becker MB, Zulch A, Bosse A, Gruss P. Irx1 and Irx2 expression in early lung development. Mech Dev. 2001;106(1–2):155–8. [DOI] [PubMed] [Google Scholar]
- 80.Zulch A, Becker MB, Gruss P. Expression pattern of Irx1 and Irx2 during mouse digit development. Mech Dev. 2001;106(1–2):159–62. [DOI] [PubMed] [Google Scholar]
- 81.Houweling AC, Dildrop R, Peters T, Mummenhoff J, Moorman AF, Ruther U, et al. Gene and cluster-specific expression of the Iroquois family members during mouse development. Mech Dev. 2001;107(1–2):169–74. [DOI] [PubMed] [Google Scholar]
- 82.Yu W, Li X, Eliason S, Romero-Bustillos M, Ries RJ, Cao H, et al. Irx1 regulates dental outer enamel epithelial and lung alveolar type II epithelial differentiation. Dev Biol. 2017;429(1):44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bennett KL, Karpenko M, Lin MT, Claus R, Arab K, Dyckhoff G, et al. Frequently methylated tumor suppressor genes in head and neck squamous cell carcinoma. Cancer Res. 2008;68(12):4494–9. [DOI] [PubMed] [Google Scholar]
- 84.Guo X, Liu W, Pan Y, Ni P, Ji J, Guo L, et al. Homeobox gene IRX1 is a tumor suppressor gene in gastric carcinoma. Oncogene. 2010;29(27):3908–20. [DOI] [PubMed] [Google Scholar]
- 85.Liu X, Zhang J, Liu L, Jiang Y, Ji J, Yan R, et al. Protein arginine methyltransferase 5-mediated epigenetic silencing of IRX1 contributes to tumorigenicity and metastasis of gastric cancer. Biochim Biophys Acta Mol Basis Dis. 2018;1864(9 Pt B):2835–44. [DOI] [PubMed] [Google Scholar]
- 86.Jiang J, Liu W, Guo X, Zhang R, Zhi Q, Ji J, et al. IRX1 influences peritoneal spreading and metastasis via inhibiting BDKRB2-dependent neovascularization on gastric cancer. Oncogene. 2011;30(44):4498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Selvarajah S, Yoshimoto M, Maire G, Paderova J, Bayani J, Squire JA, et al. Identification of cryptic microaberrations in osteosarcoma by high-definition oligonucleotide array comparative genomic hybridization. Cancer Genet Cytogenet. 2007;179(1):52–61. [DOI] [PubMed] [Google Scholar]
- 88.Lu J, Song G, Tang Q, Zou C, Han F, Zhao Z, et al. IRX1 hypomethylation promotes osteosarcoma metastasis via induction of CXCL14/NF-kappaB signaling. J Clin Invest. 2015;125(5):1839–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Julia A, Blanco F, Fernandez-Gutierrez B, Gonzalez A, Canete JD, Maymo J, et al. Identification of IRX1 as a Risk Locus for Rheumatoid Factor Positivity in Rheumatoid Arthritis in a Genome-Wide Association Study. Arthritis Rheumatol. 2016;68(6):1384–91. [DOI] [PubMed] [Google Scholar]
- 90.Park SH, Kim SK, Choe JY, Moon Y, An S, Park MJ, et al. Hypermethylation of EBF3 and IRX1 genes in synovial fibroblasts of patients with rheumatoid arthritis. Mol Cells. 2013;35(4):298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leung CH, Chan DS, Ma VP, Ma DL. DNA-binding small molecules as inhibitors of transcription factors. Med Res Rev. 2013;33(4):823–46. [DOI] [PubMed] [Google Scholar]
- 92.Sammak S, Zinzalla G. Targeting protein-protein interactions (PPIs) of transcription factors: Challenges of intrinsically disordered proteins (IDPs) and regions (IDRs). Prog Biophys Mol Biol. 2015;119(1):41–6. [DOI] [PubMed] [Google Scholar]
- 93.Fontaine F, Overman J, Moustaqil M, Mamidyala S, Salim A, Narasimhan K, et al. Small-Molecule Inhibitors of the SOX18 Transcription Factor. Cell Chem Biol. 2017;24(3):346–59. [DOI] [PubMed] [Google Scholar]
- 94.Overman J, Fontaine F, Moustaqil M, Mittal D, Sierecki E, Sacilotto N, et al. Pharmacological targeting of the transcription factor SOX18 delays breast cancer in mice. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ladam F, Sagerstrom CG. Hox regulation of transcription: more complex(es). Dev Dyn. 2014;243(1):4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alharbi RA, Pandha HS, Simpson GR, Pettengell R, Poterlowicz K, Thompson A, et al. Inhibition of HOX/PBX dimer formation leads to necroptosis in acute myeloid leukemia cells. Oncotarget. 2017;8(52):89566–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Langlet F, Haeusler RA, Linden D, Ericson E, Norris T, Johansson A, et al. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell. 2017;171(4):824–35 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agyeman A, Jha BK, Mazumdar T, Houghton JA. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget. 2014;5(12):4492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang R, Wu J, Ferrandon S, Glowacki KJ, Houghton JA. Targeting GLI by GANT61 involves mechanisms dependent on inhibition of both transcription and DNA licensing. Oncotarget. 2016;7(49):80190–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Srivastava RK, Kaylani SZ, Edrees N, Li C, Talwelkar SS, Xu J, et al. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget. 2014;5(23):12151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee SH, Park BJ. p53 activation by blocking Snail: a novel pharmacological strategy for cancer. Curr Pharm Des. 2011;17(6):610–7. [DOI] [PubMed] [Google Scholar]
- 102.Harney AS, Lee J, Manus LM, Wang P, Ballweg DM, LaBonne C, et al. Targeted inhibition of Snail family zinc finger transcription factors by oligonucleotide-Co(III) Schiff base conjugate. Proc Natl Acad Sci U S A. 2009;106(33):13667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yochum ZA, Cades J, Mazzacurati L, Neumann NM, Khetarpal SK, Chatterjee S, et al. A First-in-Class TWIST1 Inhibitor with Activity in Oncogene-Driven Lung Cancer. Mol Cancer Res. 2017;15(12):1764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]