

Abstract
In response to brain injury or infections, astrocytes become reactive, undergo striking morphological and functional changes, and secrete and respond to a spectrum of inflammatory mediators. We asked whether reactive astrocytes also display adaptive responses during sterile IL-1β-induced neuroinflammation, which may limit tissue injury associated with many disorders of the central nervous system. We found that astrocytes display days-to-weeks long specific tolerance of cytokine genes, which is coordinated by NF-κB family member, RelB. However, in contrast to innate immune cells, astrocytic tolerance does not involve epigenetic silencing of the cytokine genes. Establishment of tolerance depends on persistent higher levels of RelB in tolerant astrocytes and its phosphorylation on serine 472. Mechanistically, this phosphorylation prevents efficient removal of RelB from cytokine promoters by IκBα and helps to establish tolerance. Importantly, ablation of RelB from astrocytes in mice abolishes tolerance during experimental neuroinflammation in vivo.
Keywords: tolerance, astrocytes, neuroinflammation, RelB, IL-1β
Graphical Abstract
INTRODUCTION
The first line of defense against pathogens is activation of innate immune responses, which involve myeloid, innate lymphoid, and natural killer cells (Chaplin, 2010). These very rapid but relatively nonspecific responses are followed by engagement of the slower but highly specific adaptive immune system, which provides long-lasting memory of invading pathogens (Chaplin, 2010). However, it is now firmly established that responses of innate immune cells are also influenced by previous pathogen encounters, which can induce their polarization, priming, and tolerance, and thus allow for fine-tuning of future responses (Foster, Hargreaves, & Medzhitov, 2007; Locati, Mantovani, & Sica, 2013). This paradigm termed “innate immunity memory” depends on epigenetic reprogramming of specific genes and regulates the magnitude of subsequent responses (Foster et al., 2007; Netea et al., 2016).
The resident innate immune cells of the brain, microglia, also display adaptive responses which depend on epigenetic reprograming (Schaafsma et al., 2015; Wendeln et al., 2018). In contrast to microglia, astrocytes are the most abundant non immune glial cells in the brain. These cells provide neurotrophic and metabolic support to neurons; regulate the blood-brain barrier; control synapse formation, removal, and function; remodel the extracellular milieu; and regulate immune cells in the brain (Dong & Benveniste, 2001; Sofroniew & Vinters, 2010). These processes are critically affected during the development of reactive astrogliosis associated with brain trauma, infections, ischemia, and neurodegeneration (Pekny & Nilsson, 2005; Sofroniew & Vinters, 2010). As astrocytes become reactive, they undergo dramatic morphological and functional changes, alter their gene expression profiles, and secrete and respond to a host of inflammatory mediators, which may lead to beneficial or harmful outcomes (Sofroniew, 2009). Further, these responses of astrocytes are heterogeneous and depend on phenotypic polarization of astrocytes (Jang et al., 2013), their diversity and specialization, as well as the type and magnitude of the stimulus (Khakh & Sofroniew, 2015; Zamanian et al., 2012). The heterogeneity of astrocytes and the specificity of their responses raises the question as to whether astrocytes also display adaptive responses (tolerance or priming), which is a hallmark of innate immune cells. Although endotoxin tolerance has been reported in mouse astrocytes (Beurel, 2011), most human CNS disorders such as stroke, traumatic brain injury, or neurodegeneration are associated with sterile neuroinflammation and do not involve gram-negative bacterial infections. Sterile inflammation is initiated by recognition of the danger associated molecular patterns, which induces the production of interleukin-1β (IL-1β) mRNA, and pro-IL-1β protein (Lukens, Gross, & Kanneganti, 2012). Subsequent activation of inflammasome is then required to produce mature biologically active IL-1β (Lukens et al., 2012). The proinflammatory effects of IL-1β are primarily mediated by activation of the transcription factor nuclear factor kappa B (NF-κB) and mitogen activated protein kinases (Conze, Wu, Thomas, Landstrom, & Ashwell, 2008; Deng et al., 2000; Wang et al., 2001) coordinated by an E3 ubiquitin ligase TRAF6. Since IL-1β is a master regulator of sterile inflammation (Gadani, Walsh, Lukens, & Kipnis, 2015; Lukens et al., 2012), and increased IL-1β expression is found in most human CNS disorders associated with neuroinflammation (Allan, Tyrrell, & Rothwell, 2005), we asked whether astrocytes display adaptive responses to IL-1β. These adaptive responses may be critical to limit tissue injury in neurological disorders, including Alzheimer’s and Parkinson’s disease, or reoccurring systemic infections that induce neuroinflammation.
MATERIALS AND METHODS
Mice
RelBLoxP/LoxP mice (generated by Dr. Urlich Sibienlist, NIH) were bred with GFAP-cre mice (stock #012886, Jackson Laboratory) to generate RelBΔAST mice. RelBΔAST and RelBLoxP/LoxP littermates were used for the experiments. The mouse protocols were approved by the Institutional Animal Care and Use Committee.
LPS-induced neuroinflammation
Mice were intraperitoneally injected with 5 mg/kg LPS (Sigma-Aldrich, cat.no. L2880) or saline as a control. 24 hours later, mice brains (excluding cerebellum) were collected. The brains were snap frozen for RNA isolation. Alternatively, the brains were digested using papain dissociation system per manufacturer instructions (Worthington Biochemical). Astrocytes and microglia were isolated using anti-ACSA-2 and anti-CD11B beads exactly as recommended by Miltenyi Biotec. For LPS tolerance experiments, mice were intraperitoneally injected with 2 mg/kg LPS or saline. 7 days later mice were re-injected with LPS or saline and brains were collected 24 hours later.
Cell culture
HEK293 cells were obtained from the American Type Culture Collection. Mouse cortical astrocytes were prepared from P1 pups. Cerebral cortices were dissected, meninges removed, tissue was incubated with trypsin and DNaseI at 37°C for 30 minutes, mechanically dissociated, centrifuged, filtered through 70 μm filters, and re-centrifuged. Cells were suspended and plated on poly-d-lysine. Human cortical astrocytes were prepared from fetal tissue provided by Advanced Bioscience Resources (Alameda) and cultured as described previously (Bhardwaj et al., 2015; Kordula et al., 1998). Purity of astrocyte cultures, and their responses were analyzed by FACS and immunofluorescence (Suppl. Fig 1). The cells were stimulated with 20 ng/ml IL-1β (R&D Systems, cat. no. 201-LB). Cells were pretreated with 10 μM Rosiglitasone, 50 μM Resveratrol, or 50 μM BAY 11-7082 (Sigma-Aldrich) 1 hour before IL-1β stimulation.
Flow cytometry
Cells were trypsinized, washed, and stained with fluorescence-conjugated monoclonal antibodies against CD11b (clone M1/70, Biolegend) or an isotype control. Fluorescence data were collected on Fortessa and analyzed using FACS-Diva software (BD Biosciences).
Immunofluorescence
Cells grown on glass coverslips, were fixed with 2.5% paraformaldehyde, permeabilized in 0.1% triton-X for 5 minutes on ice, washed with PBS, blocked in 1% BSA, and incubated with primary antibodies overnight in 1% BSA. Anti-RelB (10544) and anti-GFAP (3670) (Cell Signaling), and ant-CC1 (OP80) (EMD Milipore) were used. Slides were washed with PBS, incubated with secondary antibodies in 1% BSA for 1h, washed with PBS, 0.1% triton-X100, and PBS again. Counterstaining was done using Hoescht or DAPI, and slides were mounted using vectashield mounting medium (Vector Laboratories).
Knockdown
Expression of RelB, p65, and SIRT1 was down-regulated with SmartPool siRNAs with Dharmafect 1 (Dharmacon).
qPCR
RNA was prepared with Trizol (Life Technologies) and reverse transcribed with the high-capacity cDNA kit (Applied Biosystems). SYBR Green intron-spanning qPCR primers (BioRad) were used. Gene expression levels were normalized to GAPDH and presented as percentages of IL-1β-induced expression (set at 100%).
Western Blotting
Cells were lysed in 10 mM Tris (pH 7.4), 150 mM sodium chloride, 1 mM EDTA, 1% Nonidet P-40, 1% Triton X-100, 1 mM sodium orthovanadate, 0.2 mM PMSF, and Pierce protease inhibitor mixture. Samples were separated on a 10% gel, and transferred onto nitrocellulose membranes (GE Healthcare). Anti-β-tubulin (sc-9104), anti-RelB (sc-226), anti-p65 (sc-372), anti-p105/p50 (sc-8414), anti-IκBα (sc-371), and anti-SIRT1 (sc-15404) antibodies (Santa Cruz Biotechnology); anti-Lamin A/C (2032), anti-myc (2276) anti-GAPDH (5174), and anti-phospho-p65(S536) (3031) antibodies (Cell Signaling); anti-flag (F1804) antibodies (Sigma-Aldrich); and anti-pRelB(S472) antibodies were from CliniSciences (NB-22-1004) (Nanterre, France). Antigen-antibody complexes were visualized by enhanced chemiluminescence using Immobilon Western blotting kit (Millipore).
Fractionation
Cells were washed with cold PBS and re-suspended in buffer containing 10 mM Hepes (pH 7.8), 10 mM KCl, 0.1 mM EDTA, 1 mM Na3VO4, 1 mM DTT, Pierce protease inhibitor, and 0.2 mM PMSF, and incubated on ice for 15 minutes, 1% NP-40 was added, and samples were centrifuged. The pelleted nuclei were suspended in 20 mM Hepes (pH 7.8), 0.4 M NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM DTT, and Pierce protease inhibitor. Samples were incubated on ice for 15 min, cleared by centrifugation, and stored in 10% glycerol.
Phos-tag Electrophoresis
Lysates were treated as indicated with 1 unit lambda phosphatase per 20-30 ug protein (New England Biolabs) at 30°C for 30 minutes. Gels were prepared using 0.02 mM phos-tag (Wako, cat. no. AAL-107) and 0.2 mM MnCl2 in 8% acrylamide gels, and electrophoresis was performed. Gels were washed with 1 mM EDTA for 30 minutes before transferring to membranes.
Immunoprecipitation
200-300 μg of protein lysates, were pre-cleared with 10 μl of the protein G-Sepharose beads (GE Healthcare) for 1 h. The lysates were incubated with antibodies overnight at 4°C, and then 25 μl protein G-Sepharose beads for 2 h at 4°C. The beads were washed with the lysis buffer, and proteins eluted in sample buffer. Flag-tagged RelB was immunoprecipitated with anti-Flag-M2 beads and eluted with Flag peptide (Sigma-Aldrich).
Synthetic oligonucleotides
The following oligonucleotide was used to mutate RelB: 5’-gatttcttccctgcttcaattgcgcttcctgggctggagcc-3’. The following primers were used for ChIP qPCR: IL1B forward: 5’-aatttaaaacattcttctaacgtggg-3’, reverse: 5’-ggagtagcaaactatgacacattttg-3’, and probe: 5’-[6-FAM] caactgcacaacgattgtcaggaaaa[BHQ1a-Q]-3’; IL6 forward: 5’-caccctcaccctccaacaaag-3’, reverse: 5’-ggcagaatgagcctcagacatc-3’, and probe: 5’-[6-FAM]gagtctcaatattagagtctcaaccccc[BHQ1a-Q]-3’; IL8 forward: 5’-gtgcataagttctctagtagggtgatg-3’, reverse: 5’-ggctcttgtcctagaagcttgtgt-3’, and probe: 5’-[6-FAM]cactccataaggcacaaactttcagag[BHQ1a-Q]-3’.
ChIP Assay
The cells were cross-linked with 1% formaldehyde for 10 min at 37 °C and washed with ice-cold PBS containing 125 mM glycine and 1 mM PMSF. Chromatin was sheared using a Diagenode Bioruptor (Liège, Belgium) on high setting for two 10 minute intervals (30 seconds on/off). Anti-RelB and anti-p50 (Santa Cruz), anti-acH4K16 (07329) (Millipore) and anti-2met-H3K9 (1220) (Abcam) antibodies were used. DNA was detected by qPCR using TaqMan primers. qPCR data was calculated as percent input, and values are represented as fold over control.
Transfection
The IL-8 luciferase reporter was provided by Dr. X. Fang (Virginia Commonwealth University). RelB plasmid was from Addgene (Cambridge). The site-directed mutagenesis was performed using QuikChange II site-directed mutagenesis kit (Stratagene). HEK293 were transfected using Lipofectamine 2000 (Life Technologies).
Luciferase assays
Astrocytes were transfected in 12-well dishes. 24 hours post-transfection, the luciferase assays were performed using a dual luciferase reporter assay kit (Promega Corporation). Luciferase activities were normalized to Renilla activity and expressed as fold activity relative to empty vector.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7. Values are displayed as mean +/− standard deviation for in vitro qPCR experiments, mean +/− standard error for ChIP and animal experiments. T -tests and ANOVAs were performed as indicated. Sidak’s or Tukey’s test was performed to compare multiple groups.
RESULTS
“Cytokine tolerance” in human astrocytes.
To test whether human astrocytes can adapt their responses to IL-1β, we utilized a protocol used to describe LPS tolerance in macrophages (Foster et al., 2007), and analyzed expression of classical genes undergoing tolerance. Astrocytes were preconditioned with IL-1β, washed, allowed to recover, and restimulated with IL-1β two days later. We found that expression of genes encoding proinflammatory cytokines IL-1β, IL-6, IL-8, and TNF was significantly lower in preconditioned astrocytes (Fig. 1A, Suppl. Fig. 2A), suggesting that astrocytes develop tolerance to IL-1β (hereafter termed “cytokine tolerance”). In contrast, expression of other IL-1β-dependent genes, including CHI3L1 (Bhardwaj et al., 2015) and LCN2 (Cowland, Sorensen, Sehested, & Borregaard, 2003) was enhanced in preconditioned astrocytes. These effects were long-lasting since similar results were observed in cells restimulated with IL-1β four days after the initial IL-1β exposure (Fig. 1B, Suppl. Fig. 2B). Importantly, activation of the signaling cascades known to control cytokine expression, including p65 NF-κB, p38, and JNK, was similar in naïve and tolerant astrocytes (Fig. 1C, D). Translocation of IL-1β -activated p65 to the nuclei of naïve and preconditioned astrocytes was also comparable (Fig. 1E). These adaptive responses could not be a result of signaling pathway desensitization since expression of the CHI3L1 and LCN2 genes was significantly enhanced in preconditioned astrocytes, and these genes are regulated by NF-κB. These data suggest that similar to endotoxin tolerance in innate immune cells, astrocytes can adapt their responses depending on previous encounters with IL-1β.
Fig. 1. Selective tolerance of cytokine genes is not due to desensitization of signaling pathways in astrocytes.
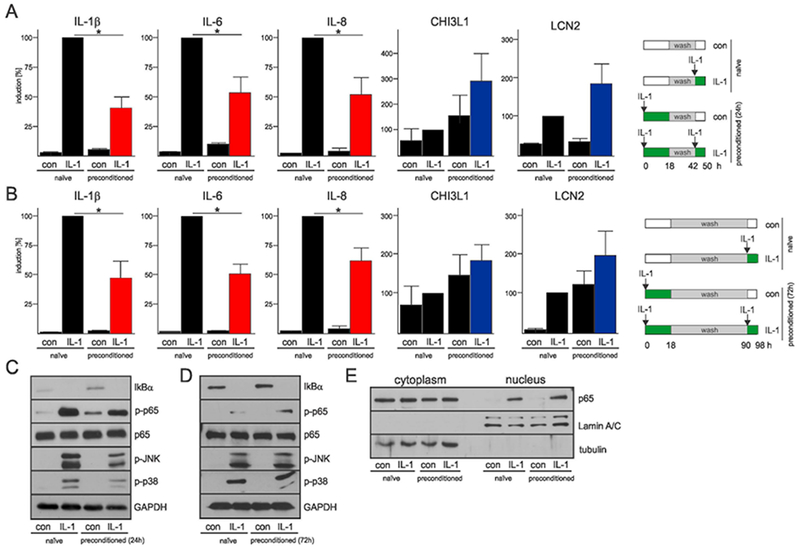
(A, B) Expression of genes encoding cytokines and other IL-1β-dependent genes in naïve and preconditioned human astrocytes stimulated with IL-1β. The expression was analyzed after 2 (A) or 4 days (B). Schemes of the experimental design are shown (right panels). qPCR analysis from three independent experiments. Error bars represent s.d., * P<0.05 (two-way ANOVA, Sidak’s test). (C, D) Activation of the signaling cascades that regulate cytokine expression in naïve and preconditioned astrocytes after 2 (C) and 4 days (D). Astrocytes were stimulated for 30 min and expression was analyzed by western blotting. Representative blots of two experiments are shown. (E) Translocation of p65 to the nucleus of both naïve and preconditioned astrocytes was analyzed 30 min after IL-1β re-stimulation. Representative western blots of two independent experiments are shown.
Cytokine expression is downregulated by RelB and SIRT1.
Establishment of LPS-induced tolerance in macrophages involves RelB and NAD+-dependent deacetylase sirtuin 1 (SIRT1) (Chen, El Gazzar, Yoza, & McCall, 2009; Liu, Yoza, El Gazzar, Vachharajani, & McCall, 2011; Millet, McCall, & Yoza, 2013). RelB is mostly known as a target of non-canonical NF-κB signaling (Millet et al., 2013) that controls development of lymphoid organs. Activation of this pathway induces processing of the RelB-binding protein p100 to p52, translocation of RelB/p52 heterodimers to the nuclei, and induction of target genes. RelB can also form RelB/p50/IκBα complexes that are activated by canonical stimuli (Shih et al., 2012). Since RelB has higher affinity for p100/p52 than for p50, canonical RelB activation is minimal in the absence of high RelB expression (Hoffmann, Natoli, & Ghosh, 2006). RelB has also been proposed to suppress cytokine expression by binding to p65 (Marienfeld et al., 2003), dimer switching (Saccani, Pantano, & Natoli, 2003), and epigenetic silencing (Chen et al., 2009; Millet et al., 2013). In myeloid cells, this epigenetic silencing involves SIRT1 that interacts with RelB and deacetylates lysine 16 of histone H4 (H4K16) (Liu et al., 2011). To test whether RelB regulates cytokine expression in astrocytes, its expression was knocked-down. p65 was also knocked-down as a control (Suppl. Fig. 3A, B). As expected, p65 was required for IL-1β-induced IL-1β, IL-6 and IL-8 mRNA expression (Fig. 2A). In contrast to p65, RelB suppressed IL-1β-induced cytokine expression but it was required for the expression of LCN2 and CHI3L1 mRNA (Fig. 2A). IL-1β induced rapid activation and nuclear translocation of p65, p50, and also rapid nuclear translocation of RelB (Fig. 2B, C). IL-1β also induced expression of p100 but did not induce its constitutive processing to p52, did not promote p52 translocation to the nuclei, or heterodimer formation with RelB (Suppl. Fig. 4A-C). Interestingly, IL-1β caused delayed induction of RelB and p50 expression, whereas SIRT1 levels were not affected (Fig. 2B, C). RelB accumulated in distinctive puncta in the nuclei of astrocytes (Fig. 2D), and its overexpression repressed p65-driven expression of the IL-8 reporter (Fig. 2E). In agreement with the findings that RelB suppresses cytokine expression and accumulates in IL-1β-stimulated astrocytes, its levels were much higher in preconditioned than naïve astrocytes (Fig. 2F), suggesting that RelB may regulate long-lasting effects in these cells.
Fig. 2. RelB suppresses expression of cytokine genes in astrocytes.
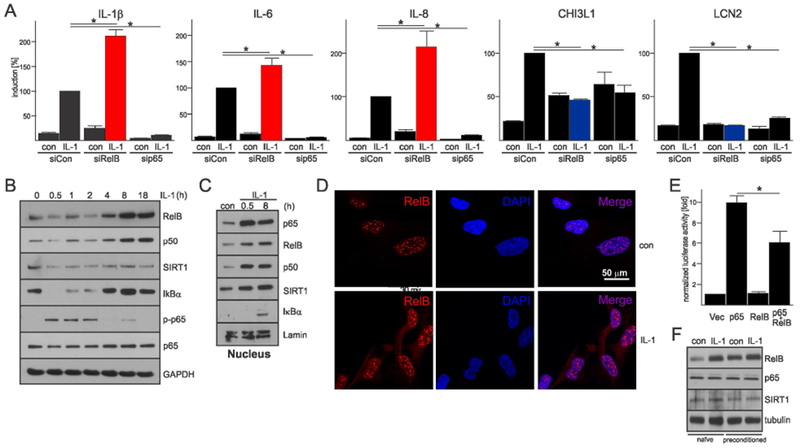
(A) RelB or p65 expression were knocked-down for 48h, cells were stimulated with IL-1β for 18h and expression was analyzed. qPCR analysis from three independent experiments. Error bars represent s.d., * P<0.05 (two-way ANOVA, Tukey’s test). (B) Time-dependent IL-1β-induced expression was analyzed by western blotting. (C) Accumulation of signaling molecules in the nuclei of astrocytes was analyzed by western blotting (three experiments). (D) Localization of RelB was analyzed by immunofluorescence in unstimulated and IL-1β stimulated astrocytes after 18h. Hoescht was used to counterstain nuclei. (E) Effect of RelB expression on p65-driven expression of IL-8 reporter. Normalized luciferase activity (triplicate samples). Error bars represent s.d., * P<0.05 (one-way ANOVA, Sidak’s test). (F) Astrocytes were untreated or preconditioned for 18h and then re-stimulated with IL-1β for 8h one day later. Representative western blots of two experiments are shown.
Since SIRT1 cooperates with RelB to establish LPS tolerance in macrophages (Liu et al., 2011), we asked whether SIRT1 also regulates cytokine expression in astrocytes. We could co-IP endogenous SIRT1 with RelB from human astrocytes (Fig. 3A, B) indicating that these proteins interact. In transfection experiments, we established that SIRT1 forms complexes with RelB and p50, but not p105 (Fig. 3C, D). Knock-down of SIRT1 (Suppl. Fig. 3C) significantly increased cytokine expression in astrocytes (Fig. 3E), but had either little or the opposite effect on Chi3L1 and LCN2 transcript levels. Conversely, activation of SIRT1 by resveratrol and rosiglitazone markedly reduced IL-1β-induced cytokine expression (Fig. 3F, G). We concluded that both RelB and SIRT1 inhibit IL-1β-induced expression of cytokines in astrocytes and thus may regulate cytokine tolerance.
Fig. 3. SIRT1 interacts with RelB/p50 complexes and suppresses cytokine gene expression in astrocytes.
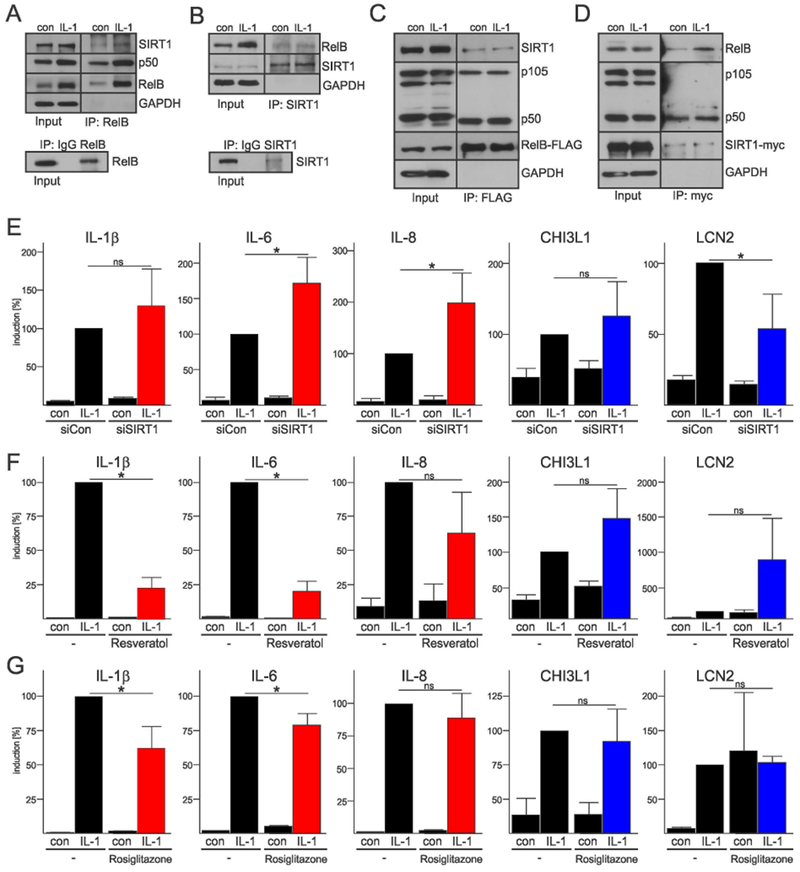
(A, B) Immunoprecipitation of endogenous complexes containing RelB and SIRT1 from human astrocytes using anti-RelB (A) and anti-SIRT1 (B) antibodies. (C, D) Immunoprecipitation of complexes from HEK293 cells expressing tagged-RelB (C), and tagged SIRT1 (D) (representative of three experiments). (E, F, G) qPCR analysis from three-four independent experiments. Error bars represent s.d., * P<0.05 (two-way ANOVA, Sidak’s test). (E) SIRT1 was knocked-down with siRNA for 48h and then cells were stimulated with IL-1β for 18h. Astrocytes were pretreated with 50 μM reseveratol (F) or 10 μM rosiglitazone (G) for 1h and then treated with IL-1β for 18h.
“Cytokine tolerance” is associated with RelB/p50 binding but not epigenetic silencing.
Recruitment of SIRT1, deacetylation of H4K16, and di-methylation of histone H3 on lysine 9 (2met-H3K9) are hallmarks of LPS-induced tolerance in macrophages (Chen et al., 2009; Liu et al., 2011; Millet et al., 2013). Surprisingly and in contrast to macrophages, both ac-H4K16 and 2met-H3K9 were comparable in naïve and tolerant astrocytes (Fig. 4A). We reasoned that putative silencing in astrocytes may involve different histone marks that should immediately impinge expression of the cytokine genes in tolerant cells. However, cytokine expression was comparable in naïve and tolerant astrocytes after re-stimulation with IL-1β for 30 minutes (Fig. 4B), suggesting that tolerance does not involve modifications of histones. Although ac-H4K16 and 2met-H3K9 at the cytokine promoters was not changed, both RelB and p50 accumulated at these promoters in response to IL-1β (Fig. 4C) pointing to their direct action. In addition, while p65 accumulated similarly in the nuclei of naïve and tolerant astrocytes 30 min after IL-1β stimulation, its levels were diminished after 8h (Fig. 4D). In contrast, RelB levels were significantly higher in the nuclei of tolerant astrocytes at 30 min but comparable at 8h. We also found comparable amounts of IκBα in the nuclei of naïve and tolerant astrocytes at 8h (Fig. 4D). This pool of newly synthesized IκBα is known to strip p65/p50 complexes off the DNA (Hoffmann, Levchenko, Scott, & Baltimore, 2002).
Fig. 4. Tolerance is associated with RelB/p50 binding but not histone modifications at the cytokine promoters.
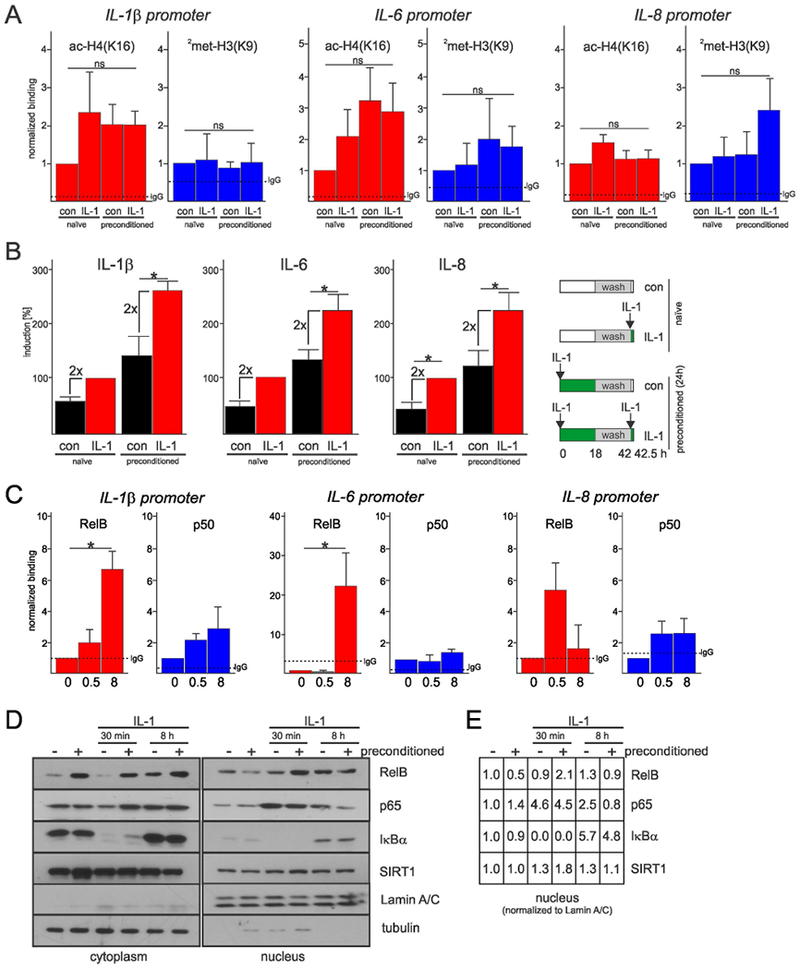
(A) Histone modifications at the cytokine promoters in naïve and preconditioned astrocytes. Cells were re-stimulated for 8h. ChIP analysis from three independent experiments. Error bars represent s.e.m., * P<0.05 (two-way ANOVA). (B) Expression of genes encoding cytokines in naïve and preconditioned astrocytes re-stimulated with IL-1β for 30 min. Scheme of the experimental design (right panel). qPCR analysis from three independent experiments. Error bars represent s.d., * P<0.05 (two-way ANOVA, Sidak’s test). (C) RelB and p50 binding at the cytokine promoters. Cells were stimulated for 0.5 or 8h. ChIP analysis from three-five independent experiments. Error bars represent s.e.m., * P<0.05 (one-way ANOVA, Tukey’s test). (D) Accumulation of signaling molecules in the nuclei of naïve and preconditioned astrocytes analyzed by western blotting (representative blots of two experiments). (E) Data shown in D (right panel) were quantified by ImageJ.
Phosphorylation of RelB on S472 persists in tolerant astrocytes and diminishes interaction with IkBα.
Since RelB did not induce epigenetic modifications in tolerant astrocytes, we reasoned that it may be posttranslationally modified. We concentrated on phosphorylation since other NF-κB members are commonly regulated by this modification. Using the Phos-tag technique (Kinoshita, Kinoshita-Kikuta, & Koike, 2009), we found that IL-1β induces long-lasting phosphorylation of RelB in astrocytes (Fig. 5A) and phospho-RelB is found in the nuclei of these cells (Fig. 5B). RelB has six putative phosphorylation sites; phosphorylation of Thr84 and Ser552 destine RelB for degradation (Marienfeld et al., 2001; Neumann et al., 2011), while phosphorylation of Ser472 is needed for the induction of genes promoting migration (Authier et al., 2014). In contrast to WT-RelB, a triple mutant (T84A/S552A/S472A) was not phosphorylated in response to IL-1β (Fig. 5C), indicating that phosphorylation is limited to these three residues. Furthermore, the T84A/S552A mutant was robustly phosphorylated, while the S472A mutant only slightly phosphorylated, indicating that S472 is the primary phosphorylation site (Fig. 5D). Indeed, IL-1β induces rapid and sustained phosphorylation of RelB on S472 in astrocytes as shown using phospho-RelB(S472) antibodies (Fig. 5E). This phosphorylation of RelB was blocked by an IκB kinase inhibitor (Fig. 5F) indicating that it is mediated by the canonical NF-κB pathway. Importantly, rapid phosphorylation of RelB on S472 in response to IL-1β occurred in both naïve and preconditioned astrocytes (Fig. 5G). Although astrocytes preconditioned for four days had high RelB expression, RelB phosphorylation was minimal (Fig. 5G) but persisted in cells preconditioned for two days (Fig. 5H). This phosphorylation was not necessary for nuclear localization of RelB since WT-RelB and the S472A mutant similarly localized to the nucleus (Fig. 5I). Phosphorylation of RelB was also not necessary to form complexes with p50 or SIRT1 (Fig. 5J). Importantly, the S472A mutant associated more efficiently with IκBα than WT-RelB, raising the possibility that phosphorylation of RelB on S472 likely prevents stripping of RelB/p50 complexes by IκBα from the DNA (Fig. 5K). Thus, in contrast to p65/p50 that is easily stripped by IκBα, RelB/p50 likely occupies cytokine promoters for more extended period of time, which provides long-lasting regulatory effects.
Fig. 5. IL-1β-induced phosphorylation of RelB on S472 diminishes interaction with IκBα.
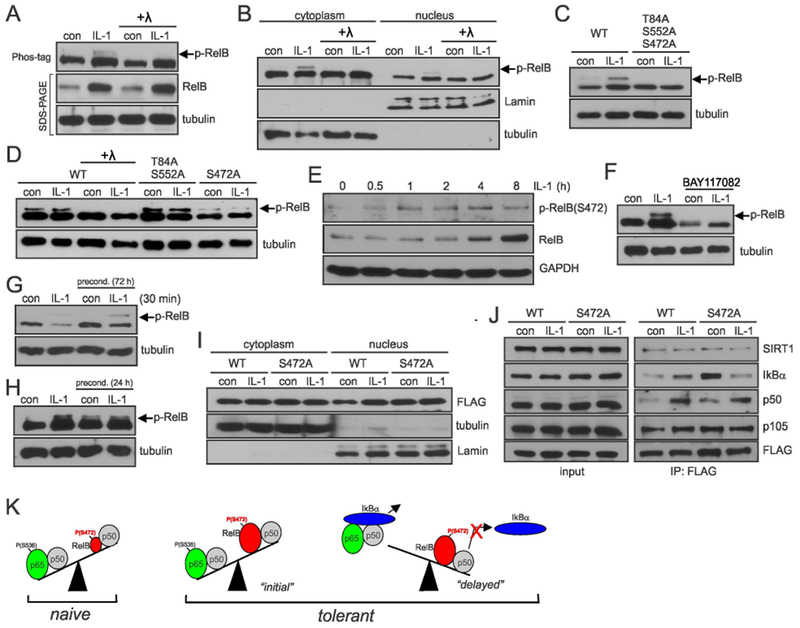
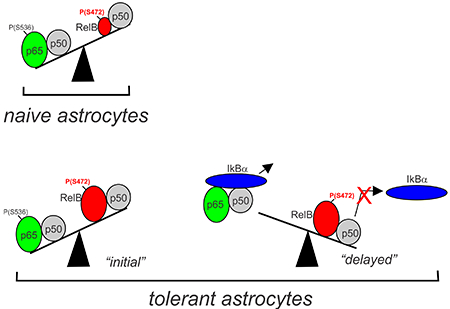
(A, B) Phosphorylation of RelB in astrocytes analyzed by Phos-Tag. Astrocytes were stimulated for 8h with IL-1β. Lysates were treated with lambda (λ) phosphatase as indicated. (C, D) Phosphorylation of RelB mutants in transfected HEK293 cells analyzed by Phos-tag. Cells were stimulated with IL-1β for 8h. (E) Phosphorylation of RelB in astrocytes was analyzed using phospho-RelB(S472) antibodies (F, G, H). Astrocytes were stimulated with IL-1β and/or BAY11-7082 and phosphorylation of RelB was analyzed by phos-tag. (I) Accumulation of overexpressed RelB in HEK293 cells analyzed by western blotting. (J) Analysis of interaction of WT-RelB and S472A mutant with binding partners in transfected HEK293 cells. (A-J) Representative blots of two-five experiments yielding similar data are shown. (K) Proposed model of tolerance: low levels of RelB ensure efficient cytokine induction by p65/p50 in naïve cells; in tolerant cells during the “initial” phase cytokine genes are activated by p65/p50 even though the presence of higher levels of RelB/p50; tolerance is established once IκBα is resynthesized and efficiently removes p65/p50 from the DNA. Phosphorylation of RelB at S472 prevents efficient removal of RelB/p50 complexes from the DNA making it inaccessible for p65/p50 to bind. Representative blots of 2-5 experiments are shown.
Astrocytic RelB regulates neuroinflammation and tolerance in vivo.
To assess the importance of astrocytic RelB in the neuroinflammatory responses in vivo, we generated astrocyte-specific RelB-deficient mice (RelBΔAST). These mice appear to be indistinguishable from their WT littermates and, in contrast to RelB-deficient mice (Barton, HogenEsch, & Weih, 2000), do not spontaneously develop any obvious inflammation. RelB expression was almost completely ablated in cultured astrocytes from RelBΔAST mice (Fig. 6A), but its expression was normal in other cell types and tissues including the spleen. We first examined whether astrocytic RelB affects cytokine production using a systemic LPS-induced model of sterile cytokine-driven neuroinflammation. Although LPS does not cross the blood-brain barrier in healthy animals (Banks & Robinson, 2010), systemic administration of LPS results in release of cytokines, which cross blood-brain barrier and induce sterile neuroinflammation (Qin et al., 2007). This model has been successfully used to examine tolerance of microglia in vivo (Schaafsma et al., 2015; Wendeln et al., 2018). To verify that proinflammatory cytokines are indeed expressed by astrocytes in vivo during LPS-induced neuroinflammation, we purified astrocytes from brains of mice. As expected, astrocytes purified from brains of LPS-treated mice expressed IL-1β, IL-6 and TNF (Suppl. Fig. 5A) and these cells were not contaminated with microglia (Suppl. Fig. 5B). Intraperitoneal administration of LPS also induced accumulation of RelB protein in the brains of WT mice, which was diminished in RelBΔAST mice (Fig. 6B). We could also detect RelB in WT astrocytes in vivo by IF (Fig. 6C, Suppl. Fig. 6A). In contrast, RelB was not expressed by astrocytes of RelBΔAST mice in vivo (Suppl. Fig. 6B); however, it was present in the other cells, including CC1+ oligodendrocytes (Suppl. Fig. 6C). These data suggested that astrocytes express RelB during cytokine-driven neuroinflammation. Although microglia and astrocytes both secrete cytokines in the brain (Norden, Trojanowski, Villanueva, Navarro, & Godbout, 2016), deletion of RelB from astrocytes in RelBΔAST mice increased cytokine production (Fig. 6D, Suppl. Fig. 7). This was particularly evident for IL-6, expression of which is strongly activated in astrocytes (Van Wagoner, Oh, Repovic, & Benveniste, 1999). IL-1β that is highly expressed by microglia (Pinteaux, Parker, Rothwell, & Luheshi, 2002) was not affected, while expression of the gene encoding TNF that is highly expressed by microglia and undergoes LPS tolerance in macrophages (Ziegler-Heitbrock, 1995), was increased although this was not statistically significant. Conversely, expression of RelB-dependent Chi3L1 was diminished in RelBΔAST mice. These data indicate that RelB in astrocytes restricts neuroinflammation in vivo by specifically suppressing cytokine production.
Fig. 6. Astrocytic RelB regulates neuroinflammation and tolerance in vivo.
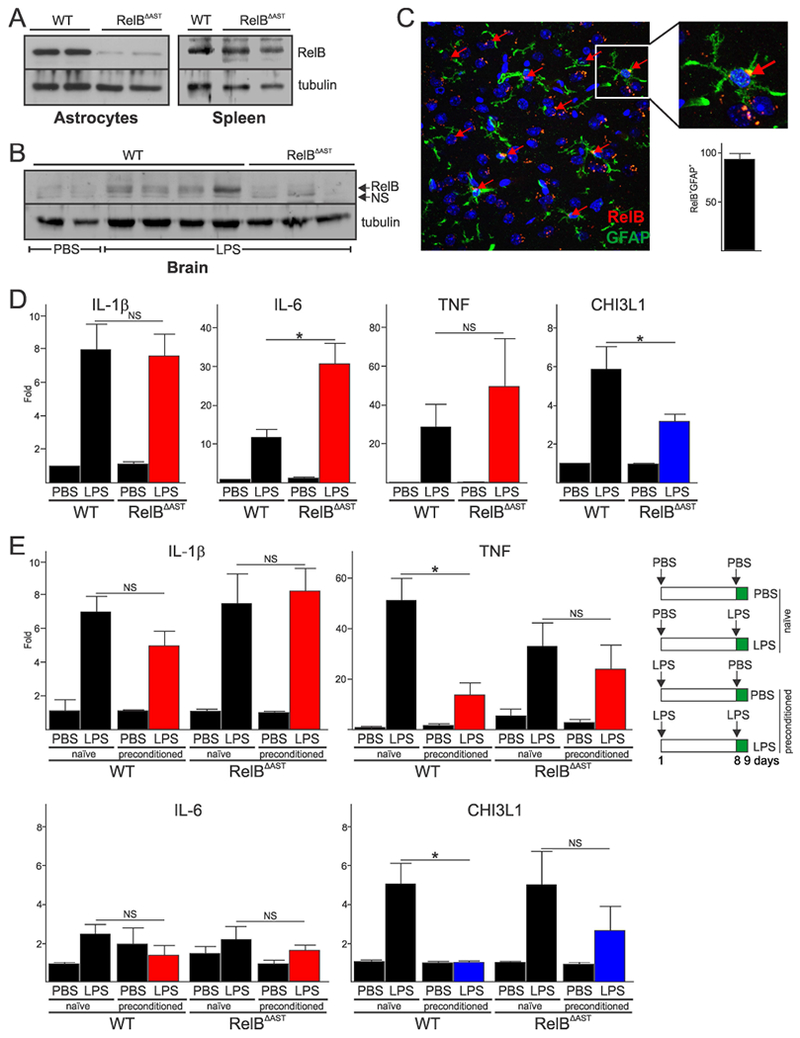
(A) Analysis of RelB expression in astrocytes of WT and RelBΔAST mice. (B) Analysis of RelB protein expression in the brains 24h post intraperitoneal injection of PBS or 5 mg/kg LPS. (A-B) Representative blots of two experiments are shown. (C) RelB protein was visualized by IF in the cortex of WT mice 24h post intraperitoneal injection of 5 mg/kg LPS. Anti-GFAP antibody was used to co-stain astrocytes, and Hoescht to visualize nuclei. Quantification of RelB+GFAP+ cells as a percentage of all GFAP+ cells is shown in lower panel (n=7). (D) Expression of mRNAs in the brains of mice receiving PBS or 5 mg/kg LPS (n=6, 11, 6, 12, respectively). (E) Expression of mRNAs in the brains of mice receiving two doses of 2 mg/kg LPS one week apart (right panel). qPCR analysis of mRNA from two independent experiment (n=4, 7, 3, 8, 3, 6, 3, 7, respectively). Error bars represent s.e.m., * P<0.05 (two-way ANOVA, Sidak’s test).
To examine whether astrocytic RelB also controls tolerance in vivo, mice were preconditioned with a lower dose of LPS and allowed to recover for 7 days. Preconditioned WT mice displayed diminished cytokine expression compared to naïve mice (Fig. 6E), which was particularly apparent for TNF. Surprisingly, expression of the Chi3L1 gene was severely diminished in preconditioned WT mice, indicating species-specificity of tolerance for some genes. In contrast to WT mice, expression of the cytokine genes and the Chi3L1 gene was similar in both naïve and preconditioned RelBΔAST mice (Fig. 6E), suggesting that RelB regulates tolerance in astrocytes. Collectively these data suggest that astrocytes display adaptive responses that depend on RelB in vitro and in vivo.
DISCUSSION
Although LPS tolerance of macrophages was recognized a decade ago (Foster et al., 2007), tolerance has only recently been found to exist in the resident innate immune cells of the brain, microglia (Schaafsma et al., 2015; Wendeln et al., 2018). Astrocyte “cytokine-tolerance” may be a general mechanism operating in sterile neuroinflammation induced by systemic infections, but likely also in a variety of pathological conditions, such as stroke or neurodegeneration. Microglia are the primary sensors of both infectious and sterile insults in the brain (Hanisch & Kettenmann, 2007) and avid producers of cytokines such as IL-1β that can subsequently stimulate surrounding cells, including astrocytes (Carpentier et al., 2005; Tarassishin, Suh, & Lee, 2014). There is growing evidence that activated reactive astrocytes are critical for both the propagation and resolution of neuroinflammation associated with human diseases (Liddelow & Barres, 2017). Our data provide evidence that in addition to epigenetically controlled tolerance in microglia, tolerance also operates in astrocytes to make these cells less reactive to repetitive activation by IL-1β. Tolerance in both microglia and astrocytes likely provides a coordinated adaptive response which may be physiologically critical in controlling the magnitude of neuroinflammation and tissue injury.
The mechanism of cytokine tolerance, we propose for astrocytes is not epigenetic as described for innate immune cells (Netea et al., 2016). Cytokine tolerance in astrocytes involves RelB and likely SIRT1, as described in macrophages and microglia; however, in contrast to these cells, deacetylation of histone H4K16 and di-methylation of H3K9 does not occur in astrocytes. The other histone modifications that could impinge expression of the cytokine gene are also not likely present in tolerant astrocytes, since initial activation of expression of the cytokine genes is comparable in naïve and tolerant cells. For this reason, the proposed sequestration of p65 by RelB (Marienfeld et al., 2003) is also not a mechanism leading to tolerance in astrocytes. Although binding of RelB to cytokine promoters is critical to establish tolerance, it is not clear why epigenetic modifications of histone tails manifest in macrophages and microglia, but not in astrocytes. The suppressive effect of SIRT1 on cytokine expression in astrocytes is likely due to SIRT1-mediated deacetylation of p65 (Yeung et al., 2004), which deactivates p65 and allows for its removal from the DNA.
We propose that low levels of RelB in naïve astrocytes ensure efficient cytokine induction by IL-1β-induced p65/p50 complexes (Fig. 5K). It should be noted that RNA-seq data indicate that astrocytes express RelB at levels lower than generally accepted cutoff of reliable expression (<5 FPKM) (Chai et al., 2017; Zhang et al., 2016). However, expression of p50 NF-κB that is needed for canonical NF-κB signaling that operates in astrocytes is also below reliable expression level (Chai et al., 2017; Zhang et al., 2016). In tolerant astrocytes, RelB levels are much higher due to the induction of RelB expression by the initial cytokine exposure; however, cytokines genes are initially activated by IL-1β-induced p65/p50 complexes that cannot be outcompeted by activated RelB/p50 complexes. Tolerance is established once p65/p50-induced IkBα is resynthesized, transported to the nucleus, and strips p65/p50 from the DNA. The previously proposed dimer switching occurs (Saccani et al., 2003) with RelB/p50 complexes substituting for p65/p50 at the cytokine promoters. We propose that, in contrast to p65/p50 complexes, RelB/p50 complexes remain bound to the DNA for much longer because of IKK-mediated phosphorylation of RelB at S472. Once RelB/p50 is bound to DNA, tolerance is established since NF-κB sites at the cytokine promoters are no longer accessible for the new wave of cytokine-induced p65/p50 complexes. Thus, phosphorylation of RelB on Ser472 is critical for the establishment of tolerance. Although this phosphorylation site has previously been identified (Authier et al., 2014), it was proposed to cause dissociation of RelB/p50 from IkBα and induce genes promoting TNF-induced migration of fibroblasts, including matrix metalloproteinase 3. Our data suggest that S472 phosphorylation of RelB diminishes association with newly synthesized IkBα, thus allowing RelB to establish silencing of cytokine genes. While RelB phosphorylation slowly diminishes with time, sustained higher levels of RelB persist in tolerant cells for days and ensure efficient establishment of tolerance once cells are re-exposed to the inflammatory stimulus.
Although it was proposed that LPS tolerance is HDAC6-dependent in mouse astrocytes and microglia (Beurel, 2011), the mechanism is not clear since LPS tolerance in microglia is coordinated by RelB, SIRT1, and KMT1C on the epigenetic level (Chen et al., 2009; Liu et al., 2011; Millet et al., 2013), whereas HDAC6 is cytosolic. Nevertheless, it is possible that HDAC6 could affect cytoplasmic-nuclear transport of RelB or IkBα.
It has not escaped our notice that the time-frames of immune memory significantly differ between adaptive immune cells, innate immune cells, and astrocytes. Memory T and B cells retain information from pathogen encounters for decades (Chaplin, 2010), innate immune cells display tolerance for months (Schaafsma et al., 2015), while astrocytes seem to display cytokine tolerance for days-weeks. These three tiers of memory likely provide fine-tuning of immune and inflammatory responses but also limit associated tissue injury.
Supplementary Material
Main points:
Astrocytes display days-to-weeks long tolerance of cytokine genes in vitro and in vivo.
This tolerance is coordinated by an NF-κB family member RelB.
Deletion of RelB from astrocytes abolishes tolerance during experimental neuroinflammation in vivo.
Acknowledgments:
This work was supported by NIH grants R21NS100698 and R01AI093718 (to T.K.) and by the Intramural Research Program of the NIAID/NIH (to U.S). ASG and MRW were supported by the F30CA203447 and F30CA200252 fellowships from the NIH. Microscopy was performed at the VCU Microscopy Facility, supported, in part, by funding from NIH-NCI Cancer Center Support Grant P30 CA016059. Authors state no conflict of interest.
REFERENCES
- Allan SM, Tyrrell PJ, & Rothwell NJ (2005). Interleukin-1 and neuronal injury. Nat Rev Immunol, 5(8), 629–640. doi: 10.1038/nri1664 [DOI] [PubMed] [Google Scholar]
- Authier H, Billot K, Derudder E, Bordereaux D, Riviere P, Rodrigues-Ferreira S, … Baud V (2014). IKK phosphorylates RelB to modulate its promoter specificity and promote fibroblast migration downstream of TNF receptors. Proc Natl Acad Sci U S A, 111(41), 14794–14799. doi: 10.1073/pnas.1410124111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, & Robinson SM (2010). Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun, 24(1), 102–109. doi: 10.1016/j.bbi.2009.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D, HogenEsch H, & Weih F (2000). Mice lacking the transcription factor RelB develop T cell-dependent skin lesions similar to human atopic dermatitis. Eur J Immunol, 30(8), 2323–2332. doi: 10.1002/1521-4141(2000)30:8<2323::AID-IMMU2323>3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- Beurel E (2011). HDAC6 regulates LPS-tolerance in astrocytes. PLoS One, 6(10), e25804. doi: 10.1371/journal.pone.0025804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj R, Yester JW, Singh SK, Biswas DD, Surace MJ, Waters MR, … Kordula T (2015). RelB/p50 Complexes Regulate Cytokine-Induced YKL-40 Expression. J Immunol. doi: 10.4049/jimmunol.1400874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, & Miller SD (2005). Differential activation of astrocytes by innate and adaptive immune stimuli. Glia, 49(3), 360–374. doi: 10.1002/glia.20117 [DOI] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, … Khakh BS (2017). Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron, 95(3), 531–549 e539. doi: 10.1016/j.neuron.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplin DD (2010). Overview of the immune response. The Journal of allergy and clinical immunology, 125(2 Suppl 2), S3–23. doi: 10.1016/j.jaci.2009.12.980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, El Gazzar M, Yoza BK, & McCall CE (2009). The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem, 284(41), 27857–27865. doi: 10.1074/jbc.M109.000950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze DB, Wu CJ, Thomas JA, Landstrom A, & Ashwell JD (2008). Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation. Mol Cell Biol, 28(10), 3538–3547. doi:MCB.02098-07 [pii] 10.1128/MCB.02098-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowland JB, Sorensen OE, Sehested M, & Borregaard N (2003). Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol, 171(12), 6630–6639. [DOI] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, … Chen ZJ (2000). Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell, 103(2), 351–361. doi:S0092-8674(00)00126-4 [pii] [DOI] [PubMed] [Google Scholar]
- Dong Y, & Benveniste EN (2001). Immune function of astrocytes. Glia, 36(2), 180–190. [DOI] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, & Medzhitov R (2007). Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature, 447(7147), 972–978. doi: 10.1038/nature05836 [DOI] [PubMed] [Google Scholar]
- Gadani SP, Walsh JT, Lukens JR, & Kipnis J (2015). Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron, 87(1), 47–62. doi: 10.1016/j.neuron.2015.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, & Kettenmann H (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci, 10(11), 1387–1394. doi: 10.1038/nn1997 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, & Baltimore D (2002). The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science, 298(5596), 1241–1245. doi: 10.1126/science.1071914 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Natoli G, & Ghosh G (2006). Transcriptional regulation via the NF-kappaB signaling module. Oncogene, 25(51), 6706–6716. doi: 10.1038/sj.onc.1209933 [DOI] [PubMed] [Google Scholar]
- Jang E, Kim JH, Lee S, Kim JH, Seo JW, Jin M, … Suk K (2013). Phenotypic polarization of activated astrocytes: the critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J Immunol, 191(10), 5204–5219. doi: 10.4049/jimmunol.1301637 [DOI] [PubMed] [Google Scholar]
- Khakh BS, & Sofroniew MV (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci, 18(7), 942–952. doi: 10.1038/nn.4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, & Koike T (2009). Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc, 4(10), 1513–1521. doi: 10.1038/nprot.2009.154 [DOI] [PubMed] [Google Scholar]
- Kordula T, Rydel RE, Brigham EF, Horn F, Heinrich PC, & Travis J (1998). Oncostatin M and the interleukin-6 and soluble interleukin-6 receptor complex regulate alpha1-antichymotrypsin expression in human cortical astrocytes. J Biol Chem, 273(7), 4112–4118. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, & Barres BA (2017). Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity, 46(6), 957–967. doi: 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, & McCall CE (2011). NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem, 286(11), 9856–9864. doi: 10.1074/jbc.M110.196790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locati M, Mantovani A, & Sica A (2013). Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol, 120, 163–184. doi: 10.1016/B978-0-12-417028-5.00006-5 [DOI] [PubMed] [Google Scholar]
- Lukens JR, Gross JM, & Kanneganti TD (2012). IL-1 family cytokines trigger sterile inflammatory disease. Front Immunol, 3, 315. doi: 10.3389/fimmu.2012.00315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marienfeld R, Berberich-Siebelt F, Berberich I, Denk A, Serfling E, & Neumann M (2001). Signal-specific and phosphorylation-dependent RelB degradation: a potential mechanism of NF-kappaB control. Oncogene, 20(56), 8142–8147. doi: 10.1038/sj.onc.1204884 [DOI] [PubMed] [Google Scholar]
- Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, & Neumann M (2003). RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem, 278(22), 19852–19860. doi: 10.1074/jbc.M301945200 [DOI] [PubMed] [Google Scholar]
- Millet P, McCall C, & Yoza B (2013). RelB: an outlier in leukocyte biology. J Leukoc Biol, 94(5), 941–951. doi: 10.1189/jlb.0513305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, … Xavier RJ (2016). Trained immunity: A program of innate immune memory in health and disease. Science, 352(6284), aaf1098. doi: 10.1126/science.aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Klar S, Wilisch-Neumann A, Hollenbach E, Kavuri S, Leverkus M, … Klingel K (2011). Glycogen synthase kinase-3beta is a crucial mediator of signal-induced RelB degradation. Oncogene, 30(21), 2485–2492. doi: 10.1038/onc.2010.580 [DOI] [PubMed] [Google Scholar]
- Norden DM, Trojanowski PJ, Villanueva E, Navarro E, & Godbout JP (2016). Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia, 64(2), 300–316. doi: 10.1002/glia.22930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, & Nilsson M (2005). Astrocyte activation and reactive gliosis. Glia, 50(4), 427–434. doi: 10.1002/glia.20207 [DOI] [PubMed] [Google Scholar]
- Pinteaux E, Parker LC, Rothwell NJ, & Luheshi GN (2002). Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J Neurochem, 83(4), 754–763. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, … Crews FT (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia, 55(5), 453–462. doi: 10.1002/glia.20467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Pantano S, & Natoli G (2003). Modulation of NF-kappaB activity by exchange of dimers. Mol Cell, 11(6), 1563–1574. [DOI] [PubMed] [Google Scholar]
- Schaafsma W, Zhang X, van Zomeren KC, Jacobs S, Georgieva PB, Wolf SA, … Eggen BJ (2015). Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav Immun, 48, 205–221. doi: 10.1016/j.bbi.2015.03.013 [DOI] [PubMed] [Google Scholar]
- Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD, … Hoffmann A (2012). Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways. Nature immunology, 13(12), 1162–1170. doi: 10.1038/ni.2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci, 32(12), 638–647. doi: 10.1016/j.tins.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, & Vinters HV (2010). Astrocytes: biology and pathology. Acta neuropathologica, 119(1), 7–35. doi: 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassishin L, Suh HS, & Lee SC (2014). LPS and IL-1 differentially activate mouse and human astrocytes: role of CD14. Glia, 62(6), 999–1013. doi: 10.1002/glia.22657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wagoner NJ, Oh JW, Repovic P, & Benveniste EN (1999). Interleukin-6 (IL-6) production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J Neurosci, 19(13), 5236–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, & Chen ZJ (2001). TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature, 412(6844), 346–351. doi: 10.1038/3508559735085597 [pii] [DOI] [PubMed] [Google Scholar]
- Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, … Neher JJ (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature, 556(7701), 332–338. doi: 10.1038/s41586-018-0023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, & Mayo MW (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J, 23(12), 2369–2380. doi: 10.1038/sj.emboj.7600244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, & Barres BA (2012). Genomic analysis of reactive astrogliosis. J Neurosci, 32(18), 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, … Barres BA (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron, 89(1), 37–53. doi: 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler-Heitbrock HW (1995). Molecular mechanism in tolerance to lipopolysaccharide. J Inflamm, 45(1), 13–26. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.