

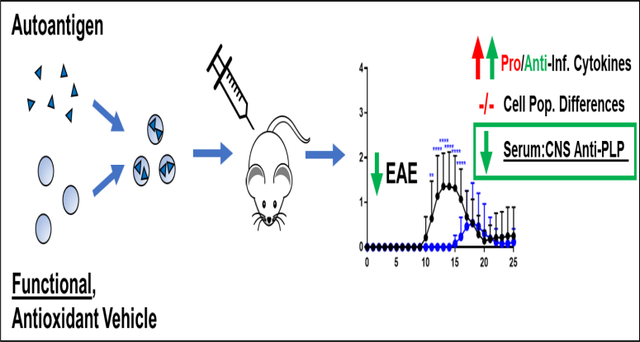
Abstract
Contemporary approaches to treating autoimmune diseases like Multiple Sclerosis broadly modulate the immune system and leave patients susceptible to severe adverse effects. Antigen-specific immunotherapies (ASIT) offer a unique opportunity to selectively suppress autoreactive cell populations, but have suffered from marginal efficacy even when employing traditional adjuvants to improve delivery. The development of immunologically active antigen delivery vehicles could potentially increase the clinical success of antigen-specific immunotherapies. An emulsion of the antioxidant tocopherol delivering an epitope of proteolipid protein autoantigen (PLP139–151) yielded significant efficacy in mice with experimental autoimmune encephalomyelitis (EAE). In vitro studies indicated tocopherol emulsions reduced oxidative stress in antigen presenting cells. Ex vivo analysis revealed that tocopherol emulsions shifted cytokines responses in EAE splenocytes. In addition, IgG responses against PLP139–151 were increased in mice treated with tocopherol emulsions delivering the antigen suggesting a possible skew in immunity. Overall, tocopherol emulsions provide a functional delivery vehicle for ASIT capable of ameliorating autoimmunity in a murine model.
Keywords: Antigen-Specific Immunotherapy, Experimental Autoimmune Encephalomyelitis (EAE), Co-Delivery, Antioxidant, Tocopherol, Proteolipid Protein (PLP139–151)
Graphical Abstract
INTRODUCTION
Current therapeutic approaches to autoimmune diseases such as multiple sclerosis (MS) are largely relegated to symptom management alone, and the most effective drugs evoke efficacy by imposing global immunosuppression [1–3]. This immunosuppression leaves patients susceptible to life-threatening side effects such as opportunistic infections and malignancies [4–7]. Indeed, there is a clear history of positive correlation between drug effectiveness and risk for adverse outcomes in the clinical treatment of MS [6, 8]. Antigen-specific immunotherapy (ASIT) has proven itself compelling as a means to more selectively disarm autoimmunity without global immunosuppression. Recent trends suggest co-delivery of autoantigen with an immunomodulatory drug may target autoreactive cells to overcome the conventional efficacy/safety trade-off in autoimmune therapeutics [9–13].
Co-delivering ASIT and an immune modulator requires spatiotemporal restriction of antigen and drug to ensure processing in the same immunological context for eliciting potent, antigen-specific outcomes [14, 15]. To accomplish this necessity of space-time proximity, formulations have historically utilized vehicles from applications such as vaccines, cancer immunotherapy, and sustained-release drug delivery [16–20]. While the polymers and metals that make up these vehicles are often touted as biocompatible and immunologically inert, co-delivery applications for autoimmunity have periodically shown negative outcomes for vehicle control groups in measures such as autoantibody titers and clinical disease severity [21, 22]. These studies each reported overall promising therapeutic results, perhaps by overcoming deleterious vehicle effects on immunity via the immunosuppression imparted through the inclusion of drug. Ultimately, works such as these have resulted in a paradigm that inclusion of immunosuppressive drugs is required to overcome inherent immunogenicity of formulations.
In recent work, co-delivery of autoantigen and dexamethasone in incomplete Freund’s adjuvant was shown to ameliorate experimental autoimmune encephalomyelitis (EAE), a murine model of relapsing-remitting multiple sclerosis [23]. Nevertheless, incomplete Freund’s adjuvant is substantially toxic in humans and has even been used to induce EAE in primates [24], suggesting poor feasibility in terms of practical therapeutic application for autoimmune diseases such as MS. It seems an efficacious ASIT to combat autoimmunity should utilize components of a co-delivery system optimally tolerable to humans. Furthermore, ASIT delivery vehicles that also exhibit desirable immunological function may simplify formulation, yielding a more translatable approach to restore immune tolerance.
Tocopherol emulsions (ETPGS) consisting of vitamin E oil and a PEGylated vitamin E surfactant (D-α-Tocopherol polyethylene glycol 1000 succinate, TPGS) in an aqueous continuous phase have been approved as safe pharmaceutical adjuvants since 2005 [25, 26]. The original application for ETPGS was the sustained delivery of paclitaxel as a chemotherapeutic agent, but since its emergence, the formulation has been pervasively modified and applied in other applications such as intestinal permeation enhancement and copolymerization [27, 28]. Because it has already been approved in humans, tolerability is known when considering it as a potential vehicle. In addition, the wealth of various ETPGS formulations explored in many discrete biomedical applications indicate the physical properties of the suspension are highly tunable. For particulate ASIT delivery approaches in cancer, autoimmunity, and vaccines, the immunotherapeutic effect is most commonly evoked through nonspecific uptake of the formulation by antigen-presenting cells (APCs), leading to downstream inflammation or tolerance by processing of antigen in the context of immunostimulatory or suppressive compounds [29–31]. Since ETPGS droplet size is versatile, suspensions can be created at mean diameters under 500 nm for optimal APC uptake [32]. Importantly, ETPGS can be tuned to have a mean diameter still greater than 100 nm to enable APC interaction to occur while increasing formulation persistence at the site of injection [33–35]. The flexibility of ETPGS for ASIT delivery is augmented by the fact vitamin E is an effective antioxidant. The propagation of an immune response is driven by a cascade of inflammatory events [36, 37]. By staving off inflammation, antioxidant compounds can skew immune responses toward tolerance [27, 38].
Herein, we report on the application of ETPGS to deliver autoantigen and ameliorate EAE in vivo. Efficacy was determined by comparing disease scores and weights of EAE mice. The immune response was characterized by quantifying cytokine responses in splenocytes and by shifts in autoantibody responses in blood and in tissues from the central nervous systems in mice. Finally, real-time measurement of oxidation in antigen presenting cells was conducted to more deeply understand the mechanism of ETPGS as an ASIT delivery vehicle.
MATERIALS AND METHODS
Materials
PLP139–151 (PLP), the antigenic peptide used, was purchased from Biomatik (Cambridge, ON, Canada). For the induction of EAE, incomplete Freund’s adjuvant (IFA) and killed Mycobacterium tuberculosis strain H37RA were purchased from Difco (Sparks, MD) as well as pertussis toxin, purchased from List Biological Laboratories (Campbell, CA). Murine RAW 264.7 cells (ATCC® TIB71™) along with Dulbecco’s Modified Eagle’s Medium (DMEM), phenol red-free DMEM, fetal bovine serum (FBS), and penicillin/streptomycin antibiotic solution were procured from American Type Culture Collection (ATCC, Manassas, VA, USA) and Atlanta Biologicals (Flowery Branch, GA, USA). Diethyldithiocarbamate (DDC), 2-methoxyestradiol (2-ME), phorbol 12-myristate 13- acetate (PMA), phosphate-buffered saline (PBS), lipopolysaccharide (LPS, from Escherichia coli 0111:B4), sodium dodecyl sulfate (SDS), and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Interferon-γ (IFN-γ) was obtained from Calbiochem (Gibbstown, NJ, USA). 4-Amino-5-methylamino-2’,7’-difluorofluorescein diacetate (DAF-FM DA) and MitoSOX Red were supplied from Life Technologies (Carlsbad, CA, USA). For flow cytometry, Alexa Fluor® 488-conjugated anti-mouse CD3, Alexa Fluor® 647-conjugated anti-mouse CD19, and Pacific Blue™-conjugated anti-mouse CD11c, and matched isotype controls were purchased from Biolegend (San Diego, CA). Goat HRP-conjugated anti-Mouse IgG for ELISAs was also purchased from Biolegend (San Diego, CA). Vitamin E and TPGS were received from Sigma Aldrich (St. Louis, MO). Other chemicals and reagents used for these studies were analytical grade and used as received.
Preparation of ETPGS Emulsions
ETPGS emulsions were prepared by adding TPGS to 1X phosphate-buffered saline (PBS), and agitating overnight at 42.5°C. If PLP was required, the peptide was then dissolved in the mixture before the aqueous phase was gently pipetted to a tube containing α-tocopherol mineral oil. Next, the tube containing α-tocopherol, TPGS and PBS (with or without PLP) was sonicated using a Fisher Scientific™ Sonic Dismembrator Model 500 with a 10 second on, 2 second off cadence (30% intensity) for 2 minutes, dipping the emulsion mixture in an ice bath during off periods to maintain temperature. After sonication, the emulsion was poured into a glass scintillation vial for storage, either on ice or at 4°C. For recipe development, amounts used of α-tocopherol, TPGS and PBS were systematically varied, though final proportions used for the ETPGS emulsions in these studies were 2:1 α-tocopherol:TPGS ultimately constituting 10% of a 10:90 oil-in-water emulsion.
Characterizing Size and Peptide Release from ETPGS Emulsions
Dynamic light scattering (DLS) was used to size ETPGS emulsions. Six ETPGS emulsions were fabricated (three with PLP, three without) and submitted to a NanoBrook Omni DLS to measure mean particle diameter and half-width for each formulation.
For imaging via transmission electron microscopy (TEM), 5 μl of ETPGS or ETPGS+PLP was placed onto 300 mesh copper grids with ultrathin carbon film. The wet grids were air-dried for several minutes prior to being examined under TEM. The emulsions were examined by bright-field and dark-field transmission electron microscopy (TEM) using an FEI Tecnai F20 transmission electron microscope at an electron acceleration voltage of 200 kV. High resolution images were captured using a standardized, normative electron dose and a constant defocus value from the carbon-coated surfaces.
To characterize peptide release, 4 mL of ETPGS emulsion containing PLP was inserted into regenerated cellulose dialysis tubing (6000–8,000 MWCO, 30 μm wall thickness, Fisherbrand Dialysis Tubing). This sealed tubing was submerged in 100 mL of PBS within a glass vessel and capped to prevent evaporation. Likewise, the same schematic was used for PBS containing PLP at the same concentration as a control. Release vessels were kept at 37°C with gentle shaking by an incubator (79 rpm, Excella E24 Incubator Shaker, New Brunswick Scientific). For sample collection, 1 mL was taken from the 100 mL bulk fluid of the release vessels and replaced with 1 mL of fresh PBS. Characterization of PLP concentration in samples was performed using reverse-phase HPLC (Waters 2796 Bioseparations Module, Waters Corp) on a C4 analytical column (Waters XBridge Protein BEH column, 300 Å, 3.5 μm, 4.6 mm x 150 mm, 10–500 K). Samples were eluted with mobile phases A (100% water with 0.05% trifluoroacetic acid (TFA)) and B (100% acetonitrile with 0.05% TFA) with a linear gradient of 95% A to 30% A over 20 minutes at a constant flow of 1 mL/min. PLP was detected at 280 nm and DEX was detected at 240 nm with a dual wavelength absorbance detector (Waters 2487 Dual λ Absorbance Detector, Waters Corp). Data was collected and processed using Empower 3 Software (Waters Corp). Concentration losses of peptide from sampling and PBS re-addition were accounted for when data were analyzed.
Measurement of ETPGS Antioxidant Power
For assessing antioxidant capabilities of ETPGS emulsions, the ferric-ion reducing antioxidant power (FRAP) assay was utilized as previously established [39]. Briefly, ETPGS with and without PLP was prepared, diluted 1:1000 and titrated at various concentrations in duplicate into FRAP reagent containing Fe(III). Fe(III) conversion to Fe(II) was measured by reading samples at a wavelength of 593 nm (Spectramax M5, Molecular Devices) over various time points.
Acute Antioxidant Effect in a Model APC
The acute effects of ETPGS on reactive nitrogen and oxygen species (RNOS) in RAW 264.7 cells were measured using a previously established, highly sensitive microfluidic assay [40, 41]. Briefly, RAW 264.7 macrophages were cultured in 25 cm3 flasks and maintained at a density of 5×106 cells/flask. The cells were stimulated with IFN-γ (600 U/mL) and LPS (100ng/mL), and incubated for 20 hours. Cells were then treated with 0.1% (v/v) ETPGS and incubated for another 60 minutes. Fluorescent probes DAF-FM DA and MitoSOX were loaded into cells at concentrations of 10 μM, each to indicate NO and superoxide species, respectively. Also included in the loading were superoxide dismutase inhibitors DDC (1 mM) and 2-ME (10 μM). After loading, cells were harvested, washed and lysed. The lysate was injected into a preconditioned PDMS/glass microfluidic device, and Labview software was used to collect and analyze data.
Induction of EAE and Therapeutic Study
Female SJL/J mice, 4–5 weeks of age were obtained from Envigo Laboratories and housed at the University of Kansas under specified, pathogen-free conditions. Female SJL/J mice are a well characterized and established strain known to exhibit a relapsing-remitting pattern of MS-like disease when immunized against the PLP139–151 epitope [42–44]. All live-mouse protocols were approved by the University of Kansas Institutional Animal Care and Use Committee. For the induction of EAE, mice were subcutaneously administered with 200 μg of PLP in a 0.2 mL emulsion of Complete Freund’s Adjuvant (CFA). The CFA mixture was comprised of equal volumes of PBS and IFA also containing killed Mycobacterium tuberculosis strain H37RA at a final concentration of 4 mg/mL. This immunization was given as four, 50 μL injections above the shoulders and the flanks. Additionally, 200 ng of pertussis toxin was given intraperitoneally on the same day of immunization (day 0) as well as day 2 post-immunization. The data presented are comprised of three independent in vivo studies; each group was comprised of 12–15 mice in total. Treatments were administered as 100 μL subcutaneous injections on days 4, 7 and 10 of the study. For the PLP and ETPGS+PLP groups, antigen was dosed at 200 nmol. ETPGS and ETPGS+PLP treatments were injected within 30 minutes of fabrication. One of the 15 PBS control mice was excluded from the study due to starting weight outside of the appropriate range. Two of 15 ETPGS+PLP mice died within 24 hours of receiving the third treatment injection due to unclear circumstances and were excluded from the study as well. Disease progression was assessed on a five-point clinical scale including: 0, no clinical evidence of disease; 1, tail weakness or limp tail; 2, paraparesis (weakness or incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with forelimb weakness or paralysis; and 5, moribund. Body weight was taken each day as well.
Tissue Harvest and Splenocyte Isolation
Mouse spleens were harvested from EAE mice on day 25 post-immunization as previously described [23, 45, 46]. Briefly, the spleens were first passed through a wire mesh using the rubber end of a sterile 1 mL syringe plunger and collected in 5 mL of RPMI 1640 media. The cellular extracts were centrifuged, and the resulting cell pellet was resuspended in 3.5 mL of 1X Gey’s lysis solution. The cells were incubated on ice for 3.5 minutes to lyse splenic red blood cells. The lysis reaction was stopped with the addition of 10 mL RPMI 1640 media containing 10% FBS and centrifuged at 1,100 × g for 5 minutes. The remaining splenocyte pellets were resuspended in fresh media (RPMI 1640 media containing 10% FBS and 1% Penicillin-Streptomycin) and plated in 96-well cell culture plates at a cell density of 1×106 cells/well in a final volume of 200 μL, and in 12-well cell culture plates at a density of 5×106 cells/well and 1 mL volume. Plain media or 25 μM PLP was immediately introduced to cells. Stimulated cell cultures were incubated for 96 hours at 37°C in a CO2 (5%) incubator.
Additionally, serum was taken from mice during sacrifice by accessing the caudal vena cava. Spinal cords were harvested, weighed, and homogenized with the Fisher Scientific™ Sonic Dismembrator Model 500 in PBS. The amount of PBS added was determined to create 50 mg homogenized central nervous tissue (hCNS) per 500 μL of PBS stocks. Serum and hCNS samples were stored at −20°C.
Measurement of Cytokines
After 96 hours of incubation, splenocytes in a 96-well culture plate were centrifuged, and supernatants were collected for cytokine analysis (GM-CSF, IFN- γ, IL-2, IL-21, IL-4, IL-10, IL-15, IL-17, IL-23, TNF-α). Marker levels were detected using a U-Plex assay kit according to manufacturer instructions (Meso Scale Discovery). Briefly, each plate was coated with 50 μL of multiplex coating solution consisting of linkers and biotinylated capture antibodies for each cytokine and incubated on a shaker at 700 rpm for 1 hour at room temperature. Following a 3-time wash step with 150 μL PBS containing 0.05% Tween 20, 25 μL of diluent and 25 μL of sample was added to each well and incubated again for 1 hour on a shaker at room temperature. Detection antibody was then added at 50 μL/well and incubated for 1 hour. Finally, each assay plate was read using the QuickPlex multiplex plate reader (Meso Scale Discovery).
Measurement of Cellular Metabolism
Resazurin (7-hydroxy-3H-phenoxazin-3-one 10-oxide) was used to determine cellular metabolism. 75 μM resazurin was introduced to splenocyte cultures and incubated for 3 hours. Metabolic reductive capacity was observed by viewing changes in fluorescence at excitation 560, emission 590 (Spectramax M5, Molecular Devices). Background fluorescence were taken using RPMI media and subtracted out from splenocyte readings for analysis.
Fluorescent Staining and Flow Cytometry
Splenocytes were collected from 12-well plates at 96 hours and stained with fluorescent antibodies. 1×106 cells were washed with 1 mL of wash buffer (RPMI 1640 media containing 5% FBS) before being centrifuged and resuspended in 50 μL of block buffer containing 20 μg/mL TruStain fcX (anti-mouse CD16/32 antibody, Biolegend) in wash buffer. Cells were incubated on ice for 30 minutes before adding the fluorescent antibodies and isotype controls in 50 μL at 2x the manufacturer recommended concentration for 1 hour. For flow cytometry data collection, 30,000 cells per sample were detected using a BD FACSFusion cytometer. Data were analyzed using Kaluza Software.
Detection of Anti-PLP IgG
PLP-specific IgG titers were assessed in an ELISA format [47]. 1μg/mL PLP was dissolved in an isoelectric (pH 9.5) solution of 50 mM NaHCO3. This coating buffer was seeded on Immulon 2HB 96-well plates and incubated overnight at 4°C. Plates were then blocked at 37°C for 1 hour with 1% (w/v) bovine serum albumin (BSA) plus 0.5% Tween 20 after washing unbound component with the same procedure as used in the multiplex assays. Serum and hCNS samples were then introduced and serially diluted 1:1 across seven concentrations. After another 1 hour incubation (37°C), plates were again washed, and HRP-conjugated anti-mouse IgG (BioLegend) was added at 0.1 μg per 100 μL and incubated again for 1 hour at 37°C. Washing the plates once more, 100 μL of TrueBlue substrate was added and plates were covered and shaken at 75 rpm and room temperature for 15 minutes. Enzymatic conversion was stopped with 100 μL 2N H2SO4 and the plate was read at 450/540 nm (Spectramax M5, Molecular Devices). For analyzing titer, linear regions across sample titration readings were fitted with linear regressions and extrapolated to their 1X concentration for comparison across samples.
Statistical Analysis
Statistical evaluation of data was performed using two-way analysis of variance (ANOVA), followed by Tukey and Sidak multiple comparison tests. The criteria for statistical significance for all analyses was set at p<0.05. All statistical analyses were performed using GraphPad Software (GraphPad Software Inc.).
RESULTS
Characterization of the ETPGS Formulation
To develop ETPGS as a functional co-delivery vehicle, a formulation recipe was empirically derived by altering proportions of vitamin E, TPGS, and phosphate-buffered saline. By measuring fabricated particle diameters by DLS, outcomes were reconciled against the design criteria for an emulsion to contain droplets between 100–500 nm. Ultimately, a stable emulsion was obtained with 10% oil in PBS (w/w) and 2:1 E:TPGS (w/w, Fig. 1A,B). The selected formulation was evaluated by fabricating three replicate emulsions, both alone and when formulated with PLP (Fig. 1B). The ETPGS recipe alone yielded a mean diameter of 263.2 nm, and with the inclusion of PLP, the emulsion increased in mean size to 303.1 nm. Droplet half-width decreased from 52.3 nm to 32.8 nm, suggesting that PLP may be surface active. The emulsions were sized again after one month at 4°C storage; negligible changes in droplet size were observed (data not shown). To image the association of PLP with ETPGS and confirm incorporation of the peptide into the formulation, transmission electron microscopy (TEM) was carried out, which suggested PLP accumulation at the oil-water interface (Fig. 1C). Peptide incorporation was further confirmed using elemental analysis to visualize nitrogen content within the oil phase of ETPGS with PLP, but not ETPGS alone (Supp. Fig. 1). A peptide release study was conducted to verify temporal restriction of peptide with the ETPGS formulation as well (Supp. Fig. 2)
Fig. 1.

Formulation of a tocopherol-based vehicle for antigen delivery. A.) Emulsion fabrication using varied proportions of Vitamin E, TPGS and PBS enabled the construction of a ternary phase diagram, where scale denotes percent composition of each component. B.) Favorable component proportions were used to fabricate emulsions both with (ETPGS+PLP) and without (ETPGS) PLP for sizing via dynamic light scattering (n = 3/group) DLS sizes and half-widths are presented in terms of average size ± standard deviation. C.) formulations of ETPGS alone (left) and ETPGS+PLP (right) were also imaged with TEM to verify antigen incorporation into the emulsion. The lower right scale bar represents 500 nm for both formulations.
ETPGS influences oxidative stress in vitro
To assess the antioxidant capacity of ETPGS emulsions, the ferric-ion reducing antioxidant power (FRAP) assay was selected as a colorimetric indicator of functionality. ETPGS with and without PLP was fabricated and titrated into FRAP reagent. After 10 minutes, a linear concentration dependence was recognized for all components (Fig. 2A). Over time, both ETPGS and ETPGS+PLP achieved similar reductions in ferric ions (Fig. 2B). Moving from these preliminary results in the simple FRAP system, acute antioxidant effects of ETPGS were measured in vitro in a model APC line. LPS-stimulated RAW 264.7 macrophages showed substantial levels of MitoSOX Red, but cells treated with ETPGS displayed markedly suppressed the analyte (Fig. 2C). Notably, while ETPGS treatment resulted in an apparent overall decrease of reactive oxygen species, a slight increase of reactive nitrogen species was observed. A third, unknown product peak formed likely as the result of a reaction between ETPGS and an indicator dye.
Fig. 2.

In vitro antioxidant power analyses for ETPGS formulations. Ferric ion reducing antioxidant power results yielded A.) linear relationships between reduction and sample dilutions (reported for t = 10 minutes), and B.) reduction kinetics over time (reported at sample:FRAP ratio 0.09). ETPGS alone is represented by green diamonds, ETPGS+PLP is represented by blue circles. C.) Acute measurement of reactive nitrogen and oxygen species in stimulated RAW 264.7 cells alone (red), and ETPGS-treated cells (green) for three repeat runs, each. Superoxide peaks are identified with blue arrows, while reactive nitrogen peaks are denoted by orange arrows.
ETPGS-facilitated autoantigen delivery delays and suppresses EAE in vivo
Next, ETPGS formulated with autoantigen was tested for efficacy against EAE in vivo. This ETPGS+PLP group was evaluated against ETPGS alone, PLP alone, and PBS control groups with treatment administrations occurring on days 4, 7 and 10 post EAE induction. Clinical scoring and weight data were collected, and are reflected in Fig. 3A–C and 3D–F, respectively. PBS treated control mice exhibited disease onset at 9 days post-induction and reached peak severity (in both weight and clinical score) by day 14. Among PLP-treated mice, disease severity was largely suppressed as reflected by both scoring and weight, but disease onset occurred at a similar time to those in the PBS control group and was seemingly prolonged when compared to PBS control mice. ETPGS vehicle treatment alone substantially delayed the onset of EAE with clinical scores not occurring until day 13 and peak severity evident on day 16 as opposed to day 14 in the PBS control mice. Treatment consisting of ETPGS+PLP delayed disease presentation even further with symptoms not appearing until day 15 and a peak at day 19. Disease severity was also suppressed, evidenced by lower clinical scores and significantly higher weights even at the end of the 25-day study. In fact, ETPGS+PLP treatment resulted in greater than 50% of mice showing no signs of EAE at all (Fig. 4A). Cumulative scores were tallied for each treatment group and are presented in Fig. 4B. ETPGS+PLP treatment indeed significantly suppressed the accumulation of clinical scores when compared to PBS control and ETPGS alone, but significance was not realized between ETPGS+PLP and PLP alone due to high variability in PLP treatment efficacy.
Fig. 3.

Clinical disease scores (A-C) and weight changes (D-F) in EAE mice given treatment components. Disease score and weight change data for the PBS control treatment are compared to those of PLP treatment (A and D, respectively), ETPGS alone treatment (B and E), and ETPGS+PLP treatment (C and F). All treatments were administered on days 4, 7 and 10 post-disease induction. Each group consisted of n = 12–15 mice combined from three independent trials (*p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001 as compared to PBS). Error bars are reported as standard deviation among samples.
Fig. 4.

Clinical scoring data were analyzed by A.) disease onset by group, as determined by proportion of mice having exhibited any clinical score by the day of each time point, and B.) area under the scoring curve for each treatment group. (n = 12–15 mice/group, *p < 0.05, **p < 0.01, ***p < 0.001). Error bars are reported as standard deviation among samples.
Ex vivo splenocyte cytokines and populations in ETPGS+PLP efficacy
On day 25 of the EAE study, all mice were sacrificed and splenocytes were isolated for ex vivo analysis. Isolated splenocytes were cultured for 96 hours either with or without a 25 μM autoantigen rechallenge. At the end of the incubation, cytokines were assessed by MSD multiplex and cell metabolism was measured via resazurin (Fig. 5). Cell populations were also probed using flow cytometry (Fig. 6).
Fig. 5.

Splenocytes were harvested on day 25 from EAE mice treated in vivo with PBS, PLP, ETPGS alone, or ETPGS+PLP. Splenocytes were incubated for 96h with or without a 25 μM PLP rechallenge. Supernatant cytokine levels of A.) GM-CSF, B.) IFN-γ, C.) Il-2, D.) Il-21, E.) Il-6, F.) Il-10, G.) Il-15, H.) Il-17, I.) Il-23, and J.) TNF-α were determined and K.) cell metabolism was measured via resazurin. (n = 5–6 mice/group, *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001). Error bars are reported as standard deviation among samples.
Fig. 6.

Splenocytes were harvested on day 25 from EAE mice treated in vivo with PBS, PLP, ETPGS alone, or ETPGS+PLP. Splenocytes were incubated for 96h with or without a 25 μM PLP rechallenge. The cells were stained with antibodies for CD3 (Alexa Fluor 488), CD19 (Alexa Fluor 647), and CD11c (Brilliant Violet 421), as well as a Rhodamine-labeled PLP. Stained splenocytes were analyzed by flow cytometry. Cell populations were evaluated for A.) T-cells (CD3+), B.) B-cells (CD19+), C.) Dendritic cells (CD11c+), D.) PLP-specific cells (PLP+), and E.) Autoimmune-associated B-cells (CD19+CD11c+). Also analyzed was F.) ratio of T cells (CD3+) to B cells (CD19+) for each group. (n = 5–6 mice/group, *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001). Error bars are reported as standard deviation among samples.
Surprisingly, a highly conserved trend was observed across many cytokines included in the study. ETPGS+PLP group splenocytes consistently exhibited elevated levels of cytokines when compared to PBS control cells, both in terms of tolerogenic and inflammatory cytokines. The most robust cytokine responses evoked in ETPGS+PLP splenocytes were in IL-21 and IL-10 (Fig. 5D and 5F), where significant elevation was recognized over all other groups. Responses in the PLP group were statistically similar to those in PBS. ETPGS group splenocytes showed higher levels of IL-2 than those from PBS control mice (Fig. 5C). Interestingly, cell metabolism was significantly elevated in splenocytes from the ETPGS and ETPGS+PLP treatment groups (Fig. 5K). This observation, combined with the conserved cytokine trend, mirrors the delayed disease presentations that were observed in clinical measures.
From cell phenotyping metrics, unchallenged splenocyte populations showed some significant differences (Fig. 6A–C). Samples from the ETPGS treatment group had slightly decreased numbers of T cells (CD3+) compared to the PLP group, and a higher B cell (CD19+) count than PBS and PLP splenocytes. However, antigen-challenged cell population differences were not present among T, B, or dendritic cells (CD3+, CD19+, and CD11c+, respectively). A rhodamine-conjugated PLP was also used to probe cells capable of interacting with autoantigen, particularly by surface recognition of PLP-specific B cells (Fig. 6D). Again, resting cells displayed significant differences with PLP, ETPGS and ETPGS+PLP populations showing lower amounts of cells triggering events in the PLP channel. Upon antigen rechallenge, all groups showed similar levels of interaction with PLP. In previous ASIT studies investigating co-delivery systems, differences in a unique population of CD19+CD11c+ “autoimmune-associated B cells” have been observed and have been believed to contribute to tolerogenic outcomes. The same population was gated in this study (Fig. 6E), but no statistically relevant differences were found. Finally, T-to-B cell ratio was assessed for each group (Fig. 6F), and ETPGS was found to show a B cell-skewed proportionality compared to other groups in unchallenged populations, but not upon rechallenge. ETPGS+PLP trended similarly to the ETPGS group, but was not significantly different than PBS or PLP splenocytes.
Autoantibodies are largely restricted to the periphery in mice treated with ETPGS+PLP
To compare evidence of immunity between the periphery and central nervous system, serum and hCNS were harvested and probed for anti-PLP IgG by ELISA (Fig. 7). In the serum (Fig. 7A) and hCNS (Fig. 7B) alone, no statistically significant differences existed between groups, though tissues from the PLP and ETPGS+PLP treated mice exhibited titers that trended higher than the PBS and ETPGS alone groups. However, when comparing the ratio of serum-to-hCNS anti-PLP titer (Fig. 7C), a stark difference was realized; ETPGS+PLP group titer ratios were markedly higher than those in any other group.
Fig. 7.

On day 25 of the in vivo study, blood and spinal cords were collected from EAE mice treated with PBS, PLP, ETPGS alone, or ETPGS+PLP. Serum was isolated from blood, and spinal cords were homogenized and centrifuged to collect hCNS supernatants. Anti-PLP IgG titers were detected by ELISA for both A.) serum and B.) hCNS. C.) Ratio of serum to hCNS titers for each mouse was also determined. (n = 5–6 mice/group, *p < 0.05, **p < 0.01, ***p<0.001). Error bars are reported as standard deviation among samples.
DISCUSSION
While autoantigen delivery as immunotherapy has shown tremendous capability to ameliorate autoimmunity across numerous disease models, past applications have pointed to the necessity for the inclusion of immunomodulatory molecules (i.e. immunosuppressant, adjuvant, etc.), such that the co-delivery of antigen and drug together can elicit maximal therapeutic effectiveness [21, 22]. Delivery vehicles have been used to restrict these components to the same immunological context, but many of these vessels can evoke adverse inflammation, or are regarded as inert at best. In this work, we present ETPGS as a functional vehicle for autoantigen delivery, potentially dispensing the need for additional drugs or adjuvants. By comparing ETPGS+PLP treatment to ETPGS and PLP alone, we evaluated this formulation’s capacity to diminish EAE in vivo.
The development of an ETPGS formulation of appropriate size and stability was empirically derived by varying recipe components (Fig. 1A). TPGS content highly influenced emulsion droplet sizes, and oil-in-water content had a large bearing on stability. Formulations with greater than 25% oil were not stable over time or temperature. A 10% oil-in-water, 2:1 ETPGS recipe created stable emulsions that were in the ~250–300 nm range (Fig 1B) and advanced for further study. PLP was found to be compatible for incorporation with ETPGS, as evidenced by TEM (Fig. 1C, Supp. Fig. 1) and peptide release (Supp. Fig. 2). ETPGS was confirmed to have antioxidant function in vitro by the FRAP assay (Fig. 2A), and the inclusion of PLP did not affect its reductive capacity (Fig. 2B). This antioxidant capability was further shown to definitively influence reactive nitrogen and oxygen species production by macrophages in vitro.
Moving to an in vivo murine model of multiple sclerosis, a synergistic effect was realized by treating EAE mice with ETPGS+PLP. PLP treatment alone led to a suppressed, but prolonged disease state (Fig. 3A, D), while ETPGS treatment resulted in a delayed, though fully severe presentation of EAE (Fig 3. B, E). Formulation of ETPGS with PLP as a treatment ultimately delayed disease even further compared to ETPGS alone, and the severity was dampened to a greater extent than in the PLP group, particularly evidenced by fewer days of clinical scores and statistical improvement in weight data at the end of the study (Fig. 3C, F). These effects were most pronounced in disease incidence, where markedly more ETPGS+PLP mice showed no clinical evidence of disease than any other group (Fig. 4A).
Undoubtedly, there was therapeutic benefit to delivering PLP with ETPGS as a vehicle, and this effect suggested that these vitamin E emulsions are functionally contributive toward combating EAE. Despite this clinical success, ex vivo mechanistic measures proved somewhat confounding. ETPGS+PLP cytokine data showed robust production of markers such as IL-10 (Fig. 5F) and IL-6 (Fig. 5E), which in combination have been shown to be productive toward ameliorating EAE [48–50]. The conserved trend of increased ETPGS+PLP cytokine production of both IFN-γ (Fig. 5B) and TNF-α (Fig. 5J) was perplexing in that each of these are known to be inversely correlated with IL-10 and IL-6, respectively [51, 52]. In this study, these cytokines among all others assessed were all elevated in the ETPGS+PLP splenocytes (Fig. 5A–J). Furthermore, cell population data were also inconclusive for discerning any induction of tolerance as a mechanism for ETPGS+PLP (Fig. 6). Significant differences between groups only existed in unchallenged splenocyte populations, and re-exposure to PLP consistently led to similar levels of T, B and dendritic cells as well as PLP-recognizing cells (Fig. 6A–D). In a previous study, therapeutic success was achieved when delivering PLP and dexamethasone with IFA, and differences in a distinct population of CD19+CD11c+ were seen [23]. These differences were believed to be mechanistically contributive, though in this work, no population differences in this subtype were observed (Fig. 6E).
Though cytokine and phenotype readouts were largely inconclusive, it remains evident that ETPGS+PLP treatment enacted therapeutic efficacy, albeit perhaps not through the antioxidant mechanism first hypothesized. Extending from the universally elevated cytokine trends, it was apparent that ETPGS+PLP group splenocytes may have been more metabolically active than those from other groups. This assertion is substantiated by resazurin data (Fig. 6K), where cellular metabolism seems to correlate with temporal differences in disease presentation and resolution. While PBS and PLP groups began showing disease symptoms on the same day, the delays in both ETPGS and ETPGS+PLP onsets may translate to incompletely resolved, or “exhausted” splenocytes on day 25 at harvest. Since ETPGS+PLP mice got sick at the latest point in the study, it is reasonable to consider the splenocytes of this group may have been in a more stimulated state, affecting elevated cytokine levels and resazurin readouts.
To reconcile observed delays in disease, we considered diversion of cells to the injection site as an explanation. One notable clinical observation of the ETPGS and ETPGS+PLP treatments was that they formed a depot at the injection site. The persistence of emulsion in the subcutaneous space was intended to draw immunological attention. This attraction of immune infiltrates could contribute to a prioritization of ETPGS and consequentially explain the slight delay in disease onset for ETPGS mice. Furthermore, the instance of encountering autoantigen in the periphery (as with the ETPGS+PLP treated mice) could trigger proliferation of PLP-specific cells before reaching the central nervous system (CNS). This off-site proliferation, preceding migration to the CNS, could result in therapeutic efficacy against the clinical onset of EAE. In fact, glatiramer acetate, one of the most established MS treatments, is characterized by injection site inflammation occurring in up to 60% of patients receiving it [53, 54]. The swollen nodules observed at the injection site of EAE mice may follow a similar mechanism.
Anti-PLP IgG titers were probed in both the periphery (serum) and central compartment (hCNS) of the mice from the study (Fig. 7). Natural antibodies are unable to cross the blood brain barrier [55], so quantities were measured in both compartments as long-lasting evidence of autoimmunity between each location. While no statistical differences were definite in serum or hCNS autoantibodies discretely, the ratio of serum-to-hCNS titer yielded a distinct separation where ETPGS+PLP treated mice exhibited a markedly higher proportion of autoantibodies restricted to the periphery. These findings give credence to the possibility that ETPGS+PLP could create a lasting immunological decoy at the injection site. It is notable, however, that compartmental antibody titers trended higher compared to other groups for ETPGS+PLP as well. This information highlights the possibility that immunity could potentially also be shifted from a T helper type-1 (cellular) response to that of Th2 (humoral) response, which is known to be beneficial toward ameliorating EAE and MS [56, 57]. Future investigation into the subclass of IgG response will be beneficial to discern skewed immunity by ETPGS+PLP, as IgG1/IgG2a ratio could provide information about the potentially protective nature of an IgG response in addition to its magnitude [58, 59]. In all, the data collected in this study suggest that both a decoy effect and immunomodulation could be potential factors in the efficacy of ETPGS+PLP.
CONCLUSIONS
The development of autoantigen delivery vehicles that also functionally direct the immune response would provide substantial benefit to ASIT for autoimmune diseases. In the past, conventional vehicles such as Freund’s adjuvant have fallen short of clinical success [14, 23]. In this work, the ability of tocopherol emulsions to act as functional vehicles for antigen delivery was investigated. PLP delivered by ETPGS was efficacious to ameliorate EAE in vivo as evidenced by a decrease in disease incidence as well as severity. While trends of skewed cytokine responses were not evident in splenocytes from ETPGS+PLP treated mice compared to controls, anti-PLP IgG was slightly elevated and significantly relegated to the periphery, potentially indicating a therapeutic shift to Th2-mediated immunity and restriction of autoimmune effectors from the CNS compartment. Overall, these data indicate that ETPGS can act as a functional delivery vehicle, making it a compelling candidate for further study. The pharmaceutical development of MF59 as an emulsion-based vaccine adjuvant has shown that large-scale manufacturing of pharmaceutical emulsions can be feasible in practice [60, 61], but it will be critical to further elucidate the mechanistic potential of ETPGS in general, as well as in the context of authentic autoimmune disorders.
Moving forward, it is pertinent to further study the mechanistic drivers of therapeutic success in ETPGS as an antigen-delivery system. Evidence such as acute in vitro inhibition of RNOS production by macrophages and robust IL-10 generation in EAE splenocytes support an antioxidant mechanism, but cytokine and cell populations should be more carefully explored in secondary lymphoid organs, CNS tissues, and at the site of administration before drawing firm conclusions. The decoy effect is substantiated by delayed disease onsets and the restriction of anti-PLP IgG to the periphery over the CNS, but more work is needed to assess the validity of this phenomenon as a mechanism to correct autoimmunity.
Supplementary Material
ACKNOWLEDGEMENTS AND FUNDING
JDG was supported by the Madison and Lila Self Graduate Fellowship at the University of Kansas. MAC was supported by Biotechnology Predoctoral Training Program at the University of Kansas. The authors would like to thank Michael Shao and Alexander Sedlacek for their assistance in executing the autoantibody detection assays and ferric ion reducing antioxidant power assays. Additional acknowledgement is due to Francisco J. Martinez-Becerra of the Kansas Vaccine Institute at the University of Kansas for resources and assistance with flow cytometry and MSD multiplex cytokine analysis, as well as Prem S. Thapa of the Microscopy and Analytical Imaging Laboratory at the University of Kansas for his assistance in executing TEM for emulsion imaging. Karen Peltier of the Tertiary Oil Recovery Program at the University of Kansas assisted ETPGS particle sizing with the NanoBrook Omni DLS. NSF grant #CHE-1411993 and COBREP20GM103638 supported authors DBW and SML to carry out the acute RNOS measurements.
Footnotes
Declarations of interest: none
SUPPORTING INFORMATION
Supporting information is available: Supp. Fig. 1 Scanning electron microscopy and elemental analysis of ETPGS formulations. Supp. Fig. 2 PLP Release from ETPGS formulation. Supp. Fig. 3 Overlaid clinical scoring and weight data from in vivo study.
REFERENCES
- 1.Lim SY and Constantinescu CS, Current and future disease-modifying therapies in multiple sclerosis. Int J Clin Pract, 2010. 64(5): p. 637–50. [DOI] [PubMed] [Google Scholar]
- 2.Loleit V, Biberacher V, and Hemmer B, Current and future therapies targeting the immune system in multiple sclerosis. Curr Pharm Biotechnol, 2014. 15(3): p. 276–96. [DOI] [PubMed] [Google Scholar]
- 3.Comi G, Radaelli M, and Soelberg Sørensen P, Evolving concepts in the treatment of relapsing multiple sclerosis. The Lancet, 2017. 389(10076): p. 1347–1356. [DOI] [PubMed] [Google Scholar]
- 4.Clifford DB, et al. , Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol, 2010. 9(4): p. 438–46. [DOI] [PubMed] [Google Scholar]
- 5.Iaffaldano P, D’Onghia M, and Trojano M, Safety profile of Tysabri: international risk management plan. Neurological Sciences, 2009. 30(2): p. 159. [DOI] [PubMed] [Google Scholar]
- 6.Rommer PS, et al. , Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clin Exp Immunol, 2014. 175(3): p. 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Havrdova E, Horakova D, and Kovarova I, Alemtuzumab in the treatment of multiple sclerosis: key clinical trial results and considerations for use. Ther Adv Neurol Disord, 2015. 8(1): p. 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minagar A, Current and future therapies for multiple sclerosis. Scientifica (Cairo), 2013 2013: p. 249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartwell BL, et al. , Multivalent nanomaterials: learning from vaccines and progressing to antigen-specific immunotherapies. J Pharm Sci, 2015. 104(2): p. 346–61. [DOI] [PubMed] [Google Scholar]
- 10.Maldonado RA, et al. , Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A, 2015. 112(2): p. E156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeste A, et al. , Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences, 2012. 109(28): p. 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tostanoski, Lisa H, et al. , Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell Reports, 2016. 16(11): p. 2940–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartwell BL, et al. , Multivalent Soluble Antigen Arrays Exhibit High Avidity Binding and Modulation of B Cell Receptor-Mediated Signaling to Drive Efficacy against Experimental Autoimmune Encephalomyelitis. Biomacromolecules, 2017. 18(6): p. 1893–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northrup L, et al. , Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Adv Drug Deliv Rev, 2016. 98: p. 86–98. [DOI] [PubMed] [Google Scholar]
- 15.Shakya AK and Nandakumar KS, Antigen-Specific Tolerization and Targeted Delivery as Therapeutic Strategies for Autoimmune Diseases. Trends Biotechnol, 2018. 36(7): p. 686–699. [DOI] [PubMed] [Google Scholar]
- 16.Moon JJ, Huang B, and Irvine DJ, Engineering Nano- and Microparticles to Tune Immunity. Advanced Materials, 2012. 24(28): p. 3724–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahdev P, Ochyl LJ, and Moon JJ, Biomaterials for Nanoparticle Vaccine Delivery Systems. Pharmaceutical Research, 2014. 31(10): p. 2563–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irvine DJ, Swartz MA, and Szeto GL, Engineering synthetic vaccines using cues from natural immunity. Nature materials, 2013. 12(11): p. 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allison AC and Byars NE, Immunological adjuvants: desirable properties and side-effects. Mol Immunol, 1991. 28(3): p. 279–84. [DOI] [PubMed] [Google Scholar]
- 20.Mohan T, Verma P, and Rao DN, Novel adjuvants & delivery vehicles for vaccines development: A road ahead. The Indian Journal of Medical Research, 2013. 138(5): p. 779–795. [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang A-H, et al. , Tolerogenic nanoparticles to induce immunologic tolerance: Prevention and reversal of FVIII inhibitor formation. Cellular Immunology, 2016. 301: p. 74–81. [DOI] [PubMed] [Google Scholar]
- 22.Yeste A, et al. , Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America, 2012. 109(28): p. 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Northrup L, et al. , Co-delivery of autoantigen and dexamethasone in incomplete Freund’s adjuvant ameliorates experimental autoimmune encephalomyelitis. Journal of Controlled Release, 2017. 266: p. 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haanstra KG, et al. , Induction of Experimental Autoimmune Encephalomyelitis With Recombinant Human Myelin Oligodendrocyte Glycoprotein in Incomplete Freund’s Adjuvant in Three Non-human Primate Species. Journal of Neuroimmune Pharmacology, 2013. 8(5): p. 1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao L and Feng SS, Enhanced oral bioavailability of paclitaxel formulated in vitamin E‐TPGS emulsified nanoparticles of biodegradable polymers: In vitro and in vivo studies. Journal of pharmaceutical sciences, 2010. 99(8): p. 3552–3560. [DOI] [PubMed] [Google Scholar]
- 26.Mu L and Feng S, A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. Journal of controlled release, 2003. 86(1): p. 33–48. [DOI] [PubMed] [Google Scholar]
- 27.Guo Y, et al. , The applications of Vitamin E TPGS in drug delivery. European Journal of Pharmaceutical Sciences, 2013. 49(2): p. 175–186. [DOI] [PubMed] [Google Scholar]
- 28.Mi Y, Liu Y, and Feng S-S, Formulation of docetaxel by folic acid-conjugated d-α-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS 2k) micelles for targeted and synergistic chemotherapy. Biomaterials, 2011. 32(16): p. 4058–4066. [DOI] [PubMed] [Google Scholar]
- 29.Steinman RM, et al. , The Induction of Tolerance by Dendritic Cells That Have Captured Apoptotic Cells. The Journal of Experimental Medicine, 2000. 191(3): p. 411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banchereau J and Steinman RM, Dendritic cells and the control of immunity. Nature, 1998. 392(6673): p. 245–52. [DOI] [PubMed] [Google Scholar]
- 31.Roche PA and Furuta K, The ins and outs of MHC class II-mediated antigen processing and presentation. Nature Reviews. Immunology, 2015. 15(4): p. 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foged C, et al. , Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. International Journal of Pharmaceutics, 2005. 298(2): p. 315–322. [DOI] [PubMed] [Google Scholar]
- 33.Manolova V, et al. , Nanoparticles target distinct dendritic cell populations according to their size. European Journal of Immunology, 2008. 38(5): p. 1404–1413. [DOI] [PubMed] [Google Scholar]
- 34.He C, et al. , Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials, 2010. 31(13): p. 3657–3666. [DOI] [PubMed] [Google Scholar]
- 35.Rice-Ficht AC, et al. , Polymeric particles in vaccine delivery. Current Opinion in Microbiology, 2010. 13(1): p. 106–112. [DOI] [PubMed] [Google Scholar]
- 36.Korn T, Mitsdoerffer M, and Kuchroo VK, Immunological basis for the development of tissue inflammation and organ-specific autoimmunity in animal models of multiple sclerosis. Results Probl Cell Differ, 2010. 51: p. 43–74. [DOI] [PubMed] [Google Scholar]
- 37.Murta V and Ferrari CC, Influence of Peripheral inflammation on the progression of multiple sclerosis: evidence from the clinic and experimental animal models. Mol Cell Neurosci, 2013. 53: p. 6–13. [DOI] [PubMed] [Google Scholar]
- 38.Huq R, et al. , Preferential uptake of antioxidant carbon nanoparticles by T lymphocytes for immunomodulation. Scientific reports, 2016. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benzie IFF and Strain JJ, The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Analytical Biochemistry, 1996. 239(1): p. 70–76. [DOI] [PubMed] [Google Scholar]
- 40.Hulvey MK, Frankenfeld CN, and Lunte SM, Separation and Detection of Peroxynitrite Using Microchip Electrophoresis with Amperometric Detection. Analytical Chemistry, 2010. 82(5): p. 1608–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caruso G, et al. , Microchip electrophoresis with laser-induced fluorescence detection for the determination of the ratio of nitric oxide to superoxide production in macrophages during inflammation. Analytical and Bioanalytical Chemistry, 2017: p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCarthy DP, Richards MH, and Miller SD, Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods in molecular biology (Clifton, N.J.), 2012. 900: p. 381–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papenfuss TL, et al. , Sex differences in experimental autoimmune encephalomyelitis in multiple murine strains. Journal of Neuroimmunology, 2004. 150(1): p. 59–69. [DOI] [PubMed] [Google Scholar]
- 44.Raine CS, et al. , Homing to central nervous system vasculature by antigen-specific lymphocytes. II. Lymphocyte/endothelial cell adhesion during the initial stages of autoimmune demyelination. Lab Invest, 1990. 63(4): p. 476–89. [PubMed] [Google Scholar]
- 45.Sestak JO, et al. , Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Molecular Therapy - Methods & Clinical Development, 2014. 1: p. 14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Northrup L, et al. , Co-delivery of autoantigen and b7 pathway modulators suppresses experimental autoimmune encephalomyelitis. AAPS J, 2014. 16(6): p. 1204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagelkerken L, Blauw B, and Tielemans M, IL-4 abrogates the inhibitory effect of IL-10 on the development of experimental allergic encephalomyelitis in SJL mice. Int Immunol, 1997. 9(9): p. 1243–51. [DOI] [PubMed] [Google Scholar]
- 48.Bettelli E, et al. , IL-10 Is Critical in the Regulation of Autoimmune Encephalomyelitis as Demonstrated by Studies of IL-10- and IL-4-Deficient and Transgenic Mice. The Journal of Immunology, 1998. 161(7): p. 3299. [PubMed] [Google Scholar]
- 49.McGeachy MJ, et al. , TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell–mediated pathology. Nature Immunology, 2007. 8: p. 1390. [DOI] [PubMed] [Google Scholar]
- 50.Kimura A and Kishimoto T, IL-6: regulator of Treg/Th17 balance. Eur J Immunol, 2010. 40(7): p. 1830–5. [DOI] [PubMed] [Google Scholar]
- 51.Tilg H, et al. , Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood, 1994. 83(1): p. 113–8. [PubMed] [Google Scholar]
- 52.Fiorentino DF, et al. , IL-10 inhibits cytokine production by activated macrophages. The Journal of Immunology, 1991. 147(11): p. 3815. [PubMed] [Google Scholar]
- 53.Soós N, Shakery K, and Mrowietz U, Localized Panniculitis and Subsequent Lipoatrophy with Subcutaneous Glatiramer Acetate (Copaxone®) Injection for the Treatment of Multiple Sclerosis. American journal of clinical dermatology, 2004. 5(5): p. 357–359. [DOI] [PubMed] [Google Scholar]
- 54.Edgar CM, et al. , Lipoatrophy in patients with multiple sclerosis on glatiramer acetate. Can J Neurol Sci, 2004. 31(1): p. 58–63. [DOI] [PubMed] [Google Scholar]
- 55.Pardridge WM, The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRX, 2005. 2(1): p. 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fletcher JM, et al. , T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol, 2010. 162(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Legroux L and Arbour N, Multiple Sclerosis and T Lymphocytes: An Entangled Story. J Neuroimmune Pharmacol, 2015. 10(4): p. 528–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uyttenhove C, et al. , Development of an anti-IL-12 p40 auto-vaccine: protection in experimental autoimmune encephalomyelitis at the expense of increased sensitivity to infection. European Journal of Immunology, 2004. 34(12): p. 3572–3581. [DOI] [PubMed] [Google Scholar]
- 59.Komiyama Y, et al. , IL-17 Plays an Important Role in the Development of Experimental Autoimmune Encephalomyelitis. The Journal of Immunology, 2006. 177(1): p. 566. [DOI] [PubMed] [Google Scholar]
- 60.Haensler J, Manufacture of Oil-in-Water Emulsion Adjuvants, in Vaccine Adjuvants: Methods and Protocols, Fox CB, Editor. 2017, Springer New York: New York, NY: p. 165–180. [DOI] [PubMed] [Google Scholar]
- 61.Schijns V and O’Hagan D, Immunopotentiators in Modern Vaccines. 2016: Academic Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.