

Abstract
Background
A newly developed drug trastuzumab emtansine (T-DM1) has improved the survival of breast cancer (BC) patients. Despite an impressive initial clinical response, a subgroup of patient develop resistance and present therapeutic challenges. The underlying resistance mechanisms are not fully investigated. We report that T-DM1 treatment modulates the expression of ROR1 (type 1 receptor tyrosine kinase-like orphan receptor) and induces self-renewal of cancer stem cells (CSCs) leading to therapeutic resistance.
Methods
Using BC patient tumor samples, and BC cell lines we gained insight into the T-DM1 treatment induced ROR1 overexpression and resistance. In vitro sphere forming assays and in vivo extreme dilution assays were employed to analyze the stemness and self-renewal capacity of the cells. A series of molecular expression and protein assays including qRT-PCR, FACS-sorting, ELISA, immunostaining, Western blotting were used to provide evidence.
Findings
Exposure of cells to T-DM1 shifted ROR1 expression from low to high, enriched within the CSC subpopulation, coincident with increased Bmi1 and stemness factors. T-DM1 induced ROR1 cells showed high spheroid and tumor forming efficiency in vitro and in an animal model exhibiting shorter tumor-free time. Mechanistically, the overexpression of ROR1 is partly induced by the activation of YAP1 and its target genes. Silencing of ROR1 and YAP1 by pharmacologic inhibitors and/or sh/siRNA inhibited spheroid formation, the initiation of tumors and the capacity for self-renewal and ROR1 overexpression.
Interpretations
The results presented here indicate that simultaneous targeting of ROR1 and YAP1 may suppress CSC self-renewal efficacy and inhibit tumor progression in BC. In this manner such treatments may overcome the T-DM1 mediated therapeutic resistance and improve clinical outcome.
Fund
This study was supported by Neurogen Technologies for interdisciplinary research.
Research in context.
Evidence before the study
T-DM1 (Trustuzumab emtansine) is an antibody-drug conjugate (ADC) consisting of trastuzumab covalently linked to DM1 through a stable linker. This newly developed drug has greatly improved the therapeutic outcome for patients overall and progression free survival. Despite all these improvements, a group of patients, either acquire or exhibit intrinsic resistance after initial response, which warrants consideration of a new therapeutic strategy to combat T-DM1 induced therapeutic resistance in order to achieve a better treatment outcome for HER2+ breast cancer patients.
Added values to this study
Our study was designed to demonstrate that resistance to T-DM1 developed due to the induction of ROR1. In addition, we showed that only cells positive for ROR1 demonstrated higher sphere forming and self-renewal efficiency in vitro, and increased resistance to T-DM1 compared to cells negative for ROR1. In the animal model we showed that only ROR1 positive cells were able to grow tumors compared ROR1-negative and bulk tumor cells. Mechanistically, the overexpression of ROR1 is partly induced by the activation of YAP1 and its target genes. Inhibition of ROR1 inhibited spheroid formation and the initiation of tumors. In addition, pharmacologic and/or siRNA mediated inhibition of YAP1 affected the capacity for self-renewal and ROR1 overexpression.
Implications of all the available evidence
Approximately 15–20% of breast cancer (BC) patients are HER2+. As of 2016, several targeted therapies, lapatinib, pertuzumab, neratinib and trastuzumab have been approved for HER2+ BC patients exhibiting improved overall and progression free survival. A newly developed drug T-DM1 has further improved survival of HER2+ metastatic BC patients. Despite all these advances and impressive clinical results, occasional drug resistance, non-responding behavior of a group of patients, disease progression and recurrence remained challenges for disease management. In this study, using fresh clinical tumor samples and BC cell lines, we describe that treatment of HER2-overexpressing BC patients by T-DM1 increased the expression of ROR1, the survival of the CSC population and treatment resistance. In HER2+ BC patients treated by T-DM1, tumors switch from low ROR1 expression to increased surface expression of ROR1 and show increased enrichment of CSCs promoting the resistance to T-DM1. We further provide evidence that the transcriptional co-activator YAP1 regulates ROR1 overexpression, and disruption of YAP1-TEAD binding limits the T-DM1 treatment-induced ROR1 overexpression and CSC self-renewal. These findings suggest an alternative therapeutic strategy for HER2+ BC patients.
Alt-text: Unlabelled Box
1. Introduction
Approximately 15–20% of breast cancer (BC) patients are HER2+. As of 2016, several targeted therapies, lapatinib, pertuzumab, neratinib and trastuzumab have been approved for HER2+ BC patients exhibiting improved overall and progression free survival [[1], [2], [3]]. A newly developed antibody-drug conjugate trastuzumab emtansine (T-DM1) has further improved survival of HER2+ metastatic BC patients [4]. Despite all these advances and impressive clinical results, occasional drug resistance, non-responding behavior of a group of patient, disease progression and recurrence remained challenges for disease management [5]. Multiple mechanisms have been proposed for T-DM1 mediated drug resistance [[6], [7], [8]], and an alternative theory that has emerged recently is the existence of a subpopulation of cells that display stem-like properties including resistance to drugs, termed as cancer stem cells (CSCs) [9]. Regrowth of these therapy resistant CSCs results in recurrence of the disease despite administration of targeted therapy followed by chemotherapies and aggressive surgical measures [10].
ROR1 (Type 1 receptor tyrosine kinase-like orphan receptor), expressed during embryogenesis and a potential CSC marker, has been implicated in many cancers [[11], [12], [13]]. ROR1 expression is highly restricted to undifferentiated tumors which assists tumors to resist and metastasize with further enrichment of epithelial-to-mesenchymal transition (EMT) features [11,14]. Zhang et al. [15] reported that ROR1 is expressed in human BC and is associated with poor prognosis. Therefore, targeting ROR1 may affect the CSC population and could be a potential target for CSC rich BC tumors in patients.
The Hippo pathway regulates gene expression alterations and plays roles in organ size determination and cell proliferation [16]. Transcriptional co-activator YAP1(Yes associated protein-1) the downstream effector of the Hippo pathway, has emerged as a regulator of cell proliferation, cancer development, progression and invasion, regulator of EMT, cancer and adult stem cells, and correlates with poor overall outcomes [16,17]. A recent study reported the mechanistic regulation of ROR1/HER3/IncRNA signaling axis in modulating the Hippo-YAP pathway [18]. YAP1 mediated EGFR overexpression and chemoresistance have recently been described in esophageal cancer [19].
Despite the studies reported above, the mechanisms of T-DM1 mediated ROR1 overexpression, CSC regulation and therapeutic resistance in HER2+ BCs are not fully understood. In this study, using fresh clinical tumor samples and BC cell lines, we describe that inhibition of HER2-amplified BC by T-DM1 treatment increased the expression of ROR1, the survival of the CSC population and treatment resistance. In HER2+ BC patients treated by T-DM1, tumors switch from low ROR1 expression to increased surface expression of ROR1 and show increased enrichment of CSCs promoting the resistance to T-DM1. We provide further evidence that the transcriptional co-activator YAP1 regulates ROR1 overexpression, and disruption of YAP1-TEAD binding limits the T-DM1 treatment-induced ROR1 overexpression and CSC self-renewal. These findings suggest an alternative therapeutic strategy for HER2+ BC patients.
2. Materials and methods
2.1. HER2+ breast cancer patients sample collection and cell isolation
This study was approved by the Institutional Review Board of the Holy Family Red Crescent Medical College and Hospital, Dhaka, Bangladesh. After obtaining patients informed consent and following local and international regulations, breast tumors were obtained from all consented BC patients at the time of surgery. Collected tumors were first minced and enzymatically dissociated with 2 mg/mL of dispase (Roche, USA), and then incubated with 0.25% Trypsin-EDTA, passed through a 21-gauge syringe and filtered through a 23 μm cell filter (Merck Millipore). Cells were either directly cultured in supplemented CSC medium or cryopreserved in 80% fetal bovine serum (FBS) and 20% dimethylsulfoxide (DMSO) until further use.
2.2. Cells and reagents
In addition to breast cancer patients' clinical samples, we used BT-474, MDA-MB-361, HCC1954 cell lines, all purchased from ATCC and maintained as described [7]. MCF-7 stably expressing HER2 (designed as MCF7-HER2+) was a kind gift from Dr. Mien-Chie Hung, The University of Texas MD Anderson Cancer Centre (Houston, Texas). Human ROR1 (AF2000, R&D System, USA), HER2 (MAB1129, R & D System, USA) and YAP1(MAB8094, R & D System, USA) antibodies were purchased from R & D System, USA. ROR1-APC conjugated antibody was purchased from Miltenyi Biotech, USA. Mouse anti-human ROR1 was purchased from BD Bioscience (cat#564464). Verteporfin and doxycycline hyclate were purchased from Sigma-Aldrich. Doxycycline inducible YAP1 lentiviral expression plasmid (PN20YAP1) [16] and lentiviral shRNA plasmid for knockdown YAP1 were previously described [16]. T-DM1 was produced by Genentech, USA and purchased through a study participating hospital.
2.3. Western blotting
Cell lysates were prepared directly in sample buffer. The primary antibodies include: human ROR1, YAP1, HER2 and GAPDH. Enhanced chemiluminescence detection kit (Thermo Fisher) was used for signal detection.
2.4. Immunostaining
ROR1 antibody was validated and tested for the specificity using immunostaining methods. Cell were cultured on cover slips, fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X for 15 min followed by 30 min blocking with 4% bovine serum albumin (BSA, Sigma). Cells were incubated with ROR1, YAP1 and HER2 primary antibody overnight at 40C, washed in PBS and incubated with secondary antibodies. Slides were mounted in Vectashield with DAPI (Vector) and images captured using the Optima fluorescence microscope. Digital images were processed using Adobe photoshop CS2.
2.5. CTGF ELISA
The human CTGF levels in the blood serum were quantitatively determined in triplicate by OmniKine ELISA kit (Assay Biotechnology).
2.6. Silencing ROR1
Silencing of ROR1 was performed by targeting the sequences of shRNA-1(5ʹ-TCCGGATTGGAATTCCCATG-3ʹ) and shRNA-2(5ʹ-CTTTACTAGGAGACGCCAATA-3ʹ). Briefly, the 293 T-FT packaging cells were transfected with either shROR1-1or shROR1–2, viral particles collected after 48 h post-transfection, and cell supernatants collected and filtered, centrifuged at 43,000 ×g and sub-confluent HCC1954 or MCF-7(HER2+ induced) cell lines transduced. Cells were selected in media containing 2 μg/mL puromycin.
2.7. Flow cytometry
Actively growing 1 × 106 patient's tumor cells or HCC1954 cells were either left untreated or treated with DMSO (control) and T-DM1 for 5 days, trypsinized, pelleted and washed once with phosphate buffered saline (PBS). Cells were resuspended in PBS wash buffer containing 2% fetal bovine serum (FBS). Cells were incubated with ROR1-APC conjugated antibody for 45 min at room temperature, washed 3 times with wash buffer, resuspended in PBS and immediately data were captured with FACSAria software and analyzed with BD FacsDIVA.
2.8. FACS cell sorting and analysis
Patient derived tumor cells, HCC1954 and MCF7-HER2+ cells were stained on ice and then isolated by FACS using BD FACSAria flow cytometer and analyzed by BD FacsDIVA. For isolation of ROR1 cell subpopulations, cells were sequentially stained with ROR1-APC conjugated antibody. Isolated ROR1+ and ROR− cells were washed and cultured in 3D in vitro sphere culture supplemented CSC medium. Cells were treated with either vehicle or T-DM1 when necessary. In addition to ROR1, samples were also stained for CD44 and ALDH.
2.9. Cell proliferation assay
Treatment naïve HER2+ breast primary tumor cells, HCC1954-HER2+ and MCF7-HER2 cells were cultured in 96-well plates and treated with increasing concentrations of T-DM1for 5 days. AlamarBlue reagent was added to the cells, incubated for 4 h and plates were read in a BioRad spectrophotometer and survival of cells was calculated.
2.10. Sphere formation assay
Sphere forming efficiency was performed according to a previously published protocol with some modification. Briefly, cells were cultured in 6-well ultra-low attachment plates at a density of 500 viable cells/well. The culture medium for sphere formation consisted of DMEM/F12 (1:1, Gibco) medium supplemented with 0.4% BSA (Sigma), 1% penicillin and streptomycin, B27, 20 ng/ml hEGF (Sigma), 5 μg/ml insulin (Sigma), 20 ng/ml FGF (Sigma), 50 ng/ml hydrocortisone (Sigma) and 4 μg/ml heparin (Sigma). To examine the effects of ROR1 knockdown, cells were plated into 60-mm plates and cells transfected with non-targeting shRNA or ROR1-shRNA. After transfection, cells were collected and re-cultured, treated with vehicle or T-DM1 for 5 days. Cells were trypsinized after 5 days and 1 × 105 single cell suspension were used for the sphere forming assay. The spent medium was changed every 72 h and number and size of spheres (>50um) were photographed, evaluated and counted every 3 days. To assess the effects on secondary and tertiary sphere formation, spheres were digested with 0.25% Trypsin into a single cell suspension and were then re-plated in ultra-low attachment plates. Percentage of sphere forming efficiency was calculated as [number of actual spheres/number of cells plated x100].
2.11. Computational analysis of TCGA RNA-Seq datasets
We downloaded the breast cancer RNA-Seq datasets (Illumina HiSeq 2000) from The Cancer Genomics Atlas (TCGA), processed and analyzed using R statistical softwire package “ggplot2”.
2.12. Quantitative real time PCR (qRT-PCR)
RNA was isolated using Qiagen RNA isolation kit (Qiagen). Real-time-PCR was performed using QuantStudio 5 System. Primers list are given below.
2.13. Extreme limiting dilution assay (ELDA assay)
Sorted ROR1+ and ROR1− cells were diluted into ultra-low attachment 6-well plates with 1 ml of DMEM/F12 (1:1) per well, supplemented with 2% B27 supplements (Invitrogen), 20 ng/ml of epidermal growth factor (EGF, Sigma), 20 ng/ml of basic fibroblast growth factor (bFGF, Sigma), 2μg/ml hydrocortisone (Sigma), 2μg/ml insulin (Sigma), 2μg/ml heparin (Stem Cell Technologies) for 20 days. Spheres were monitored every day and counted under a phase contrast microscope. Data was analyzed using Extreme Limited Dilution Analysis (ELDA) by “R- statistical software and STATMOD packages (www.https://cran-r.project.org/web/packages/statmod” to determine the stem cell frequencies.
2.14. ALDEFLUOR assay
For Aldefluor assay, the experiments were performed using aldehyde dehydrogenase-based cell detection assay kit (Stem cell technologies, Vancouver, BC, Canada) following manufacturer's instructions. Briefly, 1 × 106 cells/ml were suspended in Aldefluor assay buffer containing ALDH substrate (Bodipy-aminoacetaldehyde) and incubated for 45 min at 37 °C. As control, cells were suspended in buffer containing Aldefluor substrate in the presence of specific ALDH enzyme inhibitor diethylaminobenzaldehyde (DEAB). Desired ALDH-positive cells populations were isolated using the FACSAria flow cytometer (BD Bioscience, San Diego, USA) and the data were analyzed by the FACS DIVA software (BD Bioscience). After sorting, cells were washed twice with sterile PBS and were centrifuged for 5 min before each wash cycle.
2.15. In vivo tumor growth and limiting dilution assay
The protocol for animal study was approved by the King Faisal Specialist Hospital and Research Centre (KFSH & RC) institutional Animal Care and Use Committee. HER2-positive HCC1954 cells were either treated with vehicle (control) or T-DM1 for 5 days, trypsinized, washed in PBS and sorted by flow cytometry based on the surface ROR1 expression. ROR1− and ROR1+ or unfractionated cells were injected into the subcutaneous site of 6–8-week old Nu/J mice (n = 3/group). Tumor development was determined after 8 weeks. For limiting dilution of FACS sorted ROR1+ cells, cells were injected at a dilution of 10-fold [102, 103, 104, 105] (n = 3/group) and grown for 8 months. At the end of 8-weeks period, numbers of mice developing tumors from each group was determined and tumor initiating capacity was calculated by using Extreme Limiting Dilution Analysis (ELDA) [R-statistical packages “statmod”].
2.16. Statistics
Experiments were performed in triplicate and results were presented as mean +/− s.e.m. for comparison between mean or mean +/− s. d. and the median results as +/− IQR (Inter quartile range) for population comparisons. For independent data involving two specimens, a two-tailed t-test for equal variance, or one-way ANOVA Tukey post hoc comparison for three or more groups was applied. In all statistical analysis we used “R” Statistical software (version 3.2.1) and for graphs “ggplot2’ package in “R”. ELDA (extreme limiting dilution assay) was analyzed used to calculate the CSC frequency by using “statmod” package in “R” for in vivo limiting dilution study. Kaplan-Meier survival curves were generated and analyzed for overall survival and progression free survival using R package “survival” and “survminer 0.3.0”. The significance was calculated using log-rank and Mantel-Cox test.
3. Results
3.1. Overexpression of ROR1/YAP1 in BC is associated with poor overall survival
Given the precedent for determining their roles in different cancers we first analyzed the amplification and mutation status of ROR1 and YAP1 genes from 12 distinct cancers sequenced by TCGA. In breast cancer patients ROR1(6%) and YAP1(7%) loci are amplified (Fig. 1a, Fig. S1a; b). We then investigated the genomic alterations of ROR1 and YAP1 in BC from TCGA by using the recently developed mutation significance method (MutSigCV). MutSigCV provides a statistical metric to identify driver candidates in cancer with respect to gene nucleotide length and the background mutation rate of each cancer analyzed [20,21]. This analysis revealed that, ROR1 and YAP1 were significantly amplified as seen in their copy number alterations (CNAs) analyzed by GISTIC2.0 methods (Fig. 1b; Fig. S1c). Furthermore, our analysis of the TCGA BC cohort indicated that YAP1 mRNA (normalized z-scores) overexpression correlated with the upregulation of ROR1 (Fig. 1c). We therefore hypothesized that YAP1 and ROR1 mutually interact with each other to regulate CSC maintenance and drug resistance.
Fig. 1.

Overexpression of ROR1/YAP1 in breast cancer is associated with poor overall survival outcomes. (a) Graphical representation of cancer types in which ROR1 is most frequently altered in the TCGA dataset. (b) ROR1 and YAP1 mutation analysis of 960 breast cancer samples in the TCGA dataset. (c) Significant co-occurrences of ROR1 and YAP1 in the TCGA dataset. (d) Boxplot comparing ROR1 and YAP1 expression in ER + PR+/HER2-; ER + PR+/HER2+; ER-PR-/HER2+; subtypes samples in the TCGA dataset (one-way ANOVA). (e) Immunohistochemical detection of ROR1 and YAP1 in disease free, ER + PR+/HER2-; ER + PR+/HER2+; ER-PR-/HER2+; breast cancer subtypes from our own data cohort (one-way ANOVA). The boxes show the median ±1 quartile, with whiskers extending to the most extreme data point within 1.5 interquartile ranges from the box boundaries. (f) Survival analysis of ROR1 and YAP1 high and low expressing breast cancer patients based on expression intensity (Log-rank test; p < .0001, scale bar: 50 μM). (g) Bar graph showing the ROR1 and YAP1 expression intensity (%) in treatment naïve, sensitive, partially sensitive and resistant patient tumors (**p < .0, two-tailed Student's t-test).
Supplementary Fig. S1.

of Fig. 1: Alteration of YAP1 and ROR1 in TCGA dataset: (a) YAP1 genomic alteration in 12 different cancer types from TCGA dataset. (b) ROR1 and YAP1 alteration (each column represents an individual samples) in TCGA dataset. (c) Alteration of genes in TCGA breast cancer dataset.
Accordingly, ROR1 and YAP1 expressions were significantly upregulated in ER+/PR+ or in HER2+ subtypes in the TCGAs BC dataset as analyzed from normalized z-scores (Fig. 1d). Further confirmation was obtained by immunohistochemical (IHC) staining of BC tissues in our own samples. The results identified that ROR1 and YAP1 expressions were significantly upregulated in HER2+ patients compared to disease free individuals, correlating with poor overall survival (OS) (Fig. 1e, f). These results suggest the possible role of ROR1 and YAP1 in promoting tumor progression and therapeutic resistance with impact on the OS of BC patients.
To evaluate if both ROR1 and the YAP1 overexpression may contribute to therapy resistance, we analyzed ROR1 and YAP1 expressions in freshly resected patient's breast tumor samples. Surprisingly, most therapy resistant tumors expressed high levels of ROR1 and YAP1 compared to partially therapy sensitive and complete sensitive tumors. >80% of tumors overexpressed both ROR1 and YAP1 in the therapy resistant tumor group, while partially sensitive and sensitive tumors expressed 60% and 30% ROR1 and YAP1, respectively (Fig. 1g). These results suggest that overexpression of ROR1 and YAP1 is likely associated with the resistance to therapies.
3.2. Treatment resistant BC cells overexpress ROR1 and have a higher sphere forming efficiency
Targeted therapy reduces the risk of recurrence and increases progression free survival (PFS) in BC patient [22]. Despite these improvements, studies have reported drug resistance against targeted therapies in a subset of cancer cells known as CSCs [23]. Most CSC studies utilized cell lines that may not correctly define the biology of breast CSCs. We thus asked whether the resistance to anti-HER2 targeted therapy could be due to increased ROR1 overexpression. To examine this, we first compared the histologic expression of ROR1-positive cells in chemo-naïve (n = 6) and chemo-resistant (n = 6) HER2+ patient tumors. These patients were treated with neoadjuvant chemotherapy (combination of docetaxel and/or cyclosphoramide). We found a 3-fold increase in ROR1-positive cells in the chemo-resistant group (Fig. 2a; 21.5% vs 60.2%, p < 3.02e-02) compared to their chemo-naïve counterparts (Fig. 2a; 45.5% vs 90.25%; p < 1.628e-9).
Fig. 2.

Treatment resistant BC cells overexpress ROR1 have a higher sphere forming efficiency: (a) Administration of therapy leads to the increase of ROR1 expression intensity in HER2+ treatment naïve and treatment exposed BC patient (p = 3.021e-02. unpaired two-sided t-test). (b) Tumor cells were dissociated from treatment naïve, sensitive and resistant HER2+ BC patients and equal numbers of cells were cultured in the supplemented CSC medium and stained for ROR1 expression in the spheres. Matched HER2+ BC patient's tissues were stained for ROR1 expression by immunofluorescence (Scale bar: 50 μM). (c) FACS analysis of ROR1+ and ROR1− population in treatment naïve, sensitive and resistant cells from primary HER2+ BC patient tumors. (d) Equal number of sorted ROR1+ and ROR1− cells from primary BC patients (n = 2) were cultured in the supplemented CSC medium and analyzed for sphere forming efficiency (p < .0002, paired two-sided t-test). (e) Comparison of epithelial-to-mesenchymal transition related genes showing mRNA expression from treatment naïve, sensitive and resistant ROR1+ and ROR1− primary HER2+ BC patients cell fractions (results are mean fold change +/− SE, n = 3). (f) Comparison of ROR1 mRNA expression in primary HER2+ treatment naïve, sensitive and resistant BC patient tumors (results are mean fold change +/− SE, n = 3). (g) In vitro treatment of HER2+ primary tumors cells treated with T-DM1 (5 nM) and assessed for sphere forming efficiency (black arrowhead). Below is shown immunoblotting of ROR1 from each group of patients' protein samples (results are mean +/− SE, n = 3 run in triplicate; Scale bar: 100 μM).
To further analyze this connection and determine the status in the tumor initiating subpopulation, we isolated cells from chemo-naïve, chemo-sensitive, and chemo-resistant patients' tumors and cultured them in a supplemented CSC medium. We compared the ROR1 expression in the resulting spheres and matched tumor tissues by immunostaining. Comparatively, larger spheres were formed in cells isolated from chemo-resistant patient tumors with an increased expression of cell surface ROR1 in both chemo-resistant spheres and matched tumor tissues (Fig. 2b), suggesting a role of ROR1 in cell self-renewal capability. To examine this further, we sorted ROR1+ and ROR1− cells and analyzed their sphere forming efficiency (a surrogate assay for CSC survival) [24]. Compared to ROR1− cells, ROR1+ cells showed a high self-renewal capability and sphere forming efficiency (Fig. 2c, Table 1; Fig. 2d). These results suggest that treatment resistant patient tumors positive for ROR1 are highly self-renewal efficient and indicative of a CSC phenotype.
Table 1.
Primers list.
| Genes | Forward | Reverse |
|---|---|---|
| Bmi1 | CCAGGGCTTTTCAAAAATGA | CCGATCCAATCTGTTCTGGT |
| Nanog | TTCCTTCCTCCATGGATCTG | TCTGCTGGAGGCTGAGGTAT |
| Oct3/4 | GTACTCCTCGGTCCCTTTCC | CAAAAACCCTGGCACAAACT |
| Sox2 | ACACCAATCCCATCCACACT | GCAAACTTCCTGCAAAGCTC |
| YAP1 | GCAGTTGGGAGCTGTTTCTC | GCCATGTTGTTGTCTGATCG |
| TEAD1 | GGAAGCCTCAAACTGAGACG | GGGCTGGAACATTCTTTGAA |
| CTGF/CCN2 | GGAAAAGATTCCCACCCAAT | TGCTCCTAAAGCCACACCTT |
| CCND1 | GAGGAAGAGGAGGAGGAGGA | GAGATGGAAGGGGGAAAGAG |
A recent study indicated that ROR1 may regulate EMT and metastasis in TNBC tumors [13]. Furthermore, trastuzumab resistance in HER2+ PTEN- BC patient induces epithelial-to-mesenchymal transition (EMT) and subtype switching thereby increasing the frequency of CSCs and metastasis which require unique treatment options [25]. To explore the EMT mechanism orchestrated by ROR1+ cells, mRNA expressions of sorted ROR1+ and ROR1− cells from chemo-naïve (n = 3), sensitive (n = 3) and resistant (n = 3) HER2+ BCs were compared. Analysis of clinical samples produced differential expression of several EMT related genes; Snail1, E-cadherin, N-cadherin and ZO-1 (Fig. 2e). We then confirmed the corresponding protein expression in the isolated tumors from the same patients. Higher levels of Snail1 and N-cadherin were found in the treatment sensitive and resistant tumors, while E-cadherin was higher only in the treatment naive and resistant tumors (Fig. S2a). Interestingly, an unusual expression of EMT marker E-cadherin was observed in our study. E-cadherin was expected to be significantly downregulated in the resistant tumors, while maintaining upregulation in the treatment sensitive or naïve tumors. The explanation of this unusual finding is very difficult to explain currently, however, we suggest that this may be due to the transitioning of tumor cells from a state of MET (mesenchymal to epithelial transition).
Supplementary Fig. S2.
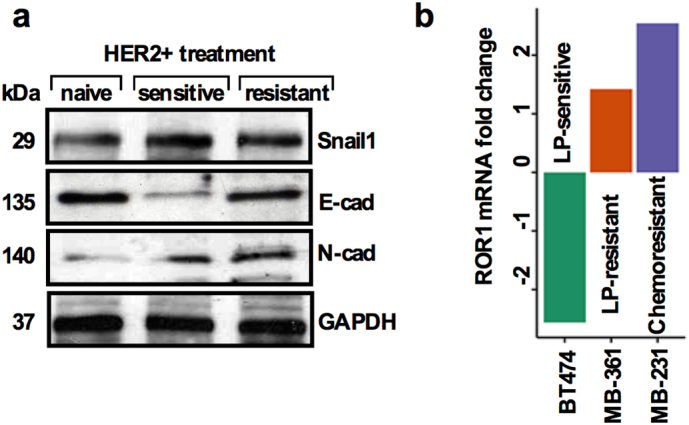
of Fig. 2: Treatment resistant BC cells show higher epithelial-to-mesenchymal transition (EMT) related proteins and overexpress ROR1 mRNA. (a) Immunoblots showing expression of EMT related markers from treatment naïve, sensitive and resistant patient's tumors. (b) Comparison of ROR1 mRNA expression in lapatinib sensitive (BT-474), lapatinib resistant (MB-361) and chemo resistant (MB-231) cells lines.
Given these findings, we postulated that ROR1+ cells might mediate drug resistance in HER2+ BC. To explore this aspect, we utilized three BC cell lines, including two HER2+ lines, BT- 474 (Lapatinib sensitive), and MDA-MB-361 (Lapatinib resistant) and assessed their ROR1 mRNA expression. Interestingly, the ROR1 mRNA level was acutely lower in BT-474 cells, while it increased in MDA-MB-361 cells (Fig. S2b). We then confirmed these results in a set of HER2+ treatment naïve (n = 3), sensitive (n = 3) and resistant (n = 3) BC patients tumor cells. Consistent with the above observation, in the treatment naïve tumors ROR1 mRNA expression was low. In contrast, treatment sensitive and treatment resistant tumors expressed moderate to higher levels of ROR1 mRNA (Fig. 2f), suggesting that ROR1 expression tended to increase when tumor cells become resistant to treatment. To directly test whether T-DM1-treatment induced ROR1 overexpression, self-renewal and survival, cells were dissociated from treatment naïve HER2+ patients' tumors, cells were first treated with T-DM1 (5 nM) for 2 days, washed with growth medium and then retreated again for remaining 3 days (total treatment time 5 days), then trypsinized and cultured in growth factors supplemented CSC medium for 10 days. T-DM1 treatment increased the sphere growth efficiency in treatment naive HER2-overexpressing cells (Fig. 2g). These results corroborate those obtained in Fig. 2a, suggesting that T-DM1 treatment induces ROR1 expression and is possibly the driving force for the resistance in treatment of HER2+ patients. Accordingly, protein extracted from spheres was subjected to ROR1 immunoblot and revealed higher expression of ROR1 in the T-DM1 treated spheres (Fig. 2g, bottom).
3.3. T-DM1 treatment alters ROR1 expression without significant changes in HER2 expression
One of the many reported resistance mechanisms for anti-HER2 therapy is the loss of HER2 expression [24,25], PIK3CA mutation [26,27] and loss of PTEN [25] and the Notch pathway [28]. Given our findings from clinical samples, T-DM1 treatment led to a graded increase in ROR1 levels (Fig. 2g). Therefore, it was essential to explore whether T-DM1 treatment could lead to an opposite reduction in HER2 expression. We therefore analyzed patient tumor samples (treated with combination of docetaxel and/or cyclosphoramide) from treatment naïve (n = 10), sensitive (n = 10) and treatment exposed (n = 10) patients and immunostained tissues with anti-HER2 antibody to detect HER2 expression. No significant changes in cell surface HER2 expression were observed in the treatment naïve and sensitive and resistant samples (Fig. 3a). Consequently, we then investigated whether T-DM1 treatment caused cells to retain HER2 expression. To investigate this, we used HCC1954 and MCF-7-HER2+ cell lines and treated cells with either vehicle or T-DM1 (5 nM) and examined HER2 expression in these cells by immunofluorescence. Cell surface HER2+ expression remained unchanged in both vehicle and T-DM1 treated cell (Fig. 3b) indicating no loss in HER2 expression after T-DM1 treatment.
Fig. 3.

T-DM1 treatment alters ROR1 expression without significant changes in HER2 expression: (a) Immunostaining of HER2 expression from treatment naïve (n = 5), sensitive (n = 5) and resistant (n = 5) HER2+ BC patient's tumor samples (Scale bar: 50 μM). (b) HCC1954 and MCF-7-HER2+ cells were cultured on a glass cover slip in 24-well plates and treated either with vehicle (DMSO) or 5 nM of T-DM1 for 5 days. Cells were fixed and immunofluorescence stained images captured for HER2 (red) and nucleus DAPI (blue) (Scale bar: 50 μM). (c) HCC1954 and MCF-7-HER2+ cells were cultured, treated and fixed as (b), and immunofluorescently stained for ROR1 (red) and nuclear DAPI (blue) expression (Scale bar: 50 μM). (d) Treatment naïve BC patient tumor cells, (e) HCC1954(HER2+), and (f) MCF-7-HER2+ cells were treated with T-DM1 (5 nM) for 5 days, trypsinized and cultured in supplemented CSC medium in sphere culture for 10 days (10 days is denoted as first generation). After 10 days, spheres were dissociated and re-cultured in CSC medium for an additional 10 days (20 days denoted as second generation) followed by dissociation of spheres and then re-cultured again in CSC medium (30 days denoted as third generation). In every 10-day culture cycle, sphere forming efficiency was examined in ROR1+ and ROR1− populations by counting the number and size of spheres. (Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = ns [not significant]; p = .05; p = .01, p = .001; one-way ANOVA). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We then tested whether ROR1 is overexpressed due to T-DM1 treatment. We treated HCC1954 and MCF-7-HER2+ cells either with vehicle or 5 nM T-DM1 (treatment time 5 days in a wash and retreat cycle) and found that both cell lines treated with T-DM1 showed an increase in ROR1 expression intensity compared to vehicle treated cells (Fig. 3c), confirming that only T-DM1 treatment induced ROR1 overexpression in these cells. To explore the sustained sphere forming efficiency of ROR1+ and ROR1− cells, treatment naïve HER2+ patients tumor cells as well as HCC1954 and MCF-7 HER2+ were treated with either vehicle or T-DM1 and sorted for ROR1+ and ROR1− cells based on cell surface ROR1 expression. We found that the ROR1+ cells sustained sphere growth efficiency at least for three generations (30 days in sphere culture), suggesting that ROR1+ cells become more resistant to T-DM1 over time (Fig. 3d, e, f), while sphere forming efficiency fells acutely in the ROR1− cells. Thus, these results confirmed that T-DM1 increased the content of surface ROR1 from low to high levels and induced long term survival and self-renewal of spheres.
3.4. T-DM1 treatment-induced ROR1+ cells show higher self-renewal efficiency and increased resistance with enriched CSC features
Thus far, we have found that T-DM1 treatment resulted in an increase in cell surface ROR1 and sphere growth efficiency (Fig. 3c, d). This led us to posit that ROR1+ cells might behave like CSCs. To test this idea, we first determined the stemness frequency of ROR1 subpopulations by using extreme limiting dilution assay (ELDA). Freshly dissociated cells from HER2+ treatment naïve tumor cells were either treated with vehicle or T-DM1 for 5 days, FACS sorted for ROR1+ and ROR1− subpopulations based on surface ROR1 expression, and then grown in the supplemented sphere culture medium for 10 days (experimental workflow Fig. 4a). The self-renewal efficiency of ROR1+ and ROR1− cells clearly indicated the differences in the stemness frequencies (Fig. 4a). Stemness frequency was 1:1.51 vs 4.21 (confidence interval: 0.95–2.04 for ROR1+ cells vs 1.97–6.41 ROR1− cells) in ROR1+ cells (Table 1). We then tested the sphere forming efficiency of T-DM1 treated unsorted bulk tumor cells and compared the results with vehicle treated cells. Unsorted bulk cells treated with T-DM1 showed higher sphere forming efficiency than vehicle counterparts (Fig. 4b). To identify if only ROR1+ cells are capable of superior sphere forming efficiency, cells were either treated with vehicle or T-DM1 for 5 days and FACS sorted based on surface ROR1 expression and cells allowed to grow in CSC medium. Vehicle treated ROR1+ cells were efficient in forming spheres, whereas ROR1− cells showed low efficiency of sphere formation (Fig. 4c). Furthermore, sphere forming efficiency was markedly increased by T-DM1 treatment-induced ROR1+ cells (Fig. 4c). In contrast, we also found that ROR1− cells failed to form spheres even after T-DM1 treatment (Fig. 4c). To confirm this result, we extended this analysis to HCC1954 and MCF-7-HER2+ cell lines. Cells were treated, sorted and cultured as describe above, and sphere forming efficiency was assessed. In vehicle treated HCC1954 cells, both ROR1+ and ROR1− cells showed significantly low efficiency of sphere formation, while T-DM1 treated ROR1+ cells were found to have higher sphere forming efficiency (Fig. 4d), with moderate sphere forming efficiency in T-DM1 treated ROR1− cells. Moreover, unsorted bulk cells showed similar results as found in patients tumor cells (Fig. 4b; d). Using a similar experimental approach identical results were achieved with the MCF-7-HER2+ cells (Fig. S3a). Taken together these data suggest a profound sphere forming efficiency by ROR1+ cells which could underlie therapeutic resistance.
Fig. 4.

T-DM1 treatment-induced ROR1+ cells enriched with CSC features show higher self-renewal efficiency and increased resistance with enriched CSC features. (a) Treatment naïve HER2+ BC patients tumor cells were enzymatically dissociated and cultured in supplemented CSC medium, treated with either vehicle (DMSO) and 5 nM T-DM1 and incubated for 10 days (follow the schematic diagram). Spheres were dissociated for single cells and subjected to FACS sorting for ROR1. Extreme limiting dilution assay (ELDA) was performed by plating sorted cells with increasing cell numbers and analyzed for stemness frequencies by ‘R' statistical package “statmod” (p < .0427; one-way ANOVA). Table 1 (right) showed the confidence interval for 1/stemness frequency. (b) Bulk cells were isolated and treated as described in (a) and analyzed for sphere forming efficiency (Bar graph represents the mean +/− SE, n = 3, run in triplicate). (c) HER2+ patient tumor cells treated with either vehicle or T-DM1 for 5 days, harvested and FACS sorted for ROR1+ and ROR1− populations, and then sorted cells were cultured in supplemented CSC medium [follow the schematic diagram] for 10 days and analyzed for the sphere forming efficiency from each cell population (black arrowhead; p = .001 for T-DM1 treatment; p = .05 for vehicle, unpaired two-sided t-test, Scale bar: 100 μM). (d) HCC1954 cells were treated and analyzed as described in (c) [black arrowhead; p = .01 for T-DM1 treatment; p = ns (not significant) for vehicle, unpaired two-sided t-test; Scale bar: 100 μM]. (e) HCC1954 cells were either treated with vehicle or T-DM1 and sorted as described in (c) and quantitatively analyzed for Bmi1, Nanog, Oct3/4. Sox2 mRNA in ROR1+ and ROR1− populations. (Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = .05; p = .01; p = .001; one-way ANOVA). (f) ROR1+ cells were more significantly resistant than ROR1− cells at individual T-DM1 treatment and showed higher EC50 in response to T-DM1 as compared to ROR1− cells. (Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = .05, p = .01).
Supplementary Fig. S3.

of Fig. 4: ROR1+ cells are resistant to T-DM1. (a) MCF-7-HER2+ cells were cultured in CSC medium and treated with either vehicle (DMSO) and/or T-DM1 for 5 days, harvested and FACS sorted for ROR1+ and ROR1− populations. Sorted cells were re-cultured in supplemented CSC medium for 10 days and analyzed for the sphere growth efficiency from each subpopulation [p = .01 for T-DM1 treatment; p = ns (not significant) for vehicle, unpaired two-sided t-test; Scale bar: 100 μM]. (b) Patients tumor cells, (c) HCC1954 and (d) MCF-7-HER2+ cells were sorted for ROR1, treated with increasing concentrations of T-DM1 for 5 days and cell survival assessed by WST-1 assay (Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = .05).
Bmi1 is implicated in CSC self-renewal through regulation of survival genes important for cell cycle control and stem cell fate determination [29,30]. We then tested if T-DM1 treatment could enhance Bmi1 expression together with stemness-regulated genes. T-DM1 treatment-induced ROR1+ cells showed an increased mRNA expression of the self-renewal marker Bmi1, together with stemness-regulated genes Nanog, Oct3/4 and Sox2, compared to ROR1− counterpart (Fig. 4e). To further characterize the ROR1+ cells and their degree of resistance, we treated both ROR1+ and ROR1− subpopulations with increasing concentrations of T-DM1. ROR1+ cells tend to become highly resistant to T-DM1 as compared to ROR1− cells (Fig. S3b–d), with a sharp increase in EC50 in the ROR1+ cells (Fig. 4f), suggesting that only ROR1+ cells resistant to T-DM1 escape from treatment and survive longer, whereas, ROR1− cells die due to T-DM1 toxicity.
3.5. T-DM1 treatment induced ROR1+ cells are enriched with expression of CD44, ALDH and YAP1 target genes
As previously reported, a group of HER2+ patients become resistant to therapies due to the presence of a CSC (CD44 and ALDH positive cells) population [31,32]. To further investigate the proportion of CSCs (CD44 and ALDH-positive cells), we first determined the proportion of the ALDH and CD44 positive cells within the ROR1+ subpopulation by FACS sorting from a pool of treatment naïve (n = 5) and treatment resistant (n = 5) HER2+ primary patient tumors. We were able to detect a comparably higher percentage of both ALDH+ and CD44+ cell fractions in the chemoresistant ROR1+ subpopulation than in the chemo-naïve ROR1− counterpart (Fig. 5a; b). We next sought to confirm if T-DM1 treatment enhances the fraction of CD44 and ALDH positive cells in a chemo-naïve tumor cell population. Accordingly, cells were treated with either vehicle or T-DM1 for 5 days and then sorted on the basis of cell surface ROR1 expression. T-DM1 treatment-induced ROR1 overexpressing cells had significantly higher percentage (81.5%) ALDH positivity as compared to the vehicle treated ROR1− counterpart (10.4%) (Fig. 5c). As supporting data, immunofluorescence staining confirmed that only cells positive for ALDH expressed ROR1 while a trace amount of ROR1 positivity was found in the ALDH-negative cells (Fig. 5c). Similarly, we found that CD44+ cells positive for ROR1 were significantly increased (71.07%) as compared to ROR1− cells (41.12%) (Fig. 5d). We then confirmed these results using the HCC1954 cell line and achieved nearly identical results as seen with treatment naïve patient tumor cells (Figs. 5e; 5f). Furthermore, expression of transcripts of YAP1 and its target genes TEAD1, CTGF and CCND1 were significantly increased in T-DM1 treated ROR1+ cells (Fig. 5g). Taken together, these results suggest that, T-DM1 treatment induces a significant increase in the ALDH and CD44 positive fractions coincident with increased expression of YAP1 and its target genes. It had been shown that YAP1-regulated CTGF plays a vital role in promoting osteolysis during bone metastasis [17]. To test whether the CTGF level in blood serum show any substantial difference between treatment naive and treatment exposed patients, an ELISA assay was performed to measure the CTGF level in the blood serum. We found an elevated CTGF level in the treatment exposed patients' blood serum compared to treatment naive patients (Fig. 5h).
Fig. 5.

T-DM1 treatment induced ROR1+ cells are enriched for CD44, ALDH and YAP1 target genes: (a-b) Freshly dissociated treatment naïve and treatment resistant HER2+ BC patients tumor cells were FACS sorted and analyzed for ROR1 subpopulation stained with ALDH and CD44 antibodies (Results are median +/− interquartile range (IQR) n = 5; unpaired two-sided t-test). (c-d. Patients tumor cells were either treated with vehicle (DMSO) and/or T-DM1 for 5 days, and sorted as (a) Analysis of ALDH and CD44 expression by FACS and immunofluorescence staining in ROR1+ and ROR1− subpopulation (Scale bar: 50 μM). (e) HCC1954 cells treated, maintained, sorted and analyzed as (d). (g) Cells were treated and sorted as (d), extracted total RNA followed by qRT-PCR for YAP1, TEAD1, CTGF and CCND1 in ROR1+ and ROR1− populations (Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = .001, 0.01, 0.05, 0.001 and 0.001). (h) Detection of CTGF from treatment naïve and treatment exposed patients' blood serum by ELISA assay (Bar graph represents the mean +/− SE, n = 3; run in triplicate; **p < .01; Students t-test).
3.6. Silencing ROR1 reduces T-DM1 treatment-induced ROR1+ spheroid growth and capacity for tumor initiation
Prior studies investigated the mechanisms of T-DM1 mediated resistance in a group of HER2 overexpressing BC patient [6,7]. A recently developed ROR1 inhibitor, cirmtuzumab, has shown encouraging results in inhibiting ROR1 enriched in breast and ovarian cancer CSCs [8,14] and is now in phase-I and -II studies. Since cirmtuzumab is currently not commercially available, we used ROR1-specific shRNA (Fig. S4a) to knockdown ROR1 expression in T-DM1 treated HCC1954 and MCF-7-HER2+ cells and assessed their sphere growth efficiency. Specificity of a set of ROR1-shRNA was tested and ROR1-shRNA efficiently reduced the ROR1 expression (Fig. S4a). ROR1-shRNA significantly inhibited the T-DM1 induced sphere growth efficiency in the bulk cells, suggesting that T-DM1 mediated CSC survival is likely dependent on ROR1 overexpression. (Fig. S4b). In addition, treatment naïve patient's primary tumor cells as well as HCC1954 and MCF-7-HER2+ cells showed that T-DM1 and ROR1-shRNA alone inhibited cell growth, with strongest inhibition observed when T-DM1 and ROR1-shRNA were applied simultaneously (Fig. S4c–e).
Supplementary Fig. S4.

for Fig. 6: Growth effects of T-DM1 on bulk HER2+ patients tumors cells and cell lines expressing HER2 (HCC1954) and MCF-HER2+) cells. (a) ROR1 was knocked down by two independent ROR1 shRNAs and Western Blot was performed to confirm the expression of ROR1. (b) HCC1954 and MCF-7-HER2+ cells were cultured and treated with either vehicle (DMSO), T-DM1 (5 nM), ROR1-shRNA alone or combination of T-DM1 and ROR1-shRNA for 5 days, trypsinized and plated in ultra-low attachment 6-well plates in a supplemented CSC medium for ten days. The spheres were counted and sphere forming efficiency was determined. Results were presented in a bar graph using ANOVA for multiple comparisons (p = .001, 0.0001, 0.01; Scale bar: 100 μM). (c) Dose response curve and growth of HER2+ treatment naïve patients tumor cells treated with a range of T-DM1 first, followed by ROR1-siRNA alone and combination of both for 5 days. AlamarBlue was added to cells and cell growth was measured by spectrophotometer (480 nm wave length). (d and e) HCC1954 and MCF-7-HER2+ cells were cultured in 96-well plates in duplicates, treated with vehicles DMSO, T-DM1 (5 nM), ROR1-shRNA and the combination of both for 5 days. AlamarBlue was added and cell growth was analyzed by spectrophotometer (480 nm wave length). Bar graph represents the mean +/− SE, n = 3 run in triplicate.
We then tested if the combination of T-DM1 and ROR1-shRNA could have any effects on the sphere forming efficiency in bulk and sorted ROR1 subpopulations. When ROR1-shRNA was added to T-DM1 treated cells, unsorted bulk tumor cells as well ROR1+ and ROR1− cells failed to regrow and reform the spheres (Fig. 6a–c). As supplementary evidence, we showed that the immunostaining of ROR1 in sphere cells gradually disappeared (Figs. 6d–f); the response was durable until the third generation (data not shown). In concordance with the in vitro results, HCC1954 and MCF-7-HER2+ cells silenced by ROR1- shRNA produced fewer numbers of spheres and lost their tumor initiating capacity in mice (Fig. 6g; h). Findings here strongly suggest that silencing ROR1 could reverse the T-DM1 induced self-renewal capability and tumor growth in vitro and in vivo.
Fig. 6.

Silencing ROR1 sensitizes T-DM1 treatment-induced ROR1+ spheroids forming efficiency and tumor initiation: (a) Patients tumor cells, (b) HCC1954, and (c) MCF-7 HER2+ cells. T-DM1 and ROR1-shRNA co-treatment resulted in eradication of sphere forming efficiency. (d-f) Loss of ROR1 expression in the spheres was analyzed by immunofluorescence staining (Scale bar: 50 μM) (red-ROR1, blue-DAPI) in HER2+ BC patients tumor cells, HCC1954 and MCF-7-HER2+ cells (Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = .05; p = .01; one-way ANOVA). (g) Average number of spheres assessed for HCC1954 and MCF-7-HER2+ cells after transfecting with control shRNA and ROR1-shRNA. (h) Representation of tumors grown in mice after transfecting HCC1954 cells with either control-shRNA or ROR-shRNA. Table below indicates the number of tumors initiated in each group with percentage. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.7. T-DM1 treatment- induced ROR1+ cells have high tumor initiating capacity in vivo
Our results thus far demonstrated that ROR1+ cells exhibit CSC features and show tumor initiating capacity (Figs. 2C, 6H). To further support our observations and to investigate if ROR1+ expressing cells show enhanced tumor growth, we injected freshly dissociated treatment naïve HER2+ patients tumor cells (n = 5) in athymic nude mice. Tumor growth of ROR1+ and ROR1- subpopulations were compared and analyzed (Fig. 7a, schematic diagram). Equal numbers (1 × 105 cells) of sorted ROR1+, ROR1− or unsorted bulk cells were subcutaneously injected and monitored for 8 weeks (Fig. 7a). All mice bearing ROR1+ cells developed tumors (5/5), while 2/5 mice in the ROR1− group and 3/5 mice in the group of unsorted HER2+ bulk cells developed tumors within 8 weeks (Fig. 7b). Significantly, in all instances, ROR1+ cells developed considerably larger tumors than those produced by ROR1− and unsorted bulk BC cells (Figs. 7b; c). The tumors were serially sectioned and immunostained for ROR1, YAP1, Ki-67, Nanog and Sox2 for ROR1+, ROR1− and bulk tumor xenografts (Fig. S5a). ROR1+ tumor xenografts showed the colocalization of both cell surface ROR1 and nuclear YAP1, with increased expression of Ki-67, Nanog and Sox2, while ROR1− and bulk tumor xenografts showed low to absence of these markers (Fig. S5).
Fig. 7.

T-DM1 treatment- induced ROR1+ cells have high tumor forming capacity in vivo: (a) Schematic representation of cell sorting and tumor initiation assay. (b) Fresh HER2+ breast tumor cells (n = 5 patient) were dissociated and FACS sorted ROR1+ and ROR1− cells as described previously. Equal number of sorted and unsorted bulk cells from each patient tumor were injected subcutaneously in Nu/J mice (n = 5) and tumor growth and incidence were monitored for 8 weeks. ROR1+ cells were capable of generating larger tumors (5 out of 5 tumors), ROR1− cells (2 out of 5 tumors) and unsorted bulk cells (3 out of 5 tumors) generated tumors but smaller in size (Scale bar 5 mm). (c) The weight distributions of tumors generated from ROR+, ROR1− and unsorted bulk tumor cells are shown in all patient's tumors. Results are median +/− IQR (Inter quartile range), p = .01 and 0.05; repeated measure ANOVA. (d) HCC1954 cells were either treated with vehicle (DMSO) and/or T-DM1 for 5 days and sorted based on T-DM1 induced cell surface ROR1 overexpression and injected to mice. Tumor growth and incidence were monitored for 8 weeks. On the basis of tumor incidence in the indicated time, a Kaplan-Meier survival curve was generated using Log-rank test (Log-rank P = .03). “R” statistical software “survival” and “survminer” packages were used to analyze the tumor-free survival curve. (e) In vivo limiting dilution assay was performed using HCC1954 cells and treatment and sorting performed as described in (a). Sorted 100, 1000, 10,000 and 100,000 cells were mixed with Matrigel and injected subcutaneously in Nu/J mice (n = 5, Scale bar 5 mm). (f) Tumor initiating capacity was monitored over 8 weeks and CSC frequency was calculated using “R” statistical software “statmod” package. The frequency of tumor-initiating cells for 3 mice is graphed. Results are median +/− IQR (repeated measure ANOVA).
Supplementary Fig. S5.

of Fig. 7: T-DM1- induced ROR1+ cells have high tumor forming capacity in vivo: (a) Representative immunofluorescence staining of ROR1, YAP1, Ki-67, Nanog and Sox2 from ROR1+, ROR1− and unsorted bulk tumor xenografts (Scale bar: 50 μM).
The results presented above clearly demonstrated that only ROR1+ cells are highly tumorigenic.
The results obtained with patient tumor cells was confirmed using a tumorigenic HCC1954 sub line. ROR1+ and ROR1− cells were sorted for surface expression of ROR1 after 5-day treatment with either vehicle or T-DM1. Equal numbers of isolated cells (1 × 105 cells) were subcutaneously injected in mice and monitored for 8 weeks. Three of five (3/5) mice in the ROR1+ group developed tumors as early as 4-weeks, whereas no tumors were developed in ROR1− mice. In comparison, using bulk cells, only 1 mouse developed small tumor foci within the 8-week time period (Fig. 7d; Log-rank p-value = .03). These results demonstrated that T-DM1 induced ROR1+ cells are enriched for tumor initiating capacity and were able to readily develop tumors in mice.
To assess the tumor initiating and self-renewal capacity of the ROR1 subpopulations in vivo, the ELDA (extreme limiting dilution assay) assay was performed and compared for each subpopulation. HCC1954 cells were treated and sorted as described above and injected subcutaneously in mice. Ten weeks after implantation of each subpopulation, tumor initiating capacity was assessed based on the tumor development in each mouse (Fig. 7e). The results from serial dilution assay demonstrate that T-DM1 induced ROR1+ cells showed significantly greater tumor initiating frequency (1/1100) than either ROR1− (1/100000) or bulk cells (1/65000) (Fig. 7f). Findings suggest that long term treatment of patients with T-DM1 might lead to the induction of the ROR1+ population, and an increased frequency of CSCs. As a consequence, only the self-renewing capable ROR1+ cells would develop resistance to therapy.
3.8. YAP1 regulates T-DM1 treatment-induced ROR1 overexpression and CSC enrichment
YAP1 mediated upregulation of epidermal growth factor receptor (EGFR) has been reported to play a role in therapy resistance [19]. YAP/TAZ influences lapatinib resistance in the HER2 amplified BC cells [33] suggesting that YAP1 confers drug resistance through CSC enrichment. To gain further insight into a potential ROR1-YAP1 relationship, we transduced doxycycline (1 μg/ml) inducible human flag-tagged YAP1S127A cDNA (PIN20 YAP1S127A) [19,34] into HCC1954 cells. The YAP1S127A transduction increased the level of ROR1 and YAP1 with substantial changes in HER2 expression (Fig. 8a). ROR1(cell surface red colour) and YAP1 (nuclear green colour) increased in doxycycline treated cells compared to doxycycline negative cells which confirms efficient transduction of YAP1 and localization of cell surface ROR1 and nuclear YAP1 (Fig. 8b). Knockdown of YAP1 by YAP-1-siRNA decreased the expression of ROR1 (Fig. 8c), suggesting that YAP1 regulates ROR1 expression and confers the resistance to therapy. To explore whether sphere forming efficiency is dependent on YAP1 activity we knocked down YAP1 by siRNA and assessed the sphere forming efficiency. Knockdown of YAP1 significantly reduced the T-DM1 induced sphere forming efficiency. When cells were sequentially treated with ROR1-siRNA-YAP1 and siRNA/T-DM1, a further reduction in sphere forming efficiency (Fig. 8d) was observed, suggesting a major role for YAP1 in the survival of CSCs.
Fig. 8.

YAP1 regulates T-DM1 treatment-induced ROR1 overexpression and CSC enrichment. (a) HCC1954 cells were transduced with full length YAP1 cDNA (PIN20YYAP1). Doxycycline (1 μg/ml) was added in the cell culture medium to induce the YAP1 expression. Western blot was performed using antibodies to YAP1, ROR1 and HER2. (b) Immunofluorescence staining for YAP1 (green; white arrow head) and ROR1 (red; white arrowhead) and DAPI (blue) confirming the transduction of YAP1 in the presence and absence of doxycycline (Scale bar: 50 μM). (c) YAP1 was knockdown by two independent YAP1 siRNA and Western Blot was performed to confirm the expression of YAP1 and ROR1 (red label box siRNA was used in our experiments). (d) HCC1954 cells were transfected with non-targeting and YAP1 specific siRNA for 48 h. Cells were then treated with T-DM1 for 48 h, followed by treatment with ROR1-shRNA for 48 h. Treated cells were trypsinized, cultured in the supplemented CSC medium, and assessed for sphere forming efficiency (black arrowhead) Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = .05; p = .01; one-way ANOVA. [Scale bar: 100 μM]. (e) Immunoblots showing the expression of YAP1, ROR1 and HER2 after treatment of HCC1954 cells with 2 μg/mL verteporfin. (f) Quantification of sphere forming efficiency (black arrowhead) after verteporfin treatment at 2 μg/ml and vehicle (DMSO) in HCC1954 cells. Bar graph represents the mean +/− SE, n = 3; run in triplicate; p = .05; p = .01; Student's t-test; Scale bar: 100 μM). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Evidence suggests that increased YAP1/TEAD activity plays a crucial role in cancer progression, metastasis and therapeutic resistance [13]. FDA approved Verteporfin, a photodynamic therapy for macular degeneration [35] has been identified as the inhibitor of YAP-TEAD binding [36]. To further explore the functional role of YAP1 mediated ROR1 overexpression and CSC properties, we utilized Verteporfin to disrupt YAP-TEAD binding and assessed the expression of ROR1 and sphere forming efficiency. Disruption of YAP-TEAD interaction by Verteporfin significantly reduced the expression of YAP1 and ROR1 proteins, leaving HER2 expression unchanged, in concert with a significant reduction in the sphere forming efficiency (Figs. 8e; 8f). Altogether these results suggest that YAP1 mediates ROR1 overexpression in the CSC subpopulation and confers therapeutic resistance.
4. Discussion
In this study, we explored the mechanisms of resistance of HER2-directed BC treatment using clinical samples, established cell lines, and an in vivo xenograft model. T-DM1 has been designed for targeted therapy, and has greatly transformed therapeutic success for HER2-overexpressing BC. Despite the promising success, a group of patients either acquire or exhibit intrinsic resistance to therapy by activating a resistance pathway, such as, factors which prevent trastuzumab binding to HER2, upregulation of HER2 downstream signaling pathways, and signaling through alternate pathways. An alternative hypothesis for the resistance mechanism is thought to be the induction of CSCs. Current BC treatment emphasis is not only on targeting differentiated tumor cells, but is intended to effectively target resistant CSCs. Here, we report that: 1) T-DM1 treatment leads to the overexpression of ROR1, in concert with increase in stemness, and 2) ROR1 overexpression then mediates therapy resistance through YAP1. These findings lead to the novel concept that overexpression of ROR1 by T-DM1 treatment contributes to treatment resistance and CSC enrichment through activation of YAP1.
HER2+ BC is a heterogeneous tumor where response to HER2-directed therapies could have different outcomes and sensitivities on the basis of different subtypes and cell populations [4]. Targeted HER2-inhibitors, trastuzumab, pertuzumab, EGFR/HER2 inhibitor lapatinib and T-DM1 have become the standard of care for HER2+ BC patients. All these drugs have been proven largely effective, however, a larger proportion of patients become refractory to treatment, or after an initial response, are followed by secondary resistance possibly due to the induction of CSCs [37,38]. CSCs are traditionally resistant to ionizing-radiation [39] and chemotherapy [40]. A recent report describes that lapatinib treatment could lead to the induction of Jagged1 and CSC enrichment [29]. To overcome these limitations, an antibody drug conjugate T-DM1 was introduced, which inhibits cell cycle division, induces cell death by blocking spindle apparatus [41]. Only a handful of reports on T-DM1 resistance have emerged since its first introduction as an anti-HER2 therapy, thus providing a limited knowledge of T-DM1 mediated drug resistance and CSC enrichment. Recent studies reported on the resistance mechanism of T-DM1 treatment as occurring through the intracellular trafficking and impaired lysosomal proteolytic activity [6], low tumor HER2 expression, poor internalization of the HER2/T-DM1 complexes, defective intracellular and endosomal trafficking of the HER2/T-DM1 complex, masking of HER2 epitope, and drug efflux pumps affecting neuregulin-HER3 signaling [42].
We now report on a newly identified resistance mechanism indicating that T-DM1 responsiveness and resistance may not only depend on factors mentioned above, but may also possibly depend on CSC enrichment. Our data from established BC cell lines, mouse xenografts and most importantly clinical samples from BC patients consistently demonstrated that long term treatment of these cells with T-DM1 could lead to an increase from a very low-level to very high-level in membrane expression of ROR1. The induction of ROR1 results in the proportional increase in CSC enrichment, thereby leading to potential resistance to T-DM1 and a further increase in tumor initiating efficiency post-therapy and potentially relapse. Results presented in this study show that, while ROR1− cells die due to T-DM1 toxicity, ROR1+ cells survive and expand possibly due to the increase in stemness and self-renewal properties.
As stated earlier, although anti-HER2 treatment showed initial sensitivity in HER2+ BC patients, the resurgence of therapy resistant disease in a group of patients prevented cures. In concert with these outcomes, our results have demonstrated that T-DM1 treatment increased surface ROR1 expression and sphere forming efficiency while an opposite effect was observed in the ROR1− population. Moreover, we established that only ROR1+ cells are highly positive for both CD44+ and ALDH+, thereby promoting an increase in sphere forming efficiency, accompanied by expression of functional characteristics of CSCs and EMT. Given the efficient tumor initiating capacity of the ROR1+ population, therapeutic strategies that can eliminate both differentiated ROR1− and especially the undifferentiated ROR1+ cells are urgently needed. We provide evidence that T-DM1 resistant ROR1+ cells could be T-DM1 sensitized by blocking overexpression of ROR1. Previously published results reported that silencing ROR1 could prevent enhancement of sphere forming capacity, and xenograft tumor growth. In contrast, anti-ROR1 mAb could enhance apoptosis, and inhibit cell proliferation and invasive behavior and prevent tumor cells becoming metastatic [13,14]. Accordingly, a ROR1 inhibitor cirmtuzumab is already in phase I and II clinical trials (NCT03420183 and NCT03088878) and has shown greater success in breast and blood cancer. Similarly, using an shRNA approach, our results also demonstrated that inhibiting ROR1 might assist in the reduction of CSC mediated treatment resistance which was caused by T-DM1 treatment. On the other hand, several promising targeted therapies for triple negative BC patients have emerged recently including use of a PI3K/AKT inhibitor [43] which promises to shift the treatment paradigm away from conventional chemotherapy. Although, a PI3K inhibitor may work for both non-TNBC and TNBC patients, nevertheless T-DM1 is particularly efficient for treatment of HER2+ BC patients.
YAP1 is a central mediator of the Hippo signaling pathway and has been reported to promote the survival and self-renewal of breast tumor-initiating cells [17]. Both YAP1 and ROR1 have been reported to be overexpressed in a variety of cancers, including BC, and increased expression of these genes has been associated with disease progression, therapeutic resistance and CSC enrichment. How YAP1 regulates ROR1 remains unclear. A recent study reported the cross-talk between ROR1-HER3 and Hippo-YAP1 pathway mediated by lncRNAs [17]. Being a member of a transcriptional coactivator, YAP1 may regulate many downstream genes. In gastric cancer, YAP1 upregulated Sox9, implying regulation of CSC properties by YAP1 [34]. In our study, we present evidence that YAP1 regulates ROR1 thereby increasing CSC properties and T-DM1 resistance in BC cells. Importantly, evidence here shows that sphere forming efficiency of resistant ROR1+ cells is dependent on YAP1 activity. This suggests that T-DM1 mediated HER2 inhibition increases expression of ROR1 through YAP1 activation, thus conferring therapy resistance in a critical subset of cells. We suggest that simultaneous inhibition of YAP1 and ROR1, together with traditional HER2-directed therapy, may improve the patient overall survival and overcome therapeutic resistance. Taken together, our results support the idea that inhibition of HER2 followed by inhibition of highly upregulated ROR1 and YAP1 could eliminate the CSC enrichment. The therapeutic strategy outlined here portends significant potential and may help to guide the future development of agents for overcoming T-DM1 resistance, and thereby achieve a new therapeutic approach for the benefit of BC patients.
The following are the supplementary data related to this article.
Funding source
This study was supported by Neurogen Technologies Research Fund.
Authors contribution
SSI, HY, WAF conceived and designed the experiments. SSI, performed the experiments and analyzed the data. HO, NJD, FU, JA, ASMN, SA, MB liaison with patients and clinicians and collected the patient's data and samples. SSI, AA, MA designed and performed the animal study, SSI, HY, WAF, AA, MA, MU wrote the manuscript. All authors approved the final version of the manuscript.
Conflict of interest
The authors declares that they have no conflict of interests.
Acknowledgements
The authors thank all patients, and staff of the hospitals and clinics. We particularly extend our sincere thanks to Dr. Mien-Chie Hung, The University of Texas MD Anderson Cancer Centre (Houston, Texas) for providing MCF-7-(HER2+ induced) cell line.
References
- 1.Martin M., Holmes F.A., Ejlertsen B. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1688—1700. doi: 10.1016/S1470-2045(17)30717-9. [DOI] [PubMed] [Google Scholar]
- 2.Moasser M.M., Krop I.E. The evolving landscape of HER2 targeting in breast cancer. JAMA Oncol. 2015;1:1154–1161. doi: 10.1001/jamaoncol.2015.2286. [DOI] [PubMed] [Google Scholar]
- 3.Michiels S., Pugliano L., Marguet S. Progression-free survival as surrogate end point for overall survival in clinical trials of HER2-targeted agents in HER2-positive metastatic breast cancer. Ann Oncol. 2016;27:1029–1033. doi: 10.1093/annonc/mdw132. [DOI] [PubMed] [Google Scholar]
- 4.Amiri-Kordestani L., Blumenthal G.M., Xu Q.C. FDA approval: ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin Cancer Res. 2014;20:4436—4441. doi: 10.1158/1078-0432.CCR-14-0012. [DOI] [PubMed] [Google Scholar]
- 5.Verma S., Miles D., Gianni L. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783—1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rios-Luci C., Garcia-Alonso S., Diaz-Rodriguez E. Resistance to the antibody-drug conjugate T-DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Res. 2017;77:4639—4651. doi: 10.1158/0008-5472.CAN-16-3127. [DOI] [PubMed] [Google Scholar]
- 7.Li G., Guo J., Shen B.Q. Mechanisms of acquired resistance to Trastuzumab Emtansine in breast Cancer cells. Mol Cancer Ther. 2018;17:1441—1453. doi: 10.1158/1535-7163.MCT-17-0296. [DOI] [PubMed] [Google Scholar]
- 8.Zhang S., Zhang H., Ghia E. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc Natl Acad Sci U S A. 2019;116:1370—1377. doi: 10.1073/pnas.1816262116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu S., Wicha M.S. Targeting breast cancer stem cells. J Clin Oncol. 2010;28:4006—4012. doi: 10.1200/JCO.2009.27.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin-Castillo B., Lopez-Bonet E., Cuyas E. Cancer stem cell-driven efficacy of trastuzumab (Herceptin): towards a reclassification of clinically HER2-positive breast carcinomas. Oncotarget. 2015;6:32317–32338. doi: 10.18632/oncotarget.6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X., Liao R., Li D., Sun J. Induced cancer stem cells generated by radiochemotherapy and their therapeutic implications. Oncotarget. 2017;8:17301–17312. doi: 10.18632/oncotarget.14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui B., Zhang S., Chen L. Targeting ROR1 inhibits epithelial-mesenchymal transition and metastasis. Cancer Res. 2013;73:3649—3660. doi: 10.1158/0008-5472.CAN-12-3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui B., Ghia E.M., Chen L. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood. 2016;128:2931—2940. doi: 10.1182/blood-2016-04-712562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S., Cui B., Lai H. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc Natl Acad Sci U S A. 2014;111:17266–17271. doi: 10.1073/pnas.1419599111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang S., Chen L., Cui B. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim T., Yang S.J., Hwang D. A basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness. Nat Commun. 2015;6 doi: 10.1038/ncomms10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamar J.M., Stern P., Liu H. The hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109 doi: 10.1073/pnas.1212021109. (E2441—2450) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C., Wang S., Xing Z. A ROR1-HER3-lncRNA signaling axis modulates the hippo-YAP pathway to regulate bone metastasis. Nat Cell Biol. 2017;19:106—119. doi: 10.1038/ncb3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song S., Honjo S., Jin J. The hippo coactivator YAP1 mediates EGFR overexpression and confers chemoresistance in esophageal cancer. Clin Cancer Res. 2015;21:2580—2590. doi: 10.1158/1078-0432.CCR-14-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin D., Degese M.S., Vitale-Cross L. Assembly and activation of the hippo signalome by FAT1 tumor suppressor. Nat Commun. 2018;9:2372. doi: 10.1038/s41467-018-04590-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawrence M.S., Stojanov P., Polak P. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214—218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiterkamp J., Ernst M.F., de Munck L. Improved survival of patients with primary distant metastatic breast cancer in the period of 1995-2008. A nationwide population-based study in the Netherlands. Breast Cancer Res Treat. 2011;128 doi: 10.1007/s10549-011-1349-x. (495—50) [DOI] [PubMed] [Google Scholar]
- 23.Korkaya H., Wicha M.S. HER2 and breast cancer stem cells: more than meets the eye. Cancer Res. 2013;73:3489—3493. doi: 10.1158/0008-5472.CAN-13-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks M.D., Burness M.L., Wicha M.S. Therapeutic implication of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17:260—271. doi: 10.1016/j.stem.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burnett J., Korkaya H., Ouzounova M.D. Trastuzumab resistance induces EMT to transform HER2(+) PTEN(−) to a triple negative breast cancer that requires unique treatment options. Sci Rep. 2015;5:15821. doi: 10.1038/srep15821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berns K., Horlings H.M., Henness B.T., Madiredjo M. A functional genetic approach identifies the Pi3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 27.Niikura N., Liu J., Hayashi N. Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. J Clin Oncol. 2012;30:593—599. doi: 10.1200/JCO.2010.33.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abravanel D.L., Belka G.K., Pan T.C. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J Clin Invest. 2015;125:2484—2496. doi: 10.1172/JCI74883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srinivasan M., Bharali D., Sudha T. Downregulation of Bmi1 in breast cancer stem cells suppress tumor growth and proliferation. Oncotarget. 2017;8:38731–38742. doi: 10.18632/oncotarget.16317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siddiqui H.R., Saleem M. Role of Bmi1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012;30:372—378. doi: 10.1002/stem.1035. [DOI] [PubMed] [Google Scholar]
- 31.Boubles D., Chouha G.B., Jin Q., Bartholomeusz C., Esteva F.J. CD44 expression contributes to trastuzumab resistance in HER2-positive breast cancer cells. Breast Cancer Res Treat. 2015;151:501—513. doi: 10.1007/s10549-015-3414-3. [DOI] [PubMed] [Google Scholar]
- 32.Shah D., Wyatt D., Baker A.T. Inhibition of HER2 increases JAGGED1-dependent breast cancer stem cells: role for membrane JAGGED1. Clin Cancer Res. 2018;24:4566—4578. doi: 10.1158/1078-0432.CCR-17-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin C.H., Pelissier F.A., Zhang H. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol Biol Cell. 2016;26:3946—3953. doi: 10.1091/mbc.E15-07-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song S., Ajani J.A., Honjo S. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014;74:4170—4182. doi: 10.1158/0008-5472.CAN-13-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michel S., Schmidt-Erfurth U. Photodynamic therapy with verteporfin: a new treatment in ophthalmology. Semin Opthalmol. 2001;14:201—206. doi: 10.1076/soph.16.4.201.10298. [DOI] [PubMed] [Google Scholar]
- 36.Liu-Chittenden Y., Huang B., Shim J.S. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slamon D.J., Leyland-Jones B., Shank S. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpress HER2. N Engl J Med. 2001;344:783—792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 38.Romond E.H., Perez E.A., Bryant J. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673—1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 39.Alexander P.B., Chen R., Gong C. Distinct receptor tyrosine subsets mediate anti-HER2 drug resistance in breast cancer. J Biol Chem. 2017;292:748—759. doi: 10.1074/jbc.M116.754960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colak S., Medema J.P. Cancer stem cells-important players in tumor therapy resistance. FEBS J. 2014;281:4779—4791. doi: 10.1111/febs.13023. [DOI] [PubMed] [Google Scholar]
- 41.Barok M., Joensuu H., Isola J. Traustuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014;16:209. doi: 10.1186/bcr3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun J.G., Chen X.W., Zhang L.P., Wang J., Diehn M. Yap1 promotes the survival and self-renewal of breast tumor initiating cells via inhibiting Smad3 signaling. Oncotarget. 2016;7:9692—9706. doi: 10.18632/oncotarget.6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vidula N., Bardia A. Trageted therapy for metastatic triple negative breast cancer: the next frontier in precision medicine. Oncotarget. 2017;8:106167–106168. doi: 10.18632/oncotarget.22580. [DOI] [PMC free article] [PubMed] [Google Scholar]