

Abstract
Background
Studies have shown that E3 ubiquitin ligase CBLL1 plays multiple roles in development and tumorigenesis. CBLL1 is over‐expressed in colon cancer and associated with cancer cell proliferation. While, the overexpression of CBLL1 inhibited the estrogenic dependent cell proliferation and migration in ER alpha dependent breast cancer cell MCF‐7.
Methods
We used an immunohistochemical method to detect CBLL1 expression in human NSCLC and corresponding normal lung tissues and analyzed its relationship with clinicopathological parameters. Moreover, we investigated the role of CBLL1 in NSCLC cell behavior by inhibiting its expression in A549 and H1299 cells.
Results
In this study, we found that CBLL1 was frequently upregulated in non‐small lung cancer (NSCLC) tissues compared to the adjacent nontumor tissues. We found that the high expression of CBLL1 was associated with the tumor size in NSCLC tissues. It has been recently reported that CBLL1 promotes cell proliferation and invasion in A549 and H460 cells. Our results confirmed that CBLL1 promoted the proliferation by promoting G1/S cell cycle transition in NSCLCs cells. Moreover, CBLL1 knockdown inhibited cell invasion via increased E‐cadherin protein expression, and decreased expression of MMP2 and MMP9 in NSCLC cell lines. The protein expression of E‐cadherin was increased after CBLL1 depletion while the E‐cadherin mRNA was not affected after knockdown of the endogenous CBLL1.
Conclusion
These results provide important insights for using CBLL1 as an oncogenic marker gene in the development and progression of non‐small cell lung cancer.
Keywords: CBLL1, E‐cadherin, E3 ubiquitin ligase, invasion, non‐small cell lung cancer, proliferation
Introduction
Lung cancer is the leading cause of cancer‐related deaths worldwide,1, 2 among which non‐small cell lung cancer (NSCLC) is the main type. The survival rate of patients with NSCLC is currently low and the prognosis of these patients is closely related to cancer metastasis. Therefore, safe and effective methods for inhibiting the growth and metastasis of lung cancer cells are needed.3
Cbl proto‐oncogene E3 ubiquitin protein ligase‐like 1, RNF188, Hakai (CBLL1) is an evolutionarily conserved E3 ubiquitin ligase containing a RING‐finger domain.4 CBLL1 contains a typical RING‐finger, short pTyr‐binding domain, and proline‐rich domain. CBLL1 is structurally and functionally related to c‐Cbl; however, CBLL1 and c‐Cbl are not homologues because the pTyr‐binding domain of c‐Cbl consists of a homodimer that forms a new structural interface.5, 6, 7, 8 E‐cadherin is a typical member of cadherin, and its loss is associated with the poor prognosis of patients with cancer. E‐cadherin contains an extracellular domain and cytoplasmic domain.9, 10, 11, 12, 13, 14 The SH2 domain of CBLL1 binds to the cytoplasmic domain of E‐cadherin and mediates its ubiquitination, endocytosis, and degradation in lysosomes.4 This disrupts cell‐cell adhesions.
Studies have shown that CBLL1 plays an important role in tumorigenesis.15, 16, 17, 18, 19, 20, 21, 22, 23, 24 First, CBLL1 expression is upregulated in human colon and gastric cancer tissues, and CBLL1 has been reported to induce anchorage‐dependent cell growth. In colorectal epithelial cells, the autocrine Slit‐Robo pathway promotes CBLL1‐mediated downregulation of E‐cadherin to promote carcinogenesis. CBLL1 expression is inversely correlated with E‐cadherin in several colon adenocarcinoma tissues. Second, knockdown of E‐cadherin in CBLL1‐overexpressing epithelial cells does not promote cusp formation, suggesting CBLL1 affects cell phenotypes in an E‐cadherin‐independent manner. In MDCK cells, CBLL1 is involved in regulating cell dynamic expansion and mobility.13 CBLL1 affects the adhesion of cells to the matrix and invasion of MDCK epithelial cells.18 Additionally, in estrogen receptor alpha‐dependent breast cancer MCF‐7 cells, CBLL1 overexpression inhibits estrogen‐dependent cell proliferation and migration and plays a negative role in the progression of estrogen‐dependent breast cancer.23 A recent study reported that inhibition of CBLL1 suppresses cell proliferation and invasion and increases the chemosensitivity to cisplatin in A549 and H460 cells.25
However, the expression level and location of CBLL1 in NSCLC tissues remain unclear. Because E‐cadherin aberrant expression plays an important role in the development of NSCLC, we predicted that CBLL1 and NSCLC progression are closely related.26, 27 Thus, we used an immunohistochemical method to detect CBLL1 expression in human NSCLC and corresponding normal lung tissues and analyzed its relationship with clinicopathological parameters. Moreover, we investigated the role of CBLL1 in NSCLC cell behavior by inhibiting its expression in A549 and H1299 cells.
Methods
Patients and tissue samples
Seventy‐nine cases of NSCLC and 24 corresponding non‐tumorous lung tissues were obtained from January 2009 to December 2013 following surgical resection at the First Affiliated Hospital of China Medical University. No patients had undergone radiation therapy or chemotherapy before surgery. The study was conducted with the approval of the Ethics Review Committee of the First Affiliated Hospital of China Medical University. Paraffin‐embedded NSCLC samples were obtained from patients admitted to the Pathological Department of the First Affiliated Hospital of China Medical University. Informed consent was obtained from all patients. Formalin‐fixed paraffin‐embedded tumor sections were stained by routine hematoxylin and eosin staining and reviewed by two senior pathologists to determine the clinicopathologic staging according to the eighth TNM Classification for Non‐Small Cell Lung Cancer. There were 39 cases of squamous cell carcinoma and 40 cases of adenocarcinoma. Forty‐one cases did not show lymph node metastasis and 38 cases showed lymph node metastasis.
Immunohistochemical staining
Tissue specimens were prepared in 4 μm sections. The expression of CBLL1 protein was detected by streptomycin‐peroxidase. Antigen retrieval was performed by pressure cooking the specimens in citric acid buffer (pH 6.0) for 1 minute 40 seconds. Rabbit anti‐human polyclonal anti‐CBLL1 antibody (1:200, Sigma, St. Louis, MO, USA) was added and incubated overnight at 4°C. Unconjugated rabbit IgG and phosphate‐buffered saline (PBS) was used as the control.
Two pathologists blinded to the clinical data semi‐quantitatively scored all slides by evaluating the staining intensity and percentages of cells stained in representative areas of each slide. Five high‐magnification fields were randomly selected from each section and 100 tumor cells in each field were counted to evaluate the intensity and range of CBLL1 staining. A negative result was given a score of 0 (no staining), light yellow staining was recorded as 1, yellow was scored as 2, and deep brown staining was recorded as 3. The percentage of stained cells was scored as follows: 0, <5%; 1, 5–25%; 2, 25–50%; 3, 51–75%; 4, >75%. Two independent scores were multiplied to obtain the patient's coloring coefficient, which was categorized as “low expression” for a coloring coefficient ≤4 and “high expression” for a coloring coefficient >4.
Cells and reagents
The immortalized human normal bronchial epithelial cell line (HBE), the human lung adenocarcinoma cell lines A549 and H1299 were purchased from the American Type Culture Collection (Manassas, VA, USA). The human NSCLC cell lines SK‐MES‐1(SK) was purchased from the Shanghai Cell Bank of the Chinese Academy of Science (Shanghai, China). Cell lines were cultured in DMEM or RPMI‐1640 medium containing 10% fetal bovine serum (Hyclone, Logan, UT, USA). All cells were incubated at 37°C in a 5% CO2 incubator.
Three pairs of CBLL1 short interfering RNA (siRNA) sequences were designed and synthesized by Gene Pharma (Shanghai, China). Considering their relative effectiveness and stability, the following siRNA sequences were selected according to our pilot experiments:: 5′‐CAC CAG ACA AGC ACC AUA UTT‐3′ and 5′‐AUA UGG UGC UUG UCU GGU GTT‐3′. The non‐silencing siRNA sequences 5′‐UUC UCC GAA CGU GUC ACG UTT‐3′ and 5′‐ACG UGA CAC GUU CGG AGA ATT‐3′ were used as negative controls. The following antibodies were used: CBLL1 (HPA021773, Sigma), cyclin D1 (sc‐717, Santa Cruz Biotechnology, Dallas, TX, USA), cyclin E (4129P, Cell Signaling Technology, Danvers, MA, USA), CDK4 (sc‐260, Santa Cruz Biotechnology), E‐cadherin (20874‐1‐AP, Proteintech, Rocky Hill, NJ, USA), matrix metalloproteinase 2 (MMP2) (4022S, Cell Signaling Technology), MMP9 (3852S, Cell Signaling Technology), GAPDH (TA‐08, ZSGB‐Bio, Beijing, China), unconjugated rabbit IgG (BA1045, Boster, Wuhan, China) and unconjugated mouse IgG (BA1046, Boster, Wuhan, China).
siRNA interference
Cells were plated into six‐ or 24‐well plates and transfected with CBLL1 siRNA and negative control siRNA. Transfection of lung cancer cell lines A549 and H1299 was performed using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Transfection efficiency was determined by quantitative RT‐PCR and western blotting.
Quantitative RT‐PCR
Total RNA isolation and cDNA preparation were carried out as previously described.28 PCR was carried out using SYBR Premix Ex Taq II (RR820A, Takara, Japan). The following primers were used for amplification. CBLL1: 5′‐GGAGTTGGATAGTAGAGGCGAGAG‐3′, 5′‐AACATCAAGACCACCCAAGGA‐3′; GAPDH: 5′‐CAGGAGGCATTGCTGATGAT‐3′, 5′‐GAAGGCTGGGGCTCATTT‐3′; and E‐cadherin: 5′‐CGAGAGCTACACGTTCACGG‐3′, 5′‐GGGTGTCGAGGGAAAAATAGG‐3′. The lengths of the PCR products were 228 base pairs (bp) (CBLL1), 138 bp (GAPDH), and 119 bp (E‐cadherin). The relative quantification of gene expression for each sample was conducted using the ΔΔCт method. Each experiment was performed in triplicate.
Western blotting
The cells were collected and lysed, and 40 μg proteins were separated by 10% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Proteins on the membranes were blocked with 5% bovine serum albumin at room temperature for two hours, incubated with primary antibody at 4°C overnight, and then incubated with secondary antibodies at room temperature for two hours. The bands were visualized by SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA) and signals were recorded with an MF‐ChemiBIS 3.2 (Bio‐Rad, Hercules, CA, USA). Protein expression levels were evaluated using GAPDH as the loading control. Each experiment was performed in triplicate.
Immunofluorescence
Cells cultured on coverslips were fixed with 4% paraformaldehyde and blocked with 5% bovine serum albumin at 37°C for one hour. The cells were incubated with primary antibody at 4°C overnight, and then followed by incubation with TRITC‐labeled secondary antibodies. The nuclei were counterstained with DAPI at 37°C for 10 minutes. Fluorescent images were recorded under the same conditions using a fluorescence microscope (BX53, Olympus, Tokyo, Japan) or confocal laser scanning microscope (FV1000, Olympus).
CCK8 assay
Cells transfected with CBLL1 siRNA and negative control siRNA were seeded into 96‐well plates. Cell proliferation was detected with a Cell Counting Kit‐8 (Beyotime, Shanghai, China) according to the manufacturer's instructions. The CCK8 assay was repeated over four consecutive days. Optical density values were measured with a microplate reader (Spectra Thermo, Männedorf, Switzerland) at an absorbance of 450 nm. Data are shown as the mean ± standard deviation (SD, n = 3). Each experiment was performed in triplicate.
Colony formation assay
A549 and H1299 cells were transfected with siRNA and negative control siRNA for 48 hours and then plated into six‐well cell culture plates (500 per well) and incubated for 12 days. The plates were washed with PBS and stained with hematoxylin. The number of colonies containing more than 50 cells was manually counted using a microscope. Each experiment was performed in triplicate.
Cell cycle assay
Cells were harvested and fixed in cold 70% ethanol 4°C overnight. After washing twice with PBS, cells were incubated with 100 uL RNaseA (KeyGen, Nanjing, China) at 37°C in a water bath for 30 minutes and then treated with 400 uL Propidium Iodide for 30 minutes. Finally, the treated cells were analysed by flow cytometry (FACSCalibur, BD Biosciences, USA). Each experiment was performed in triplicate.
Cell invasion assay
The cell invasion assay was performed using a 24‐well Transwell chamber (Costar, Corning, NY, USA). Forty‐eight hours after transfection, A549 and H1299 cell suspensions were transferred to the upper chamber with an 8 μm pore size insert precoated with Matrigel (BD Biosciences) and cultured in medium containing 2% fetal bovine serum for 24 h. Medium supplemented with 20% fetal bovine serum was added to the lower chamber as the chemoattractant. The cells were fixed in cold methanol for 15 minutes. Cells on the surface of the microporous membrane were wiped off and invaded cells were stained with hematoxylin for nuclear staining.
Statistical analysis
Statistical analysis was conducted with SPSS13.0 software (SPSS, Inc., Chicago, IL, USA). The χ2 test was used to examine the correlations between CBLL1 expression and clinicopathological factors. Independent t test, one‐way ANOVA and Kruskal‐Wallis were selected according to the distribution of the data. P < 0.05 was considered statistically significant. All reported P values were two‐tailed.
Results
CBLL1 is highly expressed in NSCLC and correlated with tumor size
We examined CBLL1 expression in 79 NSCLC tissues and 24 cases of adjacent normal lung tissues by immunohistochemistry analysis. As described above, CBLL1 immunoreactivity was graded as low and high expression. Of the 24 adjacent normal lung tissues, CBLL1 expression was low in the nuclei of alveoli, bronchi tissues and lung parenchyma. CBLL1 immunostaining was observed in both the nuclei and cytoplasm of cancer cells. High expression of CBLL1 was observed in 64 (81.01%) of the 79 NSCLC cases (Fig. 1). To explore the clinical significance of CBLL1 in NSCLC tissues, we analyzed the relationship between CBLL1 expression and clinicopathological parameters. High CBLL1 expression was positively correlated with tumor size (P = 0.010). The correlations between CBLL1 expression and age, gender, pTNM stage, differentiation, histology type, and lymph node metastasis were not significant (P > 0.05, Table 1).
Figure 1.

Expression of CBLL1 in NSCLCs and adjacent lung tissues. Immunohistochemistry was used to determine the expression of CBLL1. (a) Low CBLL1 expression in the nuclear of bronchial epithelium. (b) Low CBLL1 expression in the nuclear of alveolar epithelium. (c) High CBLL1 expression in lung squamous cell carcinoma. (d) CBLL1 was highly expressed in lung adenocarcinoma.
Table 1.
Association of CBLL1 expression with clinicopathological parameters of NSCLC patients
| CBLL1 high‐expression | ||||
|---|---|---|---|---|
| Features | Cases (n) | No | Yes | P |
| Gender | 0.565 | |||
| Male | 48 | 8 | 40 | |
| Female | 31 | 7 | 24 | |
| Age | 0.156 | |||
| <60 years | 44 | 11 | 33 | |
| ≥60 years | 35 | 4 | 31 | |
| pTNM stage | 0.363 | |||
| I | 39 | 9 | 30 | |
| II‐ III | 40 | 6 | 34 | |
| Tumor size | 0.010 | |||
| T1 | 45 | 13 | 32 | |
| T2‐T4 | 34 | 2 | 32 | |
| Differentiation | 0.388 | |||
| Well | 29 | 7 | 22 | |
| Moderate‐poor | 50 | 8 | 42 | |
| Histology type | 0.568 | |||
| SCC | 39 | 6 | 33 | |
| AC | 40 | 9 | 31 | |
| Nodal status | 0.488 | |||
| N0 | 41 | 9 | 32 | |
| N1 N2 N3 | 38 | 6 | 32 | |
CBLL1 expression in HBE and various NSCLC cell lines
The western blot results showed that CBLL1 expression in the NSCLC cell lines A549, H1299, and SK were higher than in the immortalized HBE cell line (P < 0.01, Fig 2a). We also conducted immunofluorescence staining to detect the expression and subcellular localization of CBLL1 in HBE, A549, and H1299 cells. CBLL1 was mainly expressed in the nucleus of HBE cells. CBLL1 was expressed both in the nucleus and cytoplasm of A549 and H1299 cells (Fig 2b).
Figure 2.
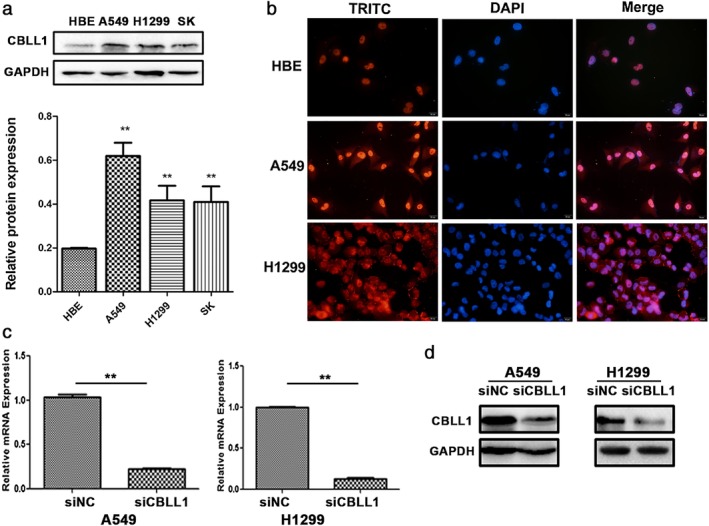
Expression and localization of CBLL1 in HBE and lung cancer cell lines A549 and H1299. CBLL1 expression levels were significantly altered after CBLL1 transient transfection and siRNA interference in A549 and H1299 cells. (a) Western blot analysis showed the protein expression of CBLL1 in HBE and NSCLC. (b) Immunofluorescence results showed that CBLL1 was expressed in both cytoplasm and nucleus in A549 and H1299 cells. (c) Realtime RT‐PCR results showed that CBLL1 mRNA significantly decreased after transfection with CBLL1 siRNA in A549 and H1299 cells (P < 0.01). (d) Western blot results showed that CBLL1 protein was decreased after the transfection of CBLL1 siRNA (P < 0.01).
To explore the roles of CBLL1 in NSCLC cell proliferation and invasion, A549 and H1299 cells were transfected with CBLL1 siRNA. The mRNA expression of CBLL1 were considerably reduced after transfection of CBLL1 siRNA in A549 and H1299 cells compared to in the negative control group (P < 0.01, Fig 2c). The western blotting results showed that CBLL1 protein was significantly decreased after transfection with CBLL1 siRNA compared to negative control siRNA in A549 and H1299 cells (P < 0.01, Fig 2d).
CBLL1 promotes cell proliferation, colony formation, and cell cycle transition in A549 and H1299 cells
A CCK8 assay was conducted to detect cell proliferation, and growth curves were drawn. Compared to the negative control group, knockdown of CBLL1 significantly inhibited cell proliferation in A549 and H1299 cells (Fig 3a). Consistent with the results of the CCK8 assay, inhibition of CBLL1 significantly reduced foci numbers and sizes in A549 and H1299 cells (P < 0.05, Fig 3b).
Figure 3.
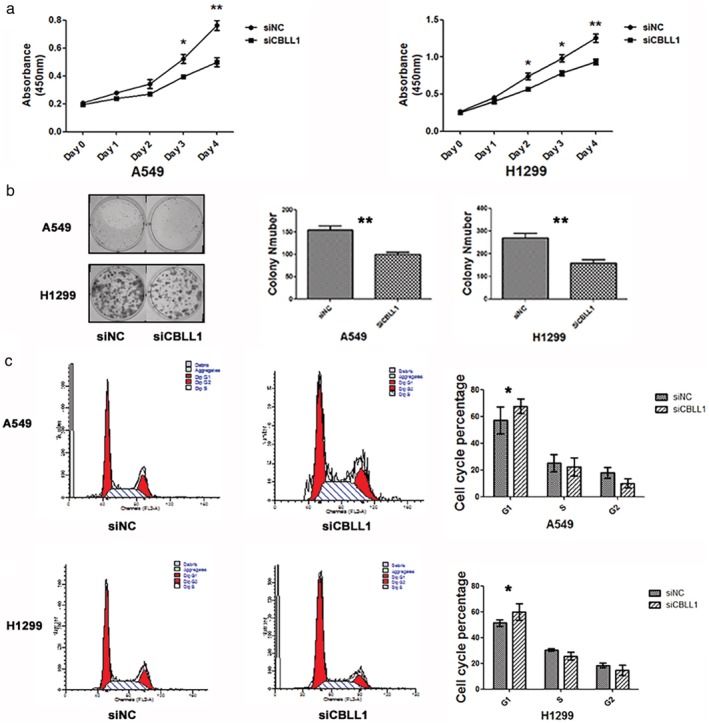
CBLL1 promotes cell proliferation and anchorage‐independent cell growth in A549 and H1299 cells. (a) CCK8 growth curves showed that the proliferation ability of A549 and H1299 cells was decreased after transfection with CBLL1 siRNA, *P < 0.05; **P < 0.01. (b) Assessment of clonogenic potential of the CBLL1‐depleted NSCLC cells. The colony formation ability decreased in the cells transfected with CBLL1 siRNA compared with the cells transfected with negative control siRNA (P < 0.01). (c) Effects of CBLL1 knockdown on A549 and H1299 cell cycle progression was analysed by flow cytometry, *P < 0.05.
We analyzed the cell cycle by flow cytometry, and found that the percentage of cells in G0/G1 phase was increased (A549, negative control vs. CBLL1 siRNA, 52.78 ± 0.90% vs. 59.42 ± 1.72%, P < 0.01; H1299 negative control vs. CBLL1 siRNA, 50.75 ± 1.88% vs. 62.00 ± 2.33%, P < 0.01; Fig. 3c), whereas the percentage of cells in S phase was decreased after treatment with CBLL1 siRNA compared to in negative control cells (A549, negative control vs. CBLL1 siRNA, 31.13 ± 2.44% vs. 25.03 ± 1.98%, P < 0.05; H1299 negative control vs. CBLL1 siRNA, 31.70 ± 2.09% vs. 25.70 ± 1.29%, P < 0.05). These results suggest that CBLL1 depletion arrests cell cycle progression at the G1/S boundary.
We further examined the effect of CBLL1 knockdown on the expression levels of several cell cycle‐related factors in A549 and H1299 cells including cyclinD1 and CDK4. Western blot analysis revealed that cyclin D1 and CDK4 expression was decreased after CBLL1 depletion (P < 0.05, Fig 3d). Taken together, these results indicate that inhibition of CBLL1 expression induces cell cycle arrest in G1 phase and suppresses NSCLC cell growth.
CBLL1 enhances the invasive ability of NSCLC cells
It has been reported that CBLL1 increased MDCK, A549, and H460 cell invasive abilities.18, 25 To explore the impact of CBLL1 on NSCLC cell invasion, we performed a Matrigel invasion assay. As shown in Fig 4a, CBLL1‐depleted A549 and H1299 cells showed weaker invasive abilities compared to negative control siRNA‐transfected cells (P < 0.001). To future explore the mechanism of how CBLL1 promoted NSCLC cell invasion, we evaluated whether CBLL1 depletion led to inhibition of cell invasion because of decreased expression of MMP2 and MMP9, which are extracellular membrane‐degrading enzymes associated with tumor invasiveness. The western blotting results showed that CBLL1 depletion significantly decreased MMP‐2 and MMP‐9 secretion (P < 0.01) (Fig 4b).
Figure 4.
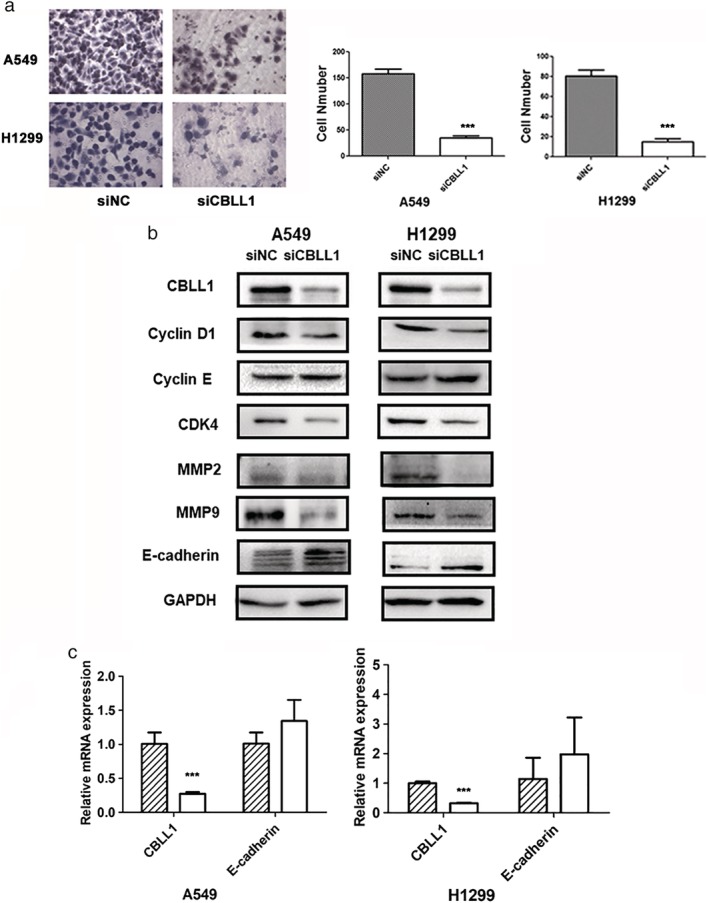
Interruption of CBLL1 decreased the cell invasive abilities of A549 and H1299 cells. (a) Cell invasion was examined using transwell chambers with Matrigel. The representative microscopic fields of invasive cells transfected with CBLL1 siRNA and negative control siRNA, respectively. Knockdown of CBLL1 greatly suppressed invasion in A549 and H1299 cells (P < 0.001). The histogram showed that the number of cells invaded (A549 and H1299) transfected with CBLL1 siRNA was significantly fewer (P < 0.001) than cells transfected with negative control siRNA (right panel),***P < 0.001. (b) Western blot analysis of cell cycle‐related and invasion‐related proteins in A549 and H1299 cells with CBLL1 depletion. (c) Realtime RT‐PCR results showed that E‐cadherin mRNA expression level was not changed after CBLL1 depletion in A549 and H1299 cells,***P < 0.001.  siNC,
siNC,  siCBLL1.
siCBLL1.
E‐cadherin was examined by qRT‐PCR and western blotting after inhibiting CBLL1 expression. The results showed that the protein level of E‐cadherin was increased after CBLL1 downregulation (P < 0.01, Fig 4b), whereas the mRNA level of E‐cadherin was not affected by downregulation of CBLL1 (Fig 4c).
Discussion
CBLL1 has been reported to be upregulated in human colon and gastric cancer tissues and its role varied in a cell type‐dependent manner.8, 13, 23 In this study, the expression of CBLL1 in NSCLC tissues and effect of CBLL1 on biological behavior were examined in A549 and H1299 cells. Analysis of CBLL1 expression in NSCLC tissues and clinicopathological factors showed that high CBLL1 protein expression was associated with tumor size in NSCLC. Thus, abnormal expression of CBLL1 in lung cancer plays an important role in the progression of NSCLC.
To further explore the effects of CBLL1 on the biological behavior of lung cancer cells, CBLL1 siRNA was transfected into cells to inhibit the expression of endogenous CBLL1. Our results showed that the cell proliferation and colony formation abilities were decreased after depletion of CBLL1 in A549 and H1299 cells, which is consistent with the MTT assay results in A549 and H460 cells.25 Cell cycle analysis showed that the cell cycle was stopped at G0/G1 after interrupting the expression of CBLL1. CBLL1 was predicted to promote cell cycle progression by inducing the G0/G1‐S transition in A549 and H1299 cells. Our results also showed that expression of the cell cycle‐related proteins cyclin D1 and CDK4 were decreased in CBLL1‐depleted A549 and H1299 cells, suggesting that CBLL1 affected the cell cycle by regulating the expression of cyclin D1 and CDK4. The protein expression of CDK4, was unchanged, while the expression of cyclin D1 was decreased after interference with CBLL1 expression in MCF7 and HEK293 cells.13
The invasive ability of A549 and H1299 cells was decreased after the expression of endogenous CBLL1 was depleted. MMP‐2 and MMP‐9 are well‐studied members of the matrix metalloproteinase (MMP) family, and play a leading role in lung cancer invasion and metastasis.29, 30 The expressions of MMP2 and MMP9 were found downregulated in CBLL1 knockdown cells, which indicated that CBLL1 probably participated in the degradation of ECM through MMP2 and MMP9 to promote the metastasis of NSCLC. In contrast, another study has found overexpression of CBLL1 decreased the breast cancer cell proliferation and migration capacity as a strong blockade of ERα in breast cancer cell line.23
In addition, CBLL1 can degrade E‐cadherin by ubiquitination to reduce cell adhesion and promote cell invasion.18, 25 In order to further clarify the effect of CBLL1 on E‐cadherin in NSCLC cells, we found that the protein expression of E‐cadherin was increased after CBLL1 depletion while the E‐cadherin mRNA was not affected after decreasing the expression of endogenous CBLL1. These findings further confirmed that CBLL1 could probably degrade E‐cadherin by protein level in lung cancer cells, which also facilitate the metastasis of NSCLC. However, the correlations between CBLL1 expression and membrane E‐cadherin degradation were not significant (data not shown) and additional NSCLC cases should be tested in the future.
In conclusion, we have demonstrated that CBLL1 is highly expressed in NSCLC cancer tissues compared with corresponding lung tissues. Our study shows that the high expression of CBLL1 is correlated with tumor size in NSCLC cases. Moreover, CBLL1 promotes the cell proliferation by inducing the G0/G1‐S transition and cell invasion by increasing degradation of ECM of NSCLC cells. These observations suggested CBLL1 as a potential target for the treatment of NSCLC. More research work is needed to elucidate the molecular functions of CBLL1 in NSCLC in the future.
Disclosure
The author reports no conflicts of interest in this work.
Acknowledgments
This work was supported by a grant from Innovation Team in Colleges and Universities of Shenyang, China 2015 (grant No.LT2015029), the National Natural Science Foundation of China (No.81101599) and the Natural Science Foundation of Liaoning Province (No. L20180551229).
References
- 1. Chen W, Zheng R, Baade PD et al. Cancer statistics in China. CA Cancer J Clin 2016; 66 (April): 115–32. [DOI] [PubMed] [Google Scholar]
- 2. Miller KD, Siegel RL, Lin CC et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016; 66 (4): 271–89. 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 3. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non‐small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc 2008; 83 (5): 584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fujita Y, Krause G, Scheffner M et al. Hakai, a c‐Cbl‐like protein, ubiquitinates and induces endocytosis of the E‐cadherin complex. Nat Cell Biol 2002; 4 (3): 222–31. [DOI] [PubMed] [Google Scholar]
- 5. Joazeiro C. The Tyrosine Kinase Negative Regulator {c‐Cbl} as a {RING‐Type,} {E2‐Dependent} {Ubiquitin‐Protein} Ligase. Science 1999; 286 (5438): 309–12. [DOI] [PubMed] [Google Scholar]
- 6. Levkowitz G, Waterman H, Ettenberg SA et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c‐Cbl/Sli‐1. Mol Cell 1999; 4 (6): 1029–40. [DOI] [PubMed] [Google Scholar]
- 7. Mukherjee M, Chow SY, Yusoff P et al. Structure of a novel phosphotyrosine‐binding domain in Hakai that targets E‐cadherin. EMBO J 2012; 31 (5): 1308–19. 10.1038/emboj.2011.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodríguez‐Rigueiro T, Valladares‐Ayerbes M, Haz‐Conde M et al. A novel procedure for protein extraction from formalin‐fixed paraffin‐embedded tissues. Proteomics 2011; 11 (12): 2555–9. [DOI] [PubMed] [Google Scholar]
- 9. Vleminckx K, Vakaet L, Mareel M, Fiers W, Van Roy F. Genetic manipulation of E‐cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991; 66 (1): 107–19. [DOI] [PubMed] [Google Scholar]
- 10. Nishimura T, Takeichi M. Chapter 2 remodeling of the adherens junctions during morphogenesis In: Current Topics in Developmental Biology, Vol. 89 Elsevier 2009; 33–54 Available from: http://www.sciencedirect.com/science/article/pii/S0070215309890029. [DOI] [PubMed] [Google Scholar]
- 11. Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell‐cell contacts. Annu Rev Cell Dev Biol 2007; 23 (1): 237–61. [DOI] [PubMed] [Google Scholar]
- 12. Palacios F, Tushir JS, Fujita Y, D'Souza‐Schorey C. Lysosomal targeting of E‐cadherin: A unique mechanism for the down‐regulation of cell‐cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol 2005; 25 (1): 389–402.http://mcb.asm.org/cgi/. 10.1128/MCB.25.1.389-402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Figueroa A, Kotani H, Toda Y et al. Novel roles of hakai in cell proliferation and oncogenesis. Mol Biol Cell 2009; 20 (15): 3533–42. 10.1091/mbc.e08-08-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hogan C, Dupré‐Crochet S, Norman M et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol 2009; 11 (4): 460–7. [DOI] [PubMed] [Google Scholar]
- 15. Zhou WJ, Geng ZH, Chi S et al. Slit‐Robo signaling induces malignant transformation through Hakai‐mediated E‐cadherin degradation during colorectal epithelial cell carcinogenesis. Cell Res 2011; 21 (4): 609–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abella V, Valladares M, Rodriguez T et al. miR‐203 regulates cell proliferation through its influence on hakai expression. PLoS One 2012; 7 (12): e52568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Figueroa A, Fujita Y, Gorospe M. Hacking RNA: Hakai promotes tumorigenesis by enhancing the RNA‐binding function of PSF. Cell Cycle 2009; 8 (22): 3648–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodríguez‐Rigueiro T, Valladares‐Ayerbes M, Haz‐Conde M, Aparicio LA, Figueroa A. Hakai reduces cell‐substratum adhesion and increases epithelial cell invasion. BMC Cancer 2011; 11: 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu M, Wu J, Hao ZW et al. Basolateral CD147 induces hepatocyte polarity loss by E‐cadherin ubiquitination and degradation in hepatocellular carcinoma progress. Hepatology 2018; 68 (1): 317–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mukherjee M, Jing‐Song F, Ramachandran S, Guy GR, Sivaraman J. Dimeric switch of Hakai‐truncated monomers during substrate recognition: Insights from solution studies and nmr structure. J Biol Chem 2014; 289 (37): 25611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Díaz‐Díaz A, Casas‐Pais A, Calamia V et al. Proteomic analysis of the E3 ubiquitin‐ligase hakai highlights a role in plasticity of the cytoskeleton dynamics and in the proteasome system. J Proteome Res 2017; 16 (8): 2773–88. 10.1021/acs.jproteome.7b00046. [DOI] [PubMed] [Google Scholar]
- 22. Shrestha H, Ryu T, Seo YW, Park SY, He Y, Dai W. Hakai, an E3‐ligase for E‐cadherin, stabilizes δ‐catenin through Src kinase. Cell Signal 2017; 31: 135–45. 10.1016/j.cellsig.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 23. Gong EY, Park E, Lee K. Hakai acts as a coregulator of estrogen receptor alpha in breast cancer cells. Cancer Sci 2010; 101 (9): 2019–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castosa R, Martinez‐Iglesias O, Roca‐Lema D et al. Hakai overexpression effectively induces tumour progression and metastasis in vivo. Sci Rep 2018; 8 (1): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Z, Wu Y, Tao Z, Ma L. E3 ubiquitin ligase Hakai regulates cell growth and invasion, and increases the chemosensitivity to cisplatin in non‐small‐cell lung cancer cells. Int J Mol Med 2018; 42 (2): 1145–51.http://www.spandidos-publications.com/. 10.3892/ijmm.2018.3683. [DOI] [PubMed] [Google Scholar]
- 26. Stewart DJ. Wnt signaling pathway in non‐small cell lung cancer. J Natl Cancer Inst 2014; 106 (1): 1–11. [DOI] [PubMed] [Google Scholar]
- 27. Yang Y‐L, Chen M‐W, Xian L. Prognostic and clinicopathological significance of downregulated e‐Cadherin expression in patients with non‐small cell lung cancer (NSCLC): A meta‐analysis. PLoS One 2014; 9 (6): e99763.http://dx.plos.org/. 10.1371/journal.pone.0099763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hui L, Zhang S, Dong X, Tian D, Cui Z, Qiu X. Prognostic significance of twist and N‐cadherin expression in NSCLC. PLoS One 2013; 8 (4): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Christofori G. New signals from the invasive front. Nature 2006; 441 (7092): 444–50. 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 30. Dong Q, Wang Y, Tang Z et al. Derlin‐1 Is overexpressed in non‐small cell lung cancer and promotes cancer cell invasion via EGFR‐ERK–mediated up‐regulation of MMP‐2 and MMP‐9. Am J Pathol 2013; 182 (3): 954–64. [DOI] [PubMed] [Google Scholar]