

Abstract
In the adult brain GABAA receptors (GABAARs) mediate the majority of synaptic inhibition that provides inhibitory balance to excitatory drive and controls neuronal output. In the immature brain GABAAR signaling is critical for neuronal development. However, the cell-autonomous role of GABAARs in synapse development remains largely unknown. We have employed the CRISPR-CAS9 technology to genetically eliminate GABAARs in individual hippocampal neurons and examined GABAergic and glutamatergic synapses. We found that development of GABAergic synapses, but not glutamatergic synapses, critically depends on GABAARs. By combining different genetic approaches, we have also removed GABAARs and two ionotropic glutamate receptors, AMPA receptors (AMPARs) and NMDA receptors (NMDARs), in single neurons and discovered a striking dichotomy. Indeed, while development of glutamatergic synapses and spines does not require signaling mediated by these receptors, inhibitory synapse formation is crucially dependent on them. Our data reveal a critical cell-autonomous role of GABAARs in inhibitory synaptogenesis and demonstrate distinct molecular mechanisms for development of inhibitory and excitatory synapses.
Keywords: GABAergic synapse, GABAergic synapse development, GABAA receptor, cell autonomous, glutamate receptor, inhibitory synapse, excitatory synapse
Introduction
GABAA receptors (GABAARs) are ligand-gated hetero-pentameric anion channels assembled from various combinations of 19 subunits: α (1-6), β (1-3), γ (1-3), δ, 𝜖, 𝜃, π, and ρ (1-3), although most GABAARs in the brain consist of two α subunits, two β subunits, and one γ or δ subunit (Macdonald and Olsen, 1994; Chang et al., 1996; Sieghart and Sperk, 2002; Mody and Pearce, 2004; Olsen and Sieghart, 2008). These receptors mediate the majority of phasic and tonic inhibition in the adult brain and are critical in the regulation of neuronal excitability and neural network function. In developing neurons, GABAAR activation can also provide membrane depolarization and increase neuronal activity, resulting from a relatively positive Cl- reversal potential due to high expression of Na+/K+/Cl- co-transporter 1 (NKCC1) and low expression of K+/Cl- co-transporter 2 (KCC2) in immature neurons (Ben-Ari et al., 1997; Owens and Kriegstein, 2002; Ben-Ari et al., 2007).
The role of GABAAR-mediated signaling in neuronal development and function has been extensively studied (Ben-Ari et al., 1997; Owens and Kriegstein, 2002; Ben-Ari et al., 2007). Early pharmacological experiments have demonstrated that GABAAR activity can modulate neurogenesis, neuronal migration and differentiation, and synapse development in the immature brain (Owens and Kriegstein, 2002; Ben-Ari et al., 2007). However, pharmacological approaches do not separate the cell-autonomous function of GABAARs from indirect neuronal network effects associated with global blockade of the receptors, and also do not address the structural role of GABAARs in the regulation of synapse development. Depolarizing GABAAR activity has also been shown to functionally interact with NMDA receptors (NMDARs) to facilitate NMDAR activation and regulate synapse development in immature neurons (Owens and Kriegstein, 2002; Ben-Ari et al., 2007). In addition, by manipulating NKCC1 or KCC2 expression, the role of depolarizing GABAAR activity in synapse, and spine development has been inferred (Chudotvorova et al., 2005; Akerman and Cline, 2006; Liu et al., 2006; Wang and Kriegstein, 2008), although recent studies have shown that the regulation of synapse development by KCC2 does not require its ion transport function (Li et al., 2007; Fiumelli et al., 2013). Furthermore, synaptic GABAARs in knockdown or conventional knockout (KO) mice of GABAAR subunits are reduced or lost, leading to the impairment of GABAergic synapse formation and maturation (Fritschy et al., 2006, 2012; Patrizi et al., 2008; Frola et al., 2013), which provides genetic evidence for the role of GABAAR subunits in synapse development. However, in these genetic models, neurons may adapt to the global absence of the GABAAR subunits and to altered neural network activities throughout their development. Thus, the cell-autonomous role of GABAARs in the regulation of synapse development remains largely unclear.
Here we have utilized the CRISPR-Cas9 approach to perform single-cell genetic deletion (Incontro et al., 2014) of all functional GABAARs and examine excitatory and inhibitory synapse development. This is achieved by targeting the β1, β2, and β3 subunits of the GABAARs in individual hippocampal neurons. These β subunits are required for the receptor assembly and agonist binding (Connolly et al., 1996; Tretter et al., 1997; Baumann et al., 2001; Olsen and Sieghart, 2008; Nguyen and Nicoll, 2018). We found that in neurons lacking GABAARs, GABAergic synapses are strongly impaired without an accompanying change of excitatory transmission and the spine density. Furthermore, combined genetic deletion of GABAARs and ionotropic glutamate receptors (iGluRs), including both AMPARs and NMDARs, reveals that signaling mediated by these receptors is critical for inhibitory, but not excitatory, synapse development.
Materials and Methods
Mouse Maintenance
All experiments using mice were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee at National Institute of Neurological Disorders and Stroke (NINDS) and National Institutes of Health (NIH). Adult C57BL/6 mice were purchased from Charles River, housed and bred under a 12-h circadian cycle. GRIA1-3fl/flGRIN1fl/fl mice were generated as described previously (Lu et al., 2013). Animals of either sex were used for the experiments.
Plasmids
Mouse GABRB-1, -2, -3 (Myc-DDK-tagged) was purchased from OriGene (Cat #: MR227185, MR 222938, and MR 222856, respectively). FUGW-Cre-mCherry plasmid was a gift from Roger Nicoll’s lab at UCSF. To screen the β1-3 single-guidance RNA (gRNA) sequences for potential off-target effects, we used the gRNA design target tool1,2 . The human codon-optimized Cas9 and chimeric gRNA expression plasmid (pSpCas9 BB-2A-GFP, or PX458) was developed by the Feng Zhang lab at MIT and obtained from Addgene (plasmid #48138). To generate (gRNA) plasmids, a pair of annealed oligos (20 bp) was ligated into the single gRNA scaffold of PX458. The primers used to design the specific gRNA targets were:
GABRB1 #5 forward (5′ to 3′) CACCg GCCGCGAGGGCTTCGGGCGT; GABRB1 #5 Reverse (5′ to 3′): AAAC ACGCCCGAAGCCCTCGCGGC c;
GABRB2 #2 forward (5′ to 3′) CACCg CAGACAGCGGCGATTATTAA; GABRB2 #2 reverse (5′ to 3′) AAAC TTAATAATCGCCGCTGTCTG c;
GABRB3#2 forward (5′ to 3′): CACCg ACGGTCGACAAGCTGTTGAA; GABRB3#2 reverse (5′ to 3′): AAAC TTCAACAGCTTGTCGACCGT c.
For β1-3 gRNAs, the GABRB1#5- GABRB2#2- GABRB3#2 triple gRNA expression unit (U6 promoter + gRNA + scaffold + PolyT tail) was de novo synthesized by Genscript and cloned into pSpCas9 BB-2A-GFP (β1-3gRNAs) via AflIII/XbaI sites. To rescue the β1-3 subunit deletion, gRNA resistant β1, 2 or 3 (β1∗, β2∗, and β3∗) plasmids were constructed. Briefly, point mutations in β1-3 gRNA-targeting sites were generated by overlapping PCR and cloned into the pCAGGS-IRES-GFP/mCherry expression plasmid. gRNA resistant β1, 2 or 3 in pCAGGS-IRES-mCherry was co-transfected with β1-3gRNAs. Neurons with both GFP and mCherry fluorescence were used for recording and imaging. All constructs were verified by DNA sequencing.
Cell Culture and Transfection
HEK293T cells were grown in DMEM (GIBCO) supplemented with 10% fetal bovine serum (FBS) (GIBCO), 1% Pen/Strep, 1% Glutamine, and 1% sodium pyruvate, in a humidified atmosphere in a 37°C incubator with 5% CO2. Transfection was performed in 24-well plates with indicated cDNAs using calcium phosphate transfection.
Dissociated Hippocampal Neuronal Culture
Hippocampal cultures were prepared from E18 time-pregnant mice as previously described (Gu et al., 2016a). Briefly, the mouse hippocampi were dissected out in ice-cold Hank’s balanced salt solution, and digested with papain (Worthington, LK003176) solution at 37°C for 45 min. After centrifugation for 5 min at 800 rpm, the pellet was resuspended in DNase I-containing Hank’s solution, then was mechanically dissociated into single cells by gentle trituration using a pipette. Cells were placed on top of Hank’s solution mixed with trypsin inhibitor (10 mg/ml, Sigma T9253) and BSA (10 mg/ml, Sigma A9647), and centrifuged at 800 rpm for 10 min. The pellet was resuspended in Neurobasal plating media containing 2% B27 supplements and L-glutamine (2 mM). Neurons were plated at a density of 150,000–200,000 cells/well on poly-D-lysine (Sigma P6407)-coated 12 mm glass coverslips residing in 24-well plates for electrophysiology recording, and a lower plating density (100,000–150,000 cells/well) was adopted when neurons were used for immunocytochemistry. Culture media were changed by half volume with Neurobasal maintenance media containing 2% B27 supplements and L-glutamine (2 mM) twice a week.
Neuronal Transfection
Hippocampal neurons were transfected at day 2–3 in vitro (DIV2-3) using a modified calcium phosphate transfection as described before (Li et al., 2017). Briefly, 5 μg total cDNA was used to generate 200 μL total precipitates, which was added to each well at a 40 μL volume (5 coverslips/group). After 2 h incubation in a 37°C incubator, the transfected cells were incubated with 37°C pre-warmed, 10% CO2 pre-equilibrated Neurobasal medium, and placed in a 37°C, 5% CO2 incubator for 20 min to dissolve the calcium-phosphate particles. The coverslips were then transferred back to the original conditioned medium. The cells were cultured to DIV 14–24 before experiments.
Immunocytochemistry
Cells grown on coverslips were rinsed with PBS twice and fixed in 4% paraformaldehyde (PFA)/4% sucrose/1x PBS solution for 15 min at RT or 1% PFA in 0.1 M Na-acetate buffer for 13 min at RT, permeabilized and blocked with 0.1% Triton X-100/10% normal goat serum in 1x PBS for 1 h (for surface labeling, cells were incubated in 10% normal goat serum in 1x PBS for 1 h without Triton X-100). Cells were labeled with primary antibodies as follows: anti-β1 (1:500, MABN498, Millipore), anti-β2 (1:800, AB5561, Millipore), anti-β3 (1:1000, 75149, Neuromab), anti-Myc (1:1000, 71D10, cell signaling), anti-gephyrin (1:500, 147021, Synaptic Systems), anti-vGAT (1:1500, 131004, 131002, Synaptic Systems), anti-Neuroligin2 (1:1,000, 129511, Synaptic Systems), GluA1(1:1000, MAB2263, Millipore), anti-PSD-95 (1:1000, 75-028, Neuromab), anti-vGluT1 (1:1000, 135302, Synaptic Systems), anti-GFP (1:10000, Aves labs) in 3%NGS/1x PBS solutions, incubated overnight at 4°C. Cells were washed three times with 1x PBS and then incubated with Alexa Fluor 405, 555 or 647-conjugated IgG for 1 h. Coverslips were washed for four times with 1× PBS and mounted with Fluoromount-G (Southern Biotech).
Image Acquisition and Analysis
Fluorescence images were obtained with the Zeiss Zen acquisition software and a Zeiss LSM 880 laser scanning confocal microscope using a 63 × oil objective (numerical aperture 1.4) at room temperature. Optical sections, merged by maximum projection, were analyzed at a time using the Image J puncta analyzer program. Thresholds were set at 3 SDs above the mean staining intensity of six nearby regions in the same visual field. Thresholded images present a fixed intensity for all pixels above threshold after having removed all of those below. Labeled puncta were defined as areas containing at least four contiguous pixels after thresholding. For puncta analysis, Images from 3 dendrites (35 μm in length) per neuron were collected and quantified. For co-localization analysis, images from soma, or three secondary dendrites (35 μm in length per dendrite) per neuron were collected and quantified. For β subunit co-localization with synaptic markers, β subunits and gephyrin or vGAT were separately thresholded and confirmed visually to select appropriate clusters following a minimal size cut-off, which included all recognizable clusters. The gephyrin- or vGAT- positive β subunit puncta indicate the number of β subunit puncta per 10 mm showing at least 50% pixel overlapping with thresholded gephyrin or vGAT puncta. Synaptic vs. Total ratios was calculated by the measurement of gephyrin- or vGAT- positive β subunit puncta compared to total β subunit puncta. For gephyrin and vGAT co-localization in dendrites, gephyrin and vGAT puncta were separately thresholded and confirmed visually to select appropriate clusters following a minimal size cut-off, which included all recognizable clusters. The gephyrin-positive vGAT puncta indicate the number of vGAT puncta showing at least 50% pixel overlapping with thresholded gephyrin puncta and the co-localization percentage was calculated by the measurement of gephyrin-positive vGAT puncta compared to total number of thresholded vGAT puncta. The same procedure was used to calculate vGAT-positive gephyrin. For spine density analysis, GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence. 2–3 secondary or tertiary dendrites (50–200 μm long, 20–100 μm from the soma) from each neuron were collected, the number of dendritic protrusions were counted manually. For spine type analysis, images from three dendrites (35 μm in length per dendrite) per neuron were collected and spine types in each dendrite were quantified. Different spine types (mushroom, thin, stubby, and filopodia) were counted manually for each dendrite, and the data were combined from three dendrites to calculate the fractions of each type of spines for that neuron.
Electrophysiology
Electrophysiological recordings were performed in dissociated hippocampal neuronal cultures as described (Gu et al., 2016b). Briefly, recordings were performed in artificial cerebrospinal fluid (ACSF) containing (in mM) NaCl 119, KCl 2.5, NaHCO3 26, Na2PO4 1, glucose 11, CaCl2 2.5, MgCl2 1.3. The intracellular solution for mIPSC recording contained (in mM) CsMeSO4 70, CsCl 70, NaCl 8, EGTA 0.3, HEPES 20, MgATP 4, and Na2GTP 0.3. The intracellular solution for mEPSC recording contained (in mM) CsMeSO4 135, NaCl 8, HEPES 10, Na3GTP 0.3, MgATP 4, EGTA 0.3, QX-314 5, and spermine 0.1. Osmolality was adjusted to 285–290 Osm and pH was buffered at 7.25–7.35. For recording AMPA mEPSCs at -70 mV, both picrotoxin (0.1 mM) and TTX (0.5 μM) were added to ACSF; for recording NMDA mEPSCs at +40 mV, glycine (1 μM), NBQX (10 μM), picrotoxin (0.1 mM) and TTX (0.5 μM) were added to ACSF; for recording mIPSCs at -70 mV, NBQX (10 μM), AP-5 (50 μM), and TTX (0.5 μM) were added to ACSF. In KCl treated experiment, 15mM KCl was applied to depolarize the cultured neuron. For the recording of GABAA receptor mediated tonic current, bicuculline (20 μM) was applied for about 1–2 min in ACSF containing NBQX, AP-5, and TTX. The change of currents was measured at -60 mV. mIPSCs, or mEPSCs were semiautomatically detected by offline analysis using in-house software in Igor Pro (Wavemetrics) developed in Dr. Roger Nicoll’s laboratory at UCSF. All events were visually inspected to ensure they were mI/EPSCs during analysis and those non-mI/EPSC traces were discarded. Series resistance was monitored and not compensated, and cells in which series resistance varied by 25% during a recording session were discarded. Synaptic responses were collected with a Multiclamp 700B amplifier (Axon Instruments), filtered at 2 kHz and digitized at 10 kHz. All pharmacological reagents were purchased from Abcam, and other chemicals were purchased from Sigma.
Statistics
All data were presented as mean ± sem (standard error of mean). Direct comparisons between two groups were made using two-tailed Student’s t-test. Multiple group comparisons were made using one-way analysis of variance (ANOVA) with the Bonferroni test. The significance of cumulative probability distributions was assessed by Kolmogorov-Smirnov test. Statistical significance was defined as p < 0.05, 0.01, 0.001, or 0.0001 (indicated as ∗, ∗∗, ∗∗∗, or ∗∗∗∗, respectively). p values ≥ 0.05 were considered not significant (ns).
Results
We first examined the expression and subcellular distribution of GABAAR β subunits (GABRBs) in mouse primary hippocampal neuron cultures. In 18 days in vitro (DIV) neurons in culture, the immunolabeling of GABAAR β subunits, including β1, β2 and β3, demonstrated that all three β subunits were expressed in hippocampal neurons with substantial synaptic localization at both dendritic (Supplementary Figures S1A,B) and somatic (Supplementary Figures S1C,D) regions, consistent with a recent electron microscopy study in rat hippocampi (Kerti-Szigeti and Nusser, 2016). Our data also indicate that among the three β subunits, β2 exhibits a higher level of synaptic distribution (Supplementary Figure S1).
Expression and synaptic localization of β1-3 subunits in hippocampal neurons indicate that functional KO of GABAARs requires genetic deletion of all three β subunits. To this end, we have employed the CRISPR-Cas9 technology to develop single-guide RNAs (gRNAs) to target gene loci of three GABRBs, respectively, that encode GABAAR β subunits in mouse genome. Among several gRNA candidates for each β subunits, positive gRNAs that also expressed GFP effectively reduced the expression of co-transfected Myc-β1, 2 or 3 in HEK293T cells (Supplementary Figure S2). We thus generated a construct (hereafter β1-3 gRNAs) containing the three positive gRNAs for β1, β2, and β3, respectively (Figure 1A). To test the KO effect of β1-3 gRNAs in neurons, we transfected hippocampal neuronal cultures at DIV2-3 and performed immunocytochemical analysis at DIV13-14. We found that compared with empty gRNA vector (hereafter GFP)-transfected neurons, the immunolabeling of β1, β2, or β3 in β1-3 gRNAs expressing neurons was strongly diminished (Figure 1B–D). Furthermore, electrophysiological recordings demonstrated that miniature inhibitory postsynaptic currents (mIPSCs) was essentially lost in neurons expressing β1-3 gRNAs (Figure 1F), in agreement with a recent report (Nguyen and Nicoll, 2018). In contrast, no change of mIPSCs was observed in neurons expressing the empty gRNA vector (Figure 1E). Furthermore, we treated the cultured neurons with 15 mM K+ to enhance synaptic activity and measured GABAergic transmission. We found that although 15 mM K+ significantly increased the mIPSC frequency in control neurons, it didn’t change GABAergic transmission in neurons expressing β1-3 gRNAs (Figure 1G). Indeed, mIPSCs were barely detectable in these neurons either before or after 15 mM K+ treatment (Figure 1G). In addition, tonic inhibition generated by persistent activation of extrasynaptic GABAARs by ambient GABA has been observed in many types of neurons and has a profound effect on neuronal excitability (Farrant and Nusser, 2005; Glykys and Mody, 2007; Belelli et al., 2009). To examine whether extrasynaptic GABAAR-mediated tonic currents were diminished in neurons expressing β1-3 gRNAs, we measured tonic currents by adding the GABAAR antagonist, bicuculline, in the perfusion solution. We found that compared with control neurons whereby GABAAR-mediated tonic currents were readily detected by bicuculline, little tonic currents were observed in neurons expressing β1-3 gRNAs (Figure 1H).
FIGURE 1.
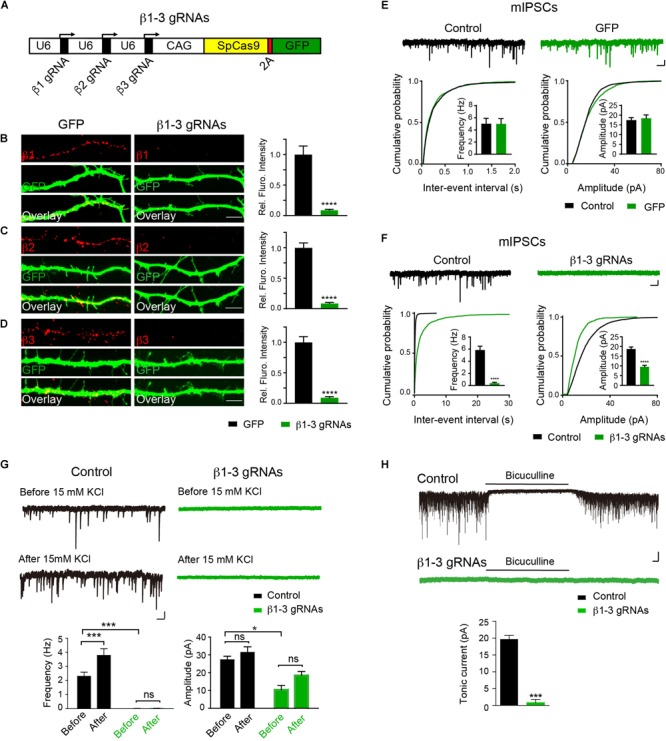
Single-cell KO of the GABAAR β1-3 subunits eliminated GABAergic synaptic transmission. (A) Schematic of β1-3 gRNA vector. (B–D) Representative images showed the loss of β1 (B), β2 (C), or β3 (D) subunits in hippocampal neurons expressing β1-3 gRNA vector as compared to neurons expressing the empty gRNA vector (GFP) (GFP, n = 15; β1-3 gRNAs, n = 15; ∗∗∗∗p < 0.0001, t-test; N = 3). Scale bar, 5 μm. (E) Expression of the empty gRNA vector (GFP) in hippocampal neurons did not change inhibitory synaptic transmission (control, n = 10; GFP, n = 9; N = 3; t-test; p > 0.05 for mIPSC frequency and amplitude; Kolmogorov-Smirnov test was used for cumulative graphs, p > 0.05 for both conditions). Scale bar, 500 ms, 20 pA. (F) Expression of β1-3 gRNA vector in hippocampal neurons essentially eliminated inhibitory synaptic transmission (control, n = 19; β1-3 gRNAs, n = 21; N = 5; t-test, ∗∗∗∗p < 0.0001 for both frequency and amplitude; Kolmogorov-Smirnov test was used for cumulative graphs, ∗∗∗∗p < 0.0001 for both conditions). Scale bar, 500 ms, 20 pA. (G) 15 mM KCl significantly increased the mIPSC frequency in control neurons but not in neurons expressing β1-3 gRNAs at DIV 12-15 (control, ∗∗∗n = 16; p < 0.001 for frequency, p > 0.05 for amplitude; β1-3 gRNAs, p > 0.05 for frequency, p > 0.05 for amplitude; n = 19; For β1-3 gRNA amplitude analysis, only 5 out of 19 cells had mIPSC events for analysis; N = 2; one-way ANOVA followed by the Bonferroni test). Scale bar, 500 ms, 20 pA. (H) GABAAR-mediated tonic currents in control and β1-3 gRNAs-expressing neurons at DIV 14-17 (Control, n = 23; β1-3 gRNAs, n = 15; N = 3; ∗∗∗p < 0.001, t-test). Scale bar, 10 s, 50 pA. n represents the number of cells analyzed and N represents the number of independent experiments.
We further performed rescue experiments to characterize the role of individual β subunits in the regulation of GABAergic transmission. To this end, we developed gRNA-resistant β1, β2 or β3 in HEK cells through immunocytochemical experiments (β1∗, β2∗, or β3∗ in Figure 2A,C,E, respectively). We then co-transfected β1∗ (Figure 2B), β2∗ (Figure 2D), or β3∗ (Figure 2F) with β1-3 gRNAs in hippocampal neuronal cultures and measured mIPSCs. We found that the loss of GABAergic transmission in neurons expressing β1-3 gRNAs could be rescued by co-expressing β1∗, β2∗, or β3∗ (Figure 2B,D,F, respectively), indicating that individual β subunits can support basal inhibitory transmission and can substitute for each other for GABAergic transmission in hippocampal neurons. Together, these data demonstrate that we have successfully eliminated GABAergic transmission in single hippocampal neurons and individual β subunits can rescue GABAergic transmission in neurons lacking all endogenous β subunits.
FIGURE 2.
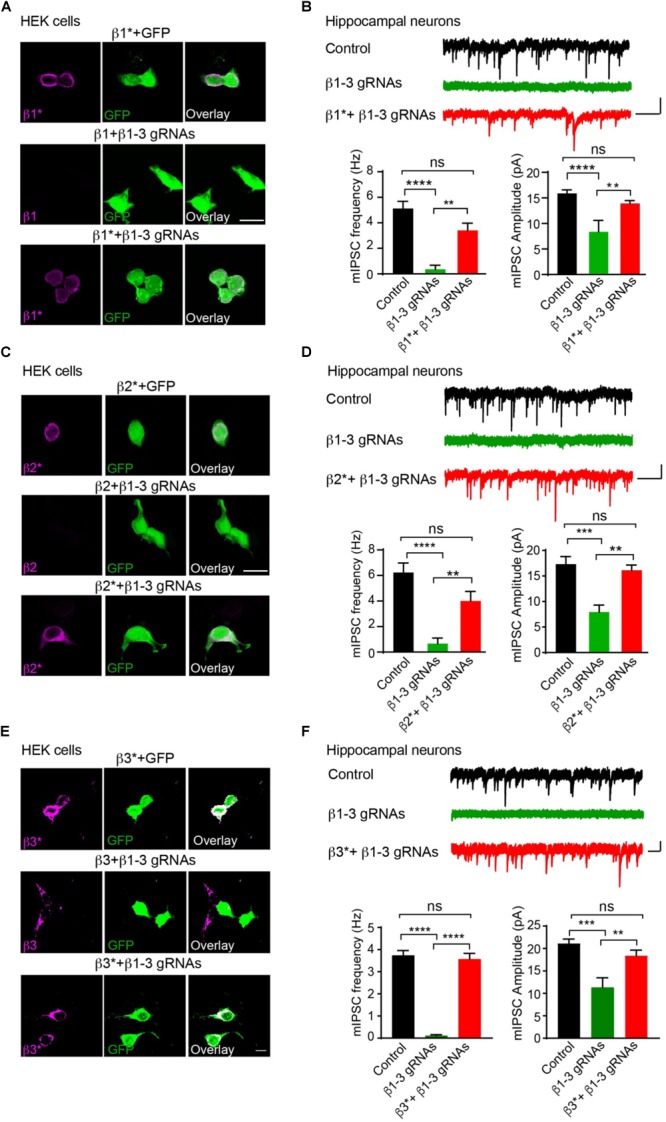
Rescue of GABAergic transmission in neurons expressing β1-3 gRNAs by gRNA resistant β1∗, β2∗, or β3∗. (A) Representative images showed that β1-3 gRNA failed to reduce the expression of the gRNA resistant β1 (β1∗) in HEK293T cells. Scale bar, 5 μm. (B) mIPSC recording showed that β1∗ largely rescued the loss of GABAergic transmission in neurons expressing β1-3 gRNAs (n = 18, 8, and 15 for control, β1-3 gRNAs, β1∗ + β1-3 gRNAs, respectively; N = 3; One-way ANOVA followed by the Bonferroni test, ∗∗p < 0.01, ∗∗∗∗p < 0.0001). Scale bar, 500 ms, 20 pA. (C) Representative images showed that β1-3 gRNA failed to reduce the expression of the gRNA resistant β2 (β2∗) in HEK293T cells. Scale bar, 5 μm. (D) mIPSC recording showed that β2∗ largely rescued the loss of GABAergic transmission in neurons expressing β1-3 gRNAs [n = 23, 8, and 12 for control, β1-3 gRNAs, β2∗ + β1-3 gRNAs, respectively; N = 3; One-way ANOVA followed by the Bonferroni test, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 (for frequency), ∗∗∗p < 0.001 (for amplitude)]. Scale bar, 500 ms, 20 pA. (E) Representative images showed that β1-3 gRNA failed to reduce the expression of the gRNA resistant β3 (β3∗) in HEK293T cells. Scale bar, 5 μm. (F) mIPSC recording showed that β3∗ rescued the loss of GABAergic transmission in neurons expressing β1-3 gRNAs (n = 12, 12, and 12 for control, β1-3 gRNAs, β3∗ + β1-3 gRNAs, respectively; N = 3; One-way ANOVA followed by the Bonferroni test, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Scale bar, 500 ms, 20 pA. n represents the number of cells analyzed and N represents the number of independent experiments.
The loss of functional GABAARs in individual hippocampal neurons allowed us to examine the cell-autonomous role of these receptors in the regulation of GABAergic synapse development. We found that in neurons expressing β1-3 gRNAs, the immunolabeling of vGAT and gephyrin, the pre- and post-synaptic markers of GABAergic synapses, respectively, at both somatic and dendritic regions, was significantly decreased by ∼50% (Figure 3A–C), indicating a reduction of GABAergic synapse density. Interestingly, there was no change of the puncta density of Neuroligin2 (NL2), a key synaptogenic cell adhesion molecule for GABAergic synapses (Graf et al., 2004; Varoqueaux et al., 2004; Chih et al., 2005; Poulopoulos et al., 2009; Li et al., 2017), in neurons lacking GABAARs (Figure 3D), suggesting that NL2 clustering is independent of GABAARs and may be an upstream event of GABAAR-mediated signaling for inhibitory synapse development. Taken together, these data demonstrate that genetic deletion of GABAARs at the single-cell level leads to a substantial reduction of GABAergic synapses.
FIGURE 3.
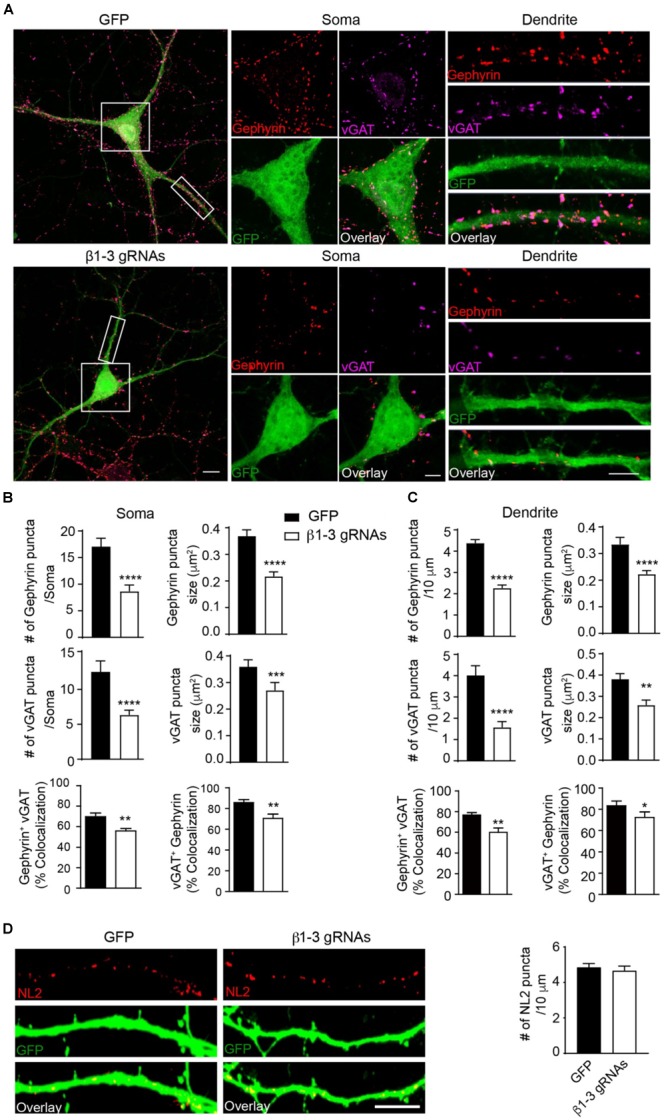
Loss of GABAARs strongly reduced inhibitory synapse density in hippocampal neurons. (A–C) Single-cell KO of GABAARs significantly reduced gephyrin (red) and vGAT (magenta) puncta as well as co-localization of gephyrin and vGAT in hippocampal neurons. (A) Representative images of gephyrin and vGAT-immunolabeling in neurons expressing GFP (top) or β1-3 gRNAs (bottom). (B) Bar graphs showed the quantitation of gephyrin (top), vGAT (middle), and co-localization of gephyrin and vGAT (bottom) in neuronal somata (gephyrin density and size: GFP, n = 22, β1-3 gRNAs, n = 24; ∗∗∗∗p < 0.0001; vGAT density and size: GFP, n = 22, β1-3 gRNAs, n = 24, ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001; co-localization: percentage of gephyrin puncta colocalized with vGAT (gephyrin+ vGAT) (GFP, n = 22, β1-3 gRNAs, n = 24; ∗∗p < 0.01) and percentage of vGAT puncta colocalized with gephyrin (vGAT+ gephyrin) (GFP, n = 22, β1-3 gRNAs, n = 24; ∗∗p < 0.01); t-test; N = 5). (C) Bar graphs showed the quantitation of gephyrin (top), vGAT (middle), and co-localization of gephyrin and vGAT (bottom) in neuronal dendrites (gephyrin density and size: GFP, n = 63, β1-3 gRNAs, n = 66, ∗∗∗∗p < 0.0001; vGAT density and size: GFP, n = 22, β1-3 gRNAs, n = 24, ∗∗∗∗p < 0.0001, ∗∗p < 0.01; co-localization: percentage of gephyrin puncta colocalized with vGAT (gephyrin+ vGAT) (GFP, n = 35, β1-3 gRNAs, n = 36; ∗∗p < 0.01) and percentage of vGAT puncta colocalized with gephyrin (vGAT+ gephyrin) (GFP, n = 35, β1-3 gRNAs, n = 36; ∗p < 0.05); t-test; N = 6). GFP was not immunolabeled with anti-GFP antibodies. Scale bar, 10 μm for whole cell, 5 μm for somata and dendrite. (D) Neuroligin2 (NL2) puncta were not changed in hippocampal neurons expressing β1-3 gRNAs (GFP, n = 19, β1-3 gRNAs, n = 21, p > 0.05, t-test; N = 3). GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence (green). Scale bar, 5 μm. n represents the number of cells analyzed and N represents the number of independent experiments.
Pharmacological blockade of GABAARs can induce homeostatic adaptation of excitatory synaptic transmission in neurons (Turrigiano et al., 1998; Shepherd et al., 2006; Anggono et al., 2011; Diering et al., 2014). Specifically, chronic inhibition of GABAARs reduces AMPAR-mediated excitatory transmission. We thus examined how excitatory synapses adapted to the single-cell silencing of GABAergic inhibitory transmission. Surprisingly, recording of miniature excitatory postsynaptic currents (mEPSCs) in the presence of TTX in hippocampal neurons expressing β1-3 gRNAs showed that there was no change of frequency and amplitude of mEPSCs (Figure 4A). In addition, immunocytochemical analysis demonstrated that the surface expression of GluA1, a key AMPAR subunit in hippocampal neurons (Lu et al., 2009), and the puncta of vesicular glutamate transporter 1 (vGluT1) did not change in β1-3 gRNA-expressing neurons (Figure 4B,C), suggesting that AMPAR trafficking to the neuronal surface and excitatory synapse density were not altered in these neurons lacking functional GABAARs. Furthermore, we measured the spine density by immunolabeling GFP and found that the spine density or type was not significantly changed in neurons expressing β1-3 gRNAs (Figure 4D,E). Collectively, these data show that single-cell elimination of GABAergic transmission does not induce a homeostatic reduction of excitatory synaptic transmission and also leads to little change of cell biological properties of excitatory synapses, including surface AMPAR expressing levels, and the density of excitatory synapses.
FIGURE 4.
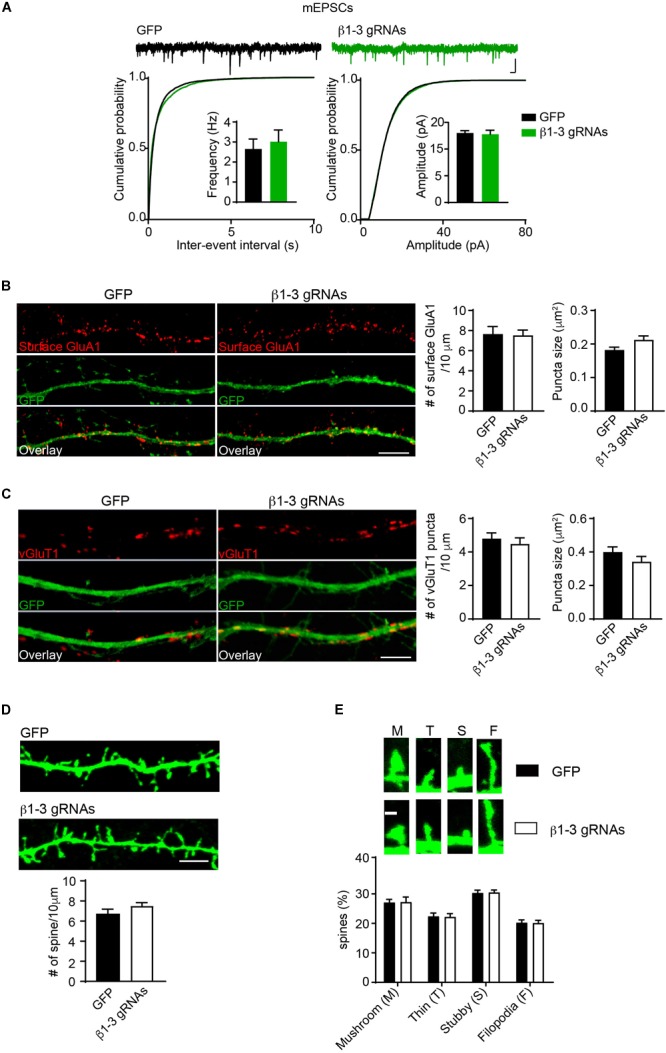
Loss of GABAergic transmission in individual neurons did not change glutamatergic transmission. (A) mEPSCs recording showed loss of GABAARs in individual neurons did not change glutamatergic transmission. Hippocampal neurons were transfected with β1-3 gRNAs at DIV3 and recorded at DIV14-17 (n = 25 for GFP and β1-3 gRNAs; p > 0.05 for mEPSC frequency and amplitude, t-test; Kolmogorov-Smirnov test was used for cumulative graphs, p > 0.05 for both conditions, N = 4). Scale bar, 100 ms, 20 pA. (B) Single-cell genetic deletion of GABAARs did not change the expression levels of surface GluA1 (GFP, n = 17; β1-3 gRNAs, n = 19; p > 0.05 for both conditions, t-test; N = 3). GFP was not immunolabeled with anti-GFP antibodies. Scale bar, 5 μm. (C) Single-cell genetic deletion of GABAARs did not change the vGluT1 puncta (GFP, n = 24; β1-3 gRNAs, n = 28; p > 0.05 for both conditions, t-test; N = 3). GFP was not immunolabeled with anti-GFP antibodies. Scale bar, 5 μm. (D) The density of dendritic spines was not altered in hippocampal neuron expressing β1-3 gRNAs at DIV 18 (GFP, n = 26; β1-3 gRNAs, n = 35; p > 0.05, t-test; N = 3). GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence (green). Scale bar, 5 μm. (E) Normal spine types in hippocampal neurons expressing β1-3 gRNAs (GFP, n = 26; β1-3 gRNAs, n = 28; p > 0.05, t-test; N = 3). GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence (green). Scale bar 1 μm. n represents the number of cells analyzed and N represents the number of independent experiments.
We previously employed a Cre-LoxP system to genetically delete both AMPARs and NMDARs in single hippocampal neurons and found that these iGluRs were dispensable for spinogenesis (Lu et al., 2013). One possibility was that in neurons lacking both AMPARs and NMDARs, remaining GABAARs could generate depolarizing drive in developing neurons and thus might provide activity necessary for spine development. We thus combined β1-3 gRNAs with the conditional KO of both AMPARs and NMDARs to genetically remove functional GABAARs, AMPARs and NMDARs in individual neurons and examined excitatory and inhibitory synapses. In hippocampal neuronal cultures prepared from GRIA1-3fl/flGRIN1fl/fl in which three genes encoding AMPAR subunits (GluA1, GluA2 and GluA3) and the gene encoding the NMDAR obligatory subunit, GluN1, are all conditional alleles (Lu et al., 2013), we co-expressed Cre-mCherry and β1-3 gRNAs through transfection. About 2 weeks after transfection, we performed mIPSCs and mEPSCs recordings in transfected neurons and found the loss of inhibitory synaptic transmission and both AMPAR- and NMDAR-mediated excitatory synaptic transmission in these neurons (Figure 5A,B).
FIGURE 5.
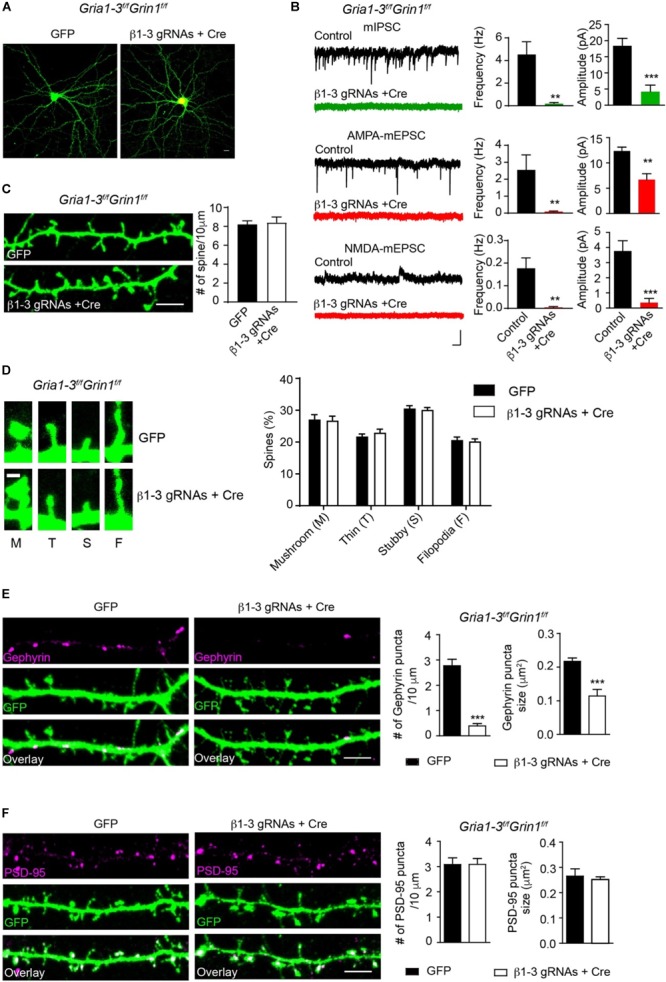
Genetic deletion of GABAARs, AMPARs, and NMDARs impaired inhibitory, but not excitatory synapses. (A) Representative images showed neurons cultured from GRIA1-3fl/flGRIN1fl/fl mice expressing empty gRNA vector (GFP, left) or expressing both Cre-mCherry/ β1-3 gRNAs(right). Scale bar, 10 μm. (B) Representative traces and bar graph showed the loss of GABAAR mIPSC, AMPAR-, or NMDAR- mediated mEPSCs in Cre/GFP-positive neurons (mIPSC: control, n = 7; Cre + β1-3 gRNAs, n = 7; frequency: ∗∗p < 0.01; amplitude: ∗∗∗p < 0.001; AMPA mEPSC: control, n = 9; Cre + β1-3 gRNAs, n = 10; frequency: ∗∗p < 0.01; amplitude: ∗∗p < 0.01; NMDA mEPSC: control, n = 7; Cre + β1-3 gRNAs, n = 7; frequency: ∗∗p < 0.01; amplitude: ∗∗∗p < 0.001; t-test; N = 2). Scale bar, 500 ms, 20 pA. (C) Spine density in GRIA1-3fl/flGRIN1fl/fl hippocampal neurons expressing either GFP or β1-3 gRNAs plus Cre-mCherry (GFP, n = 20; β1-3 gRNAs + Cre, n = 20; p > 0.05, t-test; N = 3). GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence (green). Scale bar, 5 μm. (D) Normal spine types in GRIA1-3fl/flGRIN1fl/fl hippocampal neurons expressing β1-3 gRNAs and Cre (GFP, n = 25; β1-3 gRNAs + Cre, n = 26; p > 0.05, t-test; N = 3). GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence (green). Scale bar 1 μm. (E) Single-cell genetic deletion of GABAARs, AMPARs, and NMDARs dramatically reduced the gephyrin puncta for about 90% (GFP, n = 28; β1-3 gRNAs + Cre, n = 24; ∗∗∗p < 0.0001 for puncta density; ∗∗∗p < 0.0001 for puncta size, t-test; N = 3). GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence (green). Scale bar, 5 μm. (F) Single-cell genetic deletion of GABAARs, AMPARs, and NMDARs did not change the PSD-95 puncta (GFP, n = 22; β1-3 gRNAs + Cre, n = 31; p > 0.05 for both comparisons, t-test; N = 3). GFP was immunolabeled with anti-GFP antibodies to boost the fluorescence (green). Scale bar, 5 μm. n represents the number of cells analyzed and N represents the number of independent experiments.
We then measured the spine density in neuronal dendrites and observed no change of spine density or type in Cre-positive, β1-3 gRNA-expressing neurons (Figure 5C,D), as compared to control neurons expressing the empty gRNA vector. We also examined inhibitory and excitatory synapses by the measurement of gephyrin and PSD-95 puncta. We found that compared to neurons expressing the empty gRNA vector, co-expression of both Cre and β1-3 gRNAs in GRIA1-3fl/flGRIN1fl/fl neurons led to a large reduction of the density of gephyrin puncta (∼90%) (Figure 5E). However, there was no difference of the density of PSD-95 puncta between control neurons and neurons lacking GABAARs, AMPARs and NMDARs (Figure 5F). Therefore, genetic deletion of both GABAARs and iGluRs impairs the development of GABAergic synapses but does not change the density of spines or glutamatergic synapses.
Discussion
To study the role of ionotropic GABAARs in the regulation of GABAergic synapse development, we have utilized the CRISPR-Cas9 technology to genetically delete all three β subunits of GABAARs in hippocampal neurons. GABAAR β subunits are required for the receptor assembly and GABA binding (Connolly et al., 1996; Tretter et al., 1997; Baumann et al., 2001; Olsen and Sieghart, 2008; Nguyen and Nicoll, 2018), and, consistently, we found that single-cell genetic deletion of three β subunits leads to a loss of GABAergic transmission, in agreement with a recent report (Nguyen and Nicoll, 2018). The lack of inhibitory synaptic transmission at the level of individual neurons allowed us to investigate the cell-autonomous role of GABAARs in the regulation of GABAergic synapse development.
Our data demonstrate that GABAARs are critical for GABAergic synapse development at the level of single neurons. Indeed, in hippocampal neurons lacking functional GABAARs in a mosaic fashion, gephyrin, and vGAT puncta at both somatic and dendritic areas are significantly reduced. Our data are consistent with previous reports in which germline KO of α1 subunit of GABAARs abolished GABAergic transmission in Purkinje cells, and consequently impaired GABAergic synapse formation (Fritschy et al., 2006; Patrizi et al., 2008). Similarly, knockdown or KO of the GABAAR subunit, γ2, which is important for synaptic clustering of GABAARs (Essrich et al., 1998), impaired GABAergic innervation and reduced GABAergic synapse density (Schweizer et al., 2003; Li et al., 2005; Frola et al., 2013). In addition, GABAAR activity has been shown to be important for GABAergic synapse formation (Chattopadhyaya et al., 2007; Arama et al., 2015; Oh et al., 2016; Lin et al., 2018). It is worth noting that broad genetic deletion or widespread pharmacological inhibition of GABAAR subunits alter neural network activity, and thus does not separate the cell-autonomous function of GABAARs from the indirect effects on network activity in the regulation of synapse development. Our data thus provide the genetic evidence of a critical cell-autonomous role of GABAARs for GABAergic synapse development. Currently, the molecular mechanisms underlying the regulation of GABAergic synapse development by GABAARs remain largely unclear. It has been reported that the GABAARs interact with the synaptic adhesion molecule, neurexins (Zhang et al., 2010). In addition, GABAARs may induce Ca2+ influx through NMDARs (Owens and Kriegstein, 2002; Ben-Ari et al., 2007) or voltage-gated calcium channels (Oh et al., 2016) to regulate GABAergic synaptogenesis, and may also play a synaptogenic role in GABAergic synapse development (Fuchs et al., 2013; Brown et al., 2016). Interestingly, the puncta density of NL2, a key synaptogenic cell adhesion molecule for GABAergic synapses (Lu et al., 2016), is not altered in neurons lacking GABAARs, similar to a previous report (Patrizi et al., 2008), suggesting that NL2 may act upstream of GABAAR signaling for GABAergic synaptogenesis. Recent studies have identified that NL2 is crucial for synaptic anchorage of GABAARs through binding to the GABAAR-interacting protein, GARLH/LHFPL4 (Davenport et al., 2017; Yamasaki et al., 2017; Wu et al., 2018) and is critical for GABAergic synapse development (Poulopoulos et al., 2009; Li et al., 2017; Panzanelli et al., 2017). Thus, it is plausible that during development NL2 may regulate GABAAR clustering, which in turn modulates GABAergic synapse formation and maturation. Interestingly, in GARLH/LHFPL4 KO neurons, both NL2 and GABAAR clustering are impaired (Davenport et al., 2017; Yamasaki et al., 2017; Wu et al., 2018), indicating that GARLH/LHFPL4 may function upstream of both NL2 and GABAARs in the regulation of GABAergic synapse development.
A recent elegant study has also employed the CRISPR-Cas9 technique to eliminate GABAergic transmission in hippocampal neurons, although this work did not examine the role of GABAARs in inhibitory synapse development (Nguyen and Nicoll, 2018). In this work, both β2 and β3, but not β1, subunits could rescue the loss of GABAergic transmission (Nguyen and Nicoll, 2018). Specifically, β1 subunit rescued ∼45% GABAergic transmission (Nguyen and Nicoll, 2018). In contrast, in our study all three β subunits rescued inhibitory synaptic currents in neurons expressing β1-3 gRNAs with β1 restoring ∼70% mIPSC frequency. Possibilities to explain this discrepancy include the expression levels of recombinant β subunits through different expression techniques (gene-gun mediated transfection vs. calcium phosphate transfection in our study) and different experimental preparations (hippocampal organotypic cultures vs. dissociated neuronal cultures in our study).
One surprising observation from our study is that excitatory synaptic transmission is normal in hippocampal neurons lacking GABAARs. It has been well-established that chronic pharmacological inhibition of GABAARs induces homeostatic adaptation of excitatory synapses and reduces AMPAR-mediated synaptic transmission (Turrigiano et al., 1998; Shepherd et al., 2006; Anggono et al., 2011; Diering et al., 2014). However, the role of GABAAR-mediated signaling at the single-cell level in the regulation of glutamatergic transmission remained unclear. Our data now demonstrate that at the level of individual neurons, the complete loss of functional GABAARs does not impair AMPAR-mediated excitatory transmission. Indeed, glutamatergic transmission, the expression levels of surface GluA1, a major AMPAR subunit in hippocampus (Lu et al., 2009), and the number of vGluT1 puncta are not altered in neurons lacking GABAARs. In addition, our work reveals that GABAARs are not absolutely required for spinogenesis in hippocampal neurons in vitro, as the spine density in neurons lacking GABAARs is indistinguishable from that in control neurons (Figure 4D). Our data are consistent with early work in α1 KO mice in which IPSCs are lost in cerebellar Purkinje cells, but EPSCs and excitatory synapses are largely intact (Fritschy et al., 2006; Patrizi et al., 2008). In addition, in GARLH/LHFPL4 KO neurons, GABAergic transmission is severely reduced without an accompanying change of glutamatergic transmission (Davenport et al., 2017; Yamasaki et al., 2017). Similarly, single-cell ablation of gephyrin in hippocampal neurons strongly reduces GABAergic transmission, but does not change glutamatergic transmission (Gross et al., 2016). However, it is important to point out that our data do not exclude the possibility that GABAAR activation can sufficiently induce spine formation (Oh et al., 2016) and can modulate glutamatergic synapse development (Ben-Ari et al., 2007).
Previous studies have also indicated a role of NKCC1 and KCC2 in the regulation of excitatory synapse and spine development (Akerman and Cline, 2006; Liu et al., 2006; Wang and Kriegstein, 2008). During development, NKCC1 and KCC2 play fundamental roles in determining the neuronal intracellular Cl- concentration and polarity of GABAAR action (Owens and Kriegstein, 2002; Payne et al., 2003; Ben-Ari et al., 2007), and regulate GABAAR subunit expression (Succol et al., 2012). Through manipulation of NKCC1 or KCC2 expression in neurons, the role of the depolarizing GABA in excitatory synapse development has been proposed (Akerman and Cline, 2006; Liu et al., 2006; Wang and Kriegstein, 2008). Interestingly, recent studies have indicated that the regulation of synapse or dendritic development by KCC2 is independent of its ion transport function (Li et al., 2007; Fiumelli et al., 2013), suggesting that the effect of KCC2 on neuronal development may be independent of GABA action. Future work toward a more complete understanding of depolarizing GABA in excitatory synapse development will be important to our understanding of the molecular mechanisms underlying synaptogenesis.
We have previously employed a Cre-LoxP system to genetically remove iGluRs, AMPARs and/or NMDARs, in individual hippocampal neurons, and found that excitatory synaptic input is not necessary for development of neuronal spines (Lu et al., 2009, 2013). One possibility is that in developing neurons lacking iGluRs, remaining GABAARs that are activated may generate depolarizing drives and provide activities important for neuronal morphological development. Thus, to further investigate the role of these receptors in excitatory or inhibitory synapse development, we have combined the Cre-LoxP system with the CRISPR-Cas9 approach to genetically target both iGluRs and GABAARs. We found that in individual hippocampal neurons lacking both iGluRs and GABAARs, there was no significant change of the spine density and PSD-95 immunolabeling. These data corroborate a series of recent reports that neurotransmitter release, and thus the activation of postsynaptic neurotransmitter receptors, are not required for spine and excitatory synapse development (Verhage et al., 2000; Varoqueaux et al., 2002; Lu et al., 2013; Sando et al., 2017; Sigler et al., 2017). In contrast, we found that there was a nearly 90% reduction of gephyrin puncta in these neurons lacking both iGluRs and GABAARs. Previously, we have shown that genetic deletion of both AMPARs and NMDARs led to a strong reduction of inhibitory transmission in hippocampal CA1 pyramidal neurons (Lu et al., 2013). Recently we have further shown that the NMDAR, but not the AMPAR, acts as an important molecule for controlling GABAergic synaptogenesis during development (Gu et al., 2016b; Gu and Lu, 2018). Together, these data suggest that NMDARs and GABAARs may play a synergistic role in the regulation of GABAergic synapse development. Currently, it remains unclear how NMDARs and GABAARs work together to control the development of GABAergic connections. It is conceivable that in developing, immature neurons, GABAAR activity may facilitate NMDAR activation, inducing Ca2+ influx and stimulating signaling pathways important for GABAergic synapse development (Owens and Kriegstein, 2002; Ben-Ari et al., 2007; Gu et al., 2016b). It is also possible that GABAARs and NMDARs may activate parallel pathways to regulate the formation of GABAergic connections. In the future, it will be imperative to determine the sequential action and functional interplay of signaling pathways mediated by NMDARs and GABAARs in the regulation of GABAergic synaptogenesis. It will also be important to determine how these neurotransmitter receptors functionally interact with cell surface molecules important for GABAergic synaptogenesis to regulate formation of inhibitory connections (Kuzirian and Paradis, 2011; Ko et al., 2015; Lu et al., 2016; Krueger-Burg et al., 2017).
In summary, through a single-cell genetic approach in vitro we have provided new insights into the cell-autonomous role of GABAARs in developing neurons and discovered a dichotomy in the regulation of synapse development by this prominent Cl- channel. While inhibitory synapse development is critically regulated by GABAARs, establishment of glutamatergic transmission and excitatory synapses are largely independent of GABAAR-mediated signaling at the level of individual neurons. Furthermore, we have managed to remove all AMPARs, NMDARs and GABAARs in single neurons in culture and demonstrated that iGluR- and GABAAR-mediated signaling are not essential for spinogenesis. Our data thus suggest that other developmental pathways including neurotropic factor-mediated signaling (Reichardt, 2006; Park and Poo, 2013), guidance cues, and their receptors (Klein, 2009; Shen and Cowan, 2010; Koropouli and Kolodkin, 2014), trans-synaptic cell adhesion interactions (Scheiffele, 2003; Dalva et al., 2007; Missler et al., 2012; de Wit and Ghosh, 2016), and other receptors or channels-mediated signaling (Komatsuzaki et al., 2005; Eroglu et al., 2009; Uemura et al., 2010; Lozada et al., 2012a,b; Kozorovitskiy et al., 2015; Sellers et al., 2015) may play key roles in spinogenesis and excitatory synaptogenesis. It is also worth noting that our data were collected in cultured neurons and there are limitations in using in vitro models to study synapse development. Thus, future experiments in vivo will help further establish the role of GABAARs in synaptogenesis. Nevertheless, our data demonstrate a remarkable specificity of these ionotropic receptors in mediating signaling important for synapse development in vitro. Given the prominent roles of malfunctions of these receptors in the pathogenesis of many neurodevelopmental disorders (Cellot and Cherubini, 2014; Yuan et al., 2015), our data also highlight the importance of understanding the molecular mechanisms for the regulation of GABAergic synapse development by these receptors.
Ethics Statement
All experiments using mice were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee at National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH).
Author Contributions
JD, SP, and WL designed the experiments. JD, DC, JL, and XG cloned and characterized the gRNA constructs. JD, SP, and TL performed the immunocytochemical experiments. JD performed the electrophysiological assays. QT provided critical technical design and problem solving. JD and WL wrote the manuscript. All authors read and commented on the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all members from the WL laboratory for helpful discussions.
Funding. WL was supported by the NINDS Intramural Research Program.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00217/full#supplementary-material
References
- Akerman C. J., Cline H. T. (2006). Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J. Neurosci. 26 5117–5130. 10.1523/JNEUROSCI.0319-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V., Clem R. L., Huganir R. L. (2011). PICK1 loss of function occludes homeostatic synaptic scaling. J. Neurosci. 31 2188–2196. 10.1523/JNEUROSCI.5633-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arama J., Abitbol K., Goffin D., Fuchs C., Sihra T. S., Thomson A. M., et al. (2015). GABAA receptor activity shapes the formation of inhibitory synapses between developing medium spiny neurons. Front. Cell Neurosci. 9:290. 10.3389/fncel.2015.00290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann S. W., Baur R., Sigel E. (2001). Subunit arrangement of gamma-aminobutyric acid type A receptors. J. Biol. Chem. 276 36275–36280. 10.1074/jbc.M105240200 [DOI] [PubMed] [Google Scholar]
- Belelli D., Harrison N. L., Maguire J., Macdonald R. L., Walker M. C., Cope D. W. (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J. Neurosci. 29 12757–12763. 10.1523/JNEUROSCI.3340-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y., Gaiarsa J. L., Tyzio R., Khazipov R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87 1215–1284. 10.1152/physrev.00017.2006 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y., Khazipov R., Leinekugel X., Caillard O., Gaiarsa J. L. (1997). GABAA, NMDA and AMPA receptors: a developmentally regulated ‘menage a trois’. Trends Neurosci. 20 523–529. 10.1016/s0166-2236(97)01147-8 [DOI] [PubMed] [Google Scholar]
- Brown L. E., Nicholson M. W., Arama J. E., Mercer A., Thomson A. M., Jovanovic J. N. (2016). Gamma-aminobutyric acid type A (GABAA) receptor subunits play a direct structural role in synaptic contact formation via their N-terminal extracellular domains. J. Biol. Chem. 291 13926–13942. 10.1074/jbc.M116.714790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellot G., Cherubini E. (2014). GABAergic signaling as therapeutic target for autism spectrum disorders. Front. Pediatr. 2:70. 10.3389/fped.2014.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y., Wang R., Barot S., Weiss D. S. (1996). Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 16 5415–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B., Di Cristo G., Wu C. Z., Knott G., Kuhlman S., Fu Y., et al. (2007). GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron 54 889–903. 10.1016/j.neuron.2007.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B., Engelman H., Scheiffele P. (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307 1324–1328. 10.1126/science.1107470 [DOI] [PubMed] [Google Scholar]
- Chudotvorova I., Ivanov A., Rama S., Hubner C. A., Pellegrino C., Ben-Ari Y., et al. (2005). Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physiol. 566(Pt 3) 671–679. 10.1113/jphysiol.2005.089821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly C. N., Krishek B. J., McDonald B. J., Smart T. G., Moss S. J. (1996). Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J. Biol. Chem. 271 89–96. 10.1074/jbc.271.1.89 [DOI] [PubMed] [Google Scholar]
- Dalva M. B., McClelland A. C., Kayser M. S. (2007). Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 8 206–220. 10.1038/nrn2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport E. C., Pendolino V., Kontou G., McGee T. P., Sheehan D. F., Lopez-Domenech G., et al. (2017). An essential role for the tetraspanin LHFPL4 in the cell-type-specific targeting and clustering of synaptic GABAA receptors. Cell Rep. 21 70–83. 10.1016/j.celrep.2017.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J., Ghosh A. (2016). Specification of synaptic connectivity by cell surface interactions. Nat. Rev. Neurosci. 17 22–35. 10.1038/nrn.2015.3 [DOI] [PubMed] [Google Scholar]
- Diering G. H., Gustina A. S., Huganir R. L. (2014). PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84 790–805. 10.1016/j.neuron.2014.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C., Allen N. J., Susman M. W., O’Rourke N. A., Park C. Y., Ozkan E., et al. (2009). Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139 380–392. 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essrich C., Lorez M., Benson J. A., Fritschy J. M., Luscher B. (1998). Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat. Neurosci. 1 563–571. 10.1038/2798 [DOI] [PubMed] [Google Scholar]
- Farrant M., Nusser Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6 215–229. 10.1038/nrn1625 [DOI] [PubMed] [Google Scholar]
- Fiumelli H., Briner A., Puskarjov M., Blaesse P., Belem B. J., Dayer A. G., et al. (2013). An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb. Cortex 23 378–388. 10.1093/cercor/bhs027 [DOI] [PubMed] [Google Scholar]
- Fritschy J. M., Panzanelli P., Kralic J. E., Vogt K. E., Sassoe-Pognetto M. (2006). Differential dependence of axo-dendritic and axo-somatic GABAergic synapses on GABAA receptors containing the alpha1 subunit in Purkinje cells. J. Neurosci. 26 3245–3255. 10.1523/JNEUROSCI.5118-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy J. M., Panzanelli P., Tyagarajan S. K. (2012). Molecular and functional heterogeneity of GABAergic synapses. Cell. Mol. Life Sci. 69 2485–2499. 10.1007/s00018-012-0926-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frola E., Patrizi A., Goetz T., Medrihan L., Petrini E. M., Barberis A., et al. (2013). Synaptic competition sculpts the development of GABAergic axo-dendritic but not perisomatic synapses. PLoS One 8:e56311. 10.1371/journal.pone.0056311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs C., Abitbol K., Burden J. J., Mercer A., Brown L., Iball J., et al. (2013). GABA(A) receptors can initiate the formation of functional inhibitory GABAergic synapses. Eur. J. Neurosci. 38 3146–3158. 10.1111/ejn.12331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J., Mody I. (2007). Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 56 763–770. 10.1016/j.neuron.2007.11.002 [DOI] [PubMed] [Google Scholar]
- Graf E. R., Zhang X., Jin S. X., Linhoff M. W., Craig A. M. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119 1013–1026. 10.1016/j.cell.2004.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G. G., Straub C., Perez-Sanchez J., Dempsey W. P., Junge J. A., Roberts R. W., et al. (2016). An E3-ligase-based method for ablating inhibitory synapses. Nat. Methods 13 673–678. 10.1038/nmeth.3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X., Lu W. (2018). Genetic deletion of NMDA receptors suppresses GABAergic synaptic transmission in two distinct types of central neurons. Neurosci. Lett. 668 147–153. 10.1016/j.neulet.2018.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X., Mao X., Lussier M. P., Hutchison M. A., Zhou L., Hamra F. K., et al. (2016a). GSG1L suppresses AMPA receptor-mediated synaptic transmission and uniquely modulates AMPA receptor kinetics in hippocampal neurons. Nat. Commun. 7:10873. 10.1038/ncomms10873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X., Zhou L., Lu W. (2016b). An NMDA receptor-dependent mechanism underlies inhibitory synapse development. Cell Rep. 14 471–478. 10.1016/j.celrep.2015.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incontro S., Asensio C. S., Edwards R. H., Nicoll R. A. (2014). Efficient, complete deletion of synaptic proteins using CRISPR. Neuron 83 1051–1057. 10.1016/j.neuron.2014.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerti-Szigeti K., Nusser Z. (2016). Similar GABAA receptor subunit composition in somatic and axon initial segment synapses of hippocampal pyramidal cells. eLife 5:e18426. 10.7554/eLife.18426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R. (2009). Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat. Neurosci. 12 15–20. 10.1038/nn.2231 [DOI] [PubMed] [Google Scholar]
- Ko J., Choii G., Um J. W. (2015). The balancing act of GABAergic synapse organizers. Trends Mol. Med. 21 256–268. 10.1016/j.molmed.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Komatsuzaki Y., Murakami G., Tsurugizawa T., Mukai H., Tanabe N., Mitsuhashi K., et al. (2005). Rapid spinogenesis of pyramidal neurons induced by activation of glucocorticoid receptors in adult male rat hippocampus. Biochem. Biophys. Res. Commun. 335 1002–1007. 10.1016/j.bbrc.2005.07.173 [DOI] [PubMed] [Google Scholar]
- Koropouli E., Kolodkin A. L. (2014). Semaphorins and the dynamic regulation of synapse assembly, refinement, and function. Curr. Opin. Neurobiol. 27 1–7. 10.1016/j.conb.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y., Peixoto R., Wang W., Saunders A., Sabatini B. L. (2015). Neuromodulation of excitatory synaptogenesis in striatal development. eLife 4:e10111. 10.7554/eLife.10111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger-Burg D., Papadopoulos T., Brose N. (2017). Organizers of inhibitory synapses come of age. Curr. Opin. Neurobiol. 45 66–77. 10.1016/j.conb.2017.04.003 [DOI] [PubMed] [Google Scholar]
- Kuzirian M. S., Paradis S. (2011). Emerging themes in GABAergic synapse development. Prog. Neurobiol. 95 68–87. 10.1016/j.pneurobio.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Khirug S., Cai C., Ludwig A., Blaesse P., Kolikova J., et al. (2007). KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56 1019–1033. 10.1016/j.neuron.2007.10.039 [DOI] [PubMed] [Google Scholar]
- Li J., Han W., Pelkey K. A., Duan J., Mao X., Wang Y. X., et al. (2017). Molecular dissection of neuroligin 2 and Slitrk3 reveals an essential framework for GABAergic synapse development. Neuron 96:808–826.e8. 10.1016/j.neuron.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. W., Yu W., Christie S., Miralles C. P., Bai J., Loturco J. J., et al. (2005). Disruption of postsynaptic GABA receptor clusters leads to decreased GABAergic innervation of pyramidal neurons. J. Neurochem. 95 756–770. 10.1111/j.1471-4159.2005.03426.x [DOI] [PubMed] [Google Scholar]
- Lin T. W., Tan Z., Barik A., Yin D. M., Brudvik E., Wang H., et al. (2018). Regulation of synapse development by Vgat deletion from ErbB4-positive interneurons. J. Neurosci. 38 2533–2550. 10.1523/JNEUROSCI.0669-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Neff R. A., Berg D. K. (2006). Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science 314 1610–1613. 10.1126/science.1134246 [DOI] [PubMed] [Google Scholar]
- Lozada A. F., Wang X., Gounko N. V., Massey K. A., Duan J., Liu Z., et al. (2012a). Glutamatergic synapse formation is promoted by alpha7-containing nicotinic acetylcholine receptors. J. Neurosci. 32 7651–7661. 10.1523/JNEUROSCI.6246-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada A. F., Wang X., Gounko N. V., Massey K. A., Duan J., Liu Z., et al. (2012b). Induction of dendritic spines by beta2-containing nicotinic receptors. J. Neurosci. 32 8391–8400. 10.1523/JNEUROSCI.6247-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Bromley-Coolidge S., Li J. (2016). Regulation of GABAergic synapse development by postsynaptic membrane proteins. Brain Res. Bull. 129 30–42. 10.1016/j.brainresbull.2016.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Bushong E. A., Shih T. P., Ellisman M. H., Nicoll R. A. (2013). The cell-autonomous role of excitatory synaptic transmission in the regulation of neuronal structure and function. Neuron 78 433–439. 10.1016/j.neuron.2013.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Shi Y., Jackson A. C., Bjorgan K., During M. J., Sprengel R., et al. (2009). Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62 254–268. 10.1016/j.neuron.2009.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald R. L., Olsen R. W. (1994). GABAA receptor channels. Annu. Rev. Neurosci. 17 569–602. 10.1146/annurev.ne.17.030194.003033 [DOI] [PubMed] [Google Scholar]
- Missler M., Sudhof T. C., Biederer T. (2012). Synaptic cell adhesion. Cold Spring Harb. Perspect. Biol. 4:a005694. 10.1101/cshperspect.a005694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I., Pearce R. A. (2004). Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 27 569–575. 10.1016/j.tins.2004.07.002 [DOI] [PubMed] [Google Scholar]
- Nguyen Q. A., Nicoll R. A. (2018). The GABAA receptor beta subunit is required for inhibitory transmission. Neuron 98 718–725 e713. 10.1016/j.neuron.2018.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh W. C., Lutzu S., Castillo P. E., Kwon H. B. (2016). De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 353 1037–1040. 10.1126/science.aaf5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen R. W., Sieghart W. (2008). International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60 243–260. 10.1124/pr.108.00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens D. F., Kriegstein A. R. (2002). Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 3 715–727. 10.1038/nrn919 [DOI] [PubMed] [Google Scholar]
- Panzanelli P., Fruh S., Fritschy J. M. (2017). Differential role of GABAA receptors and neuroligin 2 for perisomatic GABAergic synapse formation in the hippocampus. Brain Struct. Funct. 222 4149–4161. 10.1007/s00429-017-1462-7 [DOI] [PubMed] [Google Scholar]
- Park H., Poo M. M. (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14 7–23. 10.1038/nrn3379 [DOI] [PubMed] [Google Scholar]
- Patrizi A., Scelfo B., Viltono L., Briatore F., Fukaya M., Watanabe M., et al. (2008). Synapse formation and clustering of neuroligin-2 in the absence of GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 105 13151–13156. 10.1073/pnas.0802390105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J. A., Rivera C., Voipio J., Kaila K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26 199–206. 10.1016/s0166-2236(03)00068-7 [DOI] [PubMed] [Google Scholar]
- Poulopoulos A., Aramuni G., Meyer G., Soykan T., Hoon M., Papadopoulos T., et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63 628–642. 10.1016/j.neuron.2009.08.023 [DOI] [PubMed] [Google Scholar]
- Reichardt L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361 1545–1564. 10.1098/rstb.2006.1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sando R., Bushong E., Zhu Y., Huang M., Considine C., Phan S., et al. (2017). Assembly of excitatory synapses in the absence of glutamatergic neurotransmission. Neuron 94 312–321 e313. 10.1016/j.neuron.2017.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P. (2003). Cell-cell signaling during synapse formation in the CNS. Annu. Rev. Neurosci. 26 485–508. 10.1146/annurev.neuro.26.043002.094940 [DOI] [PubMed] [Google Scholar]
- Schweizer C., Balsiger S., Bluethmann H., Mansuy I. M., Fritschy J. M., Mohler H., et al. (2003). The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol. Cell Neurosci. 24 442–450. 10.1016/s1044-7431(03)00202-1 [DOI] [PubMed] [Google Scholar]
- Sellers K. J., Erli F., Raval P., Watson I. A., Chen D., Srivastava D. P. (2015). Rapid modulation of synaptogenesis and spinogenesis by 17beta-estradiol in primary cortical neurons. Front. Cell Neurosci. 9:137. 10.3389/fncel.2015.00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K., Cowan C. W. (2010). Guidance molecules in synapse formation and plasticity. Cold Spring Harb. Perspect. Biol. 2:a001842. 10.1101/cshperspect.a001842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd J. D., Rumbaugh G., Wu J., Chowdhury S., Plath N., Kuhl D., et al. (2006). Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52 475–484. 10.1016/j.neuron.2006.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W., Sperk G. (2002). Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr. Top. Med. Chem. 2 795–816. 10.2174/1568026023393507 [DOI] [PubMed] [Google Scholar]
- Sigler A., Oh W. C., Imig C., Altas B., Kawabe H., Cooper B. H., et al. (2017). Formation and maintenance of functional spines in the absence of presynaptic glutamate release. Neuron 94 304–311 e304. 10.1016/j.neuron.2017.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Succol F., Fiumelli H., Benfenati F., Cancedda L., Barberis A. (2012). Intracellular chloride concentration influences the GABAA receptor subunit composition. Nat. Commun. 3:738. 10.1038/ncomms1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V., Ehya N., Fuchs K., Sieghart W. (1997). Stoichiometry and assembly of a recombinant GABAA receptor subtype. J. Neurosci. 17 2728–2737. 10.1523/jneurosci.17-08-02728.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. G., Leslie K. R., Desai N. S., Rutherford L. C., Nelson S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391 892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Uemura T., Lee S. J., Yasumura M., Takeuchi T., Yoshida T., Ra M., et al. (2010). Trans-synaptic interaction of GluR delta 2 and neurexin through cbln1 mediates synapse formation in the cerebellum. Cell 141 1068–1079. 10.1016/j.cell.2010.04.035 [DOI] [PubMed] [Google Scholar]
- Varoqueaux F., Jamain S., Brose N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell. Biol. 83 449–456. 10.1078/0171-9335-00410 [DOI] [PubMed] [Google Scholar]
- Varoqueaux F., Sigler A., Rhee J. S., Brose N., Enk C., Reim K., et al. (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. U.S.A. 99 9037–9042. 10.1073/pnas.122623799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M., Maia A. S., Plomp J. J., Brussaard A. B., Heeroma J. H., Vermeer H., et al. (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287 864–869. 10.1126/science.287.5454.864 [DOI] [PubMed] [Google Scholar]
- Wang D. D., Kriegstein A. R. (2008). GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 28 5547–5558. 10.1523/JNEUROSCI.5599-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M., Tian H. L., Liu X., Lai J. H. C., Du S., Xia J. (2018). Impairment of inhibitory synapse formation and motor behavior in mice lacking the NL2 binding partner LHFPL4/GARLH4. Cell Rep. 23 1691–1705. 10.1016/j.celrep.2018.04.015 [DOI] [PubMed] [Google Scholar]
- Yamasaki T., Hoyos-Ramirez E., Martenson J. S., Morimoto-Tomita M., Tomita S. (2017). GARLH family proteins stabilize GABAA receptors at synapses. Neuron 93 1138–1152 e1136. 10.1016/j.neuron.2017.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H., Low C. M., Moody O. A., Jenkins A., Traynelis S. F. (2015). Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol. Pharmacol. 88 203–217. 10.1124/mol.115.097998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Atasoy D., Arac D., Yang X., Fucillo M. V., Robison A. J., et al. (2010). Neurexins physically and functionally interact with GABA(A) receptors. Neuron 66 403–416. 10.1016/j.neuron.2010.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.