

Abstract
Apolipoprotein (apo) E4 is the major genetic risk factor for Alzheimer’s disease (AD), increasing risk and decreasing age of disease onset. Many studies have demonstrated the detrimental effects of apoE4 in varying cellular contexts. However, the underlying mechanisms explaining how apoE4 leads to cognitive decline are not fully understood. Recently, the combination of human induced pluripotent stem cell (hiPSC) modeling of neurological diseases in vitro and electrophysiological studies in vivo have begun to unravel the intersection between apoE4, neuronal subtype dysfunction or loss, subsequent network deficits, and eventual cognitive decline. In this review, we provide an overview of the literature describing apoE4’s detrimental effects in the central nervous system (CNS), specifically focusing on its contribution to neuronal subtype dysfunction or loss. We focus on γ-aminobutyric acid (GABA)-expressing interneurons in the hippocampus, which are selectively vulnerable to apoE4-mediated neurotoxicity. Additionally, we discuss the importance of the GABAergic inhibitory network to proper cognitive function and how dysfunction of this network manifests in AD. Finally, we examine how apoE4-mediated GABAergic interneuron loss can lead to inhibitory network deficits and how this deficit results in cognitive decline. We propose the following working model: Aging and/or stress induces neuronal expression of apoE. GABAergic interneurons are selectively vulnerable to intracellularly produced apoE4, through a tau dependent mechanism, which leads to their dysfunction and eventual death. In turn, GABAergic interneuron loss causes hyperexcitability and dysregulation of neural networks in the hippocampus and cortex. This dysfunction results in learning, memory, and other cognitive deficits that are the central features of AD.
Keywords: Apolipoprotein E, Alzheimer’s disease, GABAergic interneuron, Hyperexcitability, Inhibitory network, Selective vulnerability, Tau
Background
Alzheimer’s disease (AD) is the most common form of dementia and is characterized by a progressive loss of memory and other cognitive functions [1–4]. Currently, there are 46.8 million people worldwide living with dementia, and this number is estimated to double every 20 years, reaching 74.7 million by 2030. Worldwide, AD cost $818 billion in 2015. By 2030, these costs could rise as high as $2 trillion [1]. This extreme expense combined with the growing aging population highlights the need for a better understanding of the disease mechanism and development of therapeutics.
AD is a multifactorial neurodegenerative disorder caused by interactions among multiple genetic and environmental factors. Mutations in three genes—those encoding amyloid precursor protein (APP), presenilin-1 (PS1), and presenilin-2 (PS2)—are linked to early-onset autosomal dominant AD, which accounts for less than 1% of all AD cases [2–4]. Apolipoprotein (apo) E4, an isoform of the APOE gene in humans, is the major genetic risk factor for late-onset familial and sporadic AD [4–8], which account for most AD cases. ApoE4 increases the risk and decreases the age of onset of AD in a gene dose dependent manner [4–11]. ApoE4 is present in roughly 20–25% of the human population, and apoE4 carriers account for 60–75% of AD cases in most clinical studies [11], highlighting the importance of apoE4 in AD pathogenesis.
AD is characterized by two molecular pathological hallmarks: extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) [2–4]. The accumulation of Aβ plaques and NFTs is associated with significant neuronal and synaptic loss as well as neuroinflammation. Both of these pathologies are exacerbated by the presence of apoE4 [4–7, 12]. Biochemical, cellular, transgenic animal, and clinical studies have suggested many potential explanations for apoE4’s contribution to AD pathogenesis [4–7, 12]. This review focuses on apoE4’s detrimental effects on GABAergic interneurons, the network deficits resulting from GABAergic interneuron dysfunction or loss, and the mechanisms that link these deficits to AD pathogenesis and cognitive decline.
ApoE structure, function, and expression in the CNS
ApoE is a 34-kDa protein comprised of 299 amino acids. It is a polymorphic protein with three common isoforms, apoE2, apoE3, and apoE4 in humans. Each isoform differs only by one or two amino acids [4, 6, 8, 13, 14]. The apoE3 and apoE4 amino acid sequences differ only at position 112 where apoE4 has an Arg instead of a Cys. This seemingly small difference induces significant changes to its structures and biological functions. ApoE is comprised of two domains: the amino-terminal domain and carboxyl-terminal domain. These two domains contain the receptor-binding region and the lipid-binding region, respectively, and are joined by a flexible hinge region. Multiple research groups have investigated potential interaction between the two domains, which is important to apoE’s function [15–17]. Nuclear magnetic resonance (NMR) analysis of a monomeric mutant form of apoE3 recently revealed a potential full-length structure of apoE. In this monomeric mutant apoE3, Arg-61 interacts with Thr-194 via a H-bond and Lys-95 forms a salt bridge with Glu-255 [17]. Whether this mutant form of apoE3 truthfully reflects the biophysical and biological properties of wildtype apoE3 needs to be further evaluated. An alternative model which used X-ray crystallography and circular dichroism spectroscopy to identify the structure of the amino-terminus and the carboxyl-terminus, respectively, demonstrates that Arg-112 in apoE4 interacts with Glu-109, exposing Arg-61 to interact with Glu-255. This domain interaction mediated by a salt bridge formation between Arg-61 and Glu-255 is unique to apoE4 (Fig. 1) [15]. This model of apoE4 domain interaction has been supported by Fluorescence Resonance Energy Transfer and electron paramagnetic resonance tests [18] and was observed in live neurons expressing apoE4 [19]. Importantly, this domain interaction renders apoE4 to be more susceptible to proteolytic cleavage, resulting in the generation of neurotoxic apoE4 fragments [20–22].
Fig. 1.

Model of domain interaction as a determinant of conformation of apoE. In apoE4 (left), Arg-112 orients the side chain of Arg-61 into the aqueous environment where it can interact with Glu-255, resulting in interaction between the amino- and carboxyl-terminal domains. In apoE3 (right), Arg-61 is not available to interact with residues in the carboxyl-terminal domain, resulting in a very different overall conformation
Initially, apoE was described as a lipid transport protein and was shown to play a key role in cholesterol metabolism and cardiovascular disease. However, by the mid-1980s, it had become apparent that apoE also plays significant roles in neuronal repair and remodeling as well as in neurological disease [8, 12, 13]. Astrocytes are the primary source of apoE in the brain [23, 24]. However, under aging and stress conditions, neurons also produce apoE, albeit at lower levels than astrocytes [25, 26]. Microglia also express apoE, especially under conditions of neurodegeneration and/or inflammation, and the interplay between apoE and microglia has been reviewed elsewhere [27]. Cellular origin plays a crucial role in apoE’s biophysical properties and pathological effects. Astrocytic apoE might be more heavily involved in Aβ pathology, while neuronal apoE has been shown to be more impactful on neuronal function and survival as well as on NFT formation. Clearly, more research needs to be done to completely understand how cellular origin affects apoE’s biological and pathological characteristics [27, 28]. Overall, it has been demonstrated, both in vivo and in vitro, that apoE plays major roles in AD pathogenesis in both an Aβ-dependent and independent manner, and different cellular sources of apoE4 may contribute in distinct ways to AD pathogenesis [4–8, 12–14, 21, 22].
Aβ-dependent roles of ApoE4 in AD
Accumulation of fibrillar Aβ peptides (amyloid plaques) in the brain is a requirement for an AD pathological diagnosis. Aβ accumulation can take place due to an imbalance between production and clearance in the brain. ApoE is associated with amyloid plaques and its roles in Aβ-related pathologies have been extensively reviewed elsewhere [5, 7, 29–32]. Here we only briefly overview its relationship to Aβ aggregation/deposition and clearance in the brain.
ApoE4 and Aβ seeding, aggregation, and plaque formation
The roles of apoE in Aβ seeding, aggregation, and plaque formation are still not fully understood, as research groups have shown that both increasing or decreasing apoE levels reduces plaque load [5, 7, 33–48]. These seemingly conflicting results are most likely due to the model in question, the complexity of apoE biology, and the cellular source of apoE, as lipidation status, isoform, cell source, expression level, and the aggressiveness of the Aβ production in the model can complicate results. For example, increasing apoE levels in the brain has been shown to suppress Aβ deposition, facilitating Aβ clearance, and reverse memory deficits [49–51]. However, these results were disputed by several follow-up studies. Notably, genetically decreasing apoE expression results in less Aβ deposition in amyloid mouse models, independently of apoE isoform [39, 40]. Reducing apoE through immunotherapy has also been shown to significantly reduce insoluble Aβ levels [52]. ApoE4 has also been shown to facilitate Aβ production in vitro [53]; thus, lowering apoE4 may decrease Aβ production.
Furthermore, recent studies have demonstrated that increasing or decreasing apoE levels at specific time points during Aβ plaque formation differentially affects Aβ plaque associated pathology. In an APP/PS1 mouse model where human apoE3 or apoE4 is expressed exclusively in astrocytes, apoE4 accelerated amyloid pathology. More specifically, increased expression of astrocytic apoE4 during the early seeding stage of amyloid plaque formation increased amyloid deposition and neuronal pathology [54]. In APP/PS1–21 mice with either the human apoE3 or apoE4 allele homozygously knocked-in (apoE-KI), apoE levels were reduced at different ages using antisense oligonucleotides (ASO) in order to better understand how the timing of apoE expression impacts Aβ accumulation and pathology. ASO treatment directly after birth led to a significant decrease in Aβ pathology opposed to treatment starting at 6-weeks of age (when significant amyloidosis has occurred due to the aggressive nature of amyloid pathology in these mice). Lowering apoE4 levels at 6-weeks of age led to an increase in Aβ plaque size and reduction in plaque-associated neuritic dystrophy with no change in overall plaque load [55]. Taken together, these results indicate that apoE plays a significant role in the initiation of Aβ pathology; however, after Aβ pathology has been initiated, lowering apoE modulates plaque size and toxicity.
ApoE4 and Aβ clearance
The role that apoE plays in clearing Aβ has been heavily investigated as well [29, 30, 32, 56–60]. Multiple pathways exist to clear Aβ, including proteolytic degradation, cellular clearance, and the cerebrovascular clearance, all of which have been reviewed elsewhere [7]. It has been suggested that apoE facilitates Aβ degradation by converting its structure into one that is more recognizable by proteolytic enzymes. ApoE assists in Aβ clearance in an isoform-dependent manner wherein apoE2 > apoE3 > apoE4 [29, 30, 59]. Strikingly, C-terminally truncated apoE4 clears Aβ inefficiently and acts in concert with Aβ to elicit neuronal and behavioral deficits in transgenic mice [61]. Astrocytes have been shown to internalize and degrade Aβ in an apoE dependent manner [31]. ApoE also promotes Aβ clearance by activating phagocytosis and migration of microglia wherein apoE3 is more effective than apoE4 [62–64]. Astrocytic apoE4 significantly increases brain Aβ half-life relative to apoE3, suggesting an impairment of Aβ clearance by astrocytic apoE4 relative to apoE3 [54, 65]. In addition to astrocytes, neurons are also capable of up taking and degrading Aβ, however more work needs to be done in order to dissect the mechanism of Aβ clearance in neurons [66]. Although apoE interacts with amyloid, it should be noted that plaque load does not correlate well with cognitive impairments, highlighted most obviously by people with substantial plaque burdens and normal cognition [67]. Therefore, it is important to also consider apoE4’s roles in AD pathology independently of Aβ as well.
Aβ-independent roles of ApoE4 in AD
ApoE4 has also been shown to affect many different pathological processes independently of Aβ. For example, both apoE4 transgenic and apoE4-KI mice show age- and sex-dependent learning and memory deficits in the absence of Aβ accumulation, as compared to apoE3 transgenic and apoE3-KI mice [68–71]. ApoE4 impairs synaptogenesis and decreases dendritic spine density in vivo and in vitro in primary neuronal cultures [72, 73]. Furthermore, it impairs adult hippocampal neurogenesis in mice and affects cortical thickness, brain activity, and mitochondrial function well before significant Aβ accumulation in the human brains [74–76]. Other non-amyloid pathways affected by apoE4 include lipid metabolism, synaptic plasticity, and most relevant to AD, tau pathology [77].
In response to injury, or stress such as normal aging, neurons express apoE, likely to facilitate transport of cholesterol and other lipids for membrane repair and/or remodeling [22]. As mentioned above, apoE4 is highly susceptible to neuron-specific proteolysis, which generates neurotoxic fragments [20, 77–79]. These fragments escape the secretory pathway and enter the cytosol, where they stimulate tau-phosphorylation and interact with mitochondria, leading to mitochondrial dysfunction and neurodegeneration [4]. In particular, GABAergic interneurons in the dentate gyrus (DG) are particularly vulnerable to apoE4 fragment-mediated neurotoxicity, and in apoE4 fragment transgenic mice, knocking out tau rescues GABAergic interneuron loss as well as learning and memory deficits, demonstrating the tau-dependent nature of apoE4-induced cognitive impairment [71]. Therefore, in order to better understand apoE4’s pathophysiology in the context of AD, it is important to study its interaction and impact on tau.
In mutant human Tau-P301S transgenic mice, expression of apoE4 led to more advanced tau pathology, brain atrophy, and neuroinflammation. Interestingly, knocking-out apoE (apoE-KO) protected the mice from Tau-P301S-induced neurodegeneration and neuroinflammation. These data strongly support apoE4’s gain of toxic effects on tau pathology and its related neurodegeneration and neuroinflammation, all of which are independent of Aβ [80]. However, a recent study using a gene delivery approach, in which adeno-associated virus (AAV) expressing human tau protein containing the P301L mutation (AAV-TauP301L) was injected into the cerebral lateral ventricles of neonatal apoE2-KI, apoE3-KI, and apoE4-KI mice, resulted in contradictory findings. Specifically, 6-month old apoE2-KI mice injected with the AAV-TauP301L construct at postnatal day 0 had significantly higher levels of hyperphosphorylated and aggregated tau, as well as more severe behavioral abnormalities than did 6-month old apoE3-KI and apoE4-KI mice under the same conditions [81]. Strikingly, in humans, APOE2 is associated with increased risk of two tauopathies: progressive supranuclear palsy and corticobasal degeneration [81]. The discrepancies between these studies could be the result of differences between model systems, such as cell type specificity and the overall level of tau expression, differences in toxicity between Tau-P301S and Tau-P301L mutations, and age of the mice. It should be noted that clinical manifestations of Tau-P301S and Tau-P301L are related to frontotemporal dementia (FTD) but not AD. In order to fully understand apoE isoform-dependent role in tau pathology in the context of AD, more in-depth research and new animal models are required.
Inhibitory system dysfunction in AD
In recent years, it has become clear that neurodegenerative diseases target specific neuronal populations [82]. GABAergic interneuron dysfunction, in particular, is found in a range of neurological and psychiatric disorders, including schizophrenia, autism, Fragile X syndrome, epilepsy, migraines, depression, bipolar disorder, and AD [83]. Loss of GABA, the primary inhibitory neurotransmitter in the brain, is a key component of AD. Post-mortem tissue from AD patients shows reduced GABA level throughout the brain, particularly in temporal, parietal, and frontal cortices [84, 85]. Post-mortem cortices from AD patients contain reduced GABAergic terminals, particularly near amyloid plaques [86, 87]. AD patients show reduced cortical GABA as measured by positron emission tomography, especially in the temporal cortex [88, 89], and reduced GABA in cerebrospinal fluid [90–93]. Additionally, AD patients have a specific loss of somatostatin-positive interneurons in the cortex [94] and hippocampus [95]. Several other neuronal subtypes are also affected by AD pathology, including cholinergic and glutamatergic neurons, whose loss and dysfunction in turn contribute to cognitive impairment [96]. This review will focus on the consequences of GABAergic interneuron loss and dysfunction, which have broad consequences at the network and behavioral level.
Loss of GABA and GABAergic interneurons in AD patients may be responsible for network hyperactivity manifesting as seizures. Substantial evidence shows that loss of GABAergic tone leads to seizures [97]. 10–22% of AD patients exhibit seizures [98–100], as do hAPPFAD mice [101], and the onset of these seizures precedes cognitive decline [102]. Levetiracetam, an anti-epileptic drug, successfully reverses hyperexcitability and learning and memory deficits in an hAPPFAD mouse model of AD [103, 104] and in aged mice [105–107]. Cognitively normal elderly, amnestic mild cognitive impairment (MCI), and AD patients all show cognitive improvement following chronic levetiracetam administration [108–110]. Thus, GABAergic dysfunction contributes to network-wide deficits in AD, which may in turn harm cognition.
GABAergic inhibitory interneurons make up a minority of neurons within the brain but play an outsized role in coordinating activity [111]. Inhibitory interneurons regulate network oscillations, which synchronize neuronal activity to rhythms that are crucial to learning and memory [112–116]. Inhibition also prevents hyperactivity of excitatory principal cells, which disrupts normally sparse neural coding and leads to decreased signal-to-noise ratio [117–119]. Furthermore, reducing hippocampal GABA levels impairs learning and memory [120, 121], and silencing inhibitory interneurons in the dentate gyrus prevents both encoding of new memories and recall of old memories [122]. Given their importance to proper learning and memory, it is crucial to better understand GABAergic inhibitory interneuron dysfunction and/or loss in the context of AD. As apoE4 is the major genetic risk factor for AD, understanding its effect on GABAergic interneurons, a population that is particularly vulnerable to apoE4 pathology, is essential. ApoE is expressed in neurons during periods of stress or normal aging. The neuronally expressed apoE4 is more susceptible to proteolytic cleavage and cytotoxic fragment generation. In the following sections, evidence for GABAergic interneuron susceptibility to apoE4 and the subsequent network deficits that result of inhibitory neuron loss, culminating in learning and memory deficits will be discussed.
GABAergic interneuron susceptibility to ApoE4
In vivo studies
Many lines of evidence from in vivo studies contribute to the hypothesis that GABAergic interneurons in the hippocampus are disproportionately susceptible to apoE4-mediated toxicity. For example, apoE4-KI mice display an age- and tau-dependent decrease in hilar GABAergic somatostatin-positive interneurons in the hippocampus [71]. The extent of this inhibitory interneuron loss correlates with both decreased adult hippocampal neurogenesis and with learning and memory deficits [70, 74]. The adverse effects of apoE4 are prevented by tau removal, indicating a direct link between tau pathology, apoE4, and GABAergic interneuron death [71]. Interestingly, the cellular source of apoE is critical to its pathological effect on GABAergic interneurons. ApoE4 undergoes proteolytic cleavage which generates neurotoxic fragments only when produced in neurons, but not when produced in astrocytes [20]. When expressed in neurons, apoE3 is excitoprotective whereas apoE4 is not; however, when expressed in astrocytes, apoE3 and apoE4 are equally excitoprotective [123]. Likewise, when expressed in neurons, apoE4 decreases dendrite arborization and spine density whereas apoE4 expressed in astrocytes does not show similar effects [124]. Importantly, deletion of apoE4 in GABAergic interneurons, but not deletion of apoE4 in astrocytes, is sufficient to protect aged mice from apoE4-induced GABAergic interneuron loss and learning and memory deficits [125]. These findings suggest that, although the majority of apoE is produced in astrocytes, it is apoE4 produced within GABAergic interneurons that is detrimental to their survival in vivo which leads to deficits in both learning and memory in AD models. Strikingly, bolstering inhibitory function, either through systemic GABA-agonist treatment [126] or through transplant of mouse derived inhibitory interneuron progenitors directly into the hippocampus [127], restores learning and memory in aged apoE4-KI mice without or with mutant hAPPFAD expression.
In vitro studies
GABAergic interneuron selective vulnerability to apoE4 is also supported by a recent study in an in vitro model using hiPSC-derived neurons with different APOE genotypes [128]. These included APOE4, APOE3, gene-edited isogenic APOE3 derived from APOE4, and APOE-deficient hiPSC lines. Strikingly, much of AD pathology seen in vivo was successfully recapitulated in this hiPSC-derived neuronal model in vitro. For example, apoE4/4 neurons produced significantly more Aβ and phosphorylated tau than apoE3/3 neurons. ApoE4/4 GABAergic interneurons in particular showed degeneration and displayed significantly elevated phosphorylated tau levels compared to apoE3/3 GABAergic interneurons. Importantly, there was no significant loss of glutamatergic neurons and dopaminergic neurons in apoE4/4 hiPSC-derived neuron cultures, suggesting a preferential detrimental effect of apoE4 on GABAergic neurons. Converting APOE4 to APOE3 by gene editing rescued these pathologies, including tau hyperphosphorylation, Aβ40 and Aβ42 overproduction, and GABAergic interneuron loss, suggesting that neuronal apoE4 expression alone was sufficient to induce these interneuron pathologies. Finally, a small molecule that renders apoE4 ‘apoE3-like’ by changing the protein’s conformation to nullify apoE4’s unique domain interaction was tested. Treatment with this structure corrector significantly decreased apoE4 fragmentation, reduced the levels of hyperphosphorylated tau and Aβ40 or Aβ42 overproduction and/or secretion, and increased GABAergic interneuron survival [128], again suggesting that the specific actions of neuronal apoE are responsible for this GABAergic interneuron specific toxicity. Isogenic hiPSC lines with an apoE3/3 or apoE4/4 genotype have also been used to study transcriptomic, molecular, and cellular alterations caused by apoE4 [129]. In hiPSC-derived isogenic APOE4 neurons, genes known to control synaptic function were significantly downregulated, there was an increase in Aβ42 secretion, and an increase in hyperphosphorylated tau levels in isogenic APOE4 neurons versus APOE3 controls [129].
ApoE4-mediated GABAergic interneuron loss and inhibitory network dysfunction in AD
Given that hippocampal GABAergic interneurons are selectively vulnerable to apoE4, an intriguing question is: how does interneuron dysfunction manifest at the network and behavioral or clinical levels? At the network level, loss of GABAergic function can lead to deficits in both tonic and phasic inhibition. Loss of tonic inhibition manifests itself most prominently in AD patients as hypersynchrony, leading to epilepsy and olfactory processing deficits, as well as hyperactivity, leading to aberrantly increased activation of cortical and hippocampal networks [130]. Loss of phasic inhibition manifests as reduced hippocampal rhythms [130]. These network consequences of inhibitory deficits each contribute to learning and memory impairments [131]. The following sections will address these manifestations of inhibitory network dysfunction that occur as a result of apoE4 expression.
ApoE4 and GABAergic interneuron dysfunction leading to seizure activity in AD
The loss of GABA and GABAergic interneurons in AD patients may lead to network hyperactivity, most commonly observed through seizures. ApoE4 carriers have a higher risk [132–136] and earlier onset [137–139] of developing idiopathic or secondary temporal lobe epilepsy. It is still unclear whether these patients demonstrate a higher risk for developing AD later in life, or if indeed the proportion of AD patients with concomitant epilepsy is enriched for apoE4 carriers. In addition to increased risk, apoE4 is also associated with increased epileptic pathology. The presence of apoE4 is correlated with smaller neuron size and increased DNA damage in temporal lobes of epilepsy patients [140], and epilepsy patients with at least one APOE4 allele are six times more likely to exhibit treatment resistance [141]. Investigating the connection between apoE4 and epilepsy may shed light on its role in large-scale network dysfunction in AD.
ApoE4-mediated GABAergic interneuron dysfunction and olfactory deficits in AD
Olfactory dysfunction is also an early and common symptom of AD as well as a result of carrying apoE4 and odor identification ability predicts future cognitive decline [142–145], making olfactory acuity a potential early signal of underlying neurodegenerative processes. ApoE4 carriers show particularly marked deficits in odor identification and memory relative to non-carriers [146], and evidence suggests disrupted GABA signaling in the olfactory bulb may mediate this olfactory loss [147]. In vivo electrophysiological recordings from aged apoE4-KI mice with odor memory deficits revealed increased local field potential response to odors in both the olfactory bulb and in primary olfactory cortex [148], which was attributed to inhibitory dysfunction. These studies together suggest that apoE4-mediated odor memory impairment, a potential early biomarker of cognitive dysfunction, may be due to apoE4-induced hyperactivity.
ApoE4 and microglial dysfunction in the GABAergic inhibitory network and AD
The link between apoE, microglia, and GABAergic interneuron dysfunction is also an emerging area of interest in the context of network dysfunction and AD. ApoE expression in microglia and its roles in microglial physiology and pathology have recently been actively explored. ApoE is upregulated in primed/activated microglia [149, 150], and apoE signaling in microglia following phagocytosis of apoptotic neurons or in response to Aβ accumulation leads to a transcriptional switch from promoting homeostasis to promoting inflammation and neurodegeneration [150, 151]. Deletion of the Apoe gene suppresses microglial activation in response to Aβ accumulation and prevents migration of microglia toward amyloid plaques [150]. However, the effect of specific apoE isoforms has yet to be explored [152, 153]. It has been reported that activated microglia migrate to inhibitory synapses and displace them from excitatory neurons [154] and an increase in CX3CR1 expression in activated microglia suppresses GABAA receptor signaling in excitatory neurons [155], both of which could contribute to GABAergic inhibitory network deficits in the context of apoE4. Another avenue by which microglial dysfunction may affect GABAergic interneurons is through perineuronal nets. Perineuronal nets are extracellular matrix structures which surround synapses of highly active neuronal subtypes and are associated with microglia [156]. These structures are involved in synapse development, stabilization and remodeling, buffering ions, and regulating the synapse microenvironment [157]. AD patients have reduced perineuronal net density [158]. Strikingly, the majority of neurons surrounded by perineuronal nets are parvalbumin-expressing GABAergic interneurons [159], and these interneurons show deficits in perineuronal net density in AD model of mice [160]. Since perineuronal nets protect these interneurons from oxidative stress and other injuries [161], it is possible that their breakdown in AD, which can be triggered or exacerbated by microglial dysfunction, may lead to interneuron dysfunction or death and thus inhibitory network deficits.
ApoE4 and network hyperactivity induced by GABAergic interneuron dysfunction
Network hyperactivity is an overarching symptom of AD and is evident in human apoE4 carriers. More specifically, hyperactivity in two networks which are normally disengaged during task performance in healthy individuals has been demonstrated by multiple groups. First, cognitively normal apoE4 carriers show reduced task-induced deactivation of the default mode network (DMN) [162–164]. Higher resting state GABA levels in the DMN are associated with enhanced task-induced deactivation of this network [165–167], suggesting that this DMN hyperactivity could be the result of inhibitory deficits. Reduced ability to deactivate the DMN during memory encoding is found in AD patients [168–170] and is correlated with worse task performance [171], linking this apoE4-induced deficit to memory impairments. Second, healthy elderly apoE4 carriers show increased hippocampal and entorhinal activation during encoding task performance [172, 173]. A recent study found that aged apoE4-KI mice had increased field potential synchrony and pyramidal cell firing in the entorhinal cortex [174]. This activation is dysfunctional hyperactivity rather than task-related, as levetiracetam treatment of amnestic MCI patients both reduces hippocampal over-activation and improves cognitive performance during a recognition memory task [109, 175]. Greater hippocampal activation during encoding tasks is associated with worse task performance [176] in MCI and AD patients [172, 177], and even predicts future cognitive decline in cognitively healthy elderly [178]. Finally, aberrant activity increases in these networks are seen even prior to aging. Healthy young and middle-aged adult apoE4 carriers show increased DMN activation at rest [179] and increased hippocampal activation during encoding task performance [179–181], suggesting that apoE4-induced network hyperactivity occurs before significant Aβ accumulation in human brains.
ApoE4-mediated GABAergic interneuron loss and hippocampal network dysfunction and memory deficits
Susceptibility of GABAergic interneurons to apoE4 and subsequent loss of inhibitory function can also lead to reduced coordination of hippocampal network activity involved in memory. ApoE4-KI mice show reduced abundance of sharp-wave ripples, the local field potential of hippocampal replay events which are critical for consolidating spatial memory [182, 183]. ApoE4-KI mice also display reduced slow gamma power throughout the hippocampal circuit during ripple events, suggesting reduced accuracy of these replay events [182, 184]. Thus, apoE4 leads to reduced instances as well as accuracy of spatial memory consolidation. Notably, removing apoE4 from inhibitory interneurons specifically rescues slow gamma power and learning and memory deficits, indicating that these phenotypes are caused by intraneuronal apoE4 expressed in GABAergic interneurons. Younger mice recorded before the onset of significant interneuron loss do not show significant slow gamma power loss, further implicating inhibitory interneurons in apoE4-induced hippocampal gamma loss [182].
Conclusions and perspectives
Conclusion: working model of ApoE4-induced GABAergic interneuron deficit and network dysfunction in AD
The combination of the data presented above paints a more complete picture of the mechanism underlying apoE4 mediated cognitive decline. We present a model wherein injury or aging-related stress induces neuronal apoE expression. Due to its pathological conformation (domain interaction), apoE4 is more susceptible to proteolytic cleavage than apoE3, leading to increased levels of neurotoxic fragment generation, and through a tau-dependent mechanism, results in GABAergic interneuron dysfunction and death. The loss of hippocampal GABAergic interneurons leads to network dysfunction and hyperexcitability. The network dysfunction and hyperexcitability themselves contribute to learning and memory deficits as well as induce further stress, and therefore more neuronal expression of apoE. This process culminates in further GABAergic interneuron loss and eventual cognitive decline (Fig. 2).
Fig. 2.
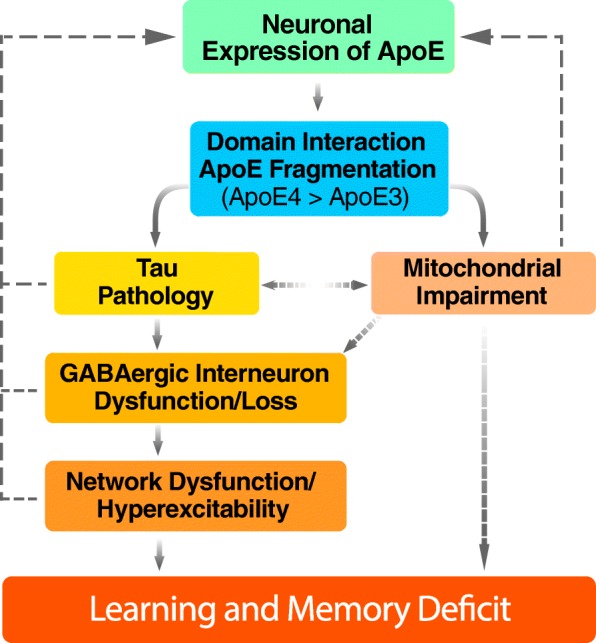
Proposed working model of apoE4-induced GABAergic interneuron deficit and network dysfunction in AD. In response to aging, stress, or injury, apoE is expressed in neurons to facilitate neuronal repair and remodeling. However, higher apoE4 fragmentation due to its pathological conformation (domain interaction) leads to tau pathology and mitochondrial impairments. GABAergic interneurons in the hippocampus are selectively vulnerable to apoE4 toxicity, resulting in dysfunction and eventual loss. The inhibitory interneuron loss leads to network dysfunction and hyperexcitability, resulting in a positive feedback loop culminating in learning and memory deficits
It is apparent that more research needs to be done on understanding apoE4’s roles in AD pathogenesis and on developing therapeutics targeted to its specific detrimental effects. This can be achieved by focusing on: 1) better understanding of the selective vulnerability of GABAergic interneurons to apoE4 and 2) better therapeutic approaches addressing apoE4’s detrimental effects at a molecular, cellular, and network level.
Perspective: better understanding of the selective vulnerability of GABAergic interneurons to ApoE4
Based on both in vivo and in vitro studies, GABAergic interneurons appear to be selectively vulnerable to apoE4 induced neurotoxicity, although the underlying molecular and cellular mechanisms are still unclear. However, a number of potential hypotheses can be put forth for experimental testing [82]. While many potential pathways could cause GABAergic interneurons to be selectively vulnerable to apoE4, we would suggest focusing on the following two. One hypothesis is that GABAergic interneurons might generate more neurotoxic apoE4 fragments due to higher expression of apoE or its cleaving protease. This increased fragment generation would lead to increased neurotoxicity and cell death [20, 123–125]. Upon identification of the apoE4 cleaving protease, a testable hypothesis would be to investigate whether GABAergic interneurons produce more of this protease and therefore generate more neurotoxic apoE4 fragments leading to their death. A second hypothesis is that the metabolic demand of GABAergic interneurons makes them selectively vulnerable to apoE4 pathology. Multiple groups have presented evidence of mitochondrial impairments in AD [185, 186]. As mentioned previously, apoE4 induces deficits in mitochondrial function [187, 188]. Interestingly, there is increasing evidence that GABAergic interneurons require a unique level of high-energy expenditure [189]. An intriguing explanation for GABAergic interneuron selective vulnerability to apoE4, then, is that they have unique demands for high energy production which, in turn, makes them vulnerable to any perturbation of mitochondrial function [189, 190]. A recent study reports that apoE4-expressing neuronal cells have 50% less reserve capacity to generate ATP than apoE3-expressing neuronal cells as well as widespread changes in mitochondrial protein production and translocation, which makes apoE4-expressing neuronal cells more vulnerable to metabolic stress [191]. Building off these data, a testable hypothesis is that apoE4-induced mitochondrial dysfunction is especially damaging to GABAergic interneurons because of their especially high demands for metabolic energy.
Perspective: better therapies targeting ApoE4’s detrimental effects on GABAergic interneurons
Several approaches could be further developed for treating apoE4-mediated pathologies or GABAergic dysfunction. First, apoE4-mediated GABAergic deficits and cognitive decline could be treated with small molecules. For example, treating apoE4-KI mice with pentobarbital early in life prevents learning and memory deficits late in life [126]. Furthermore, the use of a structure corrector has been shown in vitro to ameliorate apoE4-mediated AD pathologies in hiPSC-derived neurons, including GABAergic neuron deficits [128]. However, developing new drugs for new targets can be prohibitively expensive. Using current screening methods it is possible to find combinations of existing drugs (drug repurposing) that can correct pathological phenotypes of AD [192, 193]. In the context of apoE4, it would be especially interesting to identify existing drugs that can enhance GABAergic interneuron function or can correct gene expression signatures in apoE4/4 neurons to a more ‘apoE3/3-like’ profile.
Several treatments which enhance inhibition have been tested in animal models and in clinical trials. GABAA receptor potentiators or agonists ameliorate apoE4- or amyloid-induced toxicity and improve cognition in rodent models of AD and normal aging [126, 194]. However, across several clinical trials, these agents have produced behavioral, but not cognitive, improvements [85]. Unfortunately, these therapeutics produce undesirable side effects which limit long-term use [195, 196]. Anti-epileptic agents similarly show promise in animal models [103, 104], but have not produced cognitive improvements in clinical trials [85], with the exception of levetiracetam that improved cognition and reduced hippocampal hyperactivity in preclinical and initial clinical studies [107–110, 175, 197–199]. However, trials for both of these therapeutics used only small cohorts over short treatment periods, so further study in larger clinical trials is required. Moreover, specifically targeted therapies might be more beneficial. For instance, theta burst stimulation via transcranial magnetic stimulation has been used successfully to increase GABA within the DMN [200]. This could be used to rescue specific network pathologies rather than globally increasing inhibition.
Driving specific interneuron populations could be used to rescue network synchrony. Two foundational optogenetic studies demonstrated that optogenetically driving inhibitory interneurons specifically enhances slow gamma frequency oscillations throughout cortex, reducing circuit noise while amplifying circuit signal [201, 202]. Non-invasive stimulation can augment endogenous network oscillations to enhance learning and memory. In humans, transcranial magnetic stimulation enhances cortical slow waves and thus improve task performance [203]. In mice, slow gamma frequency visual or audio input entrains neural firing to this frequency in the cortex and hippocampus and reduces Aβ pathology and microglial abnormalities [204, 205]. Finally, enhancing activity of existing interneurons could also attenuate the network effects. For example, exogenous neuregulin 1 increases excitability of parvalbumin-positive interneurons [206] and has been used to restore hippocampal theta synchrony and fear conditioning in a mouse model of schizophrenia, which showed inhibitory impairments [207].
In addition to targeting susceptibility of GABAergic interneurons to apoE4 and the subsequent network hyperexcitability that results from inhibitory neuron loss, another potential therapy is to replace the lost population of GABAergic interneurons. Cell replacement therapy has been explored in the context of various neurodegenerative diseases [208–211]. Notably, it has been shown that GABAergic interneuron progenitor transplantation has potential to be an effective method to correct seizure activity in an epilepsy model [212]. Likewise, transplantation of mouse MGE-derived GABAergic progenitors into aged apoE4-KI mice without or with Aβ accumulation rescues learning and memory deficits [127]. Furthermore, transplanting Nav1.1-overexpressing interneurons derived from the mouse MGE into an hAPPFAD mouse model enhances behavior-dependent gamma oscillatory activity, reduces network hypersynchrony, and improves cognitive function [213]. In the future, it would be interesting to employ a similar cell therapeutic strategy, using hiPSC-derived GABAergic progenitors with an apoE3/3 genotype as donor cells for transplantation, to treat hyperexcitability and network deficits in an apoE4 model of AD.
Clearly, new hope for effective therapeutics of AD relies upon the ability of scientists to explore multiple lines of inquiry. Moving forward, it is certainly conceivable that there will be combination therapies implemented, with drugs targeting Aβ, tau, inflammation, apoE4, and apoE4-induced GABAergic interneuron impairment.
Acknowledgements
The authors would like to thank Misha Zilberter, Kelly Zalocusky, Maxine Nelson, Antara Rao, Nicole Koutsodendris, and Theodora Pak for assistance in the reviewing and editing process.
Abbreviations
- AAV
Adeno-associated virus
- AD
Alzheimer’s disease
- Apo
Apolipoprotein
- apoE-KI
ApoE knock-in
- apoE-KO
ApoE knock-out
- APP
Amyloid precursor protein
- ASO
Antisense oligonucleotides
- Aβ
Amyloid-β
- CNS
Central nervous system
- DMN
Default mode network
- GABA
γ-aminobutyric acid
- hiPSC
Human induced pluripotent stem cell
- hPSC
Human pluripotent stem cell
- MCI
Mild cognitive impairment
- MGE
Medial ganglionic eminence
- NFTs
Neurofibrillary tangles
- NMR
Nuclear magnetic resonance
- PS1
Presenilin-1
- PS2
Presenilin-2
Authors’ contributions
RN, EAJ, and YH developed the concept and structure of the review. RN and EAJ contributed equally to writing the review. YH revised and finalized the review. All authors read and approved the final manuscript.
Funding
This work was supported by grants AG048030, AG048017, AG047655, AG055421, and AG055682 to YH from the National Institutes of Health. EAJ was partially supported by a fellowship 1F31AG057150 from the National Institutes of Health.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
YH is a co-founder and SAB member of E-scape Bio, Inc. and GABAeron, Inc.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ramsey Najm and Emily A. Jones contributed equally to this work.
References
- 1.Prince M, Wimo A, Guerchet M, Ali G-C, Wu Y-T, Prina M. World Alzheimer Report 2015: The Global Impact of Dementia. Alzheimer’s Disease International. 2015.
- 2.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 3.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 4.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81(4):740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2014;72:3–12. doi: 10.1016/j.nbd.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 10.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 11.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA J Am Med Assoc. 1997;278(16):1349–1356. doi: 10.1001/jama.1997.03550160069041. [DOI] [PubMed] [Google Scholar]
- 12.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med. 2016;94(7):739–746. doi: 10.1007/s00109-016-1427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103(15):5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31(8):445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Frieden C, Garai K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci. 2012;109(28):E1972–E1979. doi: 10.1073/pnas.1208003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci. 2011;108(36):14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatters DM, Budamagunta MS, Voss JC, Weisgraber KH. Modulation of apolipoprotein E structure by domain interaction: differences in lipid-bound and lipid-free forms. J Biol Chem. 2005;280(40):34288–34295. doi: 10.1074/jbc.M506044200. [DOI] [PubMed] [Google Scholar]
- 19.Xu Q, Brecht WJ, Weisgraber KH, Mahley RW, Huang Y. Apolipoprotein E4 domain interaction occurs in living neuronal cells as determined by fluorescence resonance energy transfer. J Biol Chem. 2004;279(24):25511–25516. doi: 10.1074/jbc.M311256200. [DOI] [PubMed] [Google Scholar]
- 20.Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, et al. Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24(10):2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y. Aβ-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2010;16(6):287–294. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Mahley RW, Huang Y. Apolipoprotein E sets the stage: response to injury triggers neuropathology. Neuron. 2012;76(5):871–885. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grehan S, Tse E, Taylor JM. Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J Neurosci. 2001;21(3):812–822. doi: 10.1523/JNEUROSCI.21-03-00812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta (BBA)/Lipids Lipid Metab. 1987;917(1):148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 25.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26(19):4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu PT, Schmechel D, Rothrock-Christian T, Burkhart DS, Qiu HL, Popko B, et al. Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol Dis. 1996;3(3):229–245. doi: 10.1006/nbdi.1996.0023. [DOI] [PubMed] [Google Scholar]
- 27.Shi Yang, Holtzman David M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nature Reviews Immunology. 2018;18(12):759–772. doi: 10.1038/s41577-018-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer’s disease. J Mol Neurosci. 2004;23(3):189–204. doi: 10.1385/JMN:23:3:189. [DOI] [PubMed] [Google Scholar]
- 29.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci. 1999;96(26):15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97(6):2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10(7):719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 32.Castellano JM, Kim J, Stewart FR, Jiang H, Demattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3(89):89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golabek AA, Soto C, Vogel T, Wisniewski T. The interaction between apolipoprotein E and Alzheimer’s amyloid β-peptide is dependent on β-peptide conformation. J Biol Chem. 1996;271(18):10602–10606. doi: 10.1074/jbc.271.18.10602. [DOI] [PubMed] [Google Scholar]
- 34.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol. 2006;361(5):932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 35.Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, et al. Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29(21):6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, et al. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287(50):41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatami A, Monjazeb S, Milton S, Glabe CG. Familial Alzheimer’s disease mutations within the amyloid precursor protein Alter the aggregation and conformation of the amyloid-β peptide. J Biol Chem. 2017;292(8):3172–3185. doi: 10.1074/jbc.M116.755264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32(14):4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Jiang H, Park S, Eltorai AEM, Stewart FR, Yoon H, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011;31(49):18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irizarry MC, Rebeck GW, Cheung B, Bales K, Paul SM, Holzman D, et al. Modulation of Aβ deposition in APP transgenic mice by an apolipoprotein E null background. Ann N Y Acad Sci. 2000;920:171–178. doi: 10.1111/j.1749-6632.2000.tb06919.x. [DOI] [PubMed] [Google Scholar]
- 42.Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, et al. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer’s disease. J Clin Invest. 1999;103(6):R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, et al. Human apolipoprotein E4 alters the amyloid-β 40:42 ratio and promotes the formation of cerebral amyloid Angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25(11):2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harper JD, Lansbury PT. Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 45.Wood SJ, Chan W, Wetzel R. An ApoE-Aβ inhibition complex in Aβ fibril extension. Chem Biol. 1996;3(11):949–956. doi: 10.1016/S1074-5521(96)90183-0. [DOI] [PubMed] [Google Scholar]
- 46.Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J Neurosci. 2012;32(43):15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerf E, Gustot A, Goormaghtigh E, Ruysschaert J-M, Raussens V. High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the pathological entities responsible for Alzheimer’s disease. FASEB J. 2011;25(5):1585–1595. doi: 10.1096/fj.10-175976. [DOI] [PubMed] [Google Scholar]
- 48.Naiki H, Gejyo F, Nakakuki K. Concentration-dependent inhibitory effects of apolipoprotein E on Alzheimer’s β-amyloid fibril formation in vitro. Biochemistry. 1997;36(20):6243–6250. doi: 10.1021/bi9624705. [DOI] [PubMed] [Google Scholar]
- 49.Cramer PE, Cirrito JR, Wesson DW, Lee CYD, Karlo JC, Zinn AE, et al. ApoE-directed therapeutics rapidly clear β -amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, et al. The LXR agonist TO901317 selectively lowers hippocampal Aβ42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci. 2007;34(4):621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 51.Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson J-A, Holtzman DM, et al. Critical role of Astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Aβ phagocytosis. J Neurosci. 2011;31(19):7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J, Eltorai AEM, Jiang H, Liao F, Verghese PB, Kim J, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J Exp Med. 2012;209(12):2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vincent B, Smith JD. Astrocytes down-regulate neuronal β-amyloid precursor protein expression and modify its processing in an apolipoprotein E isoform-specific manner. Eur J Neurosci. 2001;14(2):256–266. doi: 10.1046/j.0953-816x.2001.01643.x. [DOI] [PubMed] [Google Scholar]
- 54.Liu C-C, Zhao N, Fu Y, Wang N, Linares C, Tsai C-W, et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron. 2017;96(5):1024–1032.e3. doi: 10.1016/j.neuron.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huynh T-PV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, et al. Age-dependent effects of apoE reduction using antisense oligonucleotides in a model of β-amyloidosis. Neuron. 2017;96(5):1013–1023. doi: 10.1016/j.neuron.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Irizarry MC, Deng A, Lleo A, Berezovska O, von Arnim CAF, Martin-Rehrmann M, et al. Apolipoprotein E modulates γ-secretase cleavage of the amyloid precursor protein. J Neurochem. 2004;90(5):1132–1143. doi: 10.1111/j.1471-4159.2004.02581.x. [DOI] [PubMed] [Google Scholar]
- 57.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform – specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest. 2008;118(12):4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu C-C, Hu J, Zhao N, Wang J, Wang N, Cirrito JR, et al. Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J Neurosci. 2017;37(15):4023–4031. doi: 10.1523/JNEUROSCI.3442-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma Q, Zhao Z, Sagare AP, Wu Y, Wang M, Owens NC, et al. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener. 2018;13(1):57. doi: 10.1186/s13024-018-0286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prasad H, Rao R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc Natl Acad Sci. 2018;115(28):E6640–E6649. doi: 10.1073/pnas.1801612115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Wang C, Huang Y. C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-β(Aβ) and acts in concert with Aβ to elicit neuronal and behavioral deficits in mice. Proc Natl Acad Sci U S A. 2011;108(10):4236–4241. doi: 10.1073/pnas.1018381108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baitsch D, Bock HH, Engel T, Telgmann R, Müller-Tidow C, Varga G, et al. Apolipoprotein E induces Antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. 2011;31(5):1160–1168. doi: 10.1161/ATVBAHA.111.222745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, Rebeck GW, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60(4):559–569. doi: 10.1002/glia.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cudaback E, Li X, Montine KS, Montine TJ, Keene CD. Apolipoprotein E isoform-dependent microglia migration. FASEB J. 2011;25(6):2082–2091. doi: 10.1096/fj.10-176891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fernandez CG, Hamby ME, McReynolds ML, Ray WJ. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer's Disease. Front Aging Neurosci. 2019. 10.3389/fnagi.2019.00014. [DOI] [PMC free article] [PubMed]
- 66.Kanekiyo T, Cirrito JR, Liu C-C, Shinohara M, Li J, Schuler DR, et al. Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci. 2013;33(49):19276–19283. doi: 10.1523/JNEUROSCI.3487-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60(9):1495–1500. doi: 10.1212/01.WNL.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 68.Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, et al. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci U S A. 1998;95(18):10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raber J, Wong D, Yu G-Q, Buttini M, Mahley R, Pitas R, et al. Apolipoprotein E and cognitive performance. Nature. 2000;404(6776):352–354. doi: 10.1038/35006165. [DOI] [PubMed] [Google Scholar]
- 70.Leung L, Andrews-Zwilling Y, Yoon SY, Jain S, Ring K, Dai J, et al. Apolipoprotein E4 causes age- and sex-dependent impairments of hilar GABAergic interneurons and learning and memory deficits in mice. PLoS One. 2012;7(12):e53569. doi: 10.1371/journal.pone.0053569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, et al. Apolipoprotein E4 causes age- and tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30(41):13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brodbeck J, McGuire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong DE, et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by Small-molecule structure correctors. J Biol Chem. 2011;286(19):17217–17226. doi: 10.1074/jbc.M110.217380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29(48):15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 Knockin mice. Cell Stem Cell. 2009;5(6):634–645. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, et al. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurol. 2007;6(6):494–500. doi: 10.1016/S1474-4422(07)70106-0. [DOI] [PubMed] [Google Scholar]
- 76.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 77.Uddin Md. Sahab, Kabir Md. Tanvir, Al Mamun Abdullah, Abdel-Daim Mohamed M., Barreto George E., Ashraf Ghulam Md. APOE and Alzheimer’s Disease: Evidence Mounts that Targeting APOE4 may Combat Alzheimer’s Pathogenesis. Molecular Neurobiology. 2018;56(4):2450–2465. doi: 10.1007/s12035-018-1237-z. [DOI] [PubMed] [Google Scholar]
- 78.Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci. 2001;98(15):8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao N, Liu C-C, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, et al. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat Commun. 2018;9(1):4388. doi: 10.1038/s41467-018-06783-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fu H, Hardy J, Duff KE. Selective vulnerability in neurodegenerative diseases. Nat Neurosci. 2018;21(10):1350–1358. doi: 10.1038/s41593-018-0221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramamoorthi K, Lin Y. The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol Med. 2011;17(8):452–462. doi: 10.1016/j.molmed.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Govindpani K, Calvo-Flores Guzmán B, Vinnakota C, Waldvogel H, Faull R, Kwakowsky A, et al. Towards a better understanding of GABAergic remodeling in Alzheimer’s disease. Int J Mol Sci. 2017;18(8):1813. doi: 10.3390/ijms18081813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lanctôt KL, Herrmann N, Mazzotta P, Khan LR, Ingber N. GABAergic function in Alzheimer’s disease: evidence for dysfunction and potential as a therapeutic target for the treatment of Behavioural and psychological symptoms of dementia. Can J Psychiatr. 2016;49(7):439–453. doi: 10.1177/070674370404900705. [DOI] [PubMed] [Google Scholar]
- 86.Garcia-Marin V, Blazquez-Llorca L, Rodriguez J-R, Boluda S, Muntane G, Ferrer I, et al. Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front Neuroanat. 2009;3:28. doi: 10.3389/neuro.05.028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramos-Miguel A, Hercher C, Beasley CL, Barr AM, Bayer TA, Falkai P, et al. Loss of Munc18-1 long splice variant in GABAergic terminals is associated with cognitive decline and increased risk of dementia in a community sample. Mol Neurodegener. 2015;10:65. doi: 10.1186/s13024-015-0061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Soricelli A, Postiglione A, Grivet-Fojaja MR, Mainenti PP, Discepolo A, Varrone A, et al. Reduced cortical distribution volume of iodine-123 iomazenil in Alzheimer’s disease as a measure of loss of synapses. Eur J Nucl Med. 1996;23(10):1323–1328. doi: 10.1007/BF01367587. [DOI] [PubMed] [Google Scholar]
- 89.Fukuchi K, Hashikawa K, Seike Y, Moriwaki H, Oku N, Ishida M, et al. Comparison of iodine-123-iomazenil SPECT and technetium-99m-HMPAO-SPECT in Alzheimer’s disease. J Nucl Med. 1997;38(3):467–470. [PubMed] [Google Scholar]
- 90.Bareggi SR, Franceschi M, Bonini L, Zecca L, Smirne S. Decreased CSF concentrations of Homovanillic acid and γ-aminobutyric acid in Alzheimer’s disease. Age- or disease-related modifications? Arch Neurol. 1982;39(11):709. doi: 10.1001/archneur.1982.00510230035010. [DOI] [PubMed] [Google Scholar]
- 91.Zimmer R, Teelken AW, Trieling WB, Weber W, Weihmayr T, Lauter H. γ-aminobutyric acid and Homovanillic acid concentration in the CSF of patients with senile dementia of Alzheimer’s type. Arch Neurol. 1984;41(6):602–604. doi: 10.1001/archneur.1984.04210080010005. [DOI] [PubMed] [Google Scholar]
- 92.Manyam NV, Katz L, Hare TA, Gerber JC, Grossman MH. Levels of γ-aminobutyric acid in cerebrospinal fluid in various neurologic disorders. Arch Neurol. 1980;37(6):352–355. doi: 10.1001/archneur.1980.00500550054006. [DOI] [PubMed] [Google Scholar]
- 93.Enna SJ, Stern LZ, Wastek GJ, Yamamura HI. Cerebrospinal fluid γ-aminobutyric acid variations in neurological disorders. Arch Neurol. 1977;34(11):683–685. doi: 10.1001/archneur.1977.00500230053008. [DOI] [PubMed] [Google Scholar]
- 94.Davies P, Katzman R, Terry RD. Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature. 1980;288(5788):279–280. doi: 10.1038/288279a0. [DOI] [PubMed] [Google Scholar]
- 95.Chan-Palay V. Somatostatin immunoreactive neurons in the human hippocampus and cortex shown by immunogold/silver intensification on vibratome sections: coexistence with neuropeptide Y neurons, and effects in Alzheimer-type dementia. J Comp Neurol. 1987;260(2):201–223. doi: 10.1002/cne.902600205. [DOI] [PubMed] [Google Scholar]
- 96.Palmer AM, Gershon S. Is the neuronal basis of Alzheimer’s disease cholinergic or glutamatergic ? Faseb. 1990;4(10):2745–2752. doi: 10.1096/fasebj.4.10.2165009. [DOI] [PubMed] [Google Scholar]
- 97.Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia. 2001;42(SUPPL. 3):8–12. doi: 10.1046/j.1528-1157.2001.042suppl.3008.x. [DOI] [PubMed] [Google Scholar]
- 98.Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66(4):435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13(7):812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70(9):1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55(5):697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.DiFrancesco JC, Tremolizzo L, Polonia V, Giussani G, Bianchi E, Franchi C, et al. Adult-onset epilepsy in Presymptomatic Alzheimer’s disease: a retrospective study. J Alzheimers Dis. 2017;60(4):1267–1274. doi: 10.3233/JAD-170392. [DOI] [PubMed] [Google Scholar]
- 103.Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci. 2012;109(42):E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shi J-Q, Wang B-R, Tian Y-Y, Xu J, Gao L, Zhao S-L, et al. Antiepileptics Topiramate and Levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci Ther. 2013;19(11):871–881. doi: 10.1111/cns.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Koh MT, Haberman RP, Foti S, McCown TJ, Gallagher M. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology. 2010;35(4):1016–1025. doi: 10.1038/npp.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Devi L, Ohno M. Effects of levetiracetam, an antiepileptic drug, on memory impairments associated with aging and Alzheimer’s disease in mice. Neurobiol Learn Mem. 2013;102:7–11. doi: 10.1016/j.nlm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 107.Haberman RP, Branch A, Gallagher M. Targeting neural hyperactivity as a treatment to stem progression of late-onset Alzheimer’s disease. Neurotherapeutics. 2017;14(3):662–676. doi: 10.1007/s13311-017-0541-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schoenberg MR, Rum RS, Osborn KE, Werz MA. A randomized, double-blind, placebo-controlled crossover study of the effects of levetiracetam on cognition, mood, and balance in healthy older adults. Epilepsia. 2017;58(9):1566–1574. doi: 10.1111/epi.13849. [DOI] [PubMed] [Google Scholar]
- 109.Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74(3):467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cumbo E, Ligori LD. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer’s disease. Epilepsy Behav. 2010;17(4):461–466. doi: 10.1016/j.yebeh.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 111.Moore R. Principles of synaptic transmission. Ann N Y Acad Sci. 1993;695:1–9. doi: 10.1111/j.1749-6632.1993.tb23018.x. [DOI] [PubMed] [Google Scholar]
- 112.Mongillo G, Rumpel S, Loewenstein Y. Inhibitory connectivity defines the realm of excitatory plasticity. Nat Neurosci. 2018;21(10):1463–1470. doi: 10.1038/s41593-018-0226-x. [DOI] [PubMed] [Google Scholar]
- 113.Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378(6552):75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- 114.Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562(Pt 1):9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu X, An L, Mi X, Zhang T. Impairment of cognitive function and synaptic plasticity associated with alteration of information flow in Theta and gamma oscillations in melamine-treated rats. PLoS One. 2013;8(10):e77796. doi: 10.1371/journal.pone.0077796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cardin JA. Inhibitory interneurons regulate temporal precision and correlations in cortical circuits. Trends Neurosci. 2018;41(10):689–700. doi: 10.1016/j.tins.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jones MW, Wilson MA. Theta rhythms coordinate hippocampal-prefrontal interactions in a spatial memory task. PLoS Biol. 2005;3(12):e402. doi: 10.1371/journal.pbio.0030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mann EO, Kohl MM, Paulsen O. Distinct roles of GABA(a) and GABA(B) receptors in balancing and terminating persistent cortical activity. J Neurosci. 2009;29(23):7513–7518. doi: 10.1523/JNEUROSCI.6162-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lehmann K, Steinecke A, Bolz J. GABA through the ages: regulation of cortical function and plasticity by inhibitory interneurons. Neural Plast. 2012;2012:8927841. doi: 10.1155/2012/892784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hu J-H, Ma Y-H, Jiang J, Yang N, Duan S, Jiang Z-H, et al. Cognitive impairment in mice over-expressing gamma-aminobutyric acid transporter 1 (GAT1) Neuroreport. 2004;15(1):9–12. doi: 10.1097/00001756-200401190-00003. [DOI] [PubMed] [Google Scholar]
- 121.Prut L, Prenosil G, Willadt S, Vogt K, Fritschy J-M, Crestani F. A reduction in hippocampal GABA a receptor α5 subunits disrupts the memory for location of objects in mice. Brain Behav. 2010;9(5):478–488. doi: 10.1111/j.1601-183X.2010.00575.x. [DOI] [PubMed] [Google Scholar]
- 122.Andrews-Zwilling Y, Gillespie AK, Kravitz AV, Nelson AB, Devidze N, Lo I, et al. Hilar GABAergic interneuron activity controls spatial learning and memory retrieval. PLoS One. 2012;7(7):e40555. doi: 10.1371/journal.pone.0040555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Buttini M, Masliah E, Yu G-Q, Palop JJ, Chang S, Bernardo A, et al. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am J Pathol. 2010;177(2):563–569. doi: 10.2353/ajpath.2010.090973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jain S, Yoon SY, Leung L, Knoferle J, Huang Y. Cellular source-specific effects of apolipoprotein (Apo) E4 on dendrite Arborization and dendritic spine development. PLoS One. 2013;8(3):1–14. doi: 10.1371/journal.pone.0059478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Knoferle J, Yoon SY, Walker D, Leung L, Gillespie AK, Tong LM, et al. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J Neurosci. 2014;34(42):14069–14078. doi: 10.1523/JNEUROSCI.2281-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tong LM, Yoon SY, Andrews-Zwilling Y, Yang A, Lin V, Lei H, et al. Enhancing GABA signaling during middle adulthood prevents age-dependent GABAergic interneuron decline and learning and memory deficits in ApoE4 mice. J Neurosci. 2016;36(7):2316–2322. doi: 10.1523/JNEUROSCI.3815-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tong LM, Djukic B, Arnold C, Gillespie AK, Yoon SY, Wang MM, et al. Inhibitory interneuron progenitor transplantation restores Normal learning and memory in ApoE4 Knock-in mice without or with Aβ accumulation. J Neurosci. 2014;34(29):9506–9515. doi: 10.1523/JNEUROSCI.0693-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang C, Najm R, Xu Q, Jeong D, Walker D, Balestra ME, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a Small-molecule structure corrector. Nat Med. 2018;24(5):647–657. doi: 10.1038/s41591-018-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98(6):1294. doi: 10.1016/j.neuron.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee V, Maguire J. The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front Neural Circuits. 2014;8:3. doi: 10.3389/fncir.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lucas EK, Clem RL. GABAergic interneurons: the orchestra or the conductor in fear learning and memory? Brain Res Bull. 2018;141:13–19. doi: 10.1016/j.brainresbull.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fu Y, Lv R, Jin L, Lu Q, Shao X, He J, et al. Association of apolipoprotein E polymorphisms with temporal lobe epilepsy in a Chinese Han population. Epilepsy Res. 2010;91(2–3):253–259. doi: 10.1016/j.eplepsyres.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 133.Li Z, Ding C, Gong X, Wang X, Cui T. Apolipoprotein E ε4 allele was associated with Nonlesional mesial temporal lobe epilepsy in Han Chinese population. Medicine (Baltimore) 2016;95(9):e2894. doi: 10.1097/MD.0000000000002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Diaz-Arrastia Ramon, Gong Yunhua, Fair Suzette, Scott Kristin D., Garcia Maria C., Carlile Mary C., Agostini Mark A., Van Ness Paul C. Increased Risk of Late Posttraumatic Seizures Associated With Inheritance of APOE ϵ4 Allele. Archives of Neurology. 2003;60(6):818. doi: 10.1001/archneur.60.6.818. [DOI] [PubMed] [Google Scholar]
- 135.Salzmann A, Perroud N, Crespel A, Lambercy C, Malafosse A. Candidate genes for temporal lobe epilepsy: a replication study. Neurol Sci. 2008;29(6):397–403. doi: 10.1007/s10072-008-1060-9. [DOI] [PubMed] [Google Scholar]
- 136.Johnson EL, Krauss GL, Lee AK, Schneider ALC, Dearborn JL, Kucharska-Newton AM, et al. Association between midlife risk factors and late-onset epilepsy: results from the atherosclerosis risk in communities study. JAMA Neurol. 2018;75(11):1375–1382. doi: 10.1001/jamaneurol.2018.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Briellmann RS, Torn-Broers Y, Busuttil BE, Major BJ, Kalnins RM, Olsen M, et al. APOE ε4 genotype is associated with an earlier onset of chronic temporal lobe epilepsy. Neurology. 2000;55(3):435–437. doi: 10.1212/WNL.55.3.435. [DOI] [PubMed] [Google Scholar]
- 138.Kauffman MA, Consalvo D, Moron DG, Lereis VP, Kochen S. ApoE ɛ4 genotype and the age at onset of temporal lobe epilepsy: a case–control study and meta-analysis. Epilepsy Res. 2010;90(3):234–239. doi: 10.1016/j.eplepsyres.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 139.Leal B, Chaves J, Carvalho C, Bettencourt A, Freitas J, Lopes J, et al. Age of onset of mesial temporal lobe epilepsy with hippocampal sclerosis: the effect of apolipoprotein E and febrile seizures. Int J Neurosci. 2017;127(9):800–804. doi: 10.1080/00207454.2016.1264396. [DOI] [PubMed] [Google Scholar]
- 140.Aboud O, Mrak RE, Boop F, Griffin ST. Apolipoprotein epsilon 3 alleles are associated with indicators of neuronal resilience. BMC Med. 2012;10:35. doi: 10.1186/1741-7015-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sporis D, Sertic J, Henigsberg N, Mahovic D, Bogdanovic N, Babic T. Association of refractory complex partial seizures with a polymorphism of ApoE genotype. J Cell Mol Med. 2005;9(3):698–703. doi: 10.1111/j.1582-4934.2005.tb00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schubert Carla R., Carmichael Lakeesha L., Murphy Claire, Klein Barbara E. K., Klein Ronald, Cruickshanks Karen J. Olfaction and the 5-Year Incidence of Cognitive Impairment in an Epidemiological Study of Older Adults. Journal of the American Geriatrics Society. 2008;56(8):1517–1521. doi: 10.1111/j.1532-5415.2008.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Devanand DP, Liu X, Tabert MH, Pradhaban G, Cuasay K, Bell K, et al. Combining early markers strongly predicts conversion from mild cognitive impairment to Alzheimer’s disease. Biol Psychiatry. 2008;64(10):871–879. doi: 10.1016/j.biopsych.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Olofsson JK, Rönnlund M, Nordin S, Nyberg L, Nilsson L-G, Larsson M. Odor identification deficit as a predictor of five-year global cognitive change: interactive effects with age and ApoE-ε4. Behav Genet. 2009;39(5):496–503. doi: 10.1007/s10519-009-9289-5. [DOI] [PubMed] [Google Scholar]
- 145.Olofsson JK, Josefsson M, Ekström I, Wilson D, Nyberg L, Nordin S, et al. Long-term episodic memory decline is associated with olfactory deficits only in carriers of ApoE-є4. Neuropsychologia. 2016;85:1–9. doi: 10.1016/j.neuropsychologia.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 146.Misiak MM, Hipolito MS, Ressom HW, Obisesan TO, Manaye KF, Nwulia EA. Apo E4 alleles and impaired olfaction as predictors of Alzheimer’s disease. Clin Exp Psychol. 2017;3(4):169. doi: 10.4172/2471-2701.1000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hu B, Geng C, Hou X-Y. Oligomeric amyloid-β peptide disrupts olfactory information output by impairment of local inhibitory circuits in rat olfactory bulb. Neurobiol Aging. 2017;51:113–121. doi: 10.1016/j.neurobiolaging.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 148.Peng KY, Mathews PM, Levy E, Wilson DA. Apolipoprotein E4 causes early olfactory network abnormalities and short-term olfactory memory impairments. Neuroscience. 2017;343:364–371. doi: 10.1016/j.neuroscience.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions : a co-expression meta-analysis. Acta Neuropathol. 2015;3(31):1–18. doi: 10.1186/s40478-015-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Frigerio CS, Wolfs L, Fattorelli N, Perry VH, Fiers M, De SB, et al. The Major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019;27(4):1293–1306. doi: 10.1016/j.celrep.2019.03.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Invest. 2017;127(9):3240–3249. doi: 10.1172/JCI90606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2017;217(2):459–472. doi: 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen Z, Jalabi W, Hu W, Park H, Gale JT, Kidd GJ, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun. 2014;5:4486. doi: 10.1038/ncomms5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Roseti C, Fucile S, Lauro C, Martinello K, Bertollini C, Esposito V, et al. Fractalkine/CX3CL1 modulates GABA(a) currents in human temporal lobe epilepsy. Epilepsia. 2013;5(10):1834–1844. doi: 10.1111/epi.12354. [DOI] [PubMed] [Google Scholar]
- 156.Brockner G, Brauer K, Hartg W, Wolff JR, Rickma MJ, Derouiche A, et al. Perineuronal nets provide a Polyanionic , glia-associated form of microenvironment around certain neurons in many parts of the rat brain. Glia. 1993;8(3):183–200. doi: 10.1002/glia.440080306. [DOI] [PubMed] [Google Scholar]
- 157.Kwok JCF, Dick G, Wang D, Fawcett JW. Extracellular matrix and Perineuronal nets in CNS repair. Dev Neurobiol. 2011;7(11):1073–1089. doi: 10.1002/dneu.20974. [DOI] [PubMed] [Google Scholar]
- 158.Baig S, Wilcock GK, Love S. Loss of perineuronal net N -acetylgalactosamine in Alzheimer’s disease. Acta Neuropathol. 2005;110(4):393–401. doi: 10.1007/s00401-005-1060-2. [DOI] [PubMed] [Google Scholar]
- 159.Härtig W, Brauer K. G B. Wisteria floribunda agglutinin-labelled nets surround parvalbumin- containing neurons. Neuroreport. 1992;3(10):869–872. doi: 10.1097/00001756-199210000-00012. [DOI] [PubMed] [Google Scholar]
- 160.Cattaud V, Bezzina C, Rey CC, Lejards C, Dahan L, Verret L. Early disruption of parvalbumin expression and perineuronal nets in the hippocampus of the Tg2576 mouse model of Alzheimer’s disease can be rescued by enriched environment. Neurobiol Aging. 2018;72:147–158. doi: 10.1016/j.neurobiolaging.2018.08.024. [DOI] [PubMed] [Google Scholar]
- 161.Cabungcal J, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci. 2013;110(22):9130–9135. doi: 10.1073/pnas.1300454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Persson J, Lind J, Larsson A, Ingvar M, Sleegers K, Van Broeckhoven C, et al. Altered deactivation in individuals with genetic risk for Alzheimer’s disease. Neuropsychologia. 2008;46(6):1679–1687. doi: 10.1016/j.neuropsychologia.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 163.Fleisher AS, Sherzai A, Taylor C, Langbaum JBS, Chen K, Buxton RB. Resting-state BOLD networks versus task-associated functional MRI for distinguishing Alzheimer’s disease risk groups. Neuroimage. 2009;47(4):1678–1690. doi: 10.1016/j.neuroimage.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pihlajamäki M, Sperling RA. Functional MRI assessment of task-induced deactivation of the default mode network in Alzheimer’s disease and at-risk older individuals. Behav Neurol. 2009;21(1):77–91. doi: 10.1155/2009/276384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hu Y, Chen X, Gu H, Yang Y. Resting-state glutamate and GABA concentrations predict task-induced deactivation in the default mode network. J Neurosci. 2013;33(47):18566–18573. doi: 10.1523/JNEUROSCI.1973-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kapogiannis D, Reiter DA, Willette AA, Mattson MP. Posteromedial cortex glutamate and GABA predict intrinsic functional connectivity of the default mode network. Neuroimage. 2013;64:112–119. doi: 10.1016/j.neuroimage.2012.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen Xi, Fan Xiaoying, Hu Yuzheng, Zuo Chun, Whitfield-Gabrieli Susan, Holt Daphne, Gong Qiyong, Yang Yihong, Pizzagalli Diego A, Du Fei, Ongur Dost. Regional GABA Concentrations Modulate Inter-network Resting-state Functional Connectivity. Cerebral Cortex. 2018;29(4):1607–1618. doi: 10.1093/cercor/bhy059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Buckner RL, Andrews-Hanna JR, Schacter DL. The Brain’s default network:anatomy, function and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 169.Mevel K, Chételat G, Eustache F, Desgranges B. The default mode network in healthy aging and Alzheimer’s disease. Int J Alzheimers Dis. 2011;2011:535816. doi: 10.4061/2011/535816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lustig C, Snyder AZ, Bhakta M, O’Brien KC, McAvoy M, Raichle ME, et al. Functional deactivations: change with age and dementia of the Alzheimer type. Proc Natl Acad Sci U S A. 2003;100(24):14504–14509. doi: 10.1073/pnas.2235925100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Broyd SJ, Demanuele C, Debener S, Helps SK, James CJ, Sonuga-Barke EJS. Default-mode brain dysfunction in mental disorders: a systematic review. Neurosci Biobehav Rev. 2009;33(3):279–296. doi: 10.1016/j.neubiorev.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 172.Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65(3):404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bondi MW, Houston WS, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology. 2005;64(3):501–508. doi: 10.1212/01.WNL.0000150885.00929.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY, et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun. 2017;8(1):1464. doi: 10.1038/s41467-017-01444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin. 2015;7:688–698. doi: 10.1016/j.nicl.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yassa MA, Lacy JW, Stark SM, Albert MS, Gallagher M, Stark CE. Pattern separation deficits associated with increased hippocampal CA3 and dentate gyrus activity in nondemented older adults. Hippocampus. 2011;21(9):968–979. doi: 10.1002/hipo.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Miller SL, Fenstermacher E, Bates J, Blacker D, Sperling RA, Dickerson BC. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J Neurol Neurosurg Psychiatry. 2008;79(6):630–635. doi: 10.1136/jnnp.2007.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Leal SL, Landau SM, Bell RK, Jagust WJ. Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline. Elife. 2017;6:e22978. doi: 10.7554/eLife.22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc Natl Acad Sci U S A. 2009;106(17):7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343(7):450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dennis NA, Browndyke JN, Stokes J, Need A, Burke JR, Welsh-Bohmer KA, et al. Temporal lobe functional activity and connectivity in young adult APOE ɛ4 carriers. Alzheimers Dement. 2010;6(4):303–311. doi: 10.1016/j.jalz.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gillespie AK, Jones EA, Lin Y-H, Karlsson MP, Kay K, Yoon SY, et al. Apolipoprotein E4 causes age-dependent disruption of slow gamma oscillations during hippocampal sharp-wave ripples. Neuron. 2016;90(4):740–751. doi: 10.1016/j.neuron.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Buzsáki G. Hippocampal sharp wave-ripple: a cognitive biomarker for episodic memory and planning. Hippocampus. 2015;25(10):1073–1188. doi: 10.1002/hipo.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Carr MF, Karlsson MP, Frank LM. Transient slow gamma synchrony underlies hippocampal memory replay. Neuron. 2012;75(4):700–713. doi: 10.1016/j.neuron.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Silva DF, Selfridge JE, Lu J, E L, Cardoso SM, Swerdlow RH. Mitochondrial abnormalities in Alzheimer’s disease. Possible targets for therapeutic intervention. Adv Pharmacol. 2012;64:83–126. doi: 10.1016/B978-0-12-394816-8.00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta. 2014;1842(8):1219–1231. doi: 10.1016/j.bbadis.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102(51):18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, et al. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem. 2011;286(7):5215–5221. doi: 10.1074/jbc.M110.151084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kann O, Papageorgiou IE, Draguhn A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J Cereb Blood Flow Metab. 2014;34(8):1270–1282. doi: 10.1038/jcbfm.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kann O. The interneuron energy hypothesis: implications for brain disease. Neurobiol Dis. 2016;90:75–85. doi: 10.1016/j.nbd.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 191.Orr AL, Kim C, Jimenez-morales D, Newton BW, Johnson J, Swaney D, et al. Neuronal apolipoprotein E4 expression results in proteome-wide alterations and compromises bioenergetic capacity by disrupting mitochondrial function. J Alzheimers Dis. 2019;68(3):991–1011. doi: 10.3233/JAD-181184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12(4):487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 193.Kondo T, Imamura K, Funayama M, Tsukita K, Miyake M, Ohta A, et al. iPSC-based compound screening and in vitro trials identify a synergistic anti-amyloid β combination for Alzheimer’s disease. Cell Rep. 2017;21(8):2304–2312. doi: 10.1016/j.celrep.2017.10.109. [DOI] [PubMed] [Google Scholar]
- 194.Li Y, Sun H, Chen Z, Xu H, Bu G, Zheng H. Implications of GABAergic neurotransmission in Alzheimer’s disease. Front Aging Neurosci. 2016;8:31. doi: 10.3389/fnagi.2016.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nava-Mesa MO, Jiménez-Díaz L, Yajeya J, Navarro-Lopez JD. GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer’s disease. Front Cell Neurosci. 2014;8:167. doi: 10.3389/fncel.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Calvo-Flores Guzmán B, Vinnakota C, Govindpani K, Waldvogel HJ, Faull RLM, Kwakowsky A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J Neurochem. 2018;146(6):649–669. doi: 10.1111/jnc.14345. [DOI] [PubMed] [Google Scholar]
- 197.Helmstaedter C, Witt J-A. Cognitive outcome of antiepileptic treatment with levetiracetam versus carbamazepine monotherapy: a non-interventional surveillance trial. Epilepsy Behav. 2010;18(1–2):74–80. doi: 10.1016/j.yebeh.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 198.Lippa CF, Rosso A, Hepler M, Jenssen S, Pillai J, Irwin D. Levetiracetam: a practical option for seizure management in elderly patients with cognitive impairment. Am J Alzheimer’s Dis Other Dementiasr. 2010;25(2):149–154. doi: 10.1177/1533317508325095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Haberman Rebecca P., Koh Ming Teng, Gallagher Michela. Heightened cortical excitability in aged rodents with memory impairment. Neurobiology of Aging. 2017;54:144–151. doi: 10.1016/j.neurobiolaging.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Vidal-Piñeiro D, Martín-Trias P, Falcón C, Bargalló N, Clemente IC, Valls-Solé J, et al. Neurochemical modulation in posteromedial default-mode network cortex induced by transcranial magnetic stimulation. Brain Stimul. 2015;8(5):937–944. doi: 10.1016/j.brs.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 201.Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459(7247):663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459(7247):698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ketz N, Jones AP, Bryant NB, Clark VP, Pilly PK. Closed-loop slow-wave tACS improves sleep-dependent long-term memory generalization by modulating endogenous oscillations. J Neurosci. 2018;38(33):7314–7326. doi: 10.1523/JNEUROSCI.0273-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. 2016;540(7632):230–235. doi: 10.1038/nature20587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Martorell AJ, Paulson AL, Suk H, Boyden ES, Singer AC, Tsai L. Multi-sensory gamma stimulation ameliorates Alzheimer’s-associated pathology and improves cognition. Cell. 2019;177(2):256–271. doi: 10.1016/j.cell.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li K-X, Lu Y-M, Xu Z-H, Zhang J, Zhu J-M, Zhang J-M, et al. Neuregulin 1 regulates excitability of fast-spiking neurons through Kv1.1 and acts in epilepsy. Nat Neurosci. 2011;15(2):267–273. doi: 10.1038/nn.3006. [DOI] [PubMed] [Google Scholar]
- 207.Marissal T, Salazar RF, Bertollini C, Mutel S, De Roo M, Rodriguez I, et al. Restoring wild-type-like CA1 network dynamics and behavior during adulthood in a mouse model of schizophrenia. Nat Neurosci. 2018;21(10):1412–1420. doi: 10.1038/s41593-018-0225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Srivastava D, DeWitt N. In vivo cellular reprogramming: the next generation. Cell. 2016;166(6):1386–1396. doi: 10.1016/j.cell.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tyson JA, Anderson SA. GABAergic interneuron transplants to study development and treat disease. Trends Neurosci. 2014;37(3):169–177. doi: 10.1016/j.tins.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Steinbeck JA, Studer L. Moving stem cells to the clinic: potential and limitations for brain repair. Neuron. 2015;86(1):187–206. doi: 10.1016/j.neuron.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li X, Zhu H, Sun X, Zuo F, Lei J, Wang Z, et al. Human neural stem cell transplantation rescues cognitive defects in APP/PS1 model of Alzheimer’s disease by enhancing neuronal connectivity and metabolic activity. Front Aging Neurosci. 2016;8:282. doi: 10.3389/fnagi.2016.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Cunningham M, Cho J-H, Leung A, Savvidis G, Ahn S, Moon M, et al. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell. 2014;15(5):559–573. doi: 10.1016/j.stem.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Martinez-Losa M, Tracy TE, Ma K, Verret L, Clemente-Perez A, Khan AS, et al. Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron. 2018;98(1):75–89.e5. doi: 10.1016/j.neuron.2018.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.