

Abstract
The remarkable degree of synthetic selectivity found in Nature is exemplified by the biosynthesis of paralytic shellfish toxins such as saxitoxin. The polycyclic core shared by saxitoxin and its relatives is assembled and subsequently elaborated through the installation of hydroxyl groups with exquisite precision that is not possible to replicate with traditional synthetic methods. Here, we report the identification of the enzymes that carry out a subset of C–H functionalizations involved in paralytic shellfish toxin biosynthesis. We have shown that three Rieske oxygenases mediate hydroxylation reactions with perfect site- and stereoselectivity. Specifically, the Rieske oxygenase SxtT is responsible for selective hydroxylation of a tricyclic precursor to the famous natural product saxitoxin, and a second Rieske oxygenase, GxtA, selectively hydroxylates saxitoxin to access the oxidation pattern present in gonyautoxin natural products. Unexpectedly, a third Rieske oxygenase, SxtH, does not hydroxylate tricyclic intermediates, but rather a linear substrate prior to tricycle formation, rewriting the biosynthetic route to paralytic shellfish toxins. Characterization of SxtT, SxtH, and GxtA is the first demonstration of enzymes carrying out C–H hydroxylation reactions in paralytic shellfish toxin biosynthesis. Additionally, the reactions of these oxygenases with a suite of saxitoxin-related molecules are reported, highlighting the substrate promiscuity of these catalysts and the potential for their application in the synthesis of natural and unnatural saxitoxin congeners
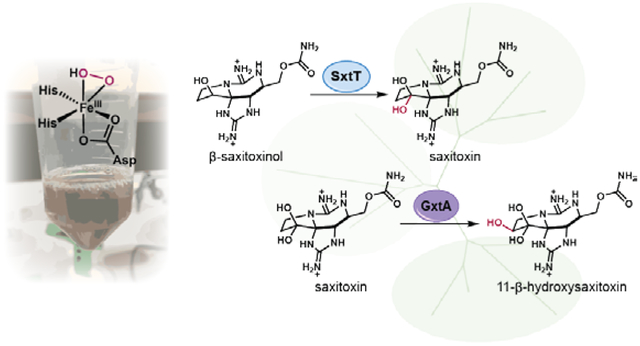
Graphical Abstract:
■ INTRODUCTION
Saxitoxin (STX, 9) has received significant attention due both to its biological activity and its remarkable architecture. STX is a paralytic shellfish toxin (PST) naturally produced by freshwater cyanobacteria as well as marine dinoflagellates.1–2 The toxic effect of STX and related PSTs stems from their high affinity for voltage-gated sodium channels (VGSC).3 This ability to control cellular action potential has made STX an indispensable tool in the fundamental study of VGSC and the lead compound in the development of pharmaceutical agents that are specific for targeted VGSC isoforms associated with disease and pain.2 A number of studies indicate that the VGSC affinity and subtype specificity of STX and related natural products are dramatically altered based on the level and pattern of oxidation of the tricyclic scaffold.1, 4–6 For example, VGSC affinity decreases as STX (9) is oxidized to neosaxitoxin (neoSTX, 4, Figure 1A), and further decreases with the addition of sulfate or sulfonamide groups at C11, which are present in gonyautoxins (GTXs, 2, 3, 5, 6 and 8, Figure 1A).1, 6 Thus, access to STX analogs that vary in their oxygenation would provide valuable tools for VGSC characterization and drug development. However, a unified synthetic strategy that provides access to such a panel of compounds challenges existing methods for selective oxygenation. To address this demand, we have identified the catalysts Nature has evolved for selective C–H hydroxylation of the STX tricyclic core.
Figure 1.

(A) Natural products derived from saxitoxin (STX, 9). The molecules with the highest and lowest Kd are outlined. *The Kd of 11-β-hydroxysaxitoxin (7) has not been determined, but its IC50 value ranks the molecule below saxitoxin in terms of affinity. (B) Previously proposed STX (9) biosynthetic pathway. (C) Saxitoxin gene cluster and potential enzymes involved in late-stage oxygenation.
Proposals on the biosynthetic route to STX are built upon feeding studies7–8, isolation of putative intermediates9–10, and bioinformatics analysis of gene clusters from STX-producing cyanobacteria.11–12 However, data verifying enzymatic transformations responsible for such proposals have been scarce. We recently demonstrated that STX biosynthesis is initiated by the conversion of arginine and malonyl-CoA to ethyl ketone 10, providing the first biochemical link between STX and the genes proposed to encode for its biosynthesis (Figure 1B) and disproving the long-standing hypothesis that acetyl-CoA is the initial substrate in STX biosynthesis.8, 13 This linear intermediate (10) is anticipated to undergo the installation of an amidino group and a number of oxidation and cyclization events to afford decarbamoylated tricycle 11.7–8, 14–17 The timing of carbamoylation is unknown as it might occur before or after oxygenation to generate dideoxysaxitoxin (ddSTX, 12) or STX (9), respectively. Finally, it has been suggested that a sequence of selective hydroxylation events of deoxygenated tricycles 11 or 12, could lead to STX (9) and higher oxidation level analogs, such as neoSTX (4) and GTXs (2, 3, 5, 6 and 8, Figure 1A)7, 9, 15–17. This series of late-stage hydroxylation events is supported by the LC-MS detection of deoxygenated tricycle 11 in cell extracts of toxic cyanobacteria and dinoflagellates9; however, these proposed oxidations have not been experimentally validated.
The annotation of four putative STX gene clusters by Neilan and coworkers includes a number of enzymes conserved across these clusters with the potential to carry out C–H bond oxygenation reactions (Figure 1C).15–17 For example, SxtD is related to fatty acid hydroxylases known to carry out hydroxylation reactions as well as dehydrogenation chemistry, while SxtS is related to α-ketoglutarate-dependent non-heme iron enzymes capable of mediating a variety of reactions, including hydroxylation and epoxidation.18 Finally, a pair of Rieske non-heme iron-dependent oxygenases, SxtH and SxtT, are conserved among the four gene clusters, with a third Rieske- type oxygenase, GxtA (previously SxtDiox), annotated in the STX gene cluster from Microseira wollei (formerly Lyngbya wollei).15 These oxygenases contain a Rieske [2Fe-2S] cluster as well as a mononuclear non-heme iron domain where the substrate binds and can undergo reactions including C–H hydroxylation, amine oxidation, dihydroxylation and oxidative cyclization.19 While several Rieske oxygenases are well studied for their role in the oxidative degradation of aromatic compounds,19–22 only four unique reactions of six Rieske oxygenases associated with secondary metabolism have been biochemically characterized. These include the RedG/McpG-catalyzed oxidative cyclizations in prodiginine biosynthesis23, the PrnD-mediated arylamine oxidation in pyrrolnitrin biosynthesis24–25, the oxidation of chlorophyll a to chlorophyll b by chlorophyllide a oxygenase26, and the hydroxylation of salvigenin by flavone-8-hydroxylase.27 The Rieske oxygenases SxtH, SxtT, and GxtA have been proposed to act exclusively on late-stage substrates in STX biosynthesis. Specifically, SxtT and SxtH have been implicated in the sequential dihydroxylation of ddSTX (12) to form STX (9). This hypothesis has been supported by the isolation of metabolites containing the C12 α-hydroxyl group (14) but lacking the C12 β-hydroxyl group (13), suggesting that one enzyme installs the α-hydroxyl group on 12 to generate 14 and a second enzyme is responsible for the oxidation to STX (9). Herein the reactions performed by SxtT, SxtH, and GxtA in paralytic shellfish toxin biosynthesis are disclosed, highlighting an unanticipated sequence of oxidation events leading to STX.
■ RESULTS AND DISCUSSION
Intrigued by the high sequence identity across the three STX Rieske oxygenases (between 82% and 87%), we hypothesized that these enzymes could be responsible for site-selective hydroxylation of the STX tricyclic core required to transform dideoxysaxitoxin (ddSTX, 12) into STX (9) and further oxygenated natural products such as the GTXs (2, 3, 5, 6 and 8) and neoSTX (4). Others have speculated that SxtH and SxtT are involved in the oxidation of the C12-methylene of ddSTX (12) to the C12 ketone hydrate moiety present in STX (9) and related natural products through two sequential oxidation events.15–17 For example, ddSTX (12) could first undergo hydroxylation at C12 to afford either β-saxitoxinol (β-STOH, 13) or α-saxitoxinol (α-STOH, 14, Figure 1B) from which a second hydroxylation could take place that would lead to STX (9). Al- ternatively, it is possible that one STX Rieske oxygenase is responsible for the hydroxylation of ddSTX (12) at C12 and that the resulting secondary alcohol could be oxidized to the ketone oxidation state by any of a number of enzymes to afford STX (9) upon hydration, mimicking established chemical methods.5 We anticipated that these Rieske oxygenases could also mediate hydroxylations at C11 or N1.
To date, there have been no reports of heterologous overexpression or functional characterization of any oxygenase associated with paralytic shellfish toxin biosynthesis. Our initial experiments focused on proteins from M. wollei.15 We achieved successful overexpression of maltose binding protein (MBP) fusions of SxtH, SxtT and GxtA in E. coli by employing established protocols for heterologous expression of iron-sulfur cluster containing proteins.22, 28–29 Based on absorbance spectrum, iron content, and gel filtration elution profiles of the purified proteins relative to known Rieske oxygenase characteristics, successful purification of each protein was achieved (Supporting Information,Fig S4–S8).
In preparation for reactions with these oxygenases, we considered options for generation of the active oxidant (Fe(III)-hydroperoxo species, Figure 2). Rieske oxygenases can have two modes of catalysis depending on the stoichiometric oxidant employed, molecular oxygen or hydrogen peroxide (Figures 2A and 2B, respectively).30–31 Natively, Rieske oxygenases utilize molecular oxygen, requiring the formation of a head-to-tail trimer complex, in which two electrons are delivered from a protein redox partner to the [2Fe-2S] cluster of one monomer and then to the mononuclear iron center of an adjacent monomer for the reduction of O2 (Figure 2A).19, 32 For initial studies, we opted for a simplified system employing hydrogen peroxide as the stoichiometric oxidant to circumvent the need for reconstituting effective electron transport (Figure 2B).
Figure 2.

Methods for Fe(III)-hydroperoxo generation in Rieske oxygenases. (A) Electrons supplied by a protein redox partner to the Rieske iron-sulfur cluster and subsequent reduction of O2 at the mononuclear iron binding site. (B) Direct generation of Fe(III)-hydroperoxo species from hydrogen peroxide.
To probe the biosynthetic hypothesis that tricycle 12 is selectively hydroxylated en route to STX (9) and other PSTs, ddSTX (12) was subjected to SxtT in the presence of H2O2. LC-MS analysis of the reaction mixture indicated that in addition to remaining starting material, a single hydroxylated product was formed, α-STOH (14), with no detectable further oxidation to STX (9, Figure 3A). Control reactions lacking SxtT or with denatured SxtT contained no detectable α-STOH (14) or other hydroxylated products (Figure 3A).
Figure 3.

Reactions of SxtT and GxtA. (A) Productive hydroxylation of ddSTX (12) with SxtT in the presence of H2O2. (B) Efficiency of the SxtT hydroxylation of ddSTX (12) with non-native redox partners. (C) Reactivity of SxtT homologs. (D) GxtA hydroxylation of STX (9).
Identifying a substrate for SxtT using H2O2 laid the groundwork for evaluating potential protein redox partners. First, we assessed the activity of SxtT with the putative 2[4Fe-4S] ferredoxin, SxtW, and L-aspartate oxidase-type flavoenzyme, SxtV, which were previously assigned the function of Rieske redox partners based on bioinformatic analysis.15–17 However, no reaction was observed when 12 was incubated with SxtT in the presence of SxtW, SxtV and relevant cofactors (Figure 3B). This result was unsurprising as SxtW and SxtV do not belong to enzyme classes known as redox partners for Rieske oxygenases.33–35 As an alternative, we considered employing redox partners of closely related Rieske oxygenases: VanB,36 NdmD,37 DdmA1 and DdmB,38 and phthalate dioxygenase reductase (PDR)22 (Supporting Information, Table S3). In reactions with SxtT, all four of the selected redox partners were effective for achieving the electron transport required for catalysis, generating varying amounts of the α-STOH (14) product (Figure 3B). The redox partner of the most closely related Rieske oxygenase system, VanB, an FMN-dependent [2Fe-2S] cluster containing enzyme (Supporting Information, Figure S9), generated the most product, while the partner from the more distantly related phthalate dioxygenase system, PDR, afforded the lowest relative yield of α-STOH (14). Using VanB as a redox partner, we demonstrated that the activity of SxtT from M. wollei was conserved across SxtT homologs from other known STX gene clusters. SxtT homologs from STX producers Cylindrospermopsis raciborskii, Aphanizomenon sp. NH-5, and Dolichospermum circinale16–17 all converted 12 exclusively to α-STOH (14, Figure 3C).
Next, we considered the role of the remaining Rieske oxygenases, SxtH and GxtA, in STX biosynthesis. Employing the conditions used in reactions with SxtT, SxtH was tested with a panel of tricyclic substrates (1, 4, 9, 11-14, and 15-16). No hydroxylated products were observed in reactions of SxtH with this panel of substrates with either VanB or H2O2, suggesting that SxtH does not play a role in the hydroxylation of the STX tricycle. Interestingly, the gene that encodes GxtA is not conserved across all identified STX gene clusters.11, 15 Thus, we anticipated that GxtA is not essential for STX biosynthesis, but rather, is involved in the production of further oxidized STX congeners. Indeed, when GxtA was combined with STX (9) under the conditions developed for SxtT reactions, a single hydroxylated product was observed. We anticipated that hydroxylation could take place at either the N1 or C11 position of STX (9), installing the hydroxyl group required for neoSTX (4) or GTX natural products (2, 3, 6 and 8), respectively. To determine the site of hydroxylation in the reaction of GxtA with STX (9), the product was first compared to a standard of the N1-hydroxylated natural product, neoSTX (4) (Supporting Information, Figure S20). LC-MS analysis indicated that the retention time of neoSTX (4) was distinct from the reaction product. Upon comparison of the reaction product to a standard containing 11-α-hydroxysaxitoxin (α-hydroxySTX, S1) and 11-β-hydroxysaxitoxin (11-β-hydroxySTX, 7) formed by hydrolysis of a GTX3/GTX2 mixture (Supporting Information, Figure S40), it was clear that the product of the reaction was identical to one of the C11-hydroxylated products (Supporting Information, Figure S20). 11-β-hydroxySTX (7) was confirmed as the product by comparison to an authentic standard of the compound formed by hydrolysis of a mixture of GTX3/GTX2 mixture and separation of the 11-α-hydroxySTX isomer (S1, Figure 3D). This assignment was further supported by MS/MS data, highlighting the identical fragmentation patterns between 11-β-hydroxySTX (7) and the reaction product (Supporting Information, Figure S32). These results support the hypothesis that 11-β-hydroxySTX (7), which has been isolated from PST-contaminated mussels39, is the biosynthetic precursor to GTX3 (3) and GTX4 (8).
The site- and stereoselectivity exhibited by SxtT and GxtA for hydroxylation of a single C–H bond on the complex STX tricycle inspired us to probe the reactivity and selectivity of these biocatalysts with a panel of substrates. Impressively, GxtA hydroxylates a number of substrates while maintaining exquisite selectivity for the C11 β C–H bond. For example, we measured a total turnover number (TTN) of 27 for the reaction of GxtA with dc-STX (1) and a TTN of 45 for the corresponding reaction with STX (9). GxtA performed a single hydroxylation reaction on a number of substrates that varied in the pattern of oxygenation at C12 and N1, converting neoSTX (4), STX (9), ddSTX (12), β-STOH (13), and α-STOH (14) to the corresponding products with a C11 β-hydroxyl group enabling access to novel STX derivatives (Figure 4B). Reactions with GxtA afford consistent results when scaled from microgram to milligram quantities and products can be isolated is sufficient purity to conduct subsequent biological assays (see Supporting Information for full details).
Figure 4.

(A) Reactions of SxtT with various substrates: hydroxylation of dc-ddSTX (11) to dc-α-STOH (15), hydroxylation of dc-β-STOH (16) to dc-STX (1), hydroxylation of ddSTX (12) to α-STOH (14), and hydroxylation of β-STOH (13) to STX (9). (B) Reactions of GxtA with various substrates: hydroxylation of dc-ddSTX (11) to decarbamoyl 11-β-saxitoxinol (17), hydroxylation of dc-α-STOH (15) to decarbamoyl 11-β-hydroxy-α-saxitoxinol (18), hydroxylation of dc-β-STOH (16) to dc-11-β-hydroxy-β-saxitoxinol (19), hydroxylation of dc-STX (1) to dc-11-β-hydroxysaxitoxin (20), hydroxylation of ddSTX (12) to 11-β-saxitoxinol (21), hydroxylation of α-STOH (14) to 11-β-hydroxy-α-saxitoxinol (22), hydroxylation of β-STOH (13) to 11-β-hydroxy-β-saxitoxinol (23), hydroxylation of STX (9) to 11-β-hydroxysaxitoxin (7), and neoSTX (4) to 11-β-hydroxy neosaxitoxin (24). TTNs were determined using reactions consisting of 5 μM SxtT or GxtA, 5 μM VanB, 200 μM substrate, 500 μM NADH, 100 μM Fe(NH4)2(SO4)2, and 50 mM TrisHCl pH 7.0 incubated at 30 °C for 2 h.
SxtT also operated on a range of tricyclic substrates while maintaining exquisite selectivity for the C12 α C–H bond (Figure 4A). For example, SxtT selectively hydroxylated decarbamoyl dideoxysaxitoxin (dc-ddSTX, 11), decarbamoyl β-saxitoxinol (dc-β-STOH, 16), and β-STOH (13) in addition to ddSTX (12), installing the C12 α-hydroxyl group to afford decarbamoyl α-saxitoxinol (dc-α-STOH, 15), decarbamoyl saxitoxin (dc-STX, 1), and STX (9), respectively (Figure 4A). Interestingly, the total turnover number (TTN) for SxtT with β-STOH (13) is significantly higher than the TTNs with ddSTX (12) and the corresponding decarbamoylated substrates, dc-ddSTX (11) and dc-β-STOH (16). Additionally, steady-state kinetic analysis comparing ddSTX (12) with β-STOH (13) revealed kinetic parameters consistent with β-STOH (13) as the preferred substrate over ddSTX (12) with a 100-fold higher specificity (kcat/KM, Table 1). These data suggest that β-STOH (13) is the native substrate for SxtT in STX biosynthesis, directly generating STX (9).
Table 1.
Steady-state kinetic parameters of SxtT with ddSTX and β-STOH.
| Substrate | Km (μM) | vmax (μm min−1) | kcat (S−1) | kcat/KM (M−1 s−1) |
|---|---|---|---|---|
| ddSTX(12) | 22 ±2 | 0.78 ± 0.02 | (1.30 ± 0.03) × 10−2 | (5.9 ±0.2) × 102 |
| β-STOH (13) | 5.4 ±0.7 | 3.1 ±0.2 | (5.2±0.3)x10−2 | (1.0±0.1) × 104 |
To answer the outstanding question of when and how the the C12 β-hydroxyl group is installed, we reconsidered the role of the third Rieske oxygenase, SxtH. With evidence that SxtH does not react with tricyclic substrates, we considered the possibility that SxtH could react with an intermediate earlier in the pathway and tested SxtH with arginine methyl ester (25), a readily available derivative of the early pathway intermediate, arginine ethyl ketone (10).13 Treatment of arginine methyl ester (25) with SxtH and VanB led to the formation of a hydroxylated product. To confirm the site of hydroxylation, we chemoenzymatically generated 26 employing a known enzymatic hydroxylation of arginine40 followed by esterification (see Supporting Information for full details). Comparison of a standard of 26 to the SxtH product by LC-MS and MS/MS analysis, confirmed the identity of the SxtH product as 26. Taken together, the lack of reactivity observed on tricyclic substrates and the selective SxtH-mediated hydroxylation of 25 to 26 suggests that SxtH is responsible for installation of C12 β-hydroxyl group present in STX (9) at an earlier stage in STX biosynthesis than previously proposed.9–10, 15–17
■ CONCLUSIONS
In summary, we have characterized the functions of three Rieske oxygenases from the STX biosynthetic pathway, that operate with excellent site- and stereoselectivity, providing new insights on the natural assembly and further elaboration of this famous molecule. These biocatalysts are each responsible for a single site- and stereoselective hydroxylation event. Contrary to previous biosynthetic proposals, our data indicates that SxtH performs a hydroxylation at an early stage in the biosynthetic pathway, calling into question previous reports that ddSTX (12) is a biosynthetic intermediate.9, 15–17 Further, two late-stage modifications of the STX tricycle have been elucidated. SxtT hydroxylates β-STOH (13) to STX, and GxtA converts STX to a GTX precursor, 7. Additionally, this study constitutes the first in vitro report of oxygenases involved in C–H bond hydroxylation within paralytic shellfish toxin biosynthesis. The impeccable site- and stereoselectivity exhibited by SxtT, SxtH and GxtA presents the opportunity to exploit these biocatalysts in the chemoenzymatic synthesis of saxitoxin analogs.
Supplementary Material
Figure 5.

(A) Reaction of SxtH with arginine methyl ester (25) to generate hydroxylated arginine methyl ester (26) and LC/MS trace compared to standard. (B) MS/MS comparison of 26 standard and product of the SxtH reaction. (C) Scheme mapping a hypothetical SxtH product (27) onto STX (9), highlighting the position of hydroxylation in the final product.
Scheme 1.

Revised biosynthetic hypothesis.
ACKNOWLEDGMENTS
This research was supported by funds from the University of Michigan Life Sciences Institute and the National Institutes of Health R35 GM124880 (to A.R.H.N.) and R01 GM117263–01A1 (to J.D.B.). A.L.L. was supported by the Graduate Assistance of Areas in National Need (GAANN P200A150164), and R.J.D. was supported by a postdoctoral fellowship from the National Institutes of Health (F32 GM112318–01A1). We thank David Ballou for donation of purified phthalate dioxygenase reductase, Ryan Summers for the gift of the plasmid containing ndmD, Professor Michael Thomas for the plasmid containing vioC, and Clark Ridge for assistance in obtaining NMR spectra. Additionally, we thank Professors Jennifer Bridwell-Rabb, John Montgomery, Melanie Sanford and David Sherman for helpful discussions.
REFERENCES
- 1.Llewellyn LE Nat. Prod. Rep 2006, 23, 200–222. [DOI] [PubMed] [Google Scholar]
- 2.Thottumkara AP; Parsons WH; Du Bois J Angew. Chem. Int. Ed 2014, 53, 5760–5784. [DOI] [PubMed] [Google Scholar]
- 3.Zhang F; Xu X; Li T; Liu Z Mar. Drugs 2013, 11, 4698–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker JR; Novick PA; Parsons WH; McGregor M; Zablocki J; Pande VS; Du Bois J Proc. Natl. Acad. Sci. U.S.A 2012, 109, 18102–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koehn FE; Ghazarossian VE; Schantz EJ; Schnoes HK; Strong FM Bioorg. Chem 1981, 10, 412–428. [Google Scholar]
- 6.Usup G; Leaw C; Cheah M; Amad A; Ng B Toxicon 2004, 44, 37–43. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu Y; Norte M; Hori A; Genenah A; Kobayashi MJ Am. Chem. Soc 1984, 106, 6433–6434. [Google Scholar]
- 8.Kellmann R; Neilan BA J. Phycol 2007, 43, 497–508. [Google Scholar]
- 9.Tsuchiya S; Cho Y; Yoshioka R; Konoki K; Nagasawa K; Oshima Y; Yotsu-Yamashita M Angew. Chem. Int. Ed 2017, 56, 5237–5331. [DOI] [PubMed] [Google Scholar]
- 10.Tsuchiya S; Cho Y; Konoki K; Nagasawa K; Oshima Y; Yotsu-Yamashita M Org. Biomol. Chem 2014, 12, 3016–3020. [DOI] [PubMed] [Google Scholar]
- 11.Soto-Liebe K; Murillo AA; Krock B; Stucken K; Fuentes-Valdes JJ; Trefault N; Cembella A; Vasquez M, Toxicon 2010, 56, 1350–61. [DOI] [PubMed] [Google Scholar]
- 12.Ballot A; Cerasino L; Hostyeva V; Cires S PLoS One 2016, 11, e0167552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chun SW; Hinze ME; Skiba MA; Narayan AR H. J. Am. Chem. Soc 2018, 140, 2430–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimizu Y Chem. Rev 1993, 93, 1685–1698. [Google Scholar]
- 15.Mihali TK; Carmichael WW; Neilan BA PLoS One 2011, 6, e14657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kellmann R; Mihali TK; Jeon YJ; Pickford R; Pomati F; Neilan BA Appl. Environ. Microbiol 2008, 74, 4044–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mihali TK; Kellmann R; Neilan BA, BMC Biochem 2009, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abu-Omar MM; Loaiza A; Hontzeas N Chem. Rev 2005, 105, 2227–2252. [DOI] [PubMed] [Google Scholar]
- 19.Barry SM; Challis GL ACS Catal. 2013, 3, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capyk JK; D’Angelo I; Strynadka NC; Eltis LD, J. Biol. Chem 2009, 284 (15), 9937–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee KG,D. T , Appl Environ Microb 1996, 62, 3101–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaganaman S; Pinto A; Tarasev M; Ballou DP Protein Expres. Purif 2007, 52, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sydor PK; Barry SM; Odulate OM; Barona-Gomez F; Haynes SW; Corre C; Song L; Challis GL Nat. Chem 2011, 3, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J; Simurdiak M; Zhao HJ Biol. Chem 2005, 280, 36719–36728. [DOI] [PubMed] [Google Scholar]
- 25.Perry C; de Los Santos ELC; Alkhalaf LM; Challis GL Nat. Prod. Rep 2018, 35, 622–632. [DOI] [PubMed] [Google Scholar]
- 26.Reinbothe C; Bartsch S; Eggink LL; Hoober JK; Brusslan J; Andrade-Paz R; Monnet J; Reinbothe S Proc. Natl. Acad. Sci. U.S.A 2006, 103, 4777–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berim A; Park JJ; Gang DR Plant J. 2014, 80, 385–395. [DOI] [PubMed] [Google Scholar]
- 28.Beinert H; Holm RH; Münck E Science 1997, 277, 653–659. [DOI] [PubMed] [Google Scholar]
- 29.Kuchenreuther JM; Grady-Smith CS; Bingham AS; George SJ; Cramer SP; Swartz JR, PLoS One 2010, 5, e15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neibergall MB; Stubna A; Mekmouche Y; Münck E; Lipscomb JD Biochemistry 2007, 46, 8004–8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfe MD; Lipscomb JD J. Biol. Chem 2003, 278, 829–835. [DOI] [PubMed] [Google Scholar]
- 32.Ashikawa Y; Fujimoto Z; Noguchi H; Habe H; Omori T; Yamane H; Nojiri H Structure 2006, 14, 1779–1789. [DOI] [PubMed] [Google Scholar]
- 33.Chakraborty J; Ghosal D; Dutta A; Dutta TK J. Biomol. Struct. Dyn 2012, 30, 419–436. [DOI] [PubMed] [Google Scholar]
- 34.Capyk JK; Eltis LD J. Biol. Inorg. Chem 2012, 17, 425–436. [DOI] [PubMed] [Google Scholar]
- 35.Kweon O; Kim S; Baek S; Chae J; Adjei MD; Baek D; Kim Y; Cerniglia CE BMC Biochem. 2008, 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Priefert H; Rabenhorst J; Steinbüchel A, J. Bacteriol 1997, 179, 2595–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Summers RM; Louie TM; Yu CL; Subramanian M Microbiology 2011, 157, 583–592. [DOI] [PubMed] [Google Scholar]
- 38.Herman PL; Behrens M; Chakraborty S; Chrastil BM; Barycki J; Weeks DP J. Biol. Chem 2005, 280, 24759–24767. [DOI] [PubMed] [Google Scholar]
- 39.Dell’ Aversano C; Walter JA; Burton IW; Stirling DJ; Fattorusso E; Quilliam MA J. Nat. Prod 2008, 71, 1518–1523. [DOI] [PubMed] [Google Scholar]
- 40.Ju J; Ozanick SG; Shen B; Thomas MG ChemBioChem 2004, 5, 1281–1285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.